Back to Journals » ImmunoTargets and Therapy » Volume 3
Antigen-based immunotherapy for the treatment of acute lymphoblastic leukemia: the emerging role of blinatumomab
Authors Litzow M ![]()
Received 15 December 2013
Accepted for publication 11 February 2014
Published 9 April 2014 Volume 2014:3 Pages 79—89
DOI https://doi.org/10.2147/ITT.S37292
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Mark R Litzow
Mayo Clinic, Rochester, MN, USA
Abstract: Acute lymphoblastic leukemia (ALL) arises from immature B and T lymphoblasts. An increasing array of cytogenetic and molecular markers have been identified in ALL, which allows for increasingly sophisticated prognostication, as well as identification of potential new targets for therapy. The treatment of ALL in children has shown astounding success in the last 50 years, with more than 90% of children now able to be cured of their ALL. In adults, these success rates have not been duplicated. However, the use of pediatric-intensive regimens in young adults has shown increasing success. The use of monoclonal antibodies conjugated to drugs, immunotoxins, and cells also has shown early success and promises to enhance the outcome of newly diagnosed patients. Blinatumomab, a bispecific T-cell engager antibody, brings a malignant B cell in proximity to a T cell with redirected lysis. This antibody construct has shown promising results in patients with relapsed and refractory disease and is entering randomized clinical trials in newly diagnosed patients. The addition of monoclonal antibody therapy to chemotherapy in adults promises to enhance outcomes while hopefully not increasing toxicity. After many years of stagnation, it appears that the therapy of adults with ALL is showing significant improvement.
Keywords: acute lymphoblastic leukemia, chemotherapy, monoclonal antibodies, CD19, blinatumomab
Introduction
Acute lymphoblastic leukemia (ALL) arises from immature hematopoietic progenitors that are destined to develop into lymphocytes but acquire somatic gene mutations, which results in altered proliferation and arrest of differentiation. Although regulation of growth and differentiation is altered in these cells, they retain many of the features of their normal lymphoid counterparts. This includes rearrangement of their immunoglobulin (Ig) and T-cell receptor (TCR) genes. Similarly, cell surface antigens characteristic of normal B and T lymphocytes are expressed on the cell surface of the malignant lymphoid blast, and the pattern of this antigen expression can help delineate where in the maturation sequence the malignant transformation occurred. The clonal nature of the malignant lymphoblasts has been established by the demonstration of identical rearrangements of Ig or TCR genes within the ALL cell population.1
ALL of B- or T-cell lineage can be further subcategorized immunophenotypically by the point in maturation when their development is interrupted and they become malignant. About 80% of ALL cases are of B-cell lineage. Most cases of B-cell ALL have an immature immunophenotype and are designated as precursor lymphoid neoplasms or lymphoblastic leukemia/lymphoma. These cases can be identified by the cell surface expression of cluster of differentiation 19 (CD19) and one other B-lineage-associated antigen, such as CD20, CD21, CD22, CD24, or CD79. These lymphoid blasts express intracytoplasmic IgM heavy chain proteins. Early B-cell blasts lack this expression but are CD10-positive, whereas the most immature subtype, pro-B, are CD10-negative. It is important to note that although leukemic lymphoblasts express antigens related to their stage of development, they may also have an aberrant immunophenotype with asynchronous gene expression related to their malignant transformation.1,2
Similarly, an ALL of T-cell origin can be classified on the basis of the sequence of expression of T-cell-associated cell surface antigens that evolve during normal thymocyte development. The earliest T-cell precursors lack expression of CD4 and CD8 and are referred to as double-negative thymocytes. They progress through a series of stages of differentiation characterized by rearrangement of the TCR genes, lose expression of CD34, and gain expression of CD1a.1 An early T-cell precursor phenotype has been identified that has a very high clinical risk and makes up 8%–15% of T-ALL in children and a higher percentage in adults. This subtype has been shown to express activating mutations of RAS, IL-7R, and FLT3, along with PTEN deletions.3
ALL can also frequently express antigens associated with cells of myeloid origin (eg, CD13, CD14, or CD33). These reflect the aberrant malignant development of these leukemic blasts. These patients were previously felt to have a poorer prognosis, but this has not been borne out with the use of chemotherapy regimens in the modern era.4
Genetic abnormalities
Genetic abnormalities play a key pathogenic role in the origin and development of ALL. These were first identified by conventional cytogenetics and can be found in up to 75% of patients with ALL. Recurring abnormalities have been identified, and the distribution of these abnormalities varies significantly between patients with pediatric ALL compared with those with adult ALL, with adult patients having a higher frequency of adverse cytogenetic abnormalities. The main adverse cytogenetic changes include the presence of t(9;22) (BCR-ABL1 or the Philadelphia chromosome), t(4;11), a complex karyotype (five or more chromosomal abnormalities), or low hypodiploidy/near triploidy. In contrast, patients with a hyperdiploid karyotype or a t(12;21) (TEL-AML1) have a favorable prognosis and are much more frequently seen in pediatric ALL, where these latter two abnormalities make up more than 50% of cases.5
The molecular revolution has led to the ability to sequence the genome of patients with ALL and identified numerous recurring genetic mutations and other alterations in the genome of patients with ALL. Some of the more common genetic alterations have included mutations in the paired box 5 (PAX5) gene.6 However, this has not been shown to have any prognostic significance. Janus kinase (JAK) 1 and 2 gene mutations are present in up to 35% of Down syndrome-associated ALL and about 10% of BCR-ABL1 ALL. In adults, JAK1 mutations are more prevalent in T-cell ALL and are associated with a poor prognosis. The Ikaros family zinc finger protein 1 (IKZF1) has been associated with high-risk ALL and poor outcomes. Mutations of IKZF1 are common in BCR-ABL1-positive ALL and in the lymphoid blast phase of chronic myeloid leukemia. The cytokine receptor-like factor 2 (CRLF2) has alterations in about 5% of adult ALL.7 Abnormal expression of CRLF2 can be detected by immunohistochemistry, and CRLF2rearranged ALL was associated with mutant JAK2 in about 50% of cases. In pediatric ALL, elevated CRLF2 expression is an adverse prognostic factor (Figure 1).
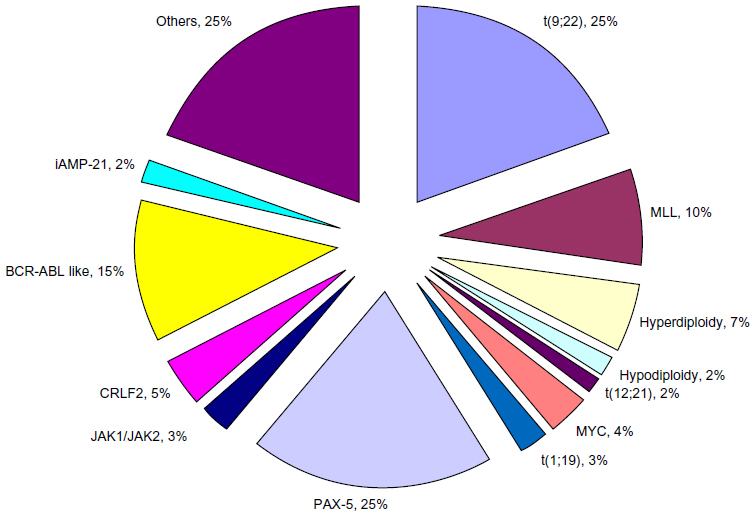 | Figure 1 Frequency of cytogenetic abnormalities in adult B-ALL. |
A new finding of great interest is the identification of a gene expression profile in BCR-ABL1-negative ALL that is similar to that seen in patients with the BCR-ABL1 translocation. This phenotype is known as the BCR-ABL1-like ALL. These cases also commonly harbor mutations of IKZF1 and have a poor prognosis.8,9 This phenotype is seen with increasing frequency in childhood ALL patients (10%–14%, and up to 26% in young adults aged 21–39 years).10 In vitro studies suggest that these cells may also be sensitive to tyrosine kinase inhibitors similar to their BCR-ABL1-positive counterparts. This obviously could have significant therapeutic implications. Intrachromosomal amplification of chromosome 21 (iAMP-21) is defined as a gain of at least five copies of the RUNX1 region of chromosome 21. In pediatrics, this abnormality has been associated with significantly inferior survival.11 Increasing identification of genetic abnormalities in ALL brings the hope that these abnormalities can translate into new therapeutic targets.12
Current treatment options and patient outcomes
ALL represents a remarkable odyssey of success in the era of cancer treatment. From the first report by Sidney Farber13 of temporary remissions induced by aminopterin in five children with ALL, there have been remarkable improvements in the treatment of this deadly leukemia. This was followed by reports in the 1960s of combination chemotherapy, including mercaptopurine and methotrexate, leading to 2-year survival rates of 20%.14 This led to the concept of using combination chemotherapy to treat malignancy. A recognition that ALL also could frequently involve the central nervous system (CNS) led to the recognition of a need for CNS-directed therapy, including cranial radiation and intrathecal methotrexate. This ultimately led to the concept of “total therapy”, which was pioneered by Donald Pinkel and colleagues at St Jude’s Research Hospital in Memphis, Tennessee. Their “total therapy” encompassed different phases of treatment, including remission induction, CNS-directed therapy, intensification (also referred to as consolidation) therapy, and continuation (or maintenance) treatment.15 Remarkably, these components of therapy remain the foundation of modern ALL therapy in both children and adults. As new chemotherapy agents were found to have activity in ALL, including anthracyclines, cytarabine, and asparaginase, they were incorporated into the treatment regimens. During this same time, it became apparent that radiation-induced complications could be severe and led to the use of triple intrathecal therapy with methotrexate, cytarabine, and hydrocortisone, along with higher doses of intravenous methotrexate to effectively prophylax the CNS and replace prophylactic cranial irradiation. Many of the advances in the treatment of childhood ALL during the 1980s and 1990s were related to optimizing the doses and schedules of existing agents, rather than simply the introduction of more new agents.14 These efforts led to remarkable improvement in survival for children with ALL, such that by the middle of the previous decade, 10-year survival estimates are at 91% (Figure 2).14
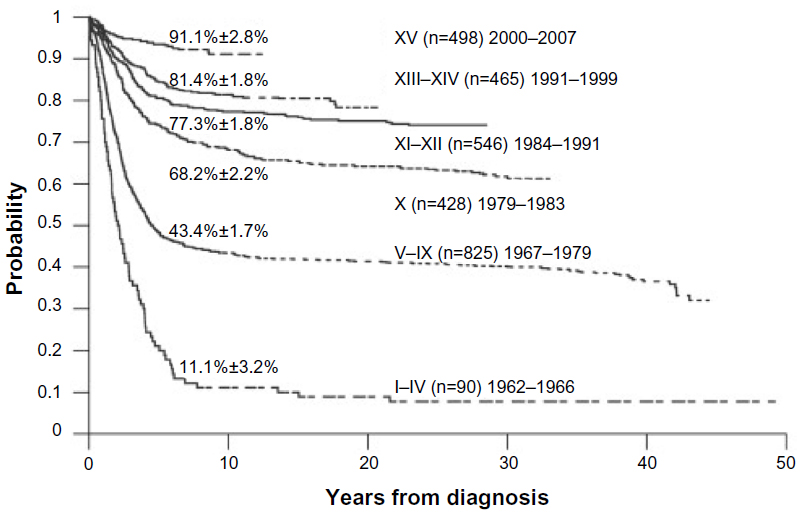 | Figure 2 A 50-year journey to cure childhood ALL. |
Paralleling these advances in treatment were advances in the understanding of the biology of ALL, as outlined in the previous section. These advances led to the ability to risk-stratify patients and alter treatment intensity on the basis of prognosis. The identification of these cytogenetic and genetic markers has also begun to result in the development of new agents effective in the treatment of ALL, including the development of tyrosine kinase inhibitors such as imatinib, which in combination with chemotherapy, has significantly improved the outcome of children and adults with BCR-ABL1 ALL (Philadelphia chromosome-positive).16,17
Unfortunately, results of treatment in adults with ALL have not paralleled the success seen with children. These poorer results can be attributed to multiple factors including the inability of older adults to tolerate the intensive chemotherapy given to pediatric patients; the relative rarity of ALL in adults compared with children, which makes it more challenging for adult oncologists to follow the complex treatment regimens developed for adult patients; and most important, the different biology of the disease in adults compared with in children.18 In particular, adults with ALL more frequently harbor adverse cytogenetic abnormalities, including the t(9;22) (Philadelphia chromosome), t(4;11), a complex karyotype (five or more chromosomal abnormalities), or low hypodiploidy/near triploidy, and less frequently have favorable cytogenetic abnormalities such as the t(12;21) (TEL-AML1) or hyperdiploidy.5 Thus, adults have increased rates of death from complications and more risk of relapse than children. Survival rates in adults at 3 years are, therefore, only in the range of 30%–40%.18
However, in the last 10 years, it has been increasingly recognized that adolescents and young adults (AYA) fare differently if they are treated with a pediatric as opposed to an adult ALL chemotherapy regimen. This was first reported by Stock et al19 in a retrospective comparison of outcome in 321 AYA (age, 16–20 years) who were treated on consecutive trials on either the Children’s Cancer Group or the Cancer and Leukemia Group B from 1988–2001. Although complete remission rates were identical in the two cohorts of patients, the Children’s Cancer Group AYA had a 67% overall survival at 7 years in contrast to the Cancer and Leukemia Group B AYA, for whom overall survival was 46% (P<0.001).19 Multiple subsequent similar comparisons from other countries comparing AYA treated on adult versus pediatric regimens showed similar results.20 The reasons for these differences in outcome are likely multiple, but an important factor is that pediatric regimens include much higher doses of nonmyelosuppressive chemotherapy drugs including corticosteroids, vincristine, and asparaginase.
Several studies have now prospectively assessed the use of pediatric-intensive regimens in adults. This was first reported by Huguet et al,21 for the French Group for Research on Adult Acute Lymphoblastic Leukemia, who gave a pediatric intensive regimen to 225 adults with a median age of 31 years but a range of 15–60 years. The overall complete remission rate was 93.5%. It was 95% in patients aged 15–45 years and 87% in patients aged 46–60 years. The overall complete response rate of 93.5% was superior to a previous trial (Leucémies Aiguěs Lymphoblastiques de l’Adulte-94 [LALA-94]), which used a traditional adult regimen and had a complete response rate of 88% (P=0.02). The overall survival rate was 66% in patients younger than 45 years, which compared favorably with the LALA-94 trial, in which the overall survival rate was 44% at 42 months of follow-up. Patients older than 45 years did not tolerate this pediatric-intensive regimen as well as patients younger than 45 years, as there was a higher cumulative incidence of chemotherapy-related deaths (23% versus 5%, respectively; P<0.001) and deaths in first complete remission (22% versus 5%, respectively; P<0.01).21 A recently published meta-analysis of trials with adult-intensive regimens has suggested that incorporation of allogeneic stem cell transplantation (SCT) into the treatment of adults with ALL in first remission results in superior outcomes compared with chemotherapy or autologous SCT.22 Thus, the question has arisen as to whether a younger adult with ALL should be treated with a pediatric intensive regimen or directed to allogeneic transplant once they achieve first remission. Many investigators feel that young adults without other high-risk features can be managed with a pediatric intensive chemotherapy regimen alone and only considered for transplant if they relapse.23
However, the therapy of older adults with ALL remains particularly challenging. They do not tolerate pediatric intensive regimens as well as younger adults but also do not tolerate intensive therapies such as allogeneic SCT either. Another option that is gaining increasing interest is the use of reduced-intensity conditioning allogenic SCT. Recent studies have suggested that the outcomes with reduced-intensity conditioning allogeneic SCT may be comparable to myeloablative conditioning allogeneic SCT, even though the patients who received reduced-intensity conditioning were older and likely had more comorbidities.24,25 The management of older adults with ALL has recently been summarized.26
Immunotherapy of cancer
The identification of the graft versus leukemia reaction after SCT and the acknowledgment that it plays a key role in the cure of hematologic malignancies led to the recognition that the immune system could play a key role in the treatment of malignancy.27 This led to the increasing study of appropriate targets for immunotherapy, including tumor-specific and/or tumor-associated antigens. These can be attacked by different cellular components of the immune system, including T-cells, natural killer cells, and dendritic cells. The advent of the development of increasing sophisticated monoclonal antibodies has led to the approval of an ever-increasing number of such antibodies for the treatment of hematologic malignancies and solid tumors.28 Conjugation of monoclonal antibodies with immunotoxins and chemotherapy drugs has also shown promise, as has the production of bispecific T-cell engaging antibodies. There is increasing interest in engineering cells that combine components of monoclonal antibodies with the T-cell to promote target-killing. Examples include chimeric antigen receptors, modified T-cells that have shown recent promise in the treatment of ALL.29,30
CD19 in immunology
During the course of B-cell ontogeny, multiple cell surface antigens are expressed, and several of them have become attractive targets for monoclonal antibody-directed therapy. These include CD20 and CD22, although these two antigens are expressed somewhat later in B-cell development.
CD19 has become one of the most attractive targets in the treatment of B-lineage ALL, as its surface expression on the cell begins around the time of Ig gene rearrangement. Its expression also increases with increasing B-cell maturity. The human CD19 antigen is a member of the Ig superfamily and is a 95 kDa transmembrane glycoprotein. The gene for CD19 is located on the short arm of chromosome 16, contains 15 exons, and produces a protein composed of 556 amino acids. The CD19 protein contains an extracellular N-terminus, a single transmembrane domain, and a cytoplasmic C-terminus. It is categorized as a type 1 transmembrane protein without significant homology with any other known proteins (Figure 3).31 CD19 serves a vital function in establishing an effective immune response, as it takes part in antigen-independent development as well as in immunoglobulin-induced activation of B-cells. Intracellular downstream targets of CD19 activation include protein kinase members of the Src family, the Ras family, Abl, Btk, and PI3K, among others. Importantly, it functions as an adaptor protein by recruiting cytoplasmic signaling proteins to the cell membrane.31
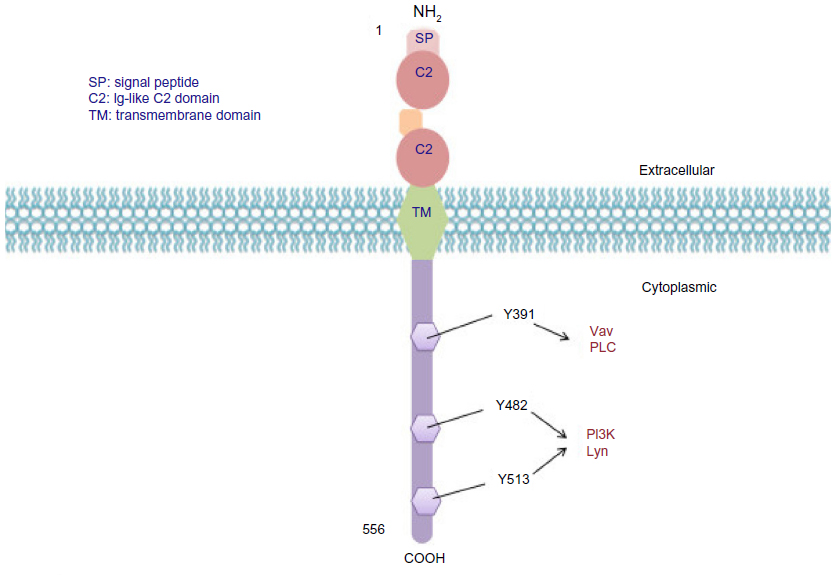 | Figure 3 CD19 molecular structure. |
Mice that are deficient in CD19 have defects in the later stages of B-cell growth and maturation. This deficiency does not affect the number of B-cell precursors in the marrow, but CD19-/- mice have marked reductions in the frequency and number of splenic and peripheral blood B cells. B-cell proliferation is also significantly reduced in response to mitogens.
Humans have been described who have homozygous frame shift mutations of the CD19 gene, which produces truncation of three key cytoplasmic tyrosine residues. These individuals have normal numbers of precursor and total B cells but decreased numbers of CD5 B cells and CD27 memory B cells. The patients develop hypogammaglobulinemia and have poor antibody responses to rabies vaccination, and they are more prone to infection.32
CD19 as an immunotherapeutic target
As noted, CD19 is an attractive target for immunotherapy because of its restricted expression on B-lineage lymphomas and leukemias, as well as normal B cells, but not on other hematopoietic cells or normal tissues. It has a broader expression profile through B-cell development compared with other antigens, such as CD20, and is more efficiently internalized.33 Despite these attractive features, conventional antibodies alone targeted against CD19 had limited activity in preclinical models, although these models had high CD19 expression and were able to internalize antibodies.34 Thus, multiple different antibody constructs and conjugations have been developed with CD19 as the target. These are summarized in Table 1 and include CD19 antibodies conjugated to a maytansine derivative; engineered anti-CD19 antibodies that enhance antibody-dependent cell-mediated cytotoxicity; antibody conjugation to diphtheria toxin; antibody conjugation to auristatin anti-CD19; the chimeric antigen receptor T-cells described earlier; dual-affinity retargeting antibodies, which are encoded by two different polypeptide chains that contain a variable heavy chain domain fused to a variable light chain domain; and a bispecific T-cell engager that comprises two single-chain antibodies (scFvs) that bind CD3 and CD19, respectively. Blinatumomab is the prototype of this last approach. These various CD19 immunotherapy approaches were recently summarized in a review.33 Blinatumomab’s name is derived from the fact that it is a B-lineage-specific antitumor mouse monoclonal antibody. It was previously referred to as bscCD19×CD3 or MT103, and now AMG103. The basic characteristics of blinatumomab, including its structure, specificity, purification, and cytotoxicity, were first reported in 2000.35
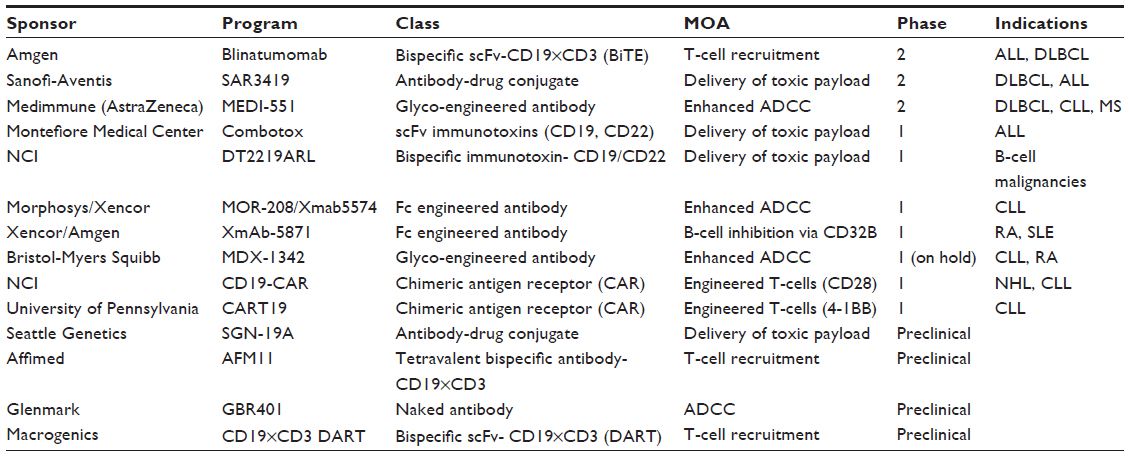 | Table 1 CD19 immunotherapy programs in development |
Blinatumomab is a construct of scFvs that forms a 55 kDa fusion protein (Figure 4). Recombinant DNA technology is used and takes the respective cDNAs, which encode the four variable domains, and three linker sequences. The scFv antibodies are generated from two longer linker sequences. A 5 amino acid linker sequence that is nonimmunogenic is used to recombinantly link the two scFvs in tandem. This is thought to give the two scFvs a significant degree of rotational ability to enhance binding of epitopes on separate cells.36
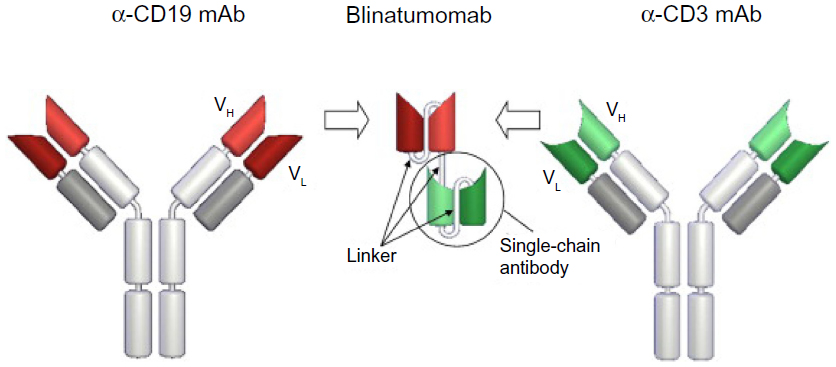 | Figure 4 Generation and structure of blinatumomab. |
Blinatumomab is produced in Chinese hamster ovary cells in a both a monomeric and dimeric form. Although both these forms are biologically active, only the monomeric form is purified for clinical use.36
In vitro, blinatumomab was found to be extremely potent, with a half-maximal concentration for redirected lysis of CD19-positive target cells by B cells in the range of 10–100 pg/mL with T cells from healthy donors.37 All T-cell populations except naive T cells showed high-level redirected lysis. This effect was seen with resting T cells that were not previously activated and was associated with upregulation of T-cell antigen expression, including CD69, CD25, and CD2, along with transient release of multiple inflammatory cytokines including interleukins 2, 6, and 10; tumor necrosis factor α, and interferon γ.38
The first clinical trials in humans began in 2001 (Table 2). Phase I studies in patients with relapsed or refractory non-Hodgkin lymphoma were carried out using doses ranging from 0.75–13 μg/m2 administered once, twice, or three times weekly as 2- or 4-hour intravenous infusions. Adverse events included fever, rigors, and fatigue. Significant neurologic events included aphasia, ataxia, disorientation, and seizures and led to discontinuation of therapy in 6 of 22 patients. Cytokine release syndrome and infections were also observed.36
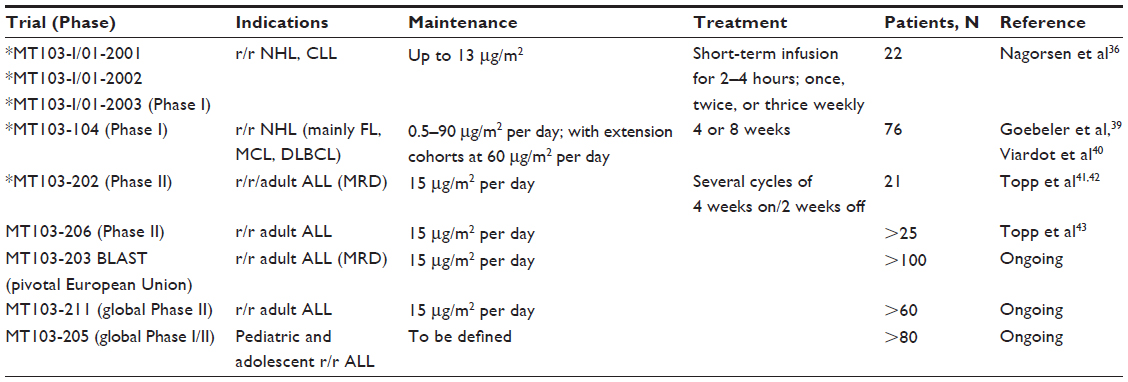 | Table 2 List of all completed and ongoing clinical trials with blinatumomab monotherapy |
Given the toxicity profile with shorter infusions, a continuous intravenous infusion over a period of 4–8 weeks was carried out in non-Hodgkin lymphoma patients. Serum levels were maintained at predictable levels during the entire infusion period. Corticosteroids were given at the start of treatment to minimize cytokine release. After an initial observation period from 3–7 days, the blinatumomab infusion could be continued as an outpatient. All trials since this initial phase I study have administered blinatumomab as a continuous infusion for 4 weeks.36
The initial clinical experience with this infusion schedule was in patients with low-grade lymphoma and mantle cell lymphoma. Dose escalation started as low as 0.5 μg/m2 per day but was escalated up to 90 μg/m2 per day. The trial was later amended to include patients with aggressive diffuse large B-cell lymphoma. The most recent update on 70 patients, of whom 93% had had prior rituximab-based regimens, showed that most patients had mild adverse events of a constitutional nature, generally of grade 1 or 2. Most of these occurred during the first few days of treatment. A transient release of inflammatory cytokines was noted in the first few days, along with a rapid decline in peripheral blood B cells. Even with the continuous infusion, CNS events were noted, again primarily during the first 3 days of treatment, and included encephalopathy, aphasia, tremor, disorientation, and convulsions. These occurred in 3%–8% of patients. The etiology of the CNS events is not entirely clear but may be related to activated T-cells that release neurotoxic cytokines into the CNS. Fortunately, these CNS events have been reversible with discontinuation of treatment.39
The maximum tolerated dose in this study was 60 μg/m2 per day. A step-wise approach to blinatumomab administration with lower doses given in the first week or 2, followed by escalation to the full dose, has lessened the incidence of neurologic toxicity.39
Complete remissions were noted with dose levels as low as 15 μg/m2 per day, and it was noted that five of six patients at this dose had clearance of lymphomatous involvement of the marrow. The overall response rate at the 60 μg/m2 per day dose was 76% (16/21 patients), with seven patients having a complete remission or unconfirmed complete remission.36,39
There is still some uncertainty related to the long-term effects of depletion of normal CD19-positive B-cells. Patients do develop hypogammaglobulinemia as expected, but long-term infectious complications have not been prominent.
In diffuse large B-cell non-Hodgkin lymphoma, experience is more limited, but of a group of 13 patients treated, eleven were evaluable, with a total of six responses and two patients with stable disease. Of the six responders, four achieved a complete remission. Patients with extranodal disease and those with prior autologous transplant also responded; these responses overall appeared to be durable. In three of six responders, allogeneic transplant was able to be performed.40
Given the activity seen in non-Hodgkin lymphoma in clearing lymphomatous cells from the marrow, it was felt that blinatumomab might be efficacious in B-lineage ALL. Because patients who are in hematologic remission, but have persistent minimal residual disease (MRD) by molecular or immunophenotypic evidence, have a poor prognosis, it was decided that a pilot study in patients in hematologic remission who were MRD-positive or who had experienced an MRD relapse would be initially carried out. MRD was measured by polymerase chain reaction for rearrangements of immunoglobulin heavy chain disease. The dose chosen was 15 μg/m2 per day by 4-week continuous intravenous infusion, as marrow clearing had been seen in lymphoma patients at this dose. Of 21 patients treated, 20 were evaluable and 16 (80%) became MRD-negative during the first 4-week cycle of therapy. The most frequent grade 3 or 4 adverse event was lymphopenia. Two patients had reversible CNS adverse events that led to discontinuation of the drug. No deaths were noted.41 Longer-term follow-up from this study has been recently reported. After a median follow-up of 33 months, the relapse-free survival was 61%. Nine patients were able to proceed to allogeneic SCT and had a relapse-free survival of 65%. Of six patients with BCR-ABL1 ALL who responded and had no further therapy after blinatumomab, four remain in ongoing hematologic and molecular remission.42 These encouraging results have led to a larger trial in patients who are MRD-positive across several countries in Europe, which was initiated in the fall of 2010. Results are eagerly awaited.
These results have also led to a trial in patients with hematologic relapse or refractory B-lineage ALL. Three dosing regimens were explored, including initiation of therapy at 15 μg/m2 per day, and continued at that dose for the entire 4-week cycle versus an initial dose of 5 μg/m2 per day for the first week and then escalation to 15 μg/m2 per day for the subsequent 3 weeks. A third group initiated therapy at 5 μg/m2 per day for the first week and then went to 15 μg/m2 per day for the second week and to 30 μg/m2 per day for the third and fourth weeks. The most recent update included 36 patients who had been treated.43 On the basis of the lowest incidence of adverse effects, the schedule selected for treatment of 18 of the 36 patients in a final cohort was the dose of 5 μg/m2 per day for the first week, followed by 15 μg/m2 per day for the subsequent 3 weeks. Of the entire cohort of patients, 26 of 36 achieved a complete remission or complete remission without hematologic recovery (72%). Of these 26 patients, 24 (92%) achieved MRD negativity within the first two cycles. Twenty of 21 (95%) patients in first relapse responded, whereas only 6 (40%) of 15 of the remaining patients achieved a hematologic complete remission or complete remission without hematologic recovery. Thirteen patients were able to proceed to allogeneic transplant. One of these patients had a relapse that was CD19-negative after their allogeneic transplant. Of the 13 patients not going to allogeneic SCT, eight have relapsed. Two of these relapses were CD19-negative, three were CD19-positive, and three were unknown. The median overall survival for all 36 treated patients is 9 months, with a median follow-up time for overall survival of 10.7 months. For patients achieving a complete remission or complete remission without hematologic recovery, the median survival is 14.1 months, whereas for patients who failed blinatumomab, the median survival is 6.6 months. As noted previously, cytokine release syndrome and CNS events were the most significant toxicities. Cytokine release syndrome could be prevented or treated by a dexamethasone regimen. The CNS events were fully reversible and were observed in six patients. Three patients had seizures, and three patients had encephalopathy.43 An international phase II trial of this regimen is currently ongoing. Further studies have also confirmed that body surface area-based dosing is not essential, as pharmacokinetic studies have not shown a significant difference in levels with weight-based versus flat dosing. Therefore, flat dosing with an initial dose of 9 μg/day for the first week followed by 28 μg/day is currently being used.
The exciting results achieved with blinatumomab in both the hematologic relapsed/refractory setting and in the MRD setting have led to the development of a US National Intergroup Trial, which will randomize patients with B-lineage ALL between the ages of 35 and 70 years to induction chemotherapy to achieve remission followed by four cycles of blinatumomab versus chemotherapy alone. Patients may proceed to transplant as indicated. This trial will be an intergroup trial led by the Eastern Cooperative Oncology Group.
Conclusion
ALL is a disorder of malignant immature B and T lymphoblasts. Approximately 80% of patients with ALL express a B lineage. Although more than 90% of children can now be cured of their ALL, these results have not been duplicated in adults. The use of pediatric intensive regimens has improved the outcome of young adults with ALL, but further progress is needed. In older adults, the limits of conventional chemotherapy have been reached, and new approaches are needed.
The CD19 antigen is nearly universally expressed on B-lineage ALL and is an attractive target for therapy. Multiple immunotherapeutic approaches using modified monoclonal antibodies have been developed as outlined in this review. One antibody construct, blinatumomab, a bispecific T-cell engager antibody, is in advanced stages of development and shows significant promise in improving outcomes of patients with B-lineage ALL. A large intergroup trial in the United States will test the efficacy of blinatumomab in newly diagnosed middle-aged and older adults with B-lineage ALL.
Disclosure
The author has received honoraria and research funding from Amgen.
References
Gutierrez A, Armstrong SA, Look AT. Pathobiology of acute lymphoblastic leukemia In: Hoffman R, Benz EJJ, Silberstein LE, Heslop HE, Weitz JI, Anastasi J, editors. Hematology: Basic Principles and Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:935–950. | |
Pui CH, Behm FG, Crist WM. Clinical and biologic relevance of immunologic marker studies in childhood acute lymphoblastic leukemia. Blood. 1993;82(2):343–362. | |
Van Vlierberghe P, Ambesi-Impiombato A, Perez-Garcia A, et al. ETV6 mutations in early immature human T cell leukemias. J Exp Med. 2011;208(13):2571–2579. | |
Pui CH, Behm FG, Singh B, et al. Myeloid-associated antigen expression lacks prognostic value in childhood acute lymphoblastic leukemia treated with intensive multiagent chemotherapy. Blood. 1990;75(1):198–202. | |
Moorman AV, Harrison CJ, Buck GA, et al; Adult Leukaemia Working Party, Medical Research Council/National Cancer Research Institute. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL):analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood. 2007;109(8):3189–3197. | |
Medvedovic J, Ebert A, Tagoh H, Busslinger M. Pax5: a master regulator of B cell development and leukemogenesis. Adv Immunol. 2011;111:179–206. | |
Moorman AV, Schwab C, Ensor HM, et al. IGH@ translocations, CRLF2 deregulation, and microdeletions in adolescents and adults with acute lymphoblastic leukemia. J Clin Oncol. 2012;30(25):3100–3108. | |
Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10(2):125–134. | |
Mullighan CG, Su X, Zhang J, et al; Children’s Oncology Group. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–480. | |
Roberts KG, Payne-Turner D, Pei D, et al. Integrated genomic and mutational profiling of adolescent and young adult all identifies a high frequency of BCR-ABL1-Like ALL with very poor outcome. Blood. 2013;122:825. | |
Harrison CJ, Moorman AV, Schwab C, et al. An international study of intrachromosomal amplification of chromosome 21 (iAMP21):cytogenetic characterization and outcome. Leukemia. Epub October 29, 2013. | |
Kenderian SS, Litzow MR. Acute lymphoblastic leukemia in adolescents and young adults – from genomics to the clinics. Clin Oncol Adolesc Young Adults. 2013;3:49–62. | |
Farber S, Diamond LK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med. 1948;238(23):787–793. | |
Pui CH, Evans WE. A 50-year journey to cure childhood acute lymphoblastic leukemia. Semin Hematol. 2013;50(3):185–196. | |
Pinkel D. Five-year follow-up of “total therapy” of childhood lymphocytic leukemia. JAMA. 1971;216(4):648–652. | |
Schultz KR, Bowman WP, Aledo A, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study. J Clin Oncol. 2009;27(31):5175–5181. | |
Fielding AK, Zakout GA. Treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia. Curr Hematol Malig Rep. 2013;8(2):98–108. | |
Jain N, Gurbuxani S, Rhee C, Stock W. Acute lymphoblastic leukemia in adults. In: Hoffman R, Benz EJJ, Silberstein LE, Heslop HE, Weitz JI, Anastasi J, editors. Hematology: Basic Principles and Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:950–980. | |
Stock W, La M, Sanford B, et al; Children’s Cancer Group; Cancer and Leukemia Group B studies. What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children’s Cancer Group and Cancer and Leukemia Group B studies. Blood. 2008;112(5):1646–1654. | |
Ramanujachar R, Richards S, Hann I, Webb D. Adolescents with acute lymphoblastic leukaemia: emerging from the shadow of paediatric and adult treatment protocols. Pediatr Blood Cancer. 2006;47(6):748–756. | |
Huguet F, Leguay T, Raffoux E, et al. Pediatric-inspired therapy in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia: the GRAALL-2003 study. J Clin Oncol. 2009;27(6):911–918. | |
Ram R, Gafter-Gvili A, Vidal L, et al. Management of adult patients with acute lymphoblastic leukemia in first complete remission: systematic review and meta-analysis. Cancer. 2010;116(14):3447–3457. | |
Isakoff MS, Freyer DR, Bleyer A. Young adults with acute lymphoblastic leukemia treated with a pediatric-inspired regimen do not need a bone marrow transplant in first remission. Blood. 2013;121(26):5253–5255. | |
Mohty M, Labopin M, Volin L, et al; Acute Leukemia Working Party of EBMT. Reduced-intensity versus conventional myeloablative conditioning allogeneic stem cell transplantation for patients with acute lymphoblastic leukemia: a retrospective study from the European Group for Blood and Marrow Transplantation. Blood. 2010;116(22):4439–4443. | |
Marks DI, Wang T, Pérez WS, et al. The outcome of full-intensity and reduced-intensity conditioning matched sibling or unrelated donor transplantation in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia in first and second complete remission. Blood 2010;116(3):366–374. | |
Gökbuget N. How I treat older patients with ALL. Blood. 2013;122(8):1366–1375. | |
Horowitz MM, Gale RP, Sondel PM, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990;75(3):555–562. | |
Wayteck L, Breckpot K, Demeester J, De Smedt SC, Raemdonck K. A personalized view on cancer immunotherapy. Cancer Lett. Epub September 17, 2013. | |
Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra38. | |
Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. | |
Wang K, Wei G, Liu D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol. 2012;1(1):36. | |
van Zelm MC, Reisli I, van der Burg M, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354(18):1901–1912. | |
Hammer O. CD19 as an attractive target for antibody-based therapy. MAbs. 2012;4(5):571–577. | |
Ingle GS, Chan P, Elliott JM, et al. High CD21 expression inhibits internalization of anti-CD19 antibodies and cytotoxicity of an anti-CD19-drug conjugate. Br J Haematol. 2008;140(1):46–58. | |
Löffler A, Kufer P, Lutterbüse R, et al. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood. 2000;95(6):2098–2103. | |
Nagorsen D, Kufer P, Baeuerle PA, Bargou R. Blinatumomab: a historical perspective. Pharmacol Ther. 2012;136(3):334–342. | |
Dreier T, Lorenczewski G, Brandl C, et al. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int J Cancer. 2002;100(6):690–697. | |
Brandl C, Haas C, d’Argouges S, et al. The effect of dexamethasone on polyclonal T cell activation and redirected target cell lysis as induced by a CD19/CD3-bispecific single-chain antibody construct. Cancer Immunol Immunother. 2007;56(10):1551–1563. | |
Goebeler M, Viardot A, Noppeney R, et al. Session 6: targeting the lymphoma cell surface: BLINATUMOMAB (CD3/CD19 Bite Antibody) Results in a High Response Rate in Patients with Relapsed Nonhodgkin Lymphoma (Nhl) Including Mcl and Dlbcl (Abstract 068). Ann Oncol. 2011;22(Suppl 4):iv104–iv105. | |
Viardot A, Goebeler M, Noppeney R, et al. Blinatumomab monotherapy shows efficacy in patients with relapsed diffuse large B cell lymphoma. Presented at: American Society of Hematology Meeting; December 10–13, 2011; San Diego, CA. ASH Annual Meeting Abstracts. 2011;118(21):1637. | |
Topp MS, Kufer P, Gökbuget N, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;29(18):2493–2498. | |
Topp MS, Gökbuget N, Zugmaier G, et al. Long-term follow-up of hematologic relapse-free survival in a phase 2 study of blinatumomab in patients with MRD in B-lineage ALL. Blood. 2012;120(26):5185–5187. | |
Topp MS, Goekbuget N, Zugmaier G, et al. Anti-CD19 BiTE blinatumomab induces high complete remission rate and prolongs overall survival in adult patients with relapsed/refractory B-precursor acute lymphoblastic leukemia (ALL). Presented at: American Society of Hematology Meeting; December 8–11, 2012; Atlanta, GA. ASH Annual Meeting Abstracts. 2012;120(21):670. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.