Back to Journals » ImmunoTargets and Therapy » Volume 7
Antibody targeting of phosphatidylserine for the detection and immunotherapy of cancer
Authors Belzile O, Huang X, Gong J, Carlson J, Schroit AJ, Brekken RA ![]() , Freimark BD
, Freimark BD
Received 6 September 2017
Accepted for publication 27 October 2017
Published 23 January 2018 Volume 2018:7 Pages 1—14
DOI https://doi.org/10.2147/ITT.S134834
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Michael Shurin
Olivier Belzile,1 Xianming Huang,2,3 Jian Gong,2,3 Jay Carlson,2,3 Alan J Schroit,1 Rolf A Brekken,1 Bruce D Freimark2,3
1Hamon Center for Therapeutic Oncology Research, University of Texas Southwestern Medical Center, Dallas, TX, 2Department of Preclinical Research, 3Department of Antibody Discovery, Peregrine Pharmaceuticals, Inc., Tustin, CA, USA
Abstract: Phosphatidylserine (PS) is a negatively charged phospholipid in all eukaryotic cells that is actively sequestered to the inner leaflet of the cell membrane. Exposure of PS on apoptotic cells is a normal physiological process that triggers their rapid removal by phagocytic engulfment under noninflammatory conditions via receptors primarily expressed on immune cells. PS is aberrantly exposed in the tumor microenvironment and contributes to the overall immunosuppressive signals that antagonize the development of local and systemic antitumor immune responses. PS-mediated immunosuppression in the tumor microenvironment is further exacerbated by chemotherapy and radiation treatments that result in increased levels of PS on dying cells and necrotic tissue. Antibodies targeting PS localize to tumors and block PS-mediated immunosuppression. Targeting exposed PS in the tumor microenvironment may be a novel approach to enhance immune responses to cancer.
Keywords: immunosuppression, tumor microenvironment, immunotherapy, imaging, phosphatidylserine, bavituximab
A Letter to the Editor has been received and published for this article.
Introduction
Major advancements to our understanding of immune response regulation have led to the development of therapeutic antibody checkpoint inhibitors for the treatment of cancer.1,2 The approval of the anti-cytotoxic T-lymphocyte antigen-4 (CTLA-4) antibody ipilimumab for the treatment of advanced melanoma in 2011 was the critical turning point to the surge in the development of antibody-based therapeutics to immune checkpoint inhibitors.3,4 The subsequent development of antibodies blocking the programmed death-1 (PD-1) pathway, first with US Food and Drug Administration-approved agents nivolumab and pembrolizumab and followed by other approved antibodies, has made a significant impact on the treatment of melanoma and other tumor types.5–11 However, many patients respond weakly or are unresponsive to antibodies targeting immune checkpoints. This has driven new treatment modalities of combining existing therapies and a search to identify other tumor-associated immunoregulatory targets.12–14
Rationale for phosphatidylserine (PS)-targeting immunotherapy for cancer
In eukaryotic cells, an asymmetric distribution of phospholipids exists across the bilayer membrane, where the positively charged phospholipids phosphatidylcholine (PC) and sphingomyelin are maintained on the outer membrane leaflet and the negatively charged amino-phospholipids PS and phosphatidylethanolamine are localized in the inner membrane leaflet.15–17 This asymmetry is actively maintained by the regulated activity of ATP-dependent and -independent enzymes, collectively known as flippases, floppases, and scramblases.18,19 Under normal physiological conditions, PS exposure serves as an “eat me” signal that attracts macrophages for the engulfment of apoptotic cells.20–22 Viable immune cells including B cells, T cells, monocytes, macrophages, and dendritic cells (DCs) transiently express PS but escape phagocytosis possibly via a PS exposure threshold.23–25 In the tumor microenvironment, various biochemical pathways associated with apoptosis result in the flipping of PS to the external membrane, including the generation of reactive oxygen species,26,27 caspase activation,28 and Ca++ influx due to cell activation.29 Moreover, the interaction between cells with exposed PS and immune cells elicits highly regulated and redundant immunological responses by triggering immunosuppressive pathways that prevent local and systemic immune activation. The immunosuppressive properties of PS exposure weaken innate and adaptive immune responses and subsequently facilitate tumor cell evasion of immune surveillance.30–32 PS exposure foreshadows, or appears “upstream” of the myriad of immunosuppressive signals that follow, and is recognized as a pharmacologically targetable immunological control point.
PS exposure in the tumor microenvironment is largely contributed to by necrotic tissue and apoptotic cells as a result of pathological conditions or therapy but is also observed on viable endothelial cells33 and extracellular vesicles derived from tumors,34 stroma, and leukocytes.34–36 PS is also exposed on the surface of infectious agents and cells infected with a variety of bacterial, viral, and parasitic pathogens and also creates noninflammatory conditions associated with diminished host immune responses,37–41 a process termed “apoptotic mimicry”.42 Multiple PS receptors have been identified. These receptors vary in structure, direct or indirect PS binding, cell type expression, and signaling pathways. Most PS receptors are involved in the stimulation of anti-inflammatory responses for quiescent removal of apoptotic cells22,30 but can contribute to proinflammatory responses under certain conditions.43 PS receptors of particular importance for PS recognition and immune suppression in the tumor microenvironment are T cell/transmembrane, immunoglobulin, and mucin (TIM)44 and Tyro, Axl, and Mertk (TAM) gene families.45,46 Tim-1, Tim-3, and Tim-4 are associated with Th2 stimulation, Th1 receptor-mediated immunosuppression, and apoptotic cell engulfment by dendritic cells and macrophages.47–51 Members of the TAM gene family are receptor tyrosine kinases (RTKs) expressed on leukocytes and on many tumor types.52 TAM RTK binding to PS occurs via γ-carboxylated bridging proteins Gas6 or Protein S. The γ-carboxylated GLA domain of Gas6 and Protein S binds directly to exposed PS, and the receptor-binding domain interacts with the TAM receptor. TAM receptor activation on macrophages triggers the engulfment of PS+ target cells and promotes an anti-inflammatory “M2” phenotype.53 The activation of TAM RTKs on tumor cells is linked to chemoresistance and epithelial plasticity.54 The blocking of vitamin K-dependent γ-carboxylation of Gas6 inhibits Axl activation on tumor cells and reduces tumor progression and metastasis in preclinical tumor models.55 PS binding to TAM RTKs on tumor cells also upregulates programmed death-ligand 1 (PD-L1) expression on tumor cells.56 Thus, blocking the PS-mediated activation of TIM and TAM RTK pathways can enhance anticancer immune responses (Figure 1).
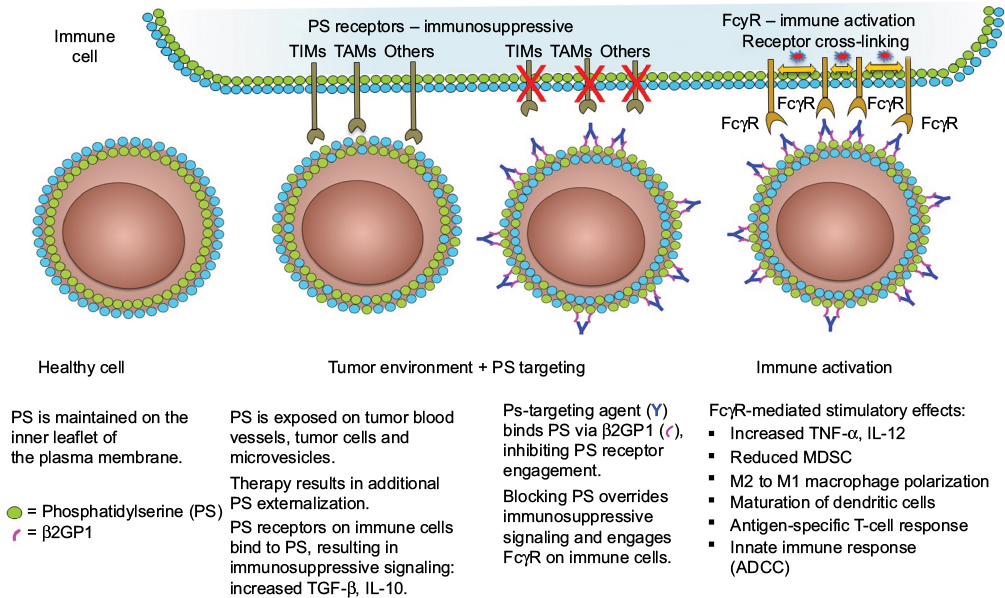 | Figure 1 PS-targeting antibodies interact with exposed PS in the tumor microenvironment to activate immune cells. Notes: PS exposure in the tumor microenvironment activates PS receptors in immune cells and causes these cells to adopt an immunosuppressive phenotype. PS-targeting antibodies 2aG4, 3G4, bavituximab, 1N11, and mch1N11 bind exposed PS via β2GP1. Binding of the PS-targeting complex blocks the interaction between PS and the PS receptors on immune cells and activates these cells via their FcγR. Abbreviations: ADCC, antibody-dependent cell-mediated cytotoxicity; β2GP1, beta 2 glycoprotein-1; FcγR, Fc gamma receptor; IL, interleukin; MDSC, myeloid-derived suppressor cell; PS, phosphatidylserine; TAMs, Tyro3, Axl, Mer receptor tyrosine kinases; TIMs, transmembranes, immunoglobulins, and mucins; TGF, transforming growth factor; TNF, tumor necrosis factor. |
The origins of PS targeting
In a study published in 1998, Ran et al57 covalently linked a monoclonal antibody to murine VCAM-1, a marker of tumor endothelium, to the extracellular domain of the human tissue factor, a protein involved in blood coagulation.57 This vascular-targeting agent, termed as “coaguligand”, was designed to induce thrombosis in tumor blood vessels, resulting in catastrophic destruction of the tumor vasculature. When administered to tumor-bearing mice, the coaguligand-induced thrombosis of VCAM-1+ tumor vasculature; however, there was no sign of thrombosis in normal organs, including VCAM-1+ vasculature of the lungs and heart. Since PS is required for tissue factor-induced coagulation,58–60 Ran et al hypothesized that PS was exposed on tumor vasculature but not on the endothelial cells of normal organs. Indeed, PS and VCAM-1 co-localize on tumor blood vessels, whereas regardless of the presence of VCAM-1, externalized PS is not detected on heart or lung vasculature.
Following up on the findings that PS is externalized on tumor vasculature, Ran et al33 generated a new monoclonal antibody, 9D2, specific to anionic phospholipids. 9D2, an immunoglobulin M, was generated by injecting rats with bEnd.3 endothelial cells that had been treated with hydrogen peroxide, a condition that induced PS externalization. The 9D2 and annexin V did not compete with one another for PS, indicating that they bind different epitopes. Moreover, unlike annexin V, the interaction of 9D2 with PS does not require calcium. The authors found that 9D2 and annexin V specifically localized to tumor blood vessels in mice but not to the endothelium of the 10 normal organs examined. The authors then investigated which factors of the tumor microenvironment might be responsible for endothelial PS externalization. In in vitro experiments, they exposed endothelial cells for 24 hours to various factors that were known to be present in the microenvironment of many tumors. The most potent factor was hydrogen peroxide, causing nearly maximal 125I-annexin V binding. Other factors such as hypoxia/reoxygenation, thrombin, and acidity were found to induce moderate PS externalization. Inflammatory mediators such as IL-1-a, IL-1-b, tumor necrosis factor-alpha (TNF-α), and interferon-gamma (IFN-g) caused a small increase in PS exposure. The cells remained attached and viable under all the above conditions, and PS asymmetry was re-established after removal of the stressor. The authors concluded by suggesting that anionic phospholipids such as PS may be a target for tumor therapy and imaging. They proposed exploring the idea of using annexin V or antibodies to anionic phospholipids for the delivery of drugs, radionuclides, or coagulants to tumor vessels.
In a subsequent study, the Thorpe Laboratory developed an immunoglobulin G (IgG)-targeting PS and hypothesized that it might exert antitumor activity by damaging tumor vasculature.61 This new antibody, a mouse IgG3 monoclonal antibody named 3G4, did not bind enzyme-linked immunosorbent assay (ELISA) plates coated with anionic phospholipids in the absence of serum. The binding was rescued when b2-glycoprotein 1 (β2GP1) was added, suggesting that this serum protein is involved in the interaction between PS and 3G4. In vitro binding of 3G4 to PS was blocked with liposomes made from anionic phospholipids but not by those made from neutral phospholipids. The authors found that 3G4 localized to tumor vessels, caused vessel destruction, and inhibited tumor growth in multiple models without causing toxicity. For example, in mice bearing orthotopic MDA-MB-231 tumors, 40% of tumor vessels were bound by 3G4; no staining was observed in normal tissues from 17 organs. A reduction in tumor growth was noted, ranging between 50 and 90%, but complete regression was not observed. The inhibition of tumor vasculature by 3G4 was shown by reduced plasma volume and decreased vascular density. The 3G4 caused monocytes/macrophages to bind to, and destroy, tumor blood vessels in an antibody-dependent manner. This was the first indication that targeting PS altered the immune microenvironment of tumors. In addition, targeting exposed PS on tumor endothelial cells resulted in tumor-associated macrophages adopting an M1-like phenotype. Based on these data, the authors hypothesized that a chimeric or humanized version of 3G4 might have anticancer activity in humans through antibody-dependent cell-mediated cytotoxicity (ADCC) of PS+ tumor endothelial cells by macrophages. The 3G4 was shown to target PS by dimerizing two molecules of b2GP1, thus stabilizing its interaction with externalized PS (Figure 2);61,62 b2GP1 (or apolipoprotein H) is a glycoprotein present in the circulation at concentrations of 100–200 mg/mL and consists of four short repeat domains and a fifth domain that contains the phospholipid-binding site.63,64 The physiological function of b2GP1 remains unknown, although it may function in homeostatic buffering of the coagulation system.
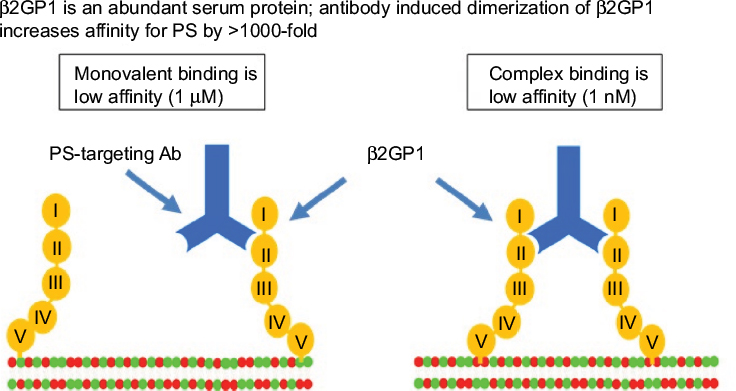 | Figure 2 PS-targeting antibodies interact with b2GP1 and PS to form a high avidity complex. Notes: PS-targeting antibodies 2aG4, 3G4, bavituximab, 1N11, and mch1N11 bind circulating monomeric b2GP1 with low affinity. When sufficient surface exposure is present on a target cell, PS-targeting antibody interacts with two molecules of b2GP1 to form a high avidity (1 nM KD) complex. Domain V of b2GP1 interacts with PS exposed on the target cell. KD, equilibrium dissociation constant. Abbreviations: Ab, antibody; β2GP1, beta 2 glycoprotein-1; PS, phosphatidylserine. |
Combining PS-targeting antibodies with chemotherapies
In the second published study on the mouse monoclonal antibody 3G4, Huang et al65 combined 3G4 with chemotherapy. First, they showed that subtoxic concentrations of docetaxel induced the externalization of anionic phospholipids in endothelial cells in vitro. Additionally, in vivo experiments demonstrated that docetaxel induced enhanced anionic phospholipid exposure in tumor blood vessels, but not in normal organs. The major finding of the study was that 3G4 significantly enhanced the therapeutic efficacy of docetaxel against the growth and dissemination to the lung of MDA-MB-435 human tumors in mice without increased toxicity. Antivascular effects of docetaxel plus 3G4 were significantly enhanced compared to the individual therapies. Similarly, the authors found more apoptotic endothelial cells in tumors treated with the combination than in those treated with single therapy. The authors concluded by suggesting that clinical evaluation of PS targeting in patients was warranted. Shortly, thereafter, a chimeric version of 3G4, bavituximab, entered clinical trials.
Additional preclinical studies in orthotopic mouse models of pancreatic cancer demonstrated that 3G4 in combination with gemcitabine was significantly more effective at controlling primary tumor growth and reducing metastatic burden than either agent alone.61 The authors also found that 3G4 alone or in combination with chemotherapy reduced microvessel density and increased macrophage recruitment into tumors. The combination treatment caused a 14-fold increase in macrophage infiltration over controls (single therapies caused 4.2- and 1.76-fold increases over controls). Second-generation PS-targeting antibodies 2aG4 and bavituximab were derived from 3G4, and separately, 1N11 was isolated for subsequent preclinical and clinical studies. Preclinical tumor studies showed that mouse PS-targeting antibodies 3G4, 2aG4, and mouse chimeric 1N11 (mch1N11) (human variable regions of 1N11 fused to mouse IgG2a-kappa constant regions) localize with the same specificity to PS-expressing tumors and tumor blood vessel endothelial cells and elicit strong antitumor effects, especially when combined with treatments that expose PS in tumors.
To better understand how PS targeting functions to reduce PS-mediated immune suppression, Yin et al32 investigated the efficacy of 2aG4, an IgG2a, class-switched version of 3G4, in prostate cancer xenografts. Similar to previous studies, the authors showed that combining PS-targeting antibody 2aG4 with docetaxel effectively inhibited tumor growth in two orthotopic models of prostate cancer. They also observed that M1-like macrophages localized to blood vessel remnants and were likely responsible for their destruction. Tumors treated with a control antibody had fewer macrophages, and the ones present exhibited an M2 phenotype and were not associated with blood vessels. Furthermore, only macrophages isolated from 2aG4-treated tumors synthesized nitric oxide, a characteristic of M1 macrophages. These macrophages were also able to drive ADCC-mediated killing of tumor cells in vitro. Additionally, the authors showed that treatment of tumors with 2aG4 transformed the tumor microenvironment from immunosuppressive to immunostimulatory by showing that tumors from 2aG4-treated mice had a decrease in myeloid-derived suppressor cells (MDSCs), an increase in macrophages and mature DCs, a shift in cytokine balance toward immune stimulation, along with the repolarization of macrophages from M2 to M1 phenotype. This macrophage repolarization could be replicated in vitro by culturing macrophages from PC3 tumors (mostly of the M2 phenotype) with 2aG4. The M1 polarization of macrophages in vitro required the Fc of 2aG4 since 2aG4 F(ab¢)2 failed to repolarize M2 to M1 macrophages. Although these studies were performed in immunocompromised mice, the authors found a significant increase in the number of mature DCs in tumors from 2aG4-treated mice compared to mice treated with the control IgG. The PS-targeting antibodies discussed earlier have been studied extensively in a variety of rodent tumor models, including breast, hepatocellular, and pancreatic cancers, showing enhanced antitumor responses when combined with standard-of-care chemotherapies utilized for human cancer (Table 1).65–67
 | Table 1 Summary of preclinical tumor models evaluating the combination of PS-targeting antibodies with chemotherapy or radiation Notes: Preclinical models were conducted in immunocompetent or immune-deficient animals with antibodies 3G4, 2aG4, or mch1N11. TGI was determined based on percent inhibition of treatment group compared to control for either the last day of measurement or when animals reached tumor volume limit. Abbreviations: biw, two times weekly; GEMM, genetically engineered mouse model; PS, phosphatidylserine; TGI, tumor growth inhibition; tiw, three times weekly. |
Combining PS-targeting antibodies with radiation
Irradiation in combination with PS-targeting antibodies also enhances tumor targeting of the antibodies and correlates with increased antitumor responses in mice bearing human lung tumors,68 an F98 glioma rat brain tumor model,69 and a B16 mouse melanoma model (Table 2).70 Combining PS targeting with radiation was first explored in a study by He et al68 where it was shown that radiation induced PS externalization on human vascular cells (HUVECs) in vitro. Similarly, they observed a 6.5-fold increase in PS-positive blood vessels in subcutaneous (SC) tumors after irradiation in severe combined immunodeficiency (SCID) mice. A therapy experiment was then conducted in nude mice bearing SC A549 human lung tumors, a relatively radio-resistant cell line. Compared to untreated mice or mice that were treated with 2aG4 alone or radiation + C44 control antibody, the mice treated with 2aG4 and radiation had significantly slower tumor growth. A similar antitumor effect was observed using the H460 human lung carcinoma cell line. Tissue sections from the treated A549 tumors had greatly reduced blood vessel density in the group that received radiation + 2aG4 compared to single agent and control groups. Additionally, tumors from combination treatment were infiltrated by macrophages to a greater extent than those from the three other groups and macrophages were seen in tumor vasculature 48 hours after treatment, suggesting that they are responsible for the decrease in vasculature density. An in vitro 51Cr release assay further supported this possible mechanism by demonstrating that 2aG4 mediates ADCC of irradiated HUVEC by RAW264.7 macrophages. This cytotoxicity was shown to depend on the Fc part of the antibody. Because no additional toxicity was detected, the authors concluded that bavituximab + radiation should be considered for use in patients.
 | Table 2 Summary of preclinical tumor models evaluating the combination of PS-targeting antibodies with ICI, OV, and ACT Note: TGIs were determined on the day that controls reached the maximum allowable tumor volumes. Abbreviations: ACT, adoptive cell therapy; a-CTLA-4, anti-cytotoxic lymphocyte-associated protein 4; ICI, immune checkpoint inhibitors; LAG-3, lymphocyte activation gene-3; ND, not determined; OV, oncolytic virus; a-PD-1, anti-programmed death-1; PDAC, pancreatic ductal adenocarcinoma; PS, phosphatidylserine; TGI, tumor growth inhibition. |
In a subsequent study, He et al69 investigated the combination of 2aG4 and radiation in an immunocompetent rat model of glioblastoma. Immunocompetent Fischer rats were injected with F98 rat glioma cells into the right caudate nuclei, and these cells developed into a solid tumor invading into the surrounding normal brain. A dose of 10 Gy of radiation to the whole brain of tumor-bearing rats caused the externalization of PS on tumor blood vessels and F98 tumor cells within 24 hours. Normal brain blood vessels were not affected. PS exposure also occurred in vitro following 10 Gy of radiation to endothelial cells. A survival experiment was performed to compare single agent and combination radiation and 2aG4 therapies. Rats receiving combination therapies had a longer median survival than the other groups, and 13% had tumor regressions up to 230 days after tumor implantation, whereas rats from all other groups survived a maximum of 8 weeks following the implantation. Strikingly, rats with regressed tumors rejected a re-challenge of F98 tumor cells into the contralateral hemisphere, suggesting that the rats were immune to F98 tumor cells. When looking at macrophages, the authors saw a large increase in these cells in tumors in the combination therapy group and this was associated with a destruction of tumor vasculature. Additionally, they showed that irradiation and 2aG4 induced hallmarks of adaptive immunity in vitro. The 2aG4 improved antigen presentation by rat DCs and triggered the generation of T cells that secreted IFN-γ in response to F98 cells and showed antigen-specific cell cytotoxicity.
Combining PS-targeting antibodies with immune activators and checkpoint inhibitors
Other novel approaches to induce PS exposure in tumors have demonstrated a combination antitumor effect with PS-targeting antibodies (Table 2). Reactivation of mutant p53 (mtp53) to a wild-type form using the small molecule “p53 reactivation and induction of massive apoptosis” (Prima-1) in nude mice bearing mtp53 human breast cell tumors restored the p53-directed apoptosis pathway and enhanced the exposure of PS in tumors.71 The mutation of the p53 tumor suppressor gene is the most common genetic alteration in human cancer, and the majority of mtp53 alleles in breast cancer cells are defective in DNA binding, cell cycle checkpoints, and the DNA damage-induced apoptosis.72,73 The combination of 2aG4 and Prima-1 resulted in greater tumor growth inhibition and tumor regressions than single agents alone.71 The oncolytic adenovirus Delta-24-RGD conditionally replicates in tumor cells with an abnormal p16/RB/E2F pathway, leading to cell lysis. PS exposure is enhanced in infected cells and increases the antitumor activity when combined with mch1N11.74
In summary, various therapies that induce tumor cell damage and subsequent PS exposure have the potential to enhance their therapeutic activity when combined with PS-targeting antibodies. Enhanced immune responses by PS-targeting antibodies are the result of direct killing by ADCC and immune reprogramming of suppressive cell types, such as MDSCs, M2-macrophages, and regulatory T cells (Tregs), with concomitant maturation of DCs and expansion of activated effector T cells. Reprogramming is thought to be mediated by the blockade of PS receptor signaling on immunosuppressive cell types and the engagement of antibody with activating Fc receptors (FcRs) on macrophages and DCs (Figure 1). The contribution of FcR activation is demonstrated by the work of DiLillo and Ravetch,75 where they have elegantly demonstrated that presentation of tumor antigen:antibody complexes engage with activating FcγRs on DCs, promoting DC maturation, the presentation of tumor antigens to T cells, and the development of long-term immunity. Further studies have demonstrated that FcR uptake of antibody:tumor immune complexes by DCs is important for T-cell cross priming and tumor rejection.76,77
In recent years, considerable attention has been given to understanding cancer immunobiology and immunotherapy, driven by the clinical success of inhibitors to immune checkpoint PD-1 and its ligands PD-L1 and PD-L2.78,79 A common signature for responsiveness has emerged, including the presence of CD8+ tumor-infiltrating lymphocytes (TILs), PD-L1 expression on tumors and immune cells, IFN-γ production and elevated expression of IFN-γ-induced genes, and high mutational load in tumor cells.80,81 Despite improvements in patient antitumor responses and survival, only a subset of patients benefits from PD-1/PD-L1 blockers.82 Several mechanisms of resistance to immune checkpoint inhibition have been identified, some of which have been integrated into decisions for next generation immunotherapies.2 IFN-γ is also capable of chronic stimulation of signaling pathways in activated T cells, leading to exhaustion83 and the production of immunosuppressive indoleamine 2,3-dioxygenase 1 (IDO).84,85 Activated TILs can coexpress multiple immune checkpoints, including CTLA-4, lymphocyte activation gene-3 (LAG-3), and Tim-3,86–88 resulting in higher immunosuppression than single-positive cells. Chemotherapies and irradiation, both inducers of PS externalization, are well documented to upregulate PD-L1.89–91 Aside from the tolerogenic signals that develop in the tumor microenvironment, genetic mutations that select for growth or “cancer immuno-editing” are recognized as an immune escape mechanism.92,93 Based on these and other observations, improvements in immunotherapy of cancer will likely include combinations of these targeted therapies.2 Recently, studies in immune-competent mice bearing EMT-6 or E0771 breast or B16 melanoma tumors revealed that the combination of PS-targeting mch1N11 and anti-PD-1 (α-PD-1) or anti-CTLA-4 (α-CTLA-4) showed greater antitumor effects than single-agent therapies (Table 2).94,95 Additionally, mch1N11-based combination therapies enhanced the levels of CD4+ and CD8+ TILs, elevated the fraction of cells expressing the proinflammatory cytokines including IL-2, IFN-γ, and TNF-α, and increased the ratio of CD8+ T cells to MDSCs and Tregs in tumors and spleens. These results are similar to the studies that demonstrated that the combined blockade of PD-1 and LAG-3 was more effective than single-agent therapy.96–98 LAG-3 is expressed on T cells where it negatively regulates effector T-cell activity. Based on the induction of LAG-3 on T cells by mch1N11 and α-PD-1, the inclusion of anti-LAG-3 to mch1N11 and α-PD-1-targeted therapies increased the tumor growth inhibition to 99% with tumor regressions occurring in 80% of mice.99 Animals with regressed tumors were resistant to a re-challenge of E0771 tumors but not syngeneic B16 tumors. Gene signatures of tumors obtained from animals treated with the mch1N11, α-PD-1, and anti-LAG-3 triple combination therapy revealed marked increases in antigen presentation pathways and a decrease in tumor growth-promoting genes. A separate strategy to include PS-targeting antibodies in combination therapies was demonstrated by the adoptive transfer of tumor-specific T cells in a mouse model of melanoma.100 Adoptive cell therapy (ACT) using ex vivo manipulated, tumor-specific T cells is also subjected to immunosuppressive conditions in the tumor microenvironment. The combination of mch1N11 with melanoma-specific (Trp1) CD4+ T cells resulted in the regression of advanced tumors in mice, with fewer side effects than mice treated with the same tumor-specific T cells combined with anti-OX40 antibodies. These results suggest that PS-targeting antibodies can reduce immunosuppression observed in ACT and minimize adverse events. ACTs such as tumor-derived T cells101 and chimeric antigen receptor T-cell therapy102 may require blockade of immunosuppressive signals, such as those induced by PS. Taken together, these data show that antibody-mediated PS blockade enhances the antitumor efficacy of multiple targets of immune checkpoint therapy.
Imaging solid tumors with PS-targeting antibodies
The monitoring of PS exposure in tumors is a potential predictor of successful therapy. The detection of exposed PS as an approach to detect tumors is compelling since many forms of treatment, including chemotherapy and radiotherapy, enhance PS exposure on cell membranes of the tumor endothelium and tumor cells.33,65,68 Histological detection of exposed PS in a biopsy is confounded by specimen collection bias and artefactual exposure of PS in the intracellular compartment. However, the imaging of PS in vivo can potentially be used as a general cancer imaging agent for detection, staging, and treatment monitoring,103 in particular with therapeutic approaches that induce cancer cell apoptosis.104,105 The early detection of PS exposure in tumors would provide guidance to continue treatment, and conversely, the lack of enhanced PS exposure could provide a basis to change a therapy. Annexin V has been evaluated as a probe to detect PS but tumor uptake is limited by a short circulating half-life (7 minutes), whereas antibody-based probes with longer half-lives offer the potential for increased tumor uptake.
Full-length PS-targeting antibodies bavituximab and PGN635 (1N11) have been evaluated in preclinical studies as imaging agents. In animal studies, 74As-labeled full-length bavituximab was successfully used to detect tumors in the R3227-AT1 rat Dunning prostate model.106 Full-length PGN635 labeled with 89Zr was evaluated by positron emission tomography (PET) imaging in mouse tumor xenograft models to measure responses to proapoptotic therapies.107 A high accumulation of 89Zr-PGN635 was observed in treated tumors undergoing apoptosis, reaching 30% injected dose/per gram tissue and tumor-to-blood ratios of up to 13. Further technologies using PS-targeting antibodies have been developed by attaching 1N11 to liposomes containing magnetic nanoparticles for magnetic resonance imaging for diagnostic evaluation or encapsulated with arsenic trioxide with therapeutic activity.108,109 The combination of a PS imaging agent together with a therapeutic drug may provide a unique opportunity to simultaneously detect, monitor, and treat tumors. PGN650, a F(ab′)2 antibody fragment derived from PGN635, has been used to image human tumor xenografts in mice with near-infrared (NIR) optical imaging and PET. NIR dye-labeled PGN650 injected in mice with SC human U87 glioma tumors had a tumor:normal tissue probe ratio (TNR) of 2.5 at 24 hours post injection.110 The treatment of SC tumors with 12 Gy irradiation enhanced tumor uptake of NIR dye-labeled PGN650 with a TNR of 4.0 when measured 24 hours following injection. The treatment of mice bearing orthotopic BT-474 human breast tumors with docetaxel enhanced the uptake of NIR dye-labeled PGN650 compared to untreated tumors.111 Compared to unlabeled PGN650, 124I-labeled PGN650 was shown to have similar binding activities in vitro and to target human PC3 SC and orthotopic tumors in mice as demonstrated by microPET (Figure 3).112 Histological evaluation of tumor-bearing mice treated with NIR-labeled PGN650 showed that the imaging agent targeted tumor vasculature and tumor cells.110,111 Based on the results from the preclinical imaging studies, a Phase 0 clinical study was conducted using 124I-labeled PGN650 as a PET imaging agent to monitor safety, pharmacokinetics, radiation dosimetry, and tumor targeting.113 Patients with solid tumors received ~140 MBq (3.8 mCi) 124I-PGN650 intravenously and underwent PET/CT ~1, 3, and either 24 or 48 hours later to establish tracer kinetics. The safety of 124I-PGN650 was established for human PET imaging; however, the tumor targeting in these patients was less than previously observed in animal studies. An example of whole-body PET imaging of a patient’s tumor is shown in a recent publication,113 which demonstrates the retention of the probe in the tumor and correlates with the targeting of 18F-deoxyglucose, a marker for high cellular metabolism.
 | Figure 3 PET imaging of PS exposed in tumors. Notes: Mice implanted with tumors (PC3-luc) are injected with 124I-labeled 1N11 F(ab´)2 (PGN650) or control F(ab´)2 probes and imaged by bioluminescence in left panels and PET in middle and right panels. The yellow circle marks the tumor uptake of 124I-PGN650. Reproduced from Stafford JH, Hao G, Best AM, Sun X, Thorpe PE. Highly specific PET imaging of prostate tumors in mice with an iodine-124-labeled antibody fragment that targets phosphatidylserine. PLoS One. 2013;8(12):e84864.112 Abbreviations: Ctrl, control; F(ab´)2, bivalent antigen-binding fragment of an antibody; I-124, iodine 124; luc, luciferase; PET, positron emission tomography; PS, phosphatidylserine. |
Clinical and translational studies of PS-targeting antibodies in cancer patients
Based on data from preclinical efficacy and safety studies, efforts were made to develop a PS-targeting antibody for human trials. Bavituximab is a chimeric monoclonal antibody constructed from the variable region of murine antibody 3G4 and fused to human IgG1 kappa constant regions. Bavituximab and 3G4 target PS in a high-affinity complex with β2GP1 in an identical manner. Bavituximab has been administered to over 700 patients in clinical trials evaluating the antibody as monotherapy and in various combination regimens in patients with multiple tumor types, chronic hepatitis C virus, and HIV infection. To date, studies have shown promising signs of activity and an acceptable safety profile (Table 3). Bavituximab has been evaluated in several investigator-sponsored trials in patients with solid tumors including late-stage clinical trials in patients with locally advanced or metastatic non-small cell lung cancer.
 | Table 3 Summary of clinical trials evaluating bavituximab in cancer Note: aPlacebo and 1 mg/kg bavituximab arms were combined for analysis and compared to 3 mg/kg bavituximab arm. |
As previously discussed, resistance to checkpoint therapy occurs in up to 70% of patients and is, in part, attributed to low levels of PD-1 and PD-L1 on TILs and tumor cells in the tumor microenvironment.82 In a translational study utilizing 3D ex vivo-cultured tumor microspheres from non-small-cell lung cancer patients, microspheres expressing low levels of PD-L1 incubated with bavituximab increased the production of immune-activating cytokines (IFN-γ, GM-CSF, and TNF-α) and decreased levels of immunosuppressive cytokine IL-10. In addition, tumor microspheres with low PD-L1 expression treated with bavituximab expressed increased IFN-γ levels in culture accompanied with an increase in M1 macrophage gene expression with a corresponding decrease in M2 macrophage gene expression.114 These translational data suggest that PS blockade may increase the proportion of patients who benefit from α-PD-1/PD-L1 checkpoint therapy.
Conclusion
PS is well-recognized as a cell surface marker of apoptotic cells which provides signals to specific receptors for non-inflammatory efferocytosis by phagocytes. The same signals are usurped in the tumor microenvironment by the exposure of PS on tumor blood vessel endothelium and tumor cells, contributing to immunosuppression and tolerance of tumor growth. Specific receptors that bind PS, including TIMs and TAMs, on immune cells and tumors, trigger these immunosuppressive pathways. The uptake of PS-targeting antibodies by tumors is readily demonstrated in preclinical models and initial clinical studies. Multiple preclinical studies serve as proof of concept that the antibody-mediated blockade of PS in tumors can reactivate innate and adaptive immune responses in the tumor microenvironment. A combination of PS-targeting antibodies with approved immune activating therapies such as chemotherapy, radiation, and immune checkpoint inhibitors (including antibodies targeting CTLA-4, PD-1, and PD-L1) and with novel therapies such as oncolytic viruses has the potential to treat a variety of different tumor types. These data support clinical trial evaluation of the PS-targeting antibody, bavituximab, in multiple oncology indications.
Acknowledgments
We acknowledge the Departments of Clinical and Regulatory Affairs and Process Sciences at Peregrine Pharmaceuticals, Inc., for current insight on the clinical development of bavituximab and for providing antibodies for preclinical studies. We also thank Steve King for helpful technical discussions and support of preclinical studies and Dave Primm for editorial assistance.
Disclosure
XH, JG, JC, and BDF are employees of Peregrine Pharmaceuticals, Inc., Tustin, CA, USA, and receive salary and stock options from the company. OB, RAB, and AJS, University of Texas Southwestern Medical Center, are collaborators of Peregrine and receive research funding from the company. The authors report no other conflicts of interest in this work.
References
Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. | ||
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. | ||
Maio M, Grob JJ, Aamdal S, et al. Five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J Clin Oncol. 2015;33(10):1191–1196. | ||
Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33(17):1889–1894. | ||
Robert C, Schachter J, Long GV, et al; KEYNOTE-006 Investigators. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521–2532. | ||
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. | ||
Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. | ||
Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. | ||
Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837–1846. | ||
Massard C, Gordon MS, Sharma S, et al. Safety and efficacy of durvalumab (MEDI4736), an anti–programmed cell death ligand-1 immune checkpoint inhibitor, in patients with advanced urothelial bladder cancer. J Clin Oncol. 2016;34(26):3119–3125. | ||
Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374–1385. | ||
Wilmott JS, Rizos H, Scolyer RA, Long GV. The “tricky business” of identifying mechanisms of resistance to anti-PD-1. Clin Cancer Res. 2017;23(12):2921–2923. | ||
Pitt Jonathan M, Vétizou M, Daillère R, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. 2016;44(6):1255–1269. | ||
De Henau O, Rausch M, Winkler D, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539(7629):443–447. | ||
Balasubramanian K, Schroit AJ. Aminophospholipid asymmetry: a matter of life and death. Annu Rev Physiol. 2003;65:701–734. | ||
Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–427. | ||
Vance JE, Steenbergen R. Metabolism and functions of phosphatidylserine. Prog Lipid Res. 2005;44(4):207–234. | ||
Pomorski T, Holthuis JC, Herrmann A, van Meer G. Tracking down lipid flippases and their biological functions. J Cell Sci. 2004;117(pt 6):805–813. | ||
Kay JG, Koivusalo M, Ma X, Wohland T, Grinstein S. Phosphatidylserine dynamics in cellular membranes. Mol Biol Cell. 2012;23(11):2198–2212. | ||
Biermann M, Maueroder C, Brauner JM, et al. Surface code – biophysical signals for apoptotic cell clearance. Phys Biol. 2013;10(6):065007. | ||
Callahan MK, Williamson P, Schlegel RA. Surface expression of phosphatidylserine on macrophages is required for phagocytosis of apoptotic thymocytes. Cell Death Differ. 2000;7(7):645–653. | ||
Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148(7):2207–2216. | ||
Fischer K, Voelkl S, Berger J, Andreesen R, Pomorski T, Mackensen A. Antigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cells. Blood. 2006;108(13):4094–4101. | ||
Dillon SR, Mancini M, Rosen A, Schlissel MS. Annexin V binds to viable B cells and colocalizes with a marker of lipid rafts upon B cell receptor activation. J Immunol. 2000;164(3):1322–1332. | ||
Appelt U, Sheriff A, Gaipl US, Kalden JR, Voll RE, Herrmann M. Viable, apoptotic and necrotic monocytes expose phosphatidylserine: cooperative binding of the ligand Annexin V to dying but not viable cells and implications for PS-dependent clearance. Cell Death Differ. 2005;12(2):194–196. | ||
Mates JM, Sanchez-Jimenez FM. Role of reactive oxygen species in apoptosis: implications for cancer therapy. Int J Biochem Cell Biol. 2000;32(2):157–170. | ||
Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5(5):415–418. | ||
Mandal D, Mazumder A, Das P, Kundu M, Basu J. Fas-, caspase 8-, and caspase 3-dependent signaling regulates the activity of the aminophospholipid translocase and phosphatidylserine externalization in human erythrocytes. J Biol Chem. 2005;280(47):39460–39467. | ||
Zwaal RF, Comfurius P, Bevers EM. Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci. 2005;62(9):971–988. | ||
Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101(4):890–898. | ||
Birge RB, Boeltz S, Kumar S, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23(6):962–978. | ||
Yin Y, Huang X, Lynn KD, Thorpe PE. Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol Res. 2013;1(4):256–268. | ||
Ran S, Downes A, Thorpe PE. Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 2002;62(21):6132–6140. | ||
Lea J, Sharma R, Yang F, Zhu H, Ward ES, Schroit AJ. Detection of phosphatidylserine-positive exosomes as a diagnostic marker for ovarian malignancies: a proof of concept study. Oncotarget. 2017;8(9):14395–14407. | ||
Kelleher RJ Jr, Balu-Iyer S, Loyall J, et al. Extracellular vesicles present in human ovarian tumor microenvironments induce a phosphatidylserine-dependent arrest in the T-cell signaling cascade. Cancer Immunol Res. 2015;3(11):1269–1278. | ||
Lima LG, Chammas R, Monteiro RQ, Moreira ME, Barcinski MA. Tumor-derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine-dependent manner. Cancer Lett. 2009;283(2):168–175. | ||
Chen YH, Du W, Hagemeijer MC, et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell. 2015;160(4):619–630. | ||
Dowall SD, Graham VA, Corbin-Lickfett K, et al. Effective binding of a phosphatidylserine-targeting antibody to Ebola virus infected cells and purified virions. J Immunol Res. 2015;2015:347903. | ||
Moody MA, Liao HX, Alam SM, et al. Anti-phospholipid human monoclonal antibodies inhibit CCR5-tropic HIV-1 and induce beta-chemokines. J Exp Med. 2010;207(4):763–776. | ||
Soares MM, King SW, Thorpe PE. Targeting inside-out phosphatidylserine as a therapeutic strategy for viral diseases. Nat Med. 2008;14(12):1357–1362. | ||
Goth SR, Stephens RS. Rapid, transient phosphatidylserine externalization induced in host cells by infection with Chlamydia spp. Infect Immun. 2001;69(2):1109–1119. | ||
Amara A, Mercer J. Viral apoptotic mimicry. Nat Rev Microbiol. 2015;13(8):461–469. | ||
Das S, Owen KA, Ly KT, et al. Brain angiogenesis inhibitor 1 (BAI1) is a pattern recognition receptor that mediates macrophage binding and engulfment of Gram-negative bacteria. Proc Natl Acad Sci U S A. 2011;108(5):2136–2141. | ||
Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 2010;235(1):172–189. | ||
Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer. 2014;14(12):769–785. | ||
Lew ED, Oh J, Burrola PG, et al. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. Elife. 2014;3:e03385. | ||
Umetsu SE, Lee WL, McIntire JJ, et al. TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat Immunol. 2005;6(5):447–454. | ||
Zhu C, Anderson AC, Schubart A, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6(12):1245–1252. | ||
Nakayama M, Akiba H, Takeda K, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009;113(16):3821–3830. | ||
Kobayashi N, Karisola P, Pena-Cruz V, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27(6):927–940. | ||
Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450(7168):435–439. | ||
Zagorska A, Traves PG, Lew ED, Dransfield I, Lemke G. Diversification of TAM receptor tyrosine kinase function. Nat Immunol. 2014;15(10):920–928. | ||
Cohen PL, Caricchio R, Abraham V, et al. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196(1):135–140. | ||
Vouri M, Hafizi S. TAM receptor tyrosine kinases in cancer drug resistance. Cancer Res. 2017;77(11):2775–2778. | ||
Kirane A, Ludwig KF, Sorrelle N, et al. Warfarin blocks Gas6-mediated Axl activation required for pancreatic cancer epithelial plasticity and metastasis. Cancer Res. 2015;75(18):3699–3705. | ||
Kasikara C, Kumar S, Kimani S, et al. Phosphatidylserine sensing by TAM receptors regulates AKT-dependent chemoresistance and PD-L1 expression. Mol Cancer Res. 2017;15(6):753–764. | ||
Ran S, Gao B, Duffy S, Watkins L, Rote N, Thorpe PE. Infarction of solid Hodgkin’s tumors in mice by antibody-directed targeting of tissue factor to tumor vasculature. Cancer Res. 1998;58(20):4646–4653. | ||
Bevers EM, Rosing J, Zwaal RF. Development of procoagulant binding sites on the platelet surface. Adv Exp Med Biol. 1985;192:359–371. | ||
Dachary-Prigent J, Toti F, Satta N, Pasquet JM, Uzan A, Freyssinet JM. Physiopathological significance of catalytic phospholipids in the generation of thrombin. Semin Thromb Hemost. 1996;22(2):157–164. | ||
Ortel TL, Devore-Carter D, Quinn-Allen M, Kane WH. Deletion analysis of recombinant human factor V. Evidence for a phosphatidylserine binding site in the second C-type domain. J Biol Chem. 1992;267(6):4189–4198. | ||
Ran S, He J, Huang X, Soares M, Scothorn D, Thorpe PE. Antitumor effects of a monoclonal antibody that binds anionic phospholipids on the surface of tumor blood vessels in mice. Clin Cancer Res. 2005;11(4):1551–1562. | ||
Luster TA, He J, Huang X, et al. Plasma protein beta-2-glycoprotein 1 mediates interaction between the anti-tumor monoclonal antibody 3G4 and anionic phospholipids on endothelial cells. J Biol Chem. 2006;281(40):29863–29871. | ||
Hunt JE, Simpson RJ, Krilis SA. Identification of a region of beta 2-glycoprotein I critical for lipid binding and anti-cardiolipin antibody cofactor activity. Proc Natl Acad Sci U S A. 1993;90(6):2141–2145. | ||
Steinkasserer A, Barlow PN, Willis AC, et al. Activity, disulphide mapping and structural modelling of the fifth domain of human beta 2-glycoprotein I. FEBS Lett. 1992;313(2):193–197. | ||
Huang X, Bennett M, Thorpe PE. A monoclonal antibody that binds anionic phospholipids on tumor blood vessels enhances the antitumor effect of docetaxel on human breast tumors in mice. Cancer Res. 2005;65(10):4408–4416. | ||
Beck AW, Luster TA, Miller AF, et al. Combination of a monoclonal anti-phosphatidylserine antibody with gemcitabine strongly inhibits the growth and metastasis of orthotopic pancreatic tumors in mice. Int J Cancer. 2006;118(10):2639–2643. | ||
Cheng X, Li L, Thorpe PE, Yopp AC, Brekken RA, Huang X. Antibody-mediated blockade of phosphatidylserine enhances the antitumor effect of sorafenib in hepatocellular carcinomas xenografts. Ann Surg Oncol. 2016;23(Suppl 5):583–591. | ||
He J, Luster TA, Thorpe PE. Radiation-enhanced vascular targeting of human lung cancers in mice with a monoclonal antibody that binds anionic phospholipids. Clin Cancer Res. 2007;13(17):5211–5218. | ||
He J, Yin Y, Luster TA, Watkins L, Thorpe PE. Antiphosphatidylserine antibody combined with irradiation damages tumor blood vessels and induces tumor immunity in a rat model of glioblastoma. Clin Cancer Res. 2009;15(22):6871–6880. | ||
Budhu S, Henau OD, Zappasodi R, et al. Abstract 574: phosphatidylserine targeting antibody in combination with tumor radiation and immune checkpoint blockade promotes anti-tumor activity in mouse B16 melanoma. Cancer Res. 2017;77(13 Suppl):574–574. | ||
Liang Y, Besch-Williford C, Benakanakere I, Thorpe PE, Hyder SM. Targeting mutant p53 protein and the tumor vasculature: an effective combination therapy for advanced breast tumors. Breast Cancer Res Treat. 2011;125(2):407–420. | ||
Kumar S, Walia V, Ray M, Elble RC. p53 in breast cancer: mutation and countermeasures. Front Biosci. 2007;12:4168–4178. | ||
Rahko E, Blanco G, Soini Y, Bloigu R, Jukkola A. A mutant TP53 gene status is associated with a poor prognosis and anthracycline-resistance in breast cancer patients. Eur J Cancer. 2003;39(4):447–453. | ||
Dai B, Roife D, Kang Y, et al. Preclinical evaluation of sequential combination of oncolytic adenovirus delta-24-RGD and phosphatidylserine-targeting antibody in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2017;16(4):662–670. | ||
DiLillo DJ, Ravetch JV. Differential Fc-receptor engagement drives an anti-tumor vaccinal effect. Cell. 2015;161(5):1035–1045. | ||
Moynihan KD, Opel CF, Szeto GL, et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med. 2016;22(12):1402–1410. | ||
Akiyama K, Ebihara S, Yada A, et al. Targeting apoptotic tumor cells to Fc gamma R provides efficient and versatile vaccination against tumors by dendritic cells. J Immunol. 2003;170(4):1641–1648. | ||
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | ||
Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. | ||
Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol. 2016;27(8):1492–1504. | ||
Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. | ||
O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. 2017;52:71–81. | ||
Ayers M, Lunceford J, Nebozhyn M, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–2940. | ||
Munn DH, Zhou M, Attwood JT, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281(5380):1191–1193. | ||
Moffett JR, Namboodiri MA. Tryptophan and the immune response. Immunol Cell Biol. 2003;81(4):247–265. | ||
Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 2013;73(23):6900–6912. | ||
Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013;73(12):3591–3603. | ||
Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2:3. | ||
Deng L, Liang H, Burnette B, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124(2):687–695. | ||
Derer A, Spiljar M, Baumler M, et al. Chemoradiation increases PD-L1 expression in certain melanoma and glioblastoma cells. Front Immunol. 2016;7:610. | ||
Derer A, Frey B, Fietkau R, Gaipl US. Immune-modulating properties of ionizing radiation: rationale for the treatment of cancer by combination radiotherapy and immune checkpoint inhibitors. Cancer Immunol Immunother. 2016;65(7):779–786. | ||
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. | ||
Matsushita H, Vesely MD, Koboldt DC, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482(7385):400–404. | ||
Freimark BD, Gong J, Ye D, et al. Antibody-mediated phosphatidylserine blockade enhances the antitumor responses to CTLA-4 and PD-1 antibodies in melanoma. Cancer Immunol Res. 2016;4(6):531–540. | ||
Gray MJ, Gong J, Hatch MM, et al. Phosphatidylserine-targeting antibodies augment the anti-tumorigenic activity of anti-PD-1 therapy by enhancing immune activation and downregulating pro-oncogenic factors induced by T-cell checkpoint inhibition in murine triple-negative breast cancers. Breast Cancer Res. 2016;18(1):50. | ||
Woo SR, Turnis ME, Goldberg MV, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–927. | ||
Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget. 2015;6(29):27359–27377. | ||
Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3 – potential mechanisms of action. Nat Rev Immunol. 2015;15(1):45–56. | ||
Gray MJ, Gong J, Hutchins J, Freimark B. Abstract B019: LAG3 is an immunotherapeutic target in murine triple negative breast cancers whose activity is significantly enhanced in combination with phosphatidylserine targeting antibodies. Cancer Immunol Res. 2016;4(11 Suppl):B019–B019. | ||
Hirschhorn-Cymerman D, Schad SS, Budhu S, et al. Abstract 1651: targeting phosphatidylserine in combination with adoptive T cell transfer eliminates advanced tumors without off-target toxicities in a melanoma preclinical model. Cancer Res. 2017;77(13 Suppl):1651. | ||
Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12(4):269–281. | ||
Maus MV, June CH. Making better chimeric antigen receptors for adoptive T-cell therapy. Clin Cancer Res. 2016;22(8):1875–1884. | ||
Belhocine T, Steinmetz N, Hustinx R, et al. Increased uptake of the apoptosis-imaging agent (99m)Tc recombinant human Annexin V in human tumors after one course of chemotherapy as a predictor of tumor response and patient prognosis. Clin Cancer Res. 2002;8(9):2766–2774. | ||
Blankenberg FG, Naumovski L, Tait JF, Post AM, Strauss HW. Imaging cyclophosphamide-induced intramedullary apoptosis in rats using 99mTc-radiolabeled annexin V. J Nucl Med. 2001;42(2):309–316. | ||
Belhocine T, Steinmetz N, Green A, Rigo P. In vivo imaging of chemotherapy-induced apoptosis in human cancers. Ann N Y Acad Sci. 2003;1010:525–529. | ||
Jennewein M, Lewis MA, Zhao D, et al. Vascular imaging of solid tumors in rats with a radioactive arsenic-labeled antibody that binds exposed phosphatidylserine. Clin Cancer Res. 2008;14(5):1377–1385. | ||
Ogasawara A, Tinianow JN, Vanderbilt AN, et al. ImmunoPET imaging of phosphatidylserine in pro-apoptotic therapy treated tumor models. Nucl Med Biol. 2013;40(1):15–22. | ||
Zhang L, Zhou H, Belzile O, Thorpe P, Zhao D. Phosphatidylserine-targeted bimodal liposomal nanoparticles for in vivo imaging of breast cancer in mice. J Control Release. 2014;183:114–123. | ||
Wang L, Habib AA, Mintz A, Li KC, Zhao D. Phosphatidylserine-targeted nanotheranostics for brain tumor imaging and therapeutic potential. Mol imaging. 2017;16:1536012117708722. | ||
Zhao D, Stafford JH, Zhou H, Thorpe PE. Near-infrared optical imaging of exposed phosphatidylserine in a mouse glioma model. Transl Oncol. 2011;4(6):355–364. | ||
Gong J, Archer R, Brown M, et al. Measuring response to therapy by near-infrared imaging of tumors using a phosphatidylserine-targeting antibody fragment. Mol Imaging. 2013;12(4):244–256. | ||
Stafford JH, Hao G, Best AM, Sun X, Thorpe PE. Highly specific PET imaging of prostate tumors in mice with an iodine-124-labeled antibody fragment that targets phosphatidylserine. PLoS One. 2013;8(12):e84864. | ||
Laforest R, Dehdashti F, Liu Y, et al. First-in-Man Evaluation of 124I-PGN650: A PET Tracer for Detecting Phosphatidylserine as a Biomarker of the Solid Tumor Microenvironment. Molecular Imaging. 2017;16:1–9. | ||
Altiok S, Mediavilla-Varela M, Kreahling J, et al. Bavituximab activates CD8+ TILs in a 3D ex vivo system of lung cancer patient derived tumors with negative PD-L1 expression. Paper presented at: World Conference on Lung Cancer; 2015; Denver, Colorado, USA. | ||
Yin Y, Huang X, Lynn KD, Thorpe PE. Cure of castration-resistant prostate cancer in TRAMP mice by reactivating tumor immunity with a phosphatidylserine-targeting antibody. Cancer Res. 2012;72(8 Suppl). Abstract nr 4395. | ||
Gerber DE, Stopeck AT, Wong L, et al. Phase I safety and pharmacokinetic study of bavituximab, a chimeric phosphatidylserine-targeting monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2011;17(21):6888–6896. | ||
Digumarti R, Bapsy P, Shan JS. A phase Ib safety and pharmacokinetic study of bavituximab plus chemotherapy in patients with refractory advanced solid tumor malignancies. J Clin Oncol. 2008;26:3038. | ||
Tabagari D, Nemsadze G, Janjalia M, Jincharadze M, Shan J. Phase II study of bavituximab plus docetaxel in locally advanced or metastatic breast cancer. J Clin Oncol. 2010;28(Suppl). | ||
Digumarti R, Bapsy PP, Suresh AV, et al. Bavituximab plus paclitaxel and carboplatin for the treatment of advanced non-small-cell lung cancer. Lung Cancer. 2014;86(2):231–236. | ||
Pandya SS, Wong L, Bullock AJ, et al. Randomized, open-label, phase II trial of gemcitabine with or without bavituximab in patients with nonresectable stage IV pancreatic adenocarcinoma. J Clin Oncol. 2013;31(Suppl):abstr4054. | ||
Shtivelband M, Spigel D, Gerber DE, et al. Randomized, blinded, placebo-controlled phase II trial of docetaxel and bavituximab as second-line therapy in locally advanced or metastatic non-squamous non-small cell lung cancer. J Clin Oncol. 2013;31:8095. | ||
Chalasani P, Marron M, Roe D, et al. A phase I clinical trial of bavituximab and paclitaxel in patients with HER2 negative metastatic breast cancer. Cancer Med. 2015;4(7):1051–1059. | ||
Grilley-Olson JE, Villaruz L, Stinchcombe TE, et al. A phase Ib study of bavituximab plus carboplatin and pemetrexed in chemotherapy naïve stage IV non-squamous non-small cell lung cancer. Paper presented at: Chicago Multidisciplinary Symposium on Thoracic Oncology; 2014; Chicago, IL. | ||
Yopp AC, Singal A, Arriaga YE, et al. A phase II study of bavituximab and sorafenib in advanced hepatocellular carcinoma (HCC). J Clin Oncol. 2015;33(Suppl):345. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.