Back to Journals » International Journal of Nanomedicine » Volume 15
Antigen-Conjugated Silica Solid Sphere as Nanovaccine for Cancer Immunotherapy
Authors Dong Y, Gao J, Pei M, Wang X ![]() , Zhang C, Du Y, Jiang Y
, Zhang C, Du Y, Jiang Y
Received 15 December 2019
Accepted for publication 8 April 2020
Published 22 April 2020 Volume 2020:15 Pages 2685—2697
DOI https://doi.org/10.2147/IJN.S242463
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Linlin Sun
Ying Dong,1 Jing Gao,1 Mengyue Pei,2 Xiaoli Wang,2 Chuangnian Zhang,2 Yingjie Du,1 Yanjun Jiang1
1School of Chemical Engineering and Technology, Hebei University of Technology, Tianjin 300130, People’s Republic of China; 2Tianjin Key Laboratory of Biomaterial Research, Institute of Biomedical Engineering, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin 300192, People’s Republic of China
Correspondence: Jing Gao; Xiaoli Wang Tel +86-22-60204945
; +86-22-87890151
Email [email protected]; [email protected]
Background: Nanocarriers could deliver significantly higher amounts of antigen to antigen-presenting cells (APCs), which have great potential to stimulate humoral and cellular response in cancer immunotherapy. Thereafter, silica solid nanosphere (SiO2) was prepared, and a model antigen (ovalbumin, OVA) was covalently conjugated on the surface of SiO2 to form nanovaccine (OVA@SiO2). And the application of OVA@SiO2 for cancer immunotherapy was evaluated.
Materials and Methods: SiO2 solid nanosphere was prepared by the Stöber method, then successively aminated by aminopropyltriethoxysilane and activated with glutaraldehyde. OVA was covalently conjugated on the surface of activated SiO2 to obtain nanovaccine (OVA@SiO2). Dynamic light scattering, scanning electron microscope, and transmission electron microscope were conducted to identify the size distribution, zeta potential and morphology of OVA@SiO2. The OVA loading capacity was investigated by varying glutaraldehyde concentration. The biocompatibility of OVA@SiO2 to DC2.4 and RAW246.7 cells was evaluated by a Cell Counting Kit-8 assay. The uptake of OVA@SiO2 by DC2.4 and its internalization pathway were evaluated in the absence or presence of different inhibitors. The activation and maturation of bone marrow-derived DC cells by OVA@SiO2 were also investigated. Finally, the in vivo transport of OVA@SiO2 and its toxicity to organs were appraised.
Results: All results indicated the successful covalent conjugation of OVA on the surface of SiO2. The as-prepared OVA@SiO2 possessed high antigen loading capacity, which had good biocompatibility to APCs and major organs. Besides, OVA@SiO2 facilitated antigen uptake by DC2.4 cells and its cytosolic release. Noteworthily, OVA@SiO2 significantly promoted the maturation of dendritic cells and up-regulation of cytokine secretion by co-administration of adjuvant CpG-ODN.
Conclusion: The as-prepared SiO2 shows promising potential for use as an antigen delivery carrier.
Keywords: antigen delivery, silica solid sphere, nanovaccine, cancer immunotherapy
Introduction
Cancer has long been a global threat and is the second leading cause of death.1 Cancer remediation using traditional strategies such as surgery, radiotherapy and chemotherapy have achieved some good results, but these treatments are not effective for all tumors, and sometimes cause serious side effects.1,2 Immunotherapy shows relatively minimal side effects, and effective control of tumor growth and metastasis has come into people’s vision gradually.3,4 Tumor vaccines consist of defined antigens, aiming to activate the patients’ immune system to recognize the tumor antigens, thus destroy tumor cells. Protein or polypeptide was widely used antigens in various vaccines.5,6 Specifically, tumor antigens are captured and degraded into short peptide by antigen-presenting cells (APCs). Then, the peptide combined with major histocompatibility complex (MHC) molecules to form a complex, which is presented to naive T cells (that is, antigen inexperienced). Therefore, an immune response is initiated by APCs.7
Tumor vaccines show significant anti-tumor potential, but there are also some shortcomings, such as easy degradation of antigen, poor uptake efficiency and weak immunogenicity, which affect their therapeutic effect. A variety of nanoscale carriers are designed to improve the efficacy of tumor vaccines.8 Nanoscale carriers loaded with antigens can delay the release of antigens, reduce their elimination rate in vivo, improve their bioavailability, and change their distribution in vivo.9 A lot of related research work has been carried out.10,11 Nanoscale carriers include organic nanoparticles (PLGA, lipoprotein coupled with antigen/adjuvants) and inorganic nanoparticles (SiO2, graphene oxide).12,13 However, there are still many problems in the basic research and the application of nanoscale carriers for cancer immunotherapy. For example, the key factors (particle size, charge, surface chemistry) that affect the targeting performance are still lack of systematic research.
Compared with organic nanoparticles, inorganic nanoparticles have the advantages of good dimensional control and large specific surface area.14 Therefore, in recent years, inorganic nanoparticles have been reported as carriers for proteins, DNA and chemical drugs. Among them, silica nanoparticles (SiO2) have developed very rapidly as drug delivery systems in cancer treatment.15 As a successful drug delivery system, some prerequisites must be met, including biodegradability, high drug loading capacity, the ability to protect loads and prevent premature leakage before reaching the target site, and controllable drug release.16,17 In addition, the toxicity and adverse effects of SiO2 can be controlled by changing its physicochemical properties and administration mode. More importantly, the surface of SiO2 is rich in silicon hydroxyl (-SiOH), which can be easily modified by silane coupling agents to design different functionalized surfaces to meet biological needs.13
In this study, SiO2 solid nanospheres were prepared, and the model antigen OVA was covalently conjugated on the surface of SiO2 to obtain nanovaccine (OVA@SiO2). The effect of SiO2 as an antigen carrier was explored via in vitro cytotoxicity assay, antigen uptake and their internalization pathways. In addition, the activation and maturation of dendritic cells (DCs), the cross-presentation of antigen, and in vivo trafficking of antigen were also been investigated. This work can provide researchers with some new design ideas about SiO2, and show unique application prospects in the field of antigen delivery.
Materials and Methods
Materials
Ovalbumin (OVA) was purchased from Sigma-Aldrich (USA). CpG oligonucleotide 1826 (5ʹ-TCC ATG ACG TTC CTG ACG TT-3ʹ) was synthesized by Sangon (China). Fetal bovine serum (FBS), phosphate-buffered saline (PBS), RPMI-1640, DMEM were purchased from Hyclone (USA). Rottlerin, chlorpromazine, Filipin III and cytochalasin D were purchased from ApexBio Technology (USA). Anhydrous dimethyl sulfoxide (DMSO), red blood cell lysis, the carbocyanine dye Dil, 4ʹ,6-Diamidino-2-phenylindole dihydrochloride (DAPI, ≥90%), fluorescein isothiocyanate (FITC) and near-Infrared Cyanine 7 dyes (Cy7 NHS ester) were purchased from Solarbio Science & Technology Co. Ltd (China). Cell Counting Kit-8 (CCK-8), NP-40 lysis buffer, X-Gal (ST912) and BCA protein detection kit were purchased from Beyotime (China). Recombinant mouse IL-4 and GM-CSF were purchased from Peprotech (USA). Anti-mouse monoclonal antibodies (CD80 (B7-1), CD86 (B7-2), CCR7, CD40, MHC I (H-2Kb) and MHC II (I-Ab)) were purchased from Biolegend (USA). None of the chemicals were further purified.
Cell Lines and Animals
Mouse dendritic cells line DC2.4 and RAW264.7 cells line were purchased from the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (Beijing, China). B3Z cells were obtained as a gift from Prof. Lianyan Wang, Institute of Process Engineering, Chinese academy of sciences (Beijing, China), and this research were approved by institutional review board of Institute of Biomedical Engineering, Chinese Academy of Medical Science and Peking Union Medical College (CAMS&PUMC). Bone marrow-derived DC cells (BMDCs) were extracted from C57BL/6 mice femur. Six–eight weeks old female C57BL/6 mice were supplied from SPF Biotechnology Co., Ltd. (China). All the protocols for animal experiments were approved by the Center of Tianjin Animal Experimental Ethics Committee and performed in compliance with the Guidelines for Care and Use of Laboratory Animals of Institute of Biomedical Engineering, CAMS&PUMC.
Synthesis of SiO2 Nanoparticles
Solution A: 9 mL of 28% ammonia, 16.25 mL of ethanol and 24.75 mL of water were stirred evenly. Solution B: 4.5 mL of tetraethyl orthosilicate (TEOS) and 45.5 mL of ethanol were mixed. Pour solution B into solution A quickly under stirring, and then the mixture was reacted at room temperature for 2 h. SiO2 nanoparticles (SiO2) were obtained after centrifugation (10,000 rpm, 5 min) and wash three times with ethanol.
Preparation of OVA@SiO2
Before antigen loading, SiO2 was first aminated by aminopropyltriethoxysilane (APTES). One hundred milligrams of SiO2, 20 mL of n-hexane and 100 mL of APTES were added into 250 mL round-bottom flask, and the mixture was refluxed for 12 h at 80°C. SiO2-NH2 were obtained after centrifugation (10,000 rpm, 5 min) and wash once with acetone and twice with ethanol. One hundred milligrams of SiO2-NH2 was mixed with 100 mL, 8% of glutaraldehyde by ultrasonic treatment, and then reacted in water bath at 25°C for 2 h. SiO2-CHO were obtained after centrifugation (10,000 rpm, 5 min) and wash three times with water. Then, 0.5 mL of OVA solution (1.5 mg mL−1) was reacted with 3 mg of SiO2-CHO for 2 h, and then OVA-loaded SiO2 (defined as OVA@SiO2) were obtained after centrifugation (10,000 rpm, 5 min) and wash three times with PBS solution.
The OVA concentration was obtained via the BCA protein detection kit, and the loading capacity was determined according to eq 1:
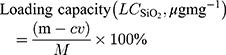
where m (mg) was the total amount of OVA added into the system; v (mL) and c (mg mL-1) were the OVA volume and concentration of the supernatant, respectively. M (mg) was the mass of OVA@SiO2.
In vitro Cytotoxicity
The cytotoxicity of OVA@SiO2 to DC2.4 and RAW246.7 cells was determined by CCK-8 assay. Cells were plated to 96-well plates at a density of 5000 cells/100 μL culture medium. After overnight culturing, the cells were incubated with different concentrations of OVA or OVA@SiO2 for 48 h. The cell viability was given as a function of concentration.
Antigen Uptake, the Uptake Pathway and Lysosome Escape
OVA was labelled with fluorescent dye FITC as follows: 10 mL, 2 mg mL-1 of OVA solution (pH 8.0, 50 mM of PBS) was mixed with 0.5 mL, 1 mg mL-1 of FITC solution (DMSO) with stirring for 45 min at ambient temperature, and then the mixture was dialyzed against buffer solution (PBS, 50 mM, pH 7.0) for 24 h and water for another 24 h in the dark. The dialysis solution was changed every 8 hours. And then FITC-labelled OVA (OVA-FITC) was loaded within SiO2 nanoparticles (OVA-FITC@SiO2). DC2.4 cells were seeded into 24-well plates with a density of 1×106 per well within 1 mL culture medium. After overnight incubation, the cells were incubated with OVA-FITC+CpG-Cy3, OVA-FITC@SiO2+CpG-Cy3 for 4 h, and the amount of OVA-FITC was 10 μg/well, the amount of CpG-Cy3 was 2.5 μg/well. Flow cytometry was used to determine the Mean fluorescence intensity (MFI) and analyze the antigen uptake by DC2.4 cells.
To determine the uptake pathway of antigen, four uptake inhibitors including rottlerin (Rot, 25 μg mL–1), chlorpromazine (Chlor, 10 μg mL–1), Filipin III (Fil III, 10 μg mL–1) and cytochalasin D (Cyt D, 10 μg mL–1) were added to DC2.4 cells and incubated for 1 hour, and then incubated with OVA-FITC@SiO2+CpG-Cy3 for another 4 h. After collection and fixation with 4% paraformaldehyde, the cells were analyzed by flow cytometry, and the antigen uptake pathway of DC2.4 was determined according to the measured MFI.
To reveal cellular localization of antigens, bone marrow-derived DC cells (BMDCs) were extracted from C57BL/6 mice according to our previously reported methods.18,19 BMDCs (1×104 cells/dish) were incubated with OVA-FITC+CpG-Cy3 and OVA-FITC@SiO2+CpG-Cy3 for 8 h and 24 h, respectively. The concentrations of OVA-FITC and CpG-Cy3 were 10 μg mL–1 and 2.5 μg mL–1, respectively. Subsequently, cells were washed with PBS and fixed/permeabilized with Cytofix/Cytoperm kit (BD Pharmingen) for 30 min. An antibody αCD107a (Lamp-1-APC, BD Pharmingen) was used to label the lysosome-associated membrane protein-1 by incubating with cells for 1.5 h at 37 oC according to the manufacturer’s instructions. Then, the cell nucleus was stained with DAPI for 2 min and the membrane was stained with the carbocyanine dye Dil after washing with PBS. Cells were then observed with confocal laser scanning microscope (CLSM, CarlZeiss LSM710).
BMDCs Maturation and Activation
BMDCs (1×106 cells/mL) were seeded in a 24-well plate and incubated with OVA+CpG and OVA@SiO2+CpG, and the final concentration of OVA and CpG were 10 μg mL–1 and 2.5 μg mL–1, respectively. After incubation for 48 h, BMDCs were collected and stained with CD11c, CD86, CD80, CD40, SIINFEKL-MHC I, MHC II, MHC I and CCR7 followed by FACS analysis. The supernatant was collected and analyzed by ELISA assay for murine TNF-α, IL-6, IL-12, IL-10, IL-4 and IL-1β, according to the manufacturer’s protocol (eBioscience).
To evaluate antigen cross-presentation, BMDCs (1×106) were cultured with OVA and OVA@SiO2 with different OVA concentrations (10, 50, 80, 100, 120, 160, 200 μg mL−1). After 24 h incubation, cells were washed with PBS and then incubated with 5×105 B3Z T cell hybridoma for 24 h. Then, 1.5 mg mL−1 of X-gal and 0.25% of NP40 lysis buffer were dissolved with PBS, and the mixture was added to the cells and incubated for another 24 h. CD8+ T cell hybridoma, B3Z cells can be specifically activated by the OVA-derived peptide SIINFEKL (OVA 257–264) cross-presented by MHC I molecules of DCs. After antigen stimulation, B3Z cells express beta-galactosidase, which can interact with X-gal substrates to make the cells appear blue. Therefore, the absorbance at 405 nm recorded by Varioskan Flash3001 (Thermo, USA) was used to evaluate antigen cross-presentation in vitro.
In vivo Trafficking
Firstly, OVA was labelled with near-Infrared fluorescent dye Cy7 as follows: 10 mL, 2 mg mL−1 of OVA solution (pH 8.0, 50 mM of PBS) was mixed with 0.5 mL, 1 mg mL−1 of Cy7 solution (DMSO) with stirring for 45 min at ambient temperature, and then the mixture was dialyzed against buffer solution (PBS, 50 mM, pH 7.0) for 24 h and water for another 24 h in the dark. The dialysis solution was changed every 8 hours. And then Cy7-labelled OVA (OVA-Cy7) was loaded within SiO2 nanoparticles (OVA-Cy7@SiO2). Subsequently, OVA-Cy7 and OVA-Cy7@SiO2 were injected subcutaneously into the tail base of C57BL/6 mice at a dose of 100 μg OVA-Cy7 dispersed in 100 μL PBS. At several time points post-vaccination, mice were anesthetized with chloral hydrate and then a small animal in vivo imaging system (Maestro, CRI USA) was used to record the fluorescence spectral cubes.
In vivo Biocompatibility Evaluation
Female C57BL/6 mice of 6–8 weeks were subcutaneously vaccinated with PBS, OVA+CpG and OVA@SiO2+CpG on days 0, 7 and 14, respectively. The doses of OVA and CpG were 100 μg per mouse and 25 μg per mouse, respectively. For analyses of the biocompatibility, main organs (heart, liver, lung and kidney) were isolated from vaccinated mice on day 21, H&E staining of these tissue sections was performed and the digital microscope images were recorded.
The tissues were washed with physiological saline and fixed in a 4% paraformaldehyde solution. Then, the tissue was removed from 4% paraformaldehyde, placed it in a petri dish containing absolute ethanol, sliced it into a suitable size with a razor blade, placed it in an embedding basket, and marked it with a pencil. The tissue was dehydrated with four concentration gradients of ethanol and xylene, and then embedded within paraffin and sliced into 5 μm slices.
H&E staining: the tissue was successively placed in different staining boxes in the following sequence: Xylene I: 10 min, Xylene II: 10 min (dewaxed); Anhydrous ethanol I: 5 min, Anhydrous ethanol II: 5 min; 90% ethanol: 5 min, 80% ethanol: 5 min, 70% ethanol: 5 min; Rinsed with water for 5 min; Hematoxylin: 5 min; Water flush: 5 min; Eosin: 3 min; Water flush: 5 min; 70% ethanol: 3 s; 80% ethanol: 3 s; 90% ethanol: 2 min; Anhydrous ethanol I: 5 min; Anhydrous ethanol II: 5min; Xylene I: 10 min; Xylene II: 10 min; Finally, the tissue slide was sealed with gum and observed with a fluorescence microscope.
Statistical Analysis
One-way ANOVA was used for multiple comparisons when more than two groups were compared, and Student’s t-test was used for two-group comparisons. All statistical analyses were implemented with Prism software (PRISM7.0, GraphPad Software).
Results and Discussions
Synthesis and Characterization of SiO2 Nanoparticles
Based on previously reported method,20 SiO2 was prepared by the Stöber method. Solution A was mixed with ammonia, ethanol and water, solution B was mixed with tetraethyl orthosilicate (TEOS) and ethanol, and solutions A and B were rapidly mixed and stirred for 2 h. The size distribution of SiO2 was observed by DLS (Figure 1A), and SiO2 had an average size of 319.5 nm with PDI of 0.074. After covalent conjugation of OVA, the particles size of OVA@SiO2 increased to 330 nm with PDI of 0.074 according to DLS results. SEM (Figure 1B) and TEM (Figure 1C) were further used to observe SiO2 particles. Obtained from SEM and TEM images, the size of SiO2 was 361±22 nm and 350±5 nm, respectively. The slight difference in particle size between DLS and electron microscope was due to the different batches of as-prepared SiO2. To sum up, SiO2 was spherical solid particle with a narrow size distribution.
 |
Figure 1 DLS size distribution of SiO2 and OVA@SiO2 (A), SEM image of SiO2 (B), TEM image of SiO2 (C), the surface zeta potential (D), the loading capacity of OVA in OVA@SiO2 as a function of glutaraldehyde concentration (E). (F) The cell viability of DC2.4 after incubating with OVA@SiO2 for 48 h, culture medium was used as control, the data are expressed as mean ± SD (n =5). |
In order to confirm the covalent conjugation of OVA onto SiO2, Fourier transform infrared spectroscopy (FTIR) was utilized to characterize the chemical composition of OVA@SiO2. As shown in Figure S1A in the Supporting Information, the spectrum of SiO2-OH clearly showed the Si-O-Si bending vibration (470 cm−1), Si-O-Si symmetric stretching (798 cm−1), Si-OH stretching vibration (957 cm−1), Si-O-Si asymmetric stretching vibration (1098 cm−1), H-O-H bending vibration (1633 cm−1) and -OH stretching (3405 cm−1) corresponding to silica. After covalent conjugation of OVA, there was also a significant reduction in the intensity of the SiO2-OH peak (957 cm-1) in OVA@SiO2 spectrum (red curve). The spectrum of OVA@SiO2 exhibited a new peak at 1650 cm-1 (C=N absorption band), which indicated the covalent conjugation of OVA on SiO2 surface. Zeta-potential measurements were also performed. According to Figure 1D, the untreated SiO2 had a negative charge (-50.9 mV), and the potential of SiO2-NH2 (-10.2 mV), SiO2-CHO (-12.0 mV), and OVA@SiO2 (-48.3 mV) changed significantly. According to the above results, the amination and glutaraldehyde activation of SiO2, and covalent attachment of OVA with SiO2 were successfully completed. As shown in Figure 1E, the loading capacity of OVA in OVA@SiO2 increased with the increase of glutaraldehyde concentration. When the concentration of glutaraldehyde was 8 v/v%, the OVA loading capacity of OVA@SiO2 can reach 458 mg g−1, which was higher than many carriers such as PLGA nanoparticles,21 chitosan/calcium phosphates nanosheet22 and layered double hydroxide nanoparticles.23
Two major specialized antigen-presenting cells (APCs) are dendritic cells and macrophages. Therefore, dendritic cells DC2.4 and macrophages RAW246.7 were used to detect the cytotoxicity of OVA@SiO2. As shown in Figure 1F and Figure S1B, the cell viabilities of DC2.4 and RAW246.7 remained above 85% when the concentration of OVA@SiO2 was in the range of 0–100 μg mL−1, which indicated the acceptable cytotoxicity of SiO2 to APCs. Therefore, according to the cytotoxicity to DC2.4 and RAW246.7, SiO2 can be used as a safe antigen carrier for tumor immunotherapy.
Antigen Uptake and the Uptake Pathway
The first step in activating an anti-tumor immune response is the uptake of antigen and adjuvant by antigen-presenting cells.24,25 At the same time, in order to study whether SiO2 can increase the bioavailability of soluble CpG, OVA@SiO2 was used in combination with soluble CpG in the following experiment. Phagocytosis of OVA@SiO2+CpG by BMDCs was determined by flow cytometry after 4 h incubation, OVA-FITC and CpG-Cy3 were used here. According to Figure 2A, compared with soluble OVA+CpG, SiO2 can increase the MFI of OVA-FITC by about 3–4 folds. Besides, SiO2 can also increase the MFI of CpG-Cy3 although CpG was in free form (Figure S1C), which ascribed that CpG was electrostatically adsorbed on OVA@SiO2. The results suggested that SiO2 can significantly enhance the internalization of antigen and adjuvant.
 |
Figure 2 The uptake of OVA (A) estimated by MFI of DC2.4 cells, and (B) their representative FACS histograms after incubation for 4 h with different formulations in the absence or presence of inhibitor including rottlerin (Rot), chlorpromazine (Chlor), filipin III (Fil III) and cytochalasin D (Cyt D). Data were the mean ± SD (n = 5), and the differences were analyzed by one-way ANOVA with Tukey’s multiple comparison test, *p < 0.05, ***p < 0.001. |
BMDCs mainly phagocytes extracellular particles through four endocytosis pathways, and their corresponding inhibitors are rottlerin26 (Rot, an inhibitor of macropinocytosis), chlorpromazine27 (Chlor, an inhibitor of clathrin-dependent endocytosis), cytochalasin D28 (Cyt D, an actin polymerization inhibitor that blocks phagocytosis and macropinocytosis), and filipin III29 (Fil III, an inhibitor of caveolae-mediated endocytosis). In order to study the uptake pathways of OVA@SiO2+CpG, the corresponding inhibitors were added to co-incubate with BMDCS. As depicted in Figure 2B, the uptake of OVA@SiO2 was obviously inhibited by all the investigated inhibitors. The result suggested that OVA@SiO2 was internalized by BMDCs through multiple pathways rather than a single pathway. As shown in Figure S1C, the uptake of CpG was also inhibited by all the investigated inhibitors, which may be ascribed to the fact that CpG was electrostatically adsorbed on the surface of OVA@SiO2. Based on the above results, SiO2 can be internalized by BMDCs in various ways.
Efficient antigen endocytosis is of great significance for activating strong CTL response and tumor immunotherapy. In order to further understand the internalization of antigen and adjuvant, CLSM was used to observe BMDCs after co-culturing with fluorescence-labeled OVA+CpG and OVA@SiO2+CpG for 8 h and 24 h. After co-culturing with soluble OVA+CpG, only weak signals of OVA-FITC (see Figure 3A and B) and CpG-Cy3 (Figure S2A and B) were observed in BMDCs. In contrast, a large amount of OVA-FITC (see Figure 3A and B) and CpG-Cy3 (Figure S2A and B) fluorescence signal was observed in BMDCs after co-culturing with OVA@SiO2+CpG. The result indicated that SiO2 could effectively deliver antigen and adjuvant.
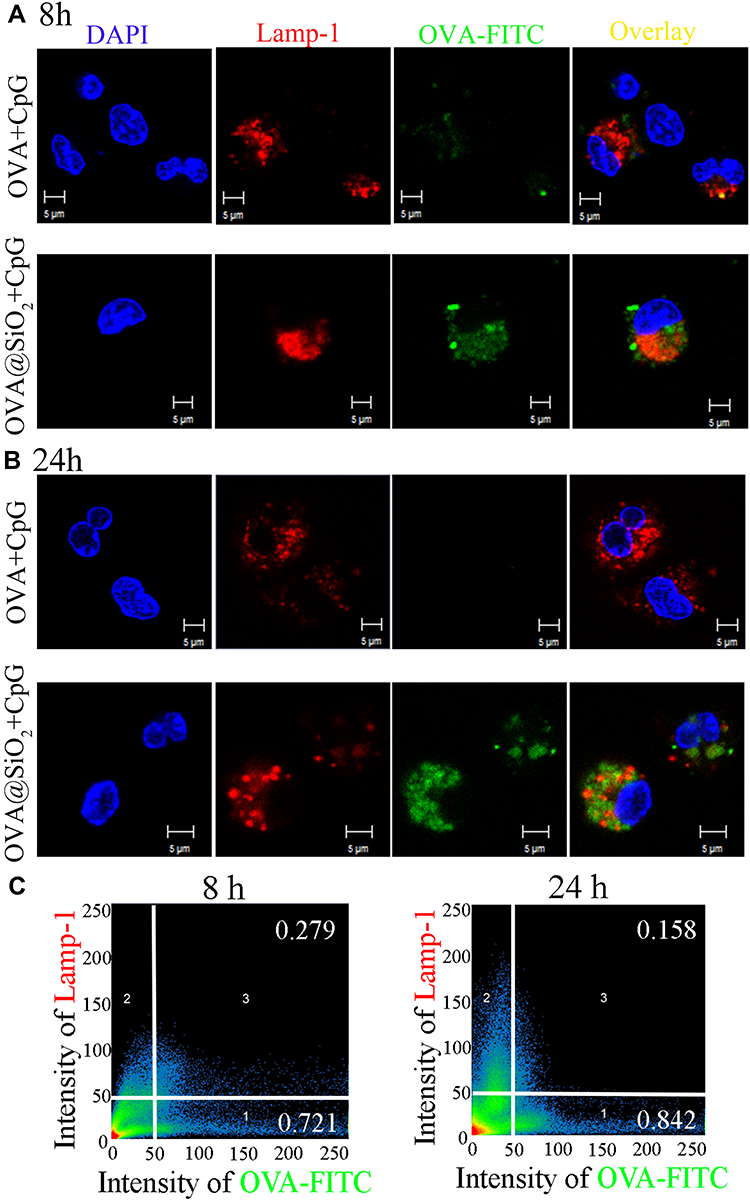 |
Figure 3 (A, B) CLSM images of BMDCs after incubating with soluble OVA+CpG, OVA@SiO2+CpG for 8 h and 24 h, the endosomes/lysosomes were stained with Lamp-1-APC, the nucleus was stained with DAPI, OVA-FITC and CpG-Cy3 were used. (C) Quantitative colocalization analysis of CLSM images was performed by Zeiss Zen 2008 Software. |
To demonstrate the location of the phagocytosed antigen by BMDCs, Lamp-1 antibody was used to mark the lysosomal membrane protein-1, DAPI was used to stain cell nucleus, then BMDCs were observed by CLSM. As shown in Figure 3A and B, only little OVA-FITC signal was co-localized with Lamp-1, suggesting that OVA@SiO2+CpG was mostly located in the cytoplasm. According to colocation analysis (Figure 3C), 15.8% of OVA-FITC was co-localized within Lamp-1 in BMDCs after co-incubation with OVA@SiO2+CpG for 24 h, which meant 84.2% of OVA-FITC was escaped into the cytoplasm. The result indicated that SiO2 could effectively deliver antigens into the cytoplasm from lysosomes. Antigen escapes into the cytoplasm and cross-presents to CD8+ T cells via the MHC I pathway to induce anti-tumor immunity.
CpG needs to move into the lysosomal membrane that also contained Toll-like receptor 9 (TLR9), in which CpG directly binds to TLR9 and signal transduction.30 Therefore, colocalization studies of CpG and lysosomal membrane can reveal the utilization efficiency of CpG to some extent. According to the colocation analysis (Figure S2C), after BMDCs co-incubating with OVA@SiO2+CpG, the amount of CpG colocalized within lysosome was increased from 4.8% to 12.2%, with the increase of incubation time from 8 h to 24 h. The result suggested that SiO2 could effectively deliver the adjuvant CpG to the lysosomal membrane.
BMDCs Activation and Antigen Cross-Presentation
After process and presentation of antigens, DCs can be induced to express costimulatory molecules and secrete cytokines, thus effectively stimulating the activation of T cells. As shown in Figure 4A-C, compared with other groups, significantly increased expression of costimulatory markers (CD86, CD80 and CD40) were observed in OVA@SiO2+CpG group, indicating that OVA@SiO2+CpG have the ability to promote BMDCs activation. As shown in Figure 4D and E, compared with other groups, OVA@SiO2+CpG also significantly increased the levels of MHC I and SIINFEKL-MHC I on BMDCs. The result suggested that SiO2 can enhance the ability of BMDCs to prime CD8+ T cells by the MHC I pathway. However, the MHC II level of BMDCs induced by OVA@SiO2+CpG was not significantly up-regulated (Figure 4F).
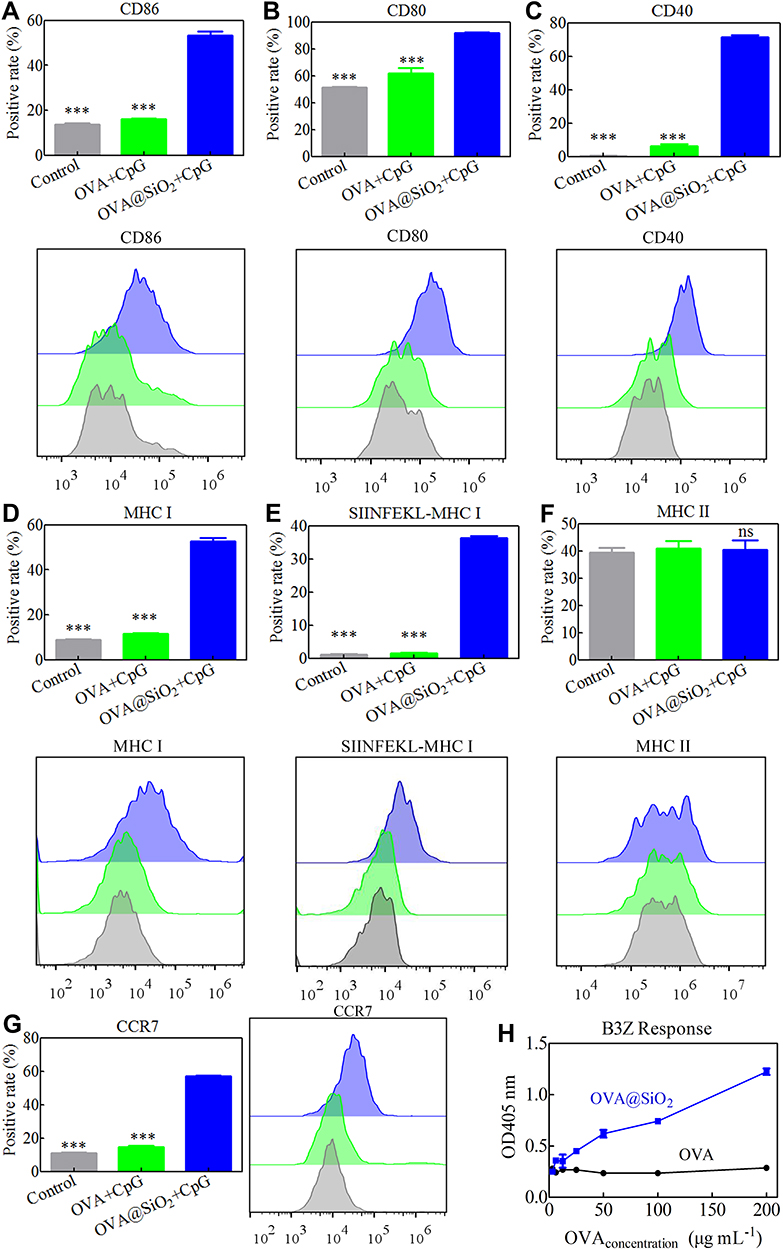 |
Figure 4 (A–G) Expression of the costimulatory markers CD86, CD80, CD40, the recognition signals (MHC I, SIINFEKL-MHC I and MHC II) and the chemokine receptor CCR7 on BMDCs stimulated with different formulations for 48 h. Data represented the mean ± SD (n = 3), the differences were analyzed using one-way ANOVA with Tukey’s multiple comparison test, ***p < 0.001. (H) Activation of SIINFEKL-specific CD8+ T cells (B3Z) after co-culturing with BMDCs. |
To investigate the migration ability of DCs, the expression of chemokine receptor CCR7 was measured. DCs enter the lymphatic system and migrate to the lymph node (LN) via the interaction between CCR7 and its ligand, CCL 19 and CCL 21.31,32 Therefore, the ability of BMDCs migrating to LN was related to the expression level of CCR7. As displayed in Figure 4G, the expression of CCR7 was significantly increased in OVA@SiO2+CpG group, indicating that SiO2 can enhance the migration ability of BMDCs.
The cross-presentation of internalized antigen is the key to inducing CD8+ T cell activation for anti-tumor immunity. To survey the ability of antigen cross-presentation by SiO2, the activation of B3Z T cells was measured by the LacZ method. B3Z T cells can be only activated by OVA-derived peptide (SINFEKL) presented by the MHC I molecule on DCs. After stimulation with antigens, B3Z T cells can express beta-galactosidase enzyme and interact with the X-Gal substrate to make the cells blue. As shown in Figure 4H, the cross-presentation of soluble OVA was low regardless of the concentration. On the contrary, in OVA@SiO2+CpG group, the cross-presentation of OVA greatly increased with the increase of OVA concentration, indicating that SiO2 can greatly promote the cross-presentation of antigen. These results indicated that SiO2 could be an effective antigen carrier.
It has been mentioned that partially mature DCs have immune tolerance, and only fully mature DCs have immunogenicity.33 High levels of MHC II, costimulatory molecules and large amounts of proinflammatory cytokines (IL-12, TNF-α, IL-1β and IL-6) can be expressed by mature DCs. However, semi-mature DCs are not considered capable of producing pro-inflammatory cytokines even they highly expressed MHC II and costimulatory molecules. OVA@SiO2+CpG stimulated DCs expressed high levels of MHC II, costimulatory molecules and large amounts of proinflammatory cytokines (Figure 5A-D), suggesting that OVA@SiO2+CpG can stimulate DCs maturation and make them immunogenic. IL-12 is an attractive tumor therapeutic cytokine.34 Moreover, IL-6 is a recruiting cytokine that recruits immune cells in the body. Therefore, under the regulation of IL-6, OVA@SiO2+CpG can recruit APCs to the vaccination site. Moreover, IL-1β contributes to the production of IFN-γ and CD8+ T cells, thereby stimulating the occurrence of an antitumor immune response. In summary, OVA@SiO2+CpG have the ability to stimulate antitumor immune responses. IL-10 can inhibit the ability of DCs to secrete cytokines and is an immunosuppressive cytokine. In addition, IL-4 can convert DCs that secrete IL-12 into DCs that can only stimulate Th2 response.35 According to Figure 5E and F, BMDCs incubated with OVA@SiO2+CpG did not secrete enhanced level of IL-10 and IL-4 cytokines, indicating that the activated DCs were not immunosuppressive. In summary, OVA@SiO2+CpG promoted the DCs maturation and cytokine production, which were beneficial to recruit APCs and prime IFN-γ-producing tumor antigen-specific CD8+ T cells.
 |
Figure 5 (A–F) The cytokines (IL-12, TNF-α, IL-6, IL-1β, IL-4 and IL-10) release of BMDCs stimulated with formulations for 48 h. Data represented the mean ± SD (n=3), the differences were analyzed using one-way ANOVA with Tukey’s multiple comparison test, *p < 0.05, **p < 0.01, ***p < 0.001. |
Activation of DCs for in vivo Transport
In vivo migration of DCs after injection of OVA@SiO2 was monitored in real time by using small animal imaging. After activation, DCs migrate to lymph nodes and initiate an immune response.36 OVA-Cy7 was first prepared by labeling OVA with the near-infrared fluorescence Cy7, then OVA-Cy7@SiO2 were prepared. OVA-Cy7 and OVA-Cy7@SiO2 were subcutaneously injected into the tail root of C57BL/6, and the injection site and adjacent LN were observed. Figure 6 shows representative images of Cy7 signals corresponding to OVA-Cy7 and OVA-Cy7@SiO2. After 6 h of injection, Cy7 signals were detected in the right dLN, while the accumulation of free OVA was also detected in adjacent tissues except for the dLN. After 8 days of injection, no fluorescence signal of soluble OVA-Cy7 was detected in LN, indicating its degradation in vivo. However, in OVA-Cy7@SiO2 group, the signal of Cy7 was still detected, indicating that SiO2 could protect the antigen from enzymatic degradation, thus extending its retention time in the body.
 |
Figure 6 The Cy7 fluorescent signal of OVA-Cy7 and OVA-Cy7@SiO2 in the vaccination site and the right inguinal LNs, all images were overlays of bright photographs with fluorescence intensity measurement, LN indicated by red arrows. |
In vivo Biocompatibility
Moreover, the in vivo biocompatibility was further evaluated by investigating the organ/tissue damages. After vaccination for three times, major organs including heart, liver, lung and kidney were excised, sectioned and stained with H&E. As shown in Figure 7, there were no observable tissue damage including apoptosis/necrosis of cardiomyocytes, cell shrinkage of liver, pulmonary interstitial fibrosis/thickened alveolar septa, or tubular dilatation/contraction of kidney among all the groups, suggesting that the subcutaneous injection of OVA@SiO2 would not cause damage to major organs/tissues. The in vivo biocompatibility of SiO2 nanoparticles indicated that SiO2 was safe as a vaccine carrier.
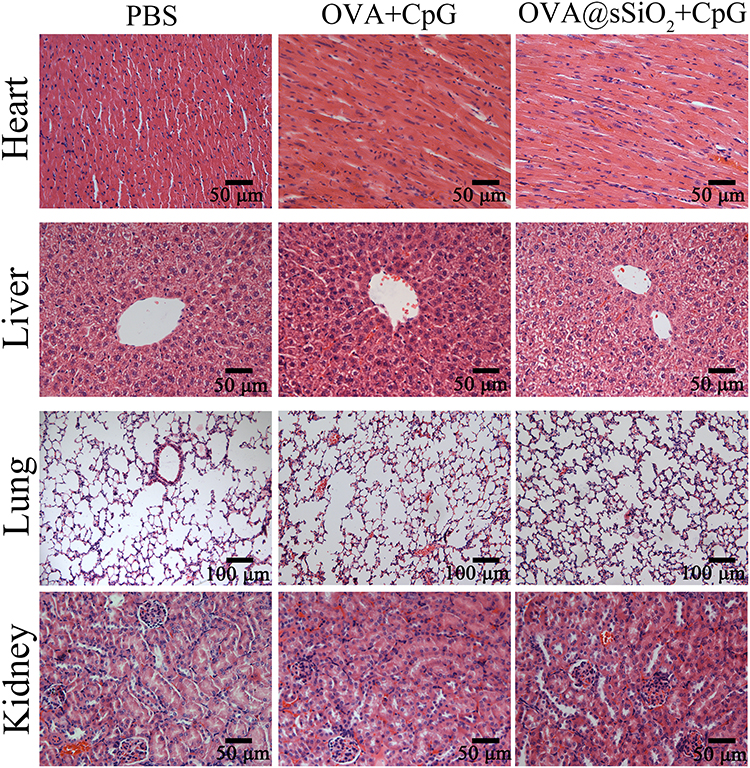 |
Figure 7 The section of mouse organs stained with hematoxylin and eosin (HE), the magnification of all the images was 40× except for the lung (20×). |
Conclusion
In the present study, a nanovaccine (OVA@SiO2) was developed by covalent conjugating antigen OVA onto the surface of SiO2 solid sphere. OVA@SiO2 with spherical structure and narrow size distribution had acceptable toxicity both in vitro and in vivo. SiO2 could significantly enhance antigen/adjuvant uptake by DCs and promote antigen cytosolic release. Moreover, OVA@SiO2+CpG induced the higher expression of co-stimulatory molecules (CD86, CD80, CD40, MHC I, CCR7 and SIINFEKL-MHC I) than that of OVA+CpG group. Specifically, OVA@SiO2 induced higher cross-presentation of exogenous antigens than free OVA in B3Z cells. In addition, OVA@SiO2 could extend antigen retention time in the draining lymph nodes. Based on these data, OVA@SiO2 manifested excellent potential as an antigen delivery carrier for cancer immunotherapy.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (81972899, 21604095), Natural Science Foundation of Tianjin City (18JCQNJC14500), CAMS Innovation Fund for Medical Sciences (2017-I2M-3-022), Specific Program for High-Tech Leader & Team of Tianjin Government, Tianjin Innovation and Promotion Plan Key Innovation Team of Immunoreactive Biomaterials.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Metelmann H-R, Seebauer C, Miller V, et al. Clinical experience with cold plasma in the treatment of locally advanced head and neck cancer. Clin Plasma Med. 2018;9:6–13. doi:10.1016/j.cpme.2017.09.001
2. Mao Q, Li L, Zhang C, Sun Y, Liu S, Cui S. Clinical effects of immunotherapy of DC-CIK combined with chemotherapy in treating patients with metastatic breast cancer. Pak J Pharm Sci. 2015;28:1055–1058.
3. Kelly PN. The cancer immunotherapy revolution. Science. 2018;359(6382):1344–1345. doi:10.1126/science.359.6382.1344
4. Couzin-Frankel J. Cancer immunotherapy. Science. 2013;342(6165):1432–1433. doi:10.1126/science.342.6165.1432
5. Cui J, De Rose R, Best JP, et al. Mechanically tunable, self-adjuvanting nanoengineered polypeptide particles. Adv Mater. 2013;25(25):3468–3472. doi:10.1002/adma.201300981
6. Schietinger A, Philip M, Krisnawan VE, et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity. 2016;45(2):389–401. doi:10.1016/j.immuni.2016.07.011
7. Santos PM, Butterfield LH. Dendritic cell–based cancer vaccines. J Immunol. 2018;200(2):443–449. doi:10.4049/jimmunol.1701024
8. DJ I, MC H, K R, T T. Synthetic nanoparticles for vaccines and immunotherapy. Chem Rev. 2015;115(19):11109–11146. doi:10.1021/acs.chemrev.5b00109
9. Luo M, Wang H, Wang Z, et al. A STING-activating nanovaccine for cancer immunotherapy. Nat Nanotechnol. 2017;12(7):648–654. doi:10.1038/nnano.2017.52
10. Voron T, Colussi O, Marcheteau E, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212(2):139–148. doi:10.1084/jem.20140559
11. Wallin JJ, Bendell JC, Funke R, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624. doi:10.1038/ncomms12624
12. Silva A, Soema P, Slütter B, Ossendorp F, Jiskoot W. PLGA particulate delivery systems for subunit vaccines: linking particle properties to immunogenicity. Hum Vaccines Immunother. 2016;12(4):1056–1069. doi:10.1080/21645515.2015.1117714
13. Gu L. Tailored silica nanomaterials for immunotherapy. ACS Cent Sci. 2018;4(5):527–529. doi:10.1021/acscentsci.8b00181
14. Tang F, Li L, Chen D. Mesoporous silica nanoparticles: synthesis, biocompatibility and drug delivery. Adv Mater. 2012;24(12):1504–1534. doi:10.1002/adma.201104763
15. Li T, Shi S, Goel S, et al. Recent advancements in mesoporous silica nanoparticles towards therapeutic applications for cancer. Acta Biomater. 2019;89:1–13. doi:10.1016/j.actbio.2019.02.031
16. Li W, Liu Z, Fontana F, et al. Tailoring porous silicon for biomedical applications: from drug delivery to cancer immunotherapy. Adv Mater. 2018;30(24):1703740. doi:10.1002/adma.201703740
17. Hudson SP, Padera RF, Langer R, Kohane DS. The biocompatibility of mesoporous silicates. Biomaterials. 2008;29(30):4045–4055. doi:10.1016/j.biomaterials.2008.07.007
18. Wang X, Chen Z, Zhang C, et al. A generic coordination assembly-enabled nanocoating of individual tumor cells for personalized immunotherapy. Adv Healthc Mater. 2019;18:1900474. doi:10.1002/adhm.201900474
19. Wang X, Cao F, Yan M, et al. Alum-functionalized graphene oxide nanocomplexes for effective anticancer vaccination. Acta Biomater. 2019;83:390–399. doi:10.1016/j.actbio.2018.11.023
20. Stöber W, Fink A, Bohn E. Controlled growth of monodisperse silica spheres in the micron size range. J Colloid Interface Sci. 1968;26(1):62–69. doi:10.1016/0021-9797(68)90272-5
21. Zhang W, Wang L, Liu Y, et al. Immune responses to vaccines involving a combined antigen–nanoparticle mixture and nanoparticle-encapsulated antigen formulation. Biomaterials. 2014;35(23):6086–6097. doi:10.1016/j.biomaterials.2014.04.022
22. Pei M, Liang J, Zhang C, et al. Chitosan/calcium phosphates nanosheet as a vaccine carrier for effective cross-presentation of exogenous antigens. Carbohydr Polym. 2019;224:115172. doi:10.1016/j.carbpol.2019.115172
23. Yan S, Rolfe BE, Zhang B, Mohammed YH, Gu W, Xu ZP. Polarized immune responses modulated by layered double hydroxides nanoparticle conjugated with CpG. Biomaterials. 2014;35(35):9508–9516. doi:10.1016/j.biomaterials.2014.07.055
24. Gao Y, Yang C, Liu X, Ma R, Kong D, Shi L. A multifunctional nanocarrier based on nanogated mesoporous silica for enhanced tumor-specific uptake and intracellular delivery. Macromol Biosci. 2012;12(2):251–259. doi:10.1002/mabi.201100208
25. Xu J, Wang H, Xu L, et al. Nanovaccine based on a protein-delivering dendrimer for effective antigen cross-presentation and cancer immunotherapy. Biomaterials. 2019;207:1–9. doi:10.1016/j.biomaterials.2019.03.037
26. Sarkar K, Kruhlak MJ, Erlandsen SL, Shaw S. Selective inhibition by rottlerin of macropinocytosis in monocyte-derived dendritic cells. Immunology. 2005;116(4):513–524. doi:10.1111/j.1365-2567.2005.02253.x
27. Wang LH, Rothberg KG, Anderson RG. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol. 1993;123(5):1107–1117. doi:10.1083/jcb.123.5.1107
28. Casaravilla C, Pittini Á, Rückerl D, et al. Unconventional maturation of dendritic cells induced by particles from the laminated layer of larval Echinococcus granulosus. Infect Immun. 2014;82(8):3164–3176. doi:10.1128/IAI.01959-14
29. Schnitzer JE, Oh P, Pinney E, Allard J. Filipin-sensitive caveolae-mediated transport in endothelium: reduced transcytosis, scavenger endocytosis, and capillary permeability of select macromolecules. J Cell Biol. 1994;127(5):1217–1232. doi:10.1083/jcb.127.5.1217
30. Latz E, Schoenemeyer A, Visintin A, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5(2):190–198. doi:10.1038/ni1028
31. Hjortø GM, Larsen O, Steen A, et al. Differential CCR7 targeting in dendritic cells by three naturally occurring CC-chemokines. Front Immunol. 2016;7:568. doi:10.3389/fimmu.2016.00568
32. Gerlach C, Moseman EA, Loughhead SM, et al. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity. 2016;45(6):1270–1284. doi:10.1016/j.immuni.2016.10.018
33. Lutz MB, Schuler G. Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol. 2002;23(9):445–449. doi:10.1016/S1471-4906(02)02281-0
34. Tugues S, Burkhard SH, Ohs I, et al. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015;22(2):237–246. doi:10.1038/cdd.2014.134
35. Wang L, Zhao Y, Liu Y, et al. IFN-γ and TNF-α synergistically induce mesenchymal stem cell impairment and tumorigenesis via NFκB signaling. Stem Cells. 2013;31(7):1383–1395. doi:10.1002/stem.1388
36. Morel PA, Butterfield LH. Dendritic cell control of immune responses. Front Immunol. 2015;6(2–3):42. doi:10.3389/fimmu.2015.00042
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.