")
Back to Journals » International Journal of Nanomedicine » Volume 13
Antibody-modified liposomes for tumor-targeting delivery of timosaponin AIII
Authors Lu L, Ding Yue, Zhang Y, Ho RJY, Zhao Y, Zhang T, Guo C
Received 3 October 2017
Accepted for publication 24 January 2018
Published 29 March 2018 Volume 2018:13 Pages 1927—1944
DOI https://doi.org/10.2147/IJN.S153107
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Lu Lu,1 Yue Ding,2 Yong Zhang,2 Rodney JY Ho,3 Yuan Zhao,4 Tong Zhang,1 Chunrong Guo2
1School of Pharmacy, 2Experiment Center for Teaching and Learning, Shanghai University of Traditional Chinese Medicine, Shanghai, China; 3Department of Pharmaceutics, University of Washington, Seattle, WA, USA; 4Center of Science and Technology, Shanghai University of Traditional Chinese Medicine, Shanghai, China
Introduction: Timosaponin AIII (TAIII), as a steroid saponin in Anemarrhena asphodeloides, has favorable potential as an antitumor candidate. However, its hydrophobicity and low bioavailability severely limit its in vivo antitumor efficacy.
Methods: To overcome this drawback, TAIII-loaded liposomes (LP) were prepared to improve TAIII solubility and extend its circulation time. Furthermore, anti-CD44 antibody-modified LP (CD44-LP) was prepared to enhance the therapeutic index of TAIII. The LP and CD44-LP were also characterized through their biological activity, target selective binding and uptake, and in vivo pharmacokinetics.
Results: Compared with free TAIII, both LP and CD44-LP possessed a desirable sustained-release profile in vitro, with ~14.2- and 10.7-fold longer TAIII half-life, respectively, and 1.7- and 1.9-fold larger area under the curve, respectively. LP and CD44-LP enhanced TAIII antitumor activity against HepG2 cells and in a xenograft mouse model without detectable toxicity. In particular, CD44-LP exhibited notably higher cytotoxicity than did LP, with a lower half-maximal inhibitory concentration (48 h). CD44-LP exhibited stronger tumor inhibition, and the tumor inhibitory effect was 1.3-fold that of LP. Furthermore, confocal laser scanning microscopy and in vivo near-infrared imaging of a xenograft mouse model revealed that compared with LP, CD44-LP could effectively enhance tumor accumulation.
Conclusion: Taken together, the results indicate that both CD44-LP and LP can considerably extend TAIII circulation time, increase tumor-targeted accumulation, and enhance antitumor activity. Thus, the anti-CD44 antibody-modified liposome is a promising candidate for treating CD44-positive cancer with considerable antitumor effects.
Keywords: timosaponin AIII, liposomes, CD44, tumor-targeting drug delivery, receptor-mediated drug targeting
Introduction
The rhizoma of Anemarrhena asphodeloides Bunge (family Liliaceae), also called rhizoma anemarrhenae and commonly known as Zhi mu, is a traditional Chinese medicinal herb that is listed in the Pharmacopoeia of the People’s Republic of China (2015). The ethnomedical uses of A. asphodeloides to treat high fever, internal heat-induced diabetes, inflammation, hemoptysis, and constipation have been recorded in China, Japan, and Korea for thousands of years.1 This herb contains various bioactive components, including flavonoids (eg, mangiferin), steroid saponins (eg, timosaponin AIII [TAIII], timosaponin BII [TBII], and sarsasapogenin [SAR]), lignin, and polysaccharides. Among steroid saponins, TAIII (Figure 1) exhibits extensive pharmacological activities, including activities against platelet aggregation,2 diabetes,3 depression,4 and inflammation;5 therapeutic effects against dementia,6 human chronic myelogenous leukemia,7 and promyelocytic leukemia;8 and amelioration of learning and memory deficits.9 TAIII also exhibits highly potent cytotoxicity against various cancer cells, such as HeLa cells,10–12 breast carcinoma cells,13,14 human colon cancer cells,15 human hepatocellular carcinoma (HCC) cells,16 PANC-1 cells,17 melanoma cells,18 and A549 human non-small-cell lung cancer cells.19 However, TAIII is nontoxic to nontransformed cells. The mechanisms underlying the antitumor effects of TAIII involve the inhibition of tumor migration and invasion,13,18,19 activation of autophagy,10,11 and induction of apoptosis.15,16 Numerous studies have revealed the potential of TAIII as an antitumor candidate. TAIII could be prepared by enzyme hydrolysis from TBII with high purity; 1 kg of TAIII can be prepared in 1 week in a laboratory,20 thus providing adequate materials for further pharmaceutical research and new drug development. However, in our previous study,21 TAIII showed hydrophobicity and low bioavailability, limiting its in vivo antitumor efficacy.
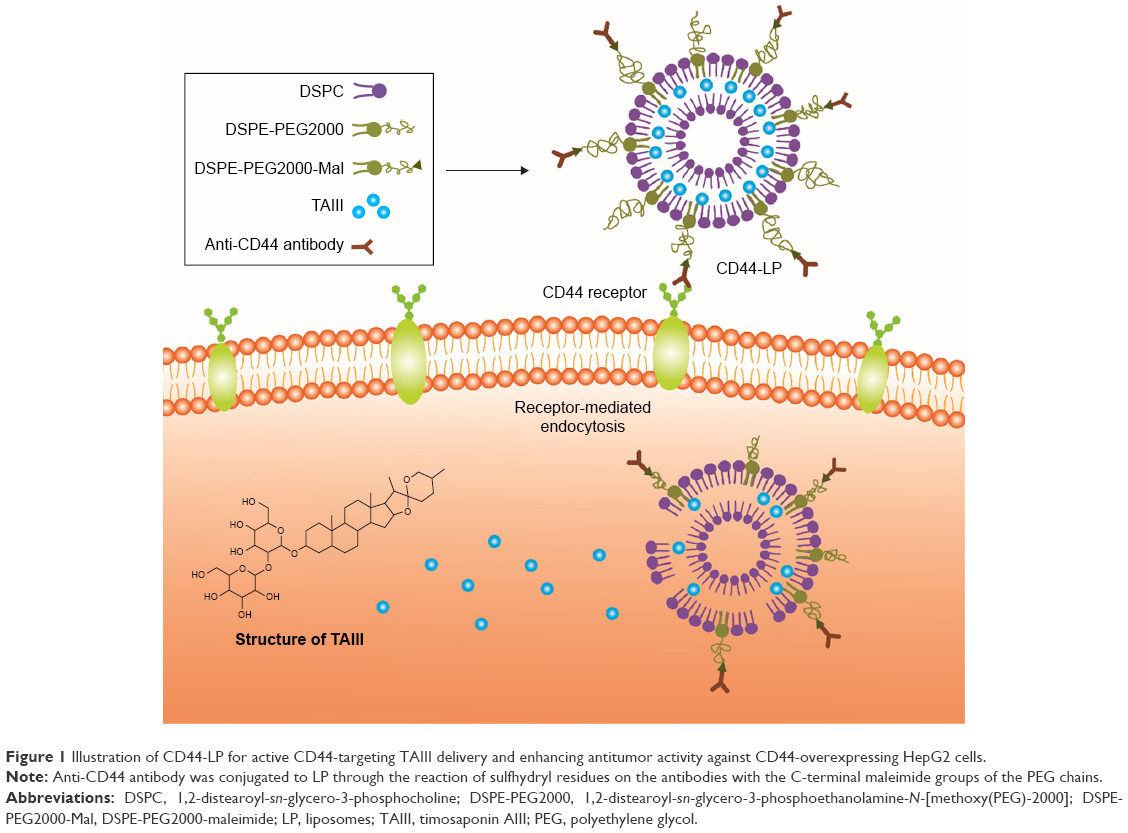 | Figure 1 Illustration of CD44-LP for active CD44-targeting TAIII delivery and enhancing antitumor activity against CD44-overexpressing HepG2 cells. |
A new TAIII delivery system, which improves TAIII solubility, reduces clearance (Cl), and increases tumor tissue penetration, is needed to enhance its antitumor efficacy. Small and stable liposomes are biocompatible nanoparticles commonly used to enhance water solubility and improve drug bioavailability and pharmacokinetics.22–24 In some cases, therapeutic effects of drugs are enhanced with few side effects.25,26 However, conventional liposomes have exhibited high plasma Cl because of rapid uptake and can be mostly captured and eliminated by the phagocytic cells of the reticuloendothelial system (RES).27 The incorporation of phospholipids with grafted polyethylene glycol (PEG) side chains into the membrane surface aids the liposomes to circulate longer in the bloodstream, thus evading the RES,28 presenting passive targeting activity by accumulating in tumors through the enhanced permeability and retention (EPR) effect.29,30 In addition, small particle-size-dependent passive accumulation of liposomes at tumor tissue could be enhanced by coating the liposomes with ligands and antibodies that can selectively target tumors.24,31–33 Various cancer cell surface markers have been identified as potent targets for specific and selective targeting of antitumor therapeutics, such as CD133,34 CD147,35 and CD44. CD44 is an extracellular protein on the cell membrane involved in tumor invasion and metastasis.36,37 Because CD44 is endogenously expressed at low levels in normal tissues,38 and its structure is modified in breast, lung, ovarian, hepatocellular, and other cancer cells,39–42 it is a potential receptor in tumor-targeting drug delivery systems. Anti-CD44 antibody, which can recognize and bind specifically to CD44 receptors, is a potent tumor-targeting ligand for improving the liposome accumulation at tumor sites and thus for enhancing cancer therapies.43,44 CD44 receptors are overexpressed in HCC and anti-CD44 antibody-conjugated drugs have shown promising results in human clinical trials,45 thus, the anti-CD44 antibody ligand-receptor system is a promising strategy for HCC therapy.
The present study was based on the potential antitumor effects of liposomes and cell-specific binding and internalization induced by antibodies. Anti-CD44 antibody-modified TAIII-loaded liposomes (CD44-LP) (Figure 1) and TAIII-loaded liposomes (LP) were prepared, and their biological activity, pharmacokinetics, target selective binding and uptake, and antitumor efficiency in vivo were characterized. The cellular uptake of CD44-LP was then investigated in comparison with that of LP containing rhodamine through confocal laser scanning microscopy (CLSM). Moreover, the in vivo tumor accumulation profile of indocyanine green (ICG)-loaded CD44-LP and LP was investigated using an IVIS imaging system. Furthermore, using the mouse-bearing HCC cell line HepG2, the therapeutic efficacies of CD44-LP were compared with those of LP and free TAIII.
Methods
Cell culture
HepG2 cell line was obtained from the Chinese Academy of Sciences Cells Bank (Shanghai, People’s Republic of China). Cells were cultured with Dulbecco’s Modified Eagle’s Medium (high glucose) containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin in 5% CO2 and 95% relative humidity at 37°C.
Animals
BALB (Bagg Albino)/c nude mice weighing 20–25 g (5–6 weeks old) and male Sprague-Dawley (SD) rats weighing 200–220 g (6–7 weeks old) were supplied by Shanghai SIPPR-Bk Lab Animal Co., Ltd. (Shanghai, People’s Republic of China). The temperature and relative humidity were maintained at 25°C and 45%–55%, respectively. All care and handling of the animals was performed in accordance with the requirements of the animal care of Shanghai University of Traditional Chinese Medicine. All experimental protocols have been reviewed and approved by the Institutional Animal Experimental Ethics Committee of Shanghai University of Traditional Chinese Medicine (approval file no SZY201611003).
Preparation of LP and CD44-LP
TAIII was prepared as described previously.20 LP was prepared through thin-film evaporation and ultrasonic technique. Briefly, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(PEG)-2000] (DSPE-PEG2000; Shanghai Advanced Vehicle Technology Pharmaceutical Co., Ltd, Shanghai, China) were dissolved in chloroform at a molar ratio of 4:1, and TAIII (2 mg) was dissolved in methanol. The solutions were mixed and evaporated using a rotary evaporator at 60°C for ~20 min to form a solid film. This process encapsulated TAIII in the lipid. The resulting thin film was hydrated in phosphate-buffered saline (PBS) at 60°C for 1 h to obtain a suspension at a 20 mM lipid concentration. The suspension was then sonicated for 30 min to form LP.
For preparing CD44-LP, liposomes composed of DSPC, DSPE-PEG2000, and DSPE-PEG2000-maleimide (DSPE-PEG2000-Mal) at a molar ratio of 4:1:0.2 were prepared. The purified anti-CD44 antibody (BD Biosciences, San Jose, CA, USA) and liposomes were mixed at a molar ratio of 1:10 (antibody/DSPE-PEG2000-Mal) for 4 h at 4°C with continuous stirring, and anti-CD44 antibody was then conjugated to these liposomes through the reaction of sulfhydryl residues on the antibodies with the C-terminal maleimide groups of the PEG chains.43 The solution was then ultracentrifuged at 55,000 rpm and 4°C for 1 h using the Optima XE-90 Ultracentrifuge (Beckman Coulter Inc., Fullerton, CA, USA) to remove the unconjugated antibody. The supernatant was collected to determine the amount of residual anti-CD44 antibody by using a BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Final LP and CD44-LP were stored at 4°C after dialysis. In detail, a 300-kDa molecular weight cutoff (MWCO) dialysis membrane (Spectrum Labs Inc., CA, USA) was used to remove free antibodies or free TAIII from liposomes by dialyzing for 2 h at 4°C.
Characterization
The entrapment efficiency (EE) and drug loading (DL) efficiency of TAIII in CD44-LP and LP were assessed with different feed molar ratios of DSPC, DSPE-PEG2000, and TAIII from 8:2:1 to 8:2:6 to obtain the optimal formulation. The particle size and zeta potential of LP and CD44-LP were measured using a Malvern Zetasizer ZEN3600 Nano ZS (Malvern Instruments, Malvern, UK) at 25°C. The surface morphology of CD44-LP was observed using a JEM-2100 transmission electron microscope (JEOL, Tokyo, Japan) with a negative stain method. To determine the EE, free TAIII in the supernatant was removed using a Sephadex G-50 gel minicolumn. TAIII was then determined through high-performance liquid chromatography equipped with an evaporative light-scattering detector (HPLC–ELSD) under the following conditions: use of an Alltima C18 column (W.R. Grace Co., Columbia, MD, USA), mobile phase of methanol–water (85:15, v/v), flow rate of 1 mL/min, and injection volume of 20 μL. The ELSD system was set to a probe temperature of 60°C, a gain of 1.0, and nebulizer N2 gas at 1.5 L/min. Briefly, 0.2 mL of CD44-LP was placed on a Sephadex G-50 gel minicolumn with 0.6 mL distilled water as the eluent. The eluent was collected after being centrifuged at 2,000 rpm for 5 min and considered to be encapsulated drug, determined through HPLC–ELSD after de-emulsification with methanol. DL efficiency (%) was calculated as (weight of encapsulated TAIII/weight of liposome) ×100%. EE (%) was calculated as (weight of encapsulated TAIII/weight of feeding TAIII) ×100%.
In vitro release of TAIII from LP and CD44-LP
The release of TAIII from LP and CD44-LP was investigated through a dialysis method. Briefly, 1 mL of LP and CD44-LP were loaded into a dialysis bag (MWCO 6,000–8,000 Da; Spectrum Labs Inc., CA, USA). The dialysis bags were immersed into release medium (PBS containing 0.5% [v:v] Tween 80) and agitated with a magnetic stirrer at 37°C for 48 h. Because of its poor solubility, the concentration of TAIII in the release medium was lower than the detection limits. Thus, liposomes in the dialysis bag were measured through HPLC–ELSD at a predetermined time.
In vivo pharmacokinetic studies
Before experimentation, 15 SD rats were fasted for 12 h and provided water ad libitum. These rats were randomly divided into three groups (n=5 per group). TAIII solution, LP, and CD44-LP (TAIII dose =10 mg/kg) were intravenously injected into Groups 1, 2, and 3, respectively. Because of its hydrophobicity, TAIII was dissolved in PBS containing 2.5% Tween 80, which is safe for parenteral administration.46 Approximately 0.3 mL of blood samples were obtained through the postorbital venous plexus veins at predetermined time points after administration (2, 5, 10, 30, 60, 120, 240, 360, 480, 720, 1,440, 2,880, and 4,320 min). The blood samples were centrifuged at 3,000 rpm for 5 min to obtain plasma, which was kept at −20°C. The plasma was treated and analyzed through HPLC–tandem mass spectrophotometry (MS/MS), as described previously.21 Briefly, 100 μL of plasma sample was deproteinized with 300 μL methanol, vortexed for 5 min, and centrifuged at 15,000 rpm for 5 min. Subsequently, the supernatant was examined through HPLC–MS/MS. Pharmacokinetic data were analyzed with DAS (Version 2.0; Mathematical Pharmacology Professional Committee of China, Shanghai, China).
In vitro cytotoxicity
The toxicity of TAIII, LP, and CD44-LP to HepG2 cells was investigated using an MTS-based assay. HepG2 cells were seeded in 96-well plates at a density of 1×104 cells/well and incubated for 24 h. The medium was replaced by different concentrations of TAIII, LP, and CD44-LP solutions for 24 and 48 h. The cells were then treated with the MTS-based CellTiter 96 AQueous One Solution Cell Proliferation Assay Reagent (Promega Corporation, Fitchburg, WI, USA) at 37°C for 4 h; hereafter, absorbance was determined at 490 nm with a microplate reader (Shanghai Bio Gene Biotech Co., Ltd, Shanghai, People’s Republic of China). Cell viability (%) was calculated as (Atreated − Ablank)/(Acontrol − Ablank) ×100%, where Ablank, Atreated, and Acontrol represent the absorbance of wells with no cells, cells treated with different TAIII solutions and control cells, respectively. The half-maximal inhibitory concentration (IC50) values were calculated using SPSS.
In vitro cellular uptake
The cellular uptake of the liposomes was evaluated in HepG2 cells by loading rhodamine (Sigma-Aldrich Co., St Louis, MO, USA) into the liposomes and tracing rhodamine fluorescence through CLSM, because rhodamine is a fluorescent marker widely used to evaluate the cellular uptake of liposomes and nanoparticles.43,47–49 Rhodamine-loaded liposomes were prepared using the same method as that used for LP. Cells were seeded onto glass-bottomed culture dishes (2×104 cells/well) and incubated for 24 h at 37°C. LP and CD44-LP (final rhodamine concentration =40 μg/mL) were then added to each dish, and the plates were incubated for 2 and 4 h. After the culture medium was removed, the cells on microscope plates were washed three times with PBS and then fixed with 4% paraformaldehyde solution for 20 min. The cytoskeleton was stained with fluorescein isothiocyanate-labeled phalloidin (phalloidin–FITC, green) for 30 min, whereas the nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI, blue) for 3 min. The cells were then rinsed three times with PBS. The fluorescence images were obtained using CLSM (Leica TCS SP8). The inhibition experiments were performed by pretreating HepG2 cells with anti-CD44 antibody for 1 h before incubation with CD44-LP.
In vivo near-infrared imaging and biodistribution
The tumor-bearing BALB/c nude mouse model was prepared through subcutaneous injection of 0.1 mL HepG2 cell suspension (6×106 cells) into the right sides of the mouse backs. As in other studies on liposomes or nanoparticles, in vivo near-infrared imaging was applied to afford a visual, time-dependent liposome biodistribution imaging with near-infrared fluorescent marker.32,50,51 Considering that hydrophobic ICG was embedded into the lipid bilayers of the liposomes,52,53 similar to TAIII, ICG-loaded liposomes were prepared and injected to evaluate the distribution and tumor accumulation of nontargeted and CD44-targeted liposomes. ICG-loaded liposomes were prepared using the same method used for LP. For in vivo distribution, tumor-bearing mice were randomly assigned to three groups, with three mice per group: PBS, LP, and CD44-LP. The ICG-loaded liposomes were administered at 200 μL through the tail vein. In vivo near-infrared imaging was performed using an IVIS Lumina XR Imaging System (PerkinElmer Inc., Waltham, MA, USA). The mice administered with PBS were used as the control. To further monitor the tumor accumulation, mice were anesthetized with 1.5% isoflurane at 1:2 O2/N2; the IVIS imaging system (excitation of 745 nm) was used to view the tumor accumulation profile. Mice were sacrificed after 24 h and the tumor, heart, liver, kidney, lung, and spleen were excised. These organs were also imaged at the aforementioned excitation wavelength.
In vivo antitumor activity
To prepare the tumor-bearing mice model, BALB/c nude mice were inoculated through subcutaneous injection with 0.1 mL HepG2 cell suspension (6×106 cells) on the right sides of their backs. When the tumor volume reached ~100 mm3, the tumor-bearing mice were randomly assigned to four groups (n=6 per group): one group was intraperitoneally injected PBS as the control and the other three groups were intraperitoneally injected TAIII solution (PBS containing 2.5% Tween 80), LP, and CD44-LP (TAIII dose =7.5 mg/kg) three times per week. The weight of the mice and tumor volumes were measured every 4 days throughout the study. Tumor volumes (mm3) were calculated as length × (width)2/2. On day 30, mice were sacrificed and the tumors were collected and weighed. Tumor inhibitory rate was calculated as (WC − WA)/WC ×100%, where WC and WA are the mean weights of tumors in the control group and each group treated with drugs, respectively. Additionally, the tumor, heart, liver, kidney, lung, and spleen were excised from the sacrificed mice of each group and fixed with 4% paraformaldehyde solution, followed by paraffin embedding. The organ tissues were then sliced and stained with hematoxylin and eosin (H&E) to assess the histological alterations through a digital microscope.
Statistical analysis
All data are presented as mean ± standard deviation (SD). One-way analysis of variance was performed using SPSS to determine the significance of differences among groups. Statistical significance was set at P<0.05.
Results
Characterization
As shown in Figure S1, TAIII could be efficiently encapsulated in the phospholipid bilayers of liposomes with higher encapsulation efficiency (>70%), with a range of molar ratios (8:2:1–8:2:4) and DL content ranging from 4.7% to 16.7%, depending on the drug feed amount. In addition, the molar ratio of DSPC, DSPE-PEG2000, and TAIII was fixed at 8:2:1, with a high EE (89.7%±4.3%) and DL (4.7%±0.2%) and an appropriate size of 39.72±0.14 nm for cancer treatment. No differences were noted in the mean diameters, polydispersity indexes, DL, and EE of the CD44-LP and LP (Table 1). The average diameters of CD44-LP were 39.73±0.05 nm, with a polydispersity index of 0.23±0.01, a suitable particle size and distribution for targeted drug delivery. Transmission electron microscopy verified that CD44-LP had a well-defined spherical shape and were homogeneously distributed (Figure 2A). The zeta potential of CD44-LP and LP was determined to be −6.79 and −5.91 mV. Moreover, conjugation efficiency of anti-CD44 antibody incorporation into CD44-LP was evaluated through ultracentrifugation to remove the unconjugated anti-CD44 antibody. The BCA protein assay demonstrated that the conjugation efficiency of anti-CD44 antibody incorporation into the liposomes was 79.5%±2.9%, similar to Arabi et al’s44 result, where 83% of anti-CD44 antibody was transferred to liposomes.
 | Table 1 Basic characterization of LP and CD44-LP |
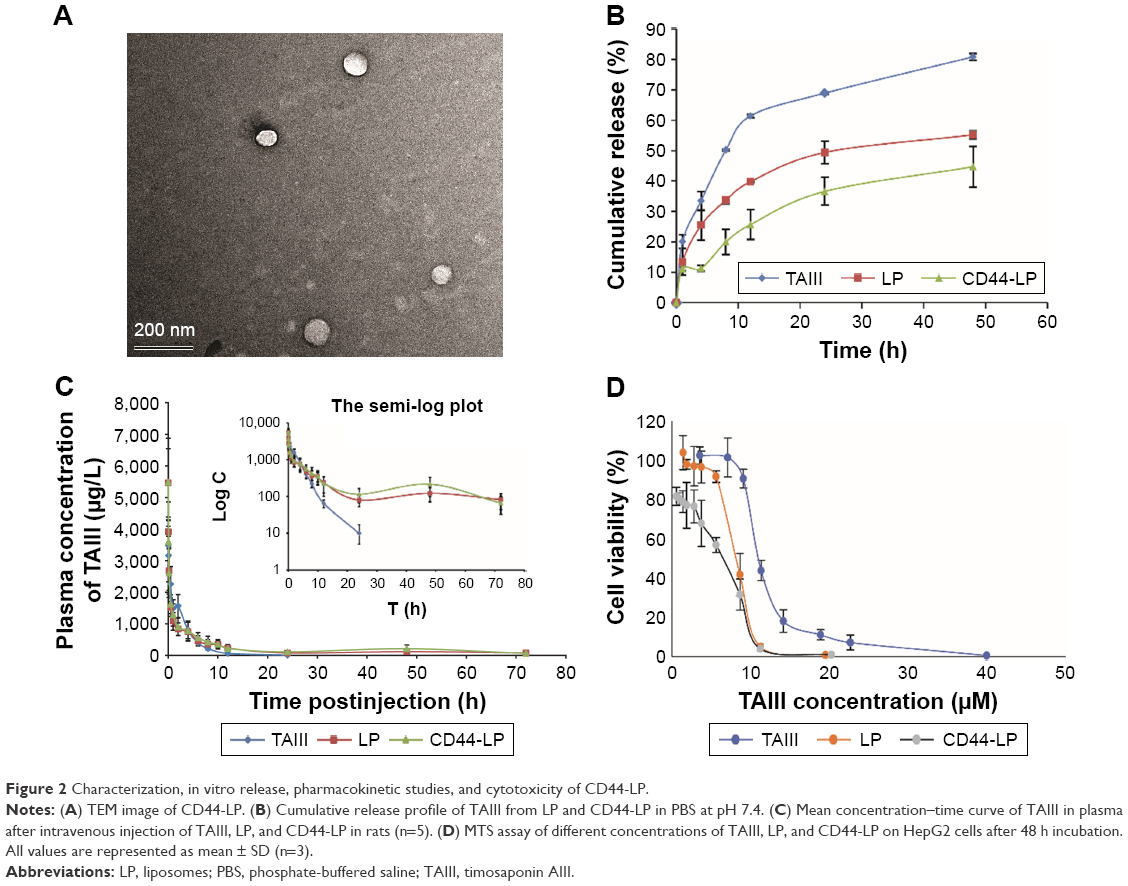 | Figure 2 Characterization, in vitro release, pharmacokinetic studies, and cytotoxicity of CD44-LP. |
In vitro release of TAIII from LP and CD44-LP
The in vitro release of TAIII from LP and CD44-LP is illustrated in Figure 2B. After the 48 h incubation period, LP and CD44-LP showed an ~55.2 and 44.7% cumulative release, respectively, significantly lower than that of free TAIII (80.8%). This was attributable to the liposomal bilayer and outer surface with hydrophilic polymers, which delay release by forming a protective shell.28 Although both the LP and CD44-LP showed favorable sustained-release capability in vitro, the release from the CD44-LP was relatively slower, possibly because of the steric protection effect endowed by anti-CD44 antibody on the surface of liposomes. Similar results were reported by studies on RGD peptide-coated liposomes,51,54 where these liposomes showed a slower drug release than did noncoated liposomes but with no statistical difference.
In vivo pharmacokinetic studies
Figure 2C and Table 2, respectively, illustrate the concentration–time curve of TAIII in plasma and pharmacokinetic parameters of TAIII after intravenous injection of TAIII, LP, and CD44-LP in rats. Compared with the LP and CD44-LP, free TAIII was cleared more rapidly, as indicated by the plasma TAIII levels reaching lower than detectable limits in the TAIII group within 24 h of administration; by contrast, TAIII could be detected in plasma of the LP- and CD44-LP-treated groups even at 72 h after administration. The highest plasma TAIII levels detected in the LP- and CD44-LP-treated rats were ~1.6-fold higher than those detected in TAIII-treated rats. Compared with TAIII alone, LP and CD44-LP increased the area under the curve (AUC) by 1.7- and 1.9-fold, respectively. The Cl rate of free TAIII was much higher (approximately twofold) than that of liposomes. In addition, LP and CD44-LP increased the half-life time of TAIII by 14.2- and 10.7-fold, respectively. All the pharmacokinetic results indicated that both the LP and CD44-LP could prolong the circulation time of TAIII in blood, thus providing significantly higher AUCs than TAIII alone. The higher AUC and mean residence time (MRT) values for LP and CD44-LP can contribute to higher plasma exposure of TAIII and greater antitumor efficiency. Notably, the TAIII volume of distribution (V) delivered by LP and CD44-LP also increased ~5.81 times and 4.47 times, compared with free TAIII. The volume of distribution data differs from the trend observed for other long circulating delivery systems,55 which sometimes reduce drug distribution and metabolism to enhance their retention effect. However, similar results have been reported,56,57 potentially because of the preferential accumulation or binding of the developed delivery system in some organs. The pharmacokinetic parameters of LP and CD44-LP did not differ significantly; thus, the conjugation of antibody fragments did not alter the liposomal stability or circulation time, in line with studies reporting that targeted and nontargeted nanoparticles have comparable pharmacokinetic parameters and biodistribution.58–62
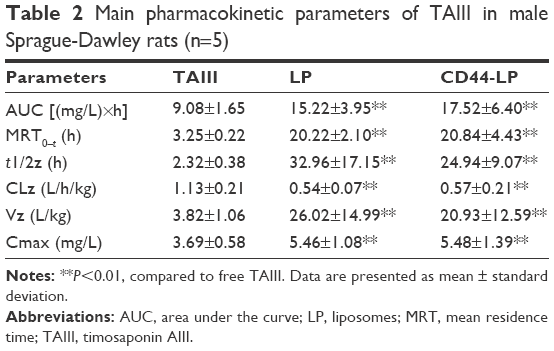 | Table 2 Main pharmacokinetic parameters of TAIII in male Sprague-Dawley rats (n=5) |
In vitro cytotoxicity
The cytotoxicity of blank liposomes (blank-LP, without DL), TAIII, LP, and CD44-LP to HepG2 cells was assessed using an MTS-based assay. Cell viability (%) was measured at various concentrations of TAIII after incubation for 24 and 48 h. As shown in Figure S2, blank-LP were nontoxic to HepG2 cells and displayed excellent biocompatibility, whereas TAIII, LP, and CD44-LP exhibited dose-dependent cytotoxic activity. Compared with the TAIII alone, LP and CD44-LP were significantly more cytotoxic (P<0.01), particularly at lower concentrations (Figure 2D). After 24 h incubation, LP and CD44-LP showed an IC50 value (11.67±0.50 and 14.05±0.61, respectively) lower than TAIII alone (20.14±0.80, P<0.01). On extending incubation time to 48 h, IC50 values became lower than those after the 24-h incubation for all groups. The CD44-LP showed the lowest IC50 (5.87±0.56) compared with the LP and TAIII alone (7.91±0.34 and 11.74±0.50, respectively), indicating a significantly enhanced antitumor activity of TAIII against HepG2 cells. The results suggest that TAIII could be efficiently released from the LP and CD44-LP in HepG2 cells, and liposome encapsulation of TAIII could effectively enhance cytotoxicity, presumably attributable to the enhanced cellular uptake of liposomes through nonspecific endocytosis and active transport.
In vitro cellular uptake
To study the targeting efficiency of CD44-targeted liposomes to CD44 high-expression tumor cells, CD44-LP carrying fluorescent rhodamine was prepared for CLSM. Figure 3 illustrates the CLSM images of HepG2 cells after 2 and 4 h incubation with rhodamine-loaded LP and CD44-LP. Considering that the fluorescence intensity of rhodamine when encapsulated into CD44-targeted liposomes is equivalent to that of nontargeted liposomes, rhodamine fluorescence observed inside HepG2 cells is correlated with the intracellular liposome levels. As expected, HepG2 cells treated with CD44-LP showed stronger red fluorescence than those treated with nontargeted LP, and the fluorescence of both groups greatly increased as incubation time increased to 4 h. The results revealed that CD44-targeted liposomes could effectively enhance cellular uptake in comparison with nontargeted liposomes. By contrast, negligible rhodamine fluorescence was observed in cells pretreated with anti-CD44 antibody, possibly because anti-CD44 antibodies blocked CD44 receptors and competed with CD44-LP in receptor binding. These results demonstrate that CD44-targeted liposomes most likely entered cells through receptor-mediated endocytosis, resulting in a higher cellular uptake.
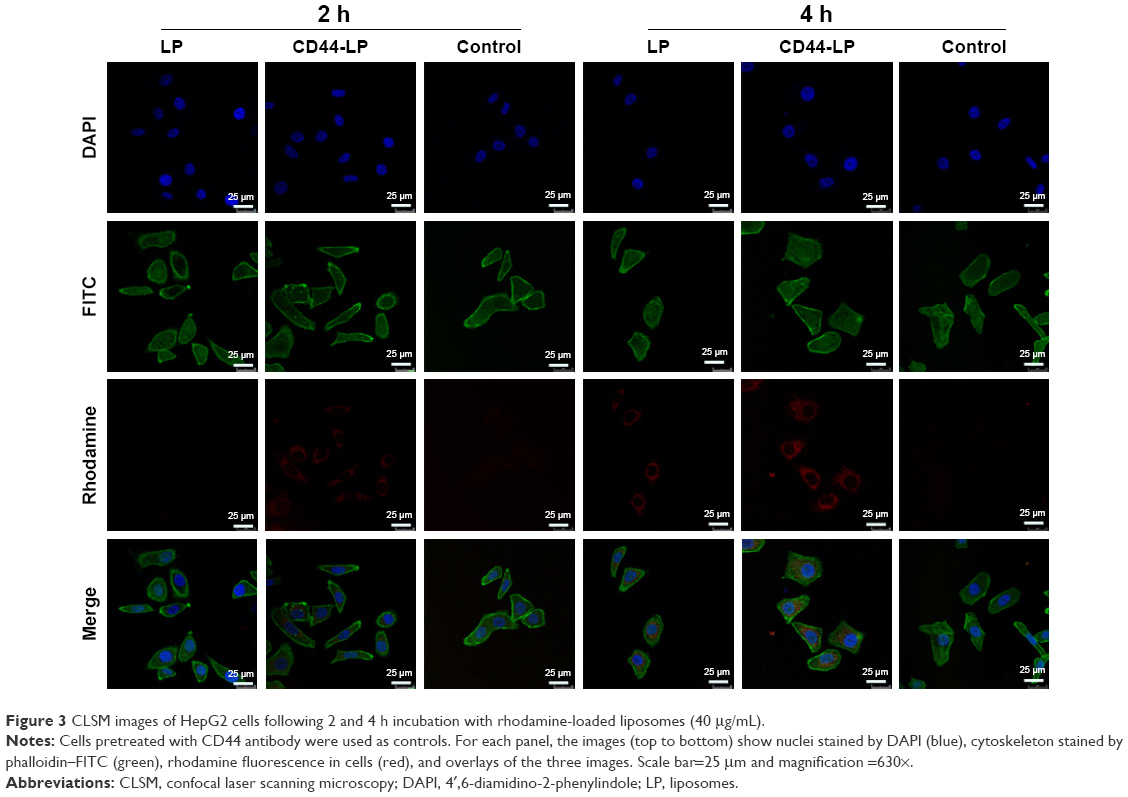 | Figure 3 CLSM images of HepG2 cells following 2 and 4 h incubation with rhodamine-loaded liposomes (40 μg/mL). |
In vivo near-infrared imaging and biodistribution
ICG-loaded liposomes were prepared and then injected to evaluate the distribution and tumor accumulation of nontargeted and CD44-targeted liposomes in tumor-bearing mice. Because the fluorescence intensity of ICG when encapsulated into CD44-targeted liposomes is equivalent to that of nontargeted liposomes, ICG fluorescence observed in the mice is correlated with the accumulation of liposomes. As shown in Figure 4A, LP and CD44-LP could be distributed through the entire body ~10 min after administration, whereas no fluorescence signal was observed in the control group. However, the range and intensity of fluorescence in mice decreased immediately, from 1 to 24 h after administration, possibly because of metabolism and elimination of ICG. Correspondingly, both LP and CD44-LP groups showed increased distribution percentage in tumors, from 10 min to 24 h after administration; furthermore, the fluorescence distribution at the tumor site in the CD44-LP group was 96.1% of the whole-body fluorescent signal at 24 h, whereas it was 69.8% in the LP group (Figure S3). Accordingly, as shown in Figure 4B, ICG fluorescence intensity at the tumor site in the CD44-LP group was stronger than LP. The subsequent comparison of quantified ICG accumulation by using fluorescence measurements (Figure 4C) was consistent with the in vitro cellular uptake on CLSM. Taken together, these collected data indicated that fluorescence efficiency at the tumor site in the CD44-LP group was 1.7-fold that in the LP group, indicating higher ICG accumulation in tumors. The tumor target efficiency of the CD44-LP is more effective, probably because of active targeting through the penetration effect mediated by anti-CD44 antibodies. However, negligible ICG fluorescence was detected in other organs in all groups: the ICG level in the liver or other organs at 24 h after administration was too weak to be detected by the imaging system, whereas the fluorescence at the tumor site was relatively stronger. Furthermore, ICG-loaded PEGylation liposomes having a smaller particle size, ~40 nm, and slight negative charge, ~-6 mV, were reported to be favorable for evading the RES and passive targeting to the tumor owing to the EPR effects, resulting in efficient tumor accumulation.63–65
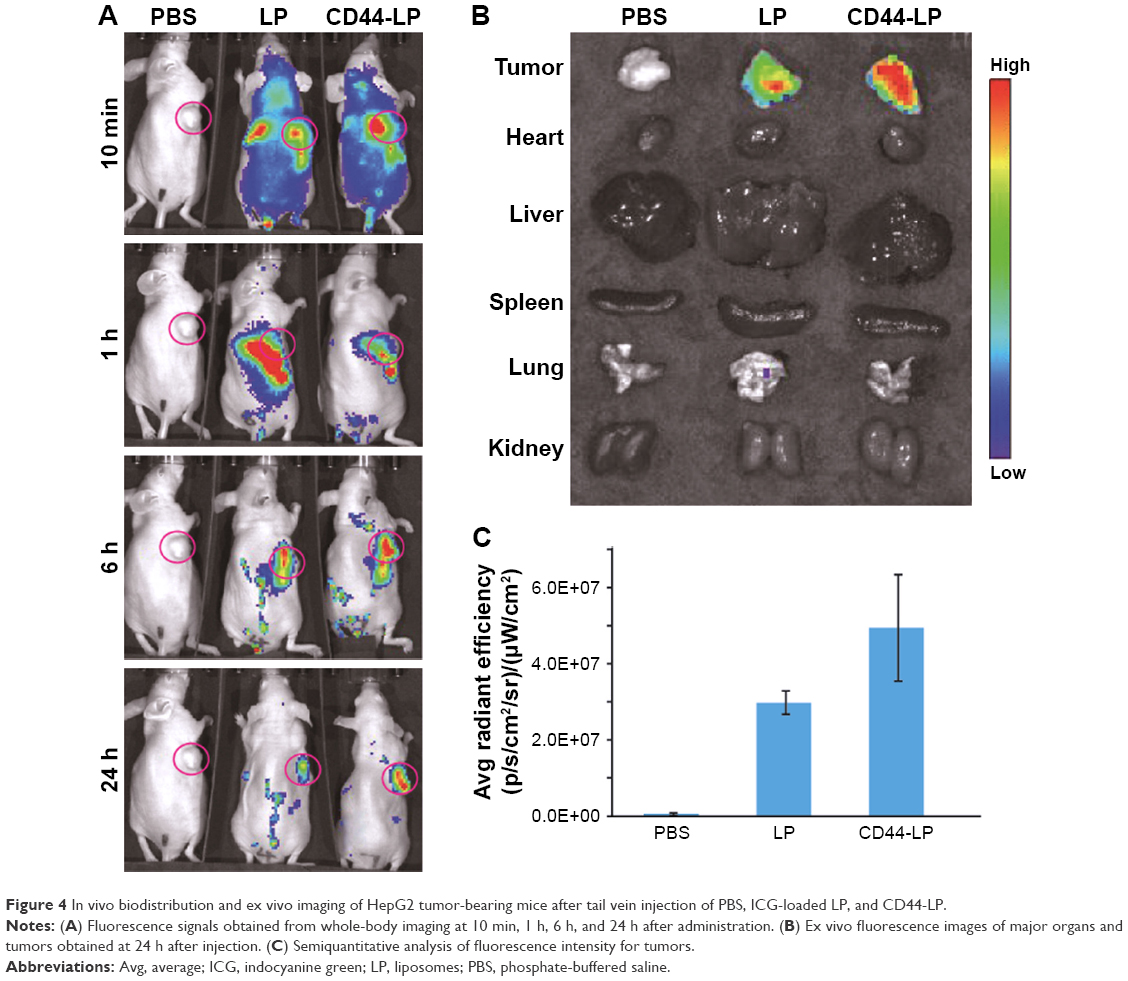 | Figure 4 In vivo biodistribution and ex vivo imaging of HepG2 tumor-bearing mice after tail vein injection of PBS, ICG-loaded LP, and CD44-LP. |
In vivo antitumor activity
In vivo antitumor study was evaluated on HepG2 tumor-bearing mice models through intraperitoneal injection with 7.5 mg/kg physiological saline, TAIII, LP, or CD44-LP. Mice treated with PBS were used as the control. As shown in Figure 5A, no serious reduction in body weight was observed in either the liposomes or the TAIII treatment groups compared with the control group, indicating good biological compatibility and innocuity of the liposomes. Figure 5B shows the change in tumor volume in all groups during the study. Both liposome formulations effectively inhibited tumor growth compared with the control group (P<0.01 for CD44-LP and P<0.05 for LP, respectively), whereas the group treated with TAIII alone exhibited negative tumor inhibition throughout the study. By contrast, intraperitoneal injection of CD44-LP and LP more efficiently inhibited tumor growth compared with TAIII alone (P<0.01 for CD44-LP and P<0.05 for LP); furthermore, CD44-LP showed more favorable tumor inhibition than did the LP at day 30 (Figure 5B). The tumor inhibitory rate of CD44-LP was calculated to be 55.2%±5.8%, which was 1.3- and 7.2-fold higher than that of the LP (40.9%±20.2%) and free TAIII (7.7%±19.6%) (Figure 5C), indicating that the encapsulation of TAIII in liposomes, particularly in CD44-LP, greatly enhanced its antitumor activity. Moreover, tumors collected from all groups (Figure 5D) visually exhibited higher antitumor efficiency of CD44-LP and LP. Notably, the higher dispersed degree of the tumor inhibitory rate value of LP (SD =20.2%) than that of CD44-LP (SD =5.8%) may be responsible for the nonsignificant difference between LP and CD44-LP, indirectly indicating that CD44-LP showed more steady and potent therapeutic efficacy compared with LP.
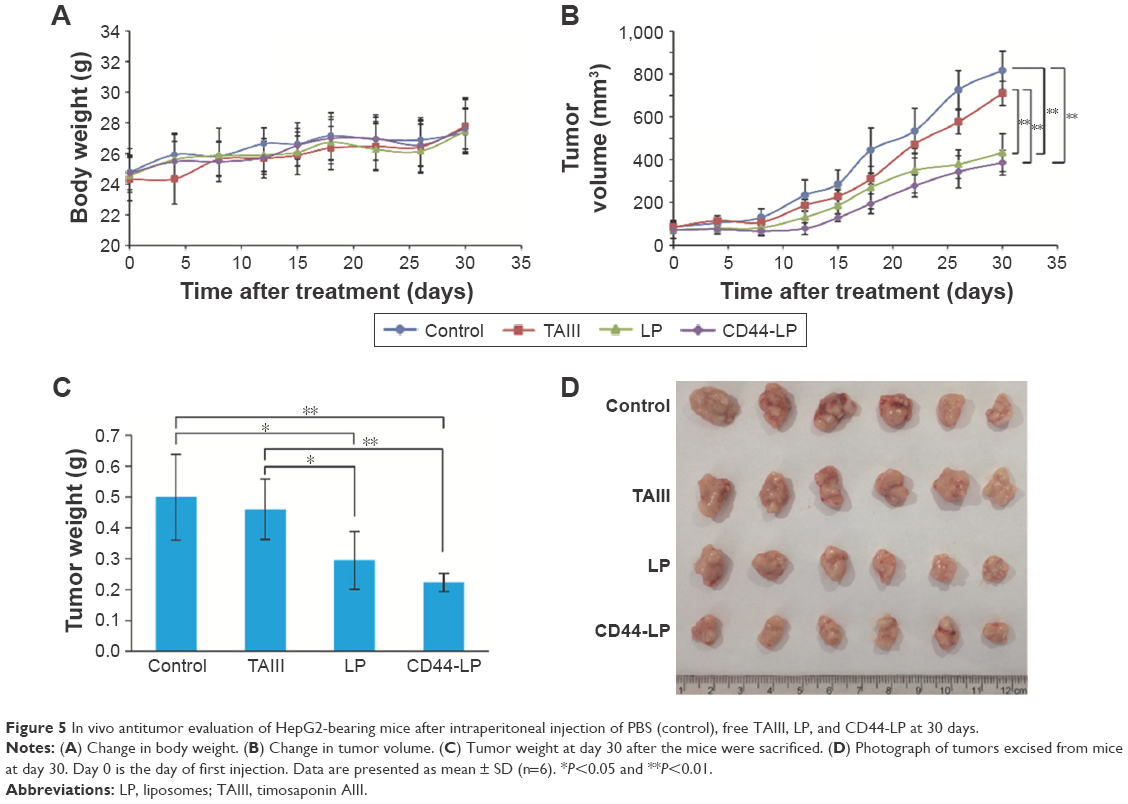 | Figure 5 In vivo antitumor evaluation of HepG2-bearing mice after intraperitoneal injection of PBS (control), free TAIII, LP, and CD44-LP at 30 days. |
Figure 6 illustrates H&E staining images of tumors and other main organs from different treatment groups. Tumors in the saline group were observed with larger, irregularly shaped nuclei, with some even being binucleolate, indicating cell proliferation in the tumors. By contrast, the tumors treated with the CD44-LP showed scattered nuclei, obvious nuclear shrinkage, and cytoplasmic vacuolation, thus indicating tumor tissue necrosis. The LP treatment group showed scattered nuclei and nuclear chromatin condensation, along with a lower inhibitory effect on tumor cell growth compared with the CD44-LP. Furthermore, free TAIII treatment led to relatively lower tumor necrosis signals. In addition, none of the treatment groups exhibited significant toxicity in the healthy organs compared with the control group (Figure 6). The integrated results imply that LP and CD44-LP not only enhance the antitumor effect of TAIII but also are tolerable in vivo.
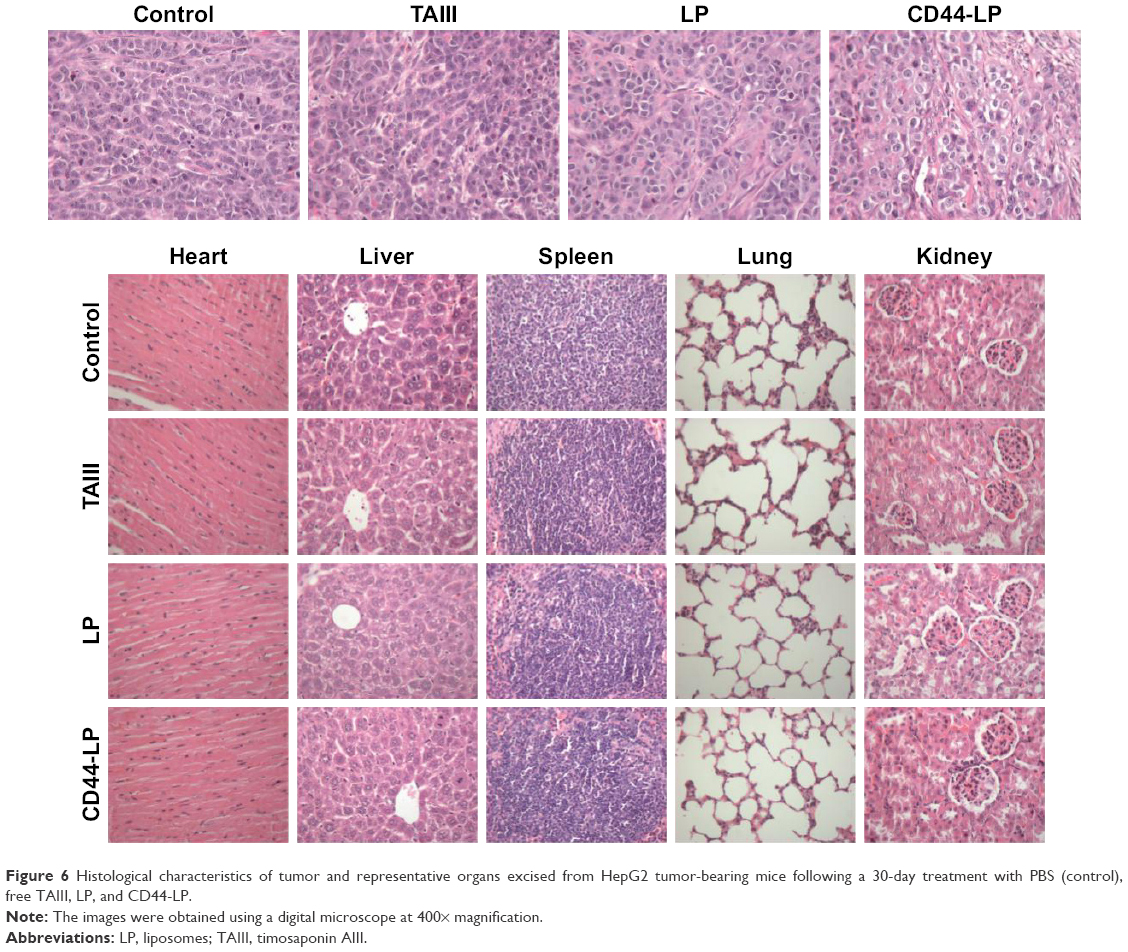 | Figure 6 Histological characteristics of tumor and representative organs excised from HepG2 tumor-bearing mice following a 30-day treatment with PBS (control), free TAIII, LP, and CD44-LP. |
Discussion
TAIII exhibits highly potent cytotoxicity against various cancer cells, showing favorable potential as a cancer therapeutic candidate. In a previous study,16 TAIII displayed potent inhibition of tumor growth and reduced cell viability in HCC cell lines HepG2, MHCC97L, PLC/PRF/5, and Hep3B through the autophagy–lysosomal pathway. However, in our initial experiment, the low aqueous solubility of TAIII (17 μg/mL) severely limited drug delivery in mice cancer models; as shown in Figure 5B, free TAIII revealed negative tumor inhibition. Liposomes have been used to encapsulate pharmacological agents in clinical trials to overcome solvent challenge, reduce toxicity but retain equal antitumor efficacy, or enhance therapeutic effects. Thus far, the US Food and Drug Administration and European Medicines Agency have approved six liposome-based products for cancer therapy, such as doxorubicin HCl stealth liposome injection (Doxil) and vincristine sulfate liposome injection (Marqibo, ONCO TCS). To the best of our knowledge, liposomal formulations of TAIII have not yet been explored for efficient drug delivery with most studies on TAIII cytotoxicity to cancer cells being mostly based on the in vitro evaluation. In this study, the hydrophobic TAIII incorporated in biocompatible PEGylated liposomes was successfully prepared for more efficient drug delivery. Anti-CD44 antibody specifically binds to CD44 receptors on the cancer cells’ surface, improving the intracellular uptake and enhancing the antitumor efficacy against CD44-positive cancers.41,43,44 In our initial experiment, the CD44 receptor was detected on a BD FACSCalibur flow cytometer and was endogenously overexpressed on the HepG2 cell surface (Figure S4); we assumed that LP expressing anti-CD44 antibody targets HepG2 cells through receptor-mediated endocytosis, resulting in high cellular accumulation and enhanced antitumor activity of TAIII.
Target recognition is sensitive to the chain length of the incorporated PEG. Furthermore, at higher molecular weights, PEG (DSPE-PEG5000) substantially prevents antibody recognition of antigens, whereas at lower molecular weight, DSPE-PEG2000 retains a similar degree of antibody target binding shown by liposomes without PEG-DSPE.66 Therefore, LP composed of DSPC and DSPE-PEG2000 were prepared using the thin-film rehydration method, followed by sonication, and DSPE-PEG2000-Mal was used for conjugation with anti-CD44 antibody. Anti-CD44 antibody was coupled to the PEG terminus by covalent binding for targeted drug delivery to tumor regions (Figure 1). This process is simple and controllable, without complicated chemical reactions. CD44-LP assumed a spherical shape with an evenly distributed diameter (39.73 nm), a weakly negative zeta potential (−6.79 mV), and an acceptable EE (87.6%). The particle size of nanoparticles of 30–100 nm is sufficient to prevent leakage into capillaries and also small enough to avoid reticuloendothelial Cl.63 Accordingly, in a previous study,64 nanoparticles with larger particle sizes (>300 nm) tended to be arrested by the RES and trapped by the hepatic sinusoid, whereas smaller nanoparticles achieved the lowest distribution percentage in the liver and spleen. In addition, Xiao et al65 showed that nanoparticles with a slightly negative charge (−8.5±1.8 mV) exhibited higher tumor-to-background fluorescence contrast ratio and decreased liver uptake compared with nanoparticles with highly positive or negative surface charge. Thus, the slightly negative charge aids in reducing the undesirable Cl by the RES, such as that in the liver and spleen, and improves blood compatibility, finally delivering the drugs more efficiently to the tumor sites. Furthermore, PEG surface coating can counteract the hydrophobic and electrostatic interactions between nanoparticles and plasma proteins or macrophages, resulting in low RES uptake and prolonged blood circulation time.63 Therefore, the small particle size and weak negative charge of LP and CD44-LP might permit favorable in vivo tumor accumulation and transport of nanoparticles through the EPR effect, as observed in Figure 4.
As shown in Figure 2, CD44-LP and LP displayed desirable sustained-release profiles in vitro and significantly higher AUCs in vivo compared with TAIII alone; thus, in vivo and in vitro results were directly correlated, and the results of the pharmacokinetic study were supported by the in vitro results. CD44-LP and LP also extended the TAIII half-life by ~10.7- and 14.2-fold, respectively, and improved its bioavailability. This may be attributed to the coating of PEG on the liposome surface, which potentially prevents the binding of the serum protein opsonin to the liposome surface and extends circulation half-lives.66 Delaying the blood Cl and prolonging the blood circulation of CD44-LP contribute to drug redistribution to target tumor sites,66 thus improving antitumor activity of TAIII.
In our previous in vivo imaging experiments, free ICG and ICG-loaded liposomes were administered to mice at an equivalent ICG dose. At all observed time points, the fluorescence at the tumor site was stronger in the LP-treated group than in the free ICG-treated group. Furthermore, in the LP-treated group, ex vivo fluorescence in tumor in the LP-treated group was strong, whereas it was negligible in the free ICG-treated group (Figure S5); thus, liposomes can alter the biodistribution of free ICG and improve the accumulation of drugs at the HepG2 tumor site, possibly because of EPR effect. These data corroborate a study,67 which reported that ICG-loaded liposomes can be selectively internalized and retained in tumor cells. In the present study, we intended to study anti-CD44 antibody modified liposomes for active tumor targeting; therefore, mice administered with unmodified liposomes, with PBS-treated mice as control, were compared to ensure data validity and reliability. In vivo near-infrared imaging of an HepG2 xenograft mouse model revealed a higher tumor accumulation of CD44-LP, in line with in vitro cellular uptake studies. These data verified that CD44-LP most likely enters cells through receptor-mediated endocytosis, contributing to high selectivity to CD44-positive tumor cells and enhanced cellular uptake.
Notably, CD44-LP significantly increased the in vitro cytotoxicity and tumor inhibitory rate of TAIII in the xenograft model by 7.2- and 1.3- fold compared with free TAIII and the LP, without detectable toxicity. This may be attributed to the effective tumor-targeting efficacy of CD44-LP through passive and active targeting. The spherical shape and 39 nm particle size make liposomes preferable for passively penetrating and accumulating on the surrounding tumor tissue through the EPR effect; furthermore, the presence of anti-CD44 antibodies on their surface enhances the cellular uptake of liposomes through CD44 receptor-mediated endocytosis. TAIII can initiate apoptosis in HeLa cells10 and colon cancer cells15 and induce caspase-dependent apoptosis in HCC cells.16 The apoptosis-inducing effect of TAIII was further confirmed in this study (Figure S6). The percentages of apoptosis in HepG2 cells incubated with TAIII alone, LP, and CD44-LP (TAIII dose =10 μM) for 24 h were 18.0±3.2, 20.3±1.9, and 23.5±1.5, respectively. In the fluorescence images of HepG2 cells after annexin V-FITC/propidium iodide (PI) dual staining (Figure S6B) in all treatment groups, early- or late-stage apoptotic cells were stained green (annexin V-FITC) and dead or later apoptotic cells were stained red (PI). Western blotting (Figure S6C) revealed that TAIII and liposomes significantly increased the active cleaved forms of caspase-9, consistent with the literature.16 Furthermore, the caspase-3 protein level was significantly upregulated in the CD44-LP treatment group (fourfold higher than the control), but only a small increase was observed in TAIII and LP treatment groups (1.29- and 1.85-fold higher than the control, respectively), potentially attributable to enhanced uptake of active targeting liposomes. TAIII can induce dose-dependent increase in caspase-3 levels in HCC cells after 24 h of treatment;16 therefore, caspase-3 activation is related to TAIII dose. Furthermore, in a xenograft mouse model, intraperitoneal injection of TAIII (7.5 mg/kg every 2 days) initiated apoptosis and cleaved caspase-3 in the xenografted tumor.16 These data are consistent and suggest that TAIII and liposomes initiate caspase-dependent apoptosis in HepG2 cells.
Although a study indicated that TAIII triggered liver injury when the rats were administered TAIII (100 mg/kg/day, po) for 14 days,68 H&E staining experiment in this study revealed that none of the treatment groups exhibited significant toxicity in the healthy organs compared with the control group. This is most likely because mice were intraperitoneally injected TAIII at a dose of 7.5 mg/kg, much lower than the dose that can cause liver injury. Moreover, the absence of obvious body weight loss indicated that TAIII and the liposomal formulations were well tolerated. Nevertheless, the potential toxicity of TAIII will remain a focus of our future studies.
Conclusion
CD44-LP and LP significantly enhanced plasma half-life, reduced systemic Cl, and improved bioavailability of TAIII. High selectivity of the CD44-LP toward CD44-positive tumor cells contributed to a more substantial uptake by HepG2 cells and a higher tumor accumulation in HepG2 xenograft mice compared with the LP, resulting in slower HepG2 xenograft growth and tumor necrosis. In particular, CD44-LP exhibited considerably higher cytotoxicity than did LP, with a lower IC50 (48 h). CD44-LP exhibited stronger tumor inhibition; the tumor inhibitory effect was 1.3-fold higher than that of the LP. Therefore, CD44-LP represents a promising drug delivery system with favorable biocompatibility and antitumor efficiency against CD44-positive tumor cells. In addition, our results can guide researchers studying the antitumor effects of TAIII and other potential compounds from natural herbs.
Acknowledgments
This work was supported by programs of the National Natural Science Foundation of China [grant numbers 81403175]; Program of Shanghai Academic/Technology Research Leader [grant number 18XD1403700]; Project of the Shanghai Municipal Commission of Health and Family Planning [grant number 2017YQ072 and 201740152]; Research Fund for the Doctoral Program of Shanghai [grant number B201703]; and Xinglin Scholar Plan.
Disclosure
The authors report no conflicts of interest in this work.
References
Wang YL, Dan Y, Yang DW, et al. The genus Anemarrhena Bunge: a review on ethnopharmacology, phytochemistry and pharmacology. J Ethnopharmacol. 2014;153(1):42–60. | ||
Zhang JY, Meng ZY, Zhang MY, Ma DS, Xu SX, Kodama H. Effect of six steroidal saponins isolated from anemarrhenae rhizoma on platelet aggregation and hemolysis in human blood. Clin Chim Acta. 1999;289(1–2):79–88. | ||
Nakashima N, Kimura I, Kimura M, Matsuura H. Isolation of pseudoprototimosaponin AIII from rhizomes of Anemarrhena asphodeloides and its hypoglycemic activity in streptozotocin-induced diabetic mice. J Nat Prod. 1993;56(3):345–350. | ||
Jiang WX, Guo J, Xue R, et al. Anti-depressive activities and biotransformation of timosaponin B-III and its derivatives. Nat Prod Res. 2014;28(18):1446–1453. | ||
Lim SM, Jeong JJ, Kang GD, Kim KA, Choi HS, Kim DH. Timosaponin AIII and its metabolite sarsasapogenin ameliorate colitis in mice by inhibiting NF-kappa B and MAPK activation and restoring Th17/Treg cell balance. Int Immunopharmacol. 2015;25(2):493–503. | ||
Tundis R, Bonesi M, Menichini F, Loizzo MR. Recent knowledge on medicinal plants as source of cholinesterase inhibitors for the treatment of dementia. Mini Rev Med Chem. 2016;16(8):605–618. | ||
Chen JR, Jia XH, Wang H, Yi YJ, Wang JY, Li YJ. Timosaponin A-III reverses multi-drug resistance in human chronic myelogenous leukemia K562/ADM cells via downregulation of MDR1 and MRP1 expression by inhibiting PI3K/Akt signaling pathway. Int J Oncol. 2016;48(5):2063–2070. | ||
Huang HL, Chiang WL, Hsiao PC, et al. Timosaponin AIII mediates caspase activation and induces apoptosis through JNK1/2 pathway in human promyelocytic leukemia cells. Tumour Biol. 2015;36(5):3489–3497. | ||
Lee B, Jung K, Kim DH. Timosaponin AIII, a saponin isolated from Anemarrhena asphodeloides, ameliorates learning and memory deficits in mice. Pharmacol Biochem Behav. 2009;93(2):121–127. | ||
Sy LK, Yan SC, Lok CN, Man RY, Che CM. Timosaponin A-III induces autophagy preceding mitochondria-mediated apoptosis in HeLa cancer cells. Cancer Res. 2008;68(24):10229–10237. | ||
Lok CN, Sy LK, Liu FL, Che CM. Activation of autophagy of aggregation-prone ubiquitinated proteins by timosaponin A-III. J Biol Chem. 2011;286(36):31684–31696. | ||
Fang M, Gu L, Gu G, Fang J. Facile synthesis and antitumor activities of timosaponin AIII and its analogs. J Carbohydr Chem. 2012;31(3):187–202. | ||
Tsai CH, Yang CW, Wang JY, et al. Timosaponin AIII suppresses hepatocyte growth factor-induced invasive activity through sustained ERK activation in breast cancer MDA-MB-231 cells. Evid Based Complement Alternat Med. 2013;2013:421051. | ||
King FW, Fong S, Griffin C, et al. Timosaponin AIII is preferentially cytotoxic to tumor cells through inhibition of mTOR and induction of ER stress. PLoS One. 2009;4(9):12. | ||
Kang YJ, Chung HJ, Nam JW, et al. Cytotoxic and antineoplastic activity of timosaponin A-III for human colon cancer cells. J Nat Prod. 2011;74(4):701–706. | ||
Wang N, Feng Y, Zhu M, Siu FM, Ng KM, Che CM. A novel mechanism of XIAP degradation induced by timosaponin AIII in hepatocellular carcinoma. Biochim Biophys Acta. 2013;1833(12):2890–2899. | ||
Pan H-J, Nie X-Q, Liu D, Bian K. Effects of four kinds of Chinese medicine monomer on growth of PANC-1 xenograft tumor and studying of molecular mechanism. China J Chin Mater Med. 2013;38(2):245–248. | ||
Kim KM, Im AR, Kim SH, Hyun JW, Chae S. Timosaponin AIII inhibits melanoma cell migration by suppressing COX-2 and in vivo tumor metastasis. Cancer Sci. 2016;107(2):181–188. | ||
Jung O, Lee J, Lee YJ, et al. Timosaponin AIII inhibits migration and invasion of A549 human non-small-cell lung cancer cells via attenuations of MMP-2 and MMP-9 by inhibitions of ERK1/2, Src/FAK and beta-catenin signaling pathways. Bioorg Med Chem Lett. 2016;26(16):3963–3967. | ||
Lu L, Liu Y, Ding Y, et al. Preparation of highly purified timosaponin AIII from rhizoma anemarrhenae through an enzymatic method combined with preparative liquid chromatography. Nat Prod Res. 2016;30(20):2364–2367. | ||
Liu Y, Pu Y, Zhang T, Ding Y, Wang B, Cai Z. Rapid and sensitive determination of timosaponin AIII in rat plasma by LC-MS/MS and its pharmacokinetic application. Int J Mol Sci. 2013;14(2):3656–3670. | ||
Li C, Cui J, Wang C, et al. Encapsulation of vinorelbine into cholesterol-polyethylene glycol coated vesicles: drug loading and pharmacokinetic studies. J Pharm Pharmacol. 2011;63(3):376–384. | ||
Guo B-H, Cheng Y, Wu W, Lin L-P, Lin D-H. HPLC assay and pharmacokinetics and tissue distribution study of glycyrrhetinic acid liposomes modified with galactosylated lipid. J Liposome Res. 2012;22(2):120–127. | ||
Chen H, Wu J, Sun M, et al. N-trimethyl chitosan chloride-coated liposomes for the oral delivery of curcumin. J Liposome Res. 2012;22(2):100–109. | ||
Li J, Chen J, Cai B-C, Yang T. Preparation, characterization and tissue distribution of brucine stealth liposomes with different lipid composition. Pharm Dev Technol. 2013;18(4):772–778. | ||
Chen J, Yan G-J, Hu R-R, et al. Improved pharmacokinetics and reduced toxicity of brucine after encapsulation into stealth liposomes: role of phosphatidylcholine. Int J Nanomedicine. 2012;2012(7):3567–3577. | ||
Huwyler J, Drewe J, Kraehenbuehl S. Tumor targeting using liposomal antineoplastic drugs. Int J Nanomedicine. 2008;3(1):21–29. | ||
Gabizon A, Catane R, Uziely B, et al. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994;54(4):987–992. | ||
Papahadjopoulos D, Allen TM, Gabizon A, et al. Sterically stabilized liposomes: improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc Natl Acad Sci U S A. 1991;88(24):11460–11464. | ||
Gabizon A, Martin F. Polyethylene glycol coated (pegylated) liposomal doxorubicin – rationale for use in solid tumours. Drugs. 1997;1997(54):15–21. | ||
Chen W-L, Yuan Z-Q, Liu Y, et al. Liposomes coated with N-trimethyl chitosan to improve the absorption of harmine in vivo and in vitro. Int J Nanomedicine. 2016;2016(11):325–336. | ||
Yu K-F, Zhang W-Q, Luo L-M, et al. The antitumor activity of a doxorubicin loaded, iRGD-modified sterically-stabilized liposome on B16-F10 melanoma cells: in vitro and in vivo evaluation. Int J Nanomedicine. 2013;2013(8):2473–2485. | ||
Meng S, Su B, Li W, et al. Integrin-targeted paclitaxel nanoliposomes for tumor therapy. Med Oncol. 2011;28(4):1180–1187. | ||
Eramo A, Lotti F, Sette G, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15(3):504–514. | ||
Tang X, Guo N, Xu L, Gou X, Mi M. CD147/EMMPRIN: an effective therapeutic target for hepatocellular carcinoma. J Drug Target. Epub 2012 Aug 29. | ||
Sironen RK, Tammi M, Tammi R, Auvinen PK, Anttila M, Kosma VM. Hyaluronan in human malignancies. Exp Cell Res. 2011;317(4):383–391. | ||
Sneath RJS, Mangham DC. The normal structure and function of CD44 and its role in neoplasia. Mol Pathol. 1998;51(4):191–200. | ||
Cichy J, Pure E. The liberation of CD44. J Cell Biol. 2003;161(5):839–843. | ||
Naor D, Wallach-Dayan SB, Zahalka MA, Sionov RV. Involvement of CD44, a molecule with a thousand faces, in cancer dissemination. Semin Cancer Biol. 2008;18(4):260–267. | ||
Liu Y, Sun J, Lian H, Cao W, Wang Y, He Z. Folate and CD44 receptors dual-targeting hydrophobized hyaluronic acid paclitaxel-loaded polymeric micelles for overcoming multidrug resistance and improving tumor distribution. J Pharm Sci. 2014;103(5):1538–1547. | ||
Bushman J, Vaughan A, Sheihet L, Zhang Z, Costache M, Kohn J. Functionalized nanospheres for targeted delivery of paclitaxel. J Control Release. 2013;171(3):315–321. | ||
Ganesh S, Iyer AK, Morrissey DV, Amiji MM. Hyaluronic acid based self-assembling nanosystems for CD44 target mediated siRNA delivery to solid tumors. Biomaterials. 2013;34(13):3489–3502. | ||
Wang L, Su W, Liu Z, et al. CD44 antibody-targeted liposomal nanoparticles for molecular imaging and therapy of hepatocellular carcinoma. Biomaterials. 2012;33(20):5107–5114. | ||
Arabi L, Badiee A, Mosaffa F, Jaafari MR. Targeting CD44 expressing cancer cells with anti-CD44 monoclonal antibody improves cellular uptake and antitumor efficacy of liposomal doxorubicin. J Control Release. 2015;220(pt A):275–286. | ||
Platt VM, Szoka FC Jr. Anticancer therapeutics: targeting macromolecules and nanocarriers to hyaluronan or CD44, a hyaluronan receptor. Mol Pharm. 2008;5(4):474–486. | ||
Park MH, Keum CG, Song JY, Kim D, Cho CW. A novel aqueous parenteral formulation of docetaxel using prodrugs. Int J Pharm. 2014;462(1–2):1–7. | ||
Alshaer W, Hillaireau H, Vergnaud J, Ismail S, Fattal E. Functionalizing liposomes with anti-CD44 aptamer for selective targeting of cancer cells. Bioconjug Chem. 2015;26(7):1307–1313. | ||
Zashikhina NN, Volokitina MV, Korzhikov-Vlakh VA, et al. Self-assembled polypeptide nanoparticles for intracellular irinotecan delivery. Eur J Pharm Sci. 2017;109:1–12. | ||
Song X, Wen YT, Zhu JL, Zhao F, Zhang ZX, Li J. Thermoresponsive delivery of paclitaxel by beta-cyclodextrin-based poly(N-isopropylacrylamide) star polymer via inclusion complexation. Biomacromolecules. 2016;17(12):3957–3963. | ||
Ling L, Du Y, Ismail M, et al. Self-assembled liposomes of dual paclitaxel-phospholipid prodrug for anticancer therapy. Int J Pharm. 2017;526(1–2):11–22. | ||
Chang M, Lu S, Zhang F, et al. RGD-modified pH-sensitive liposomes for docetaxel tumor targeting. Colloids Surf B Biointerfaces. 2015;129:175–182. | ||
Yan F, Wu H, Liu H, et al. Molecular imaging-guided photothermal/photodynamic therapy against tumor by iRGD-modified indocyanine green nanoparticles. J Control Release. 2016;224:217–228. | ||
Yoon HJ, Lee HS, Lim JY, Park JH. Liposomal indocyanine green for enhanced photothermal therapy. ACS Appl Mater Interfaces. 2017;9(7):5683–5691. | ||
Sonali S, Singh RP, Sharma G, et al. RGD-TPGS decorated theranostic liposomes for brain targeted delivery. Colloids Surf B Biointerfaces. 2016;147:129–141. | ||
Zhang S, Wang J, Pan J. Baicalin-loaded PEGylated lipid nanoparticles: characterization, pharmacokinetics, and protective effects on acute myocardial ischemia in rats. Drug Deliv. 2016;23(9):3696–3703. | ||
Liu M, Jin S, Yan H, Du S. Effect of oxymatrine HSPC liposomes on improving bioavailability, liver target distribution and hepatoprotective activity of oxymatrine. Eur J Pharm Sci. 2017;104:212–220. | ||
Zhang L, Han L, Sun X, Gao D, Qin J, Wang J. The use of PEGylated liposomes to prolong the circulation lifetime of salvianolic acid B. Fitoterapia. 2012;83(4):678–689. | ||
Chu C, Xu P, Zhao H, et al. Effect of surface ligand density on cytotoxicity and pharmacokinetic profile of docetaxel loaded liposomes. Asian J Pharm Sci. 2016;11(5):655–661. | ||
Fei W, Zhang Y, Han S, et al. RGD conjugated liposome-hollow silica hybrid nanovehicles for targeted and controlled delivery of arsenic trioxide against hepatic carcinoma. Int J Pharm. 2017;519(1–2):250–262. | ||
Zuo T, Guan Y, Chang M, et al. RGD(Arg-Gly-Asp) internalized docetaxel-loaded pH sensitive liposomes: preparation, characterization and antitumor efficacy in vivo and in vitro. Colloids Surf B Biointerfaces. 2016;147:90–99. | ||
Pirollo KF, Chang EH. Does a targeting ligand influence nanoparticle tumor localization or uptake? Trends Biotechnol. 2008;26(10):552–558. | ||
van der Meel R, Vehmeijer LJ, Kok RJ, Storm G, van Gaal EV. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: current status. Adv Drug Deliv Rev. 2013;65(10):1284–1298. | ||
Faraji AH, Wipf P. Nanoparticles in cellular drug delivery. Bioorg Med Chem. 2009;17(8):2950–2962. | ||
He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666. | ||
Xiao K, Li Y, Luo J, et al. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. 2011;32(13):3435–3446. | ||
Hansen CB, Kao GY, Moase EH, Zalipsky S, Allen TM. Attachment of antibodies to sterically stabilized liposomes – evaluation, comparison and optimization of coupling procedures. Biochim Biophys Acta Biomembr. 1995;1239(2):133–144. | ||
Zheng X, Zhou F, Wu B, Chen WR, Xing D. Enhanced tumor treatment using biofunctional indocyanine green-containing nanostructure by intratumoral or intravenous injection. Mol Pharm. 2012;9(3):514–522. | ||
Wu ZT, Qi XM, Sheng JJ, et al. Timosaponin A3 induces hepatotoxicity in rats through inducing oxidative stress and down-regulating bile acid transporters. Acta Pharmacol Sin. 2014;35(9):1188–1198. |
Supplementary materials
Characterization of timosaponin AIII (TAIII)-loaded liposomes
The drug-loading efficiency (%) and encapsulation efficiency (%) of TAIII loaded liposomes are presented in Figure S1.
Investigation of CD44 expression level
To evaluate the CD44 expression level in HepG2 cell line, 1×106 cells were resuspended in phosphate-buffered saline (PBS) containing 0.1% fetal bovine serum (FBS) and then incubated with fluorescein isothiocyanate-labeled anti-CD44 antibody (1 μL) for 20 min at 4°C. Then, cells were washed three times with cold PBS and the fluorescence intensity was measured by flow cytometry (BD FACSCalibur; BD Biosciences, San Jose, CA, USA).
According to Figure S4, CD44 receptor is endogenously overexpressed in the HepG2 cell line, which is consistent with Wang et al’s report.1
In vitro cytotoxicity
The results of in vitro cytotoxicity of TAIII, LP and CD44-LP are presented in Figure S2 and Table S1.
In vivo near infrared imaging and biodistribution of indocyanine green (ICG) and liposomes (LP)
The tumor-bearing BALB/c nude mice model was prepared through the subcutaneous injection of 0.1 mL HepG2 cell suspension (6×106) into the right sides of the mouse backs. ICG-loaded liposomes were prepared in the same manner as the TAIII liposomes. For the in vivo distribution, tumor-bearing mice were randomly assigned to two groups (ICG solution and LP). The liposomes containing ICG were injected in amounts of 200 μL through the tail vein. In vivo real-time imaging was taken using an IVIS Lumina XR Imaging System (PerkinElmer Inc., Waltham, MA, USA). Mice untreated with free ICG and liposomes were used as controls (0 h). To further monitor tumor accumulation, mice were anesthetized with 1.5% isoflurane at 1:2 O2/N2; the IVIS imaging system (excitation of 745 nm) was used to view the tumor accumulation profile. Mice were sacrificed after 24 h, and the tumor, heart, liver, kidney, lung, and spleen were excised. These organs were also imaged at the aforementioned excitation wavelength.
As shown in Figure S5, the ICG fluorescence signal in tumor gradually increased in the mice treated with ICG-loaded liposomes from the 10-min to 24-h time points, whereas weaker fluorescence signal was observed at the tumor site in the free ICG-treated group. Additionally, stronger fluorescence at the tumor site was viewed in the LP-treated group than in the free ICG-treated group at all observed time points. Furthermore, ex vivo fluorescence in tumor in the LP-treated group was strong, whereas negligible ICG fluorescence was detected in other major organs in the LP-treated group and all the tissues including tumor in the free ICG-treated group. This indicated that liposomes could alter the biodistribution of free ICG and improve the accumulation of drugs at the tumor site, possibly caused by the enhanced permeability and retention (EPR) effect, thus presenting passive targeting activity. These data are in line with the literature,2 which reported that ICG-loaded liposomes could be selectively internalized and retained in target tumor cells. Correspondingly, both LP and CD44-LP groups showed increased distribution percentage in tumors, from 10 min to 24 h after administration. Furthermore, the fluorescence distribution at the tumor site in the CD44-LP group was 96.1% of the whole-body fluorescent signal at 24 h, whereas it was 69.8% in the LP group (Figure S3), indicating the more effective active targeting efficiency of CD44-LP.
Apoptosis assays
Cell apoptosis was detected using an annexin V-FITC/propidium iodide (PI) apoptosis assay kit. Briefly, 1×106 HepG2 cells were seeded in six-well plates and incubated for 24 h. The cells were treated with TAIII, LP, and CD44-LP at a dose of 10 μM for 24 h. After incubation, the cells were washed twice with PBS, resuspended in binding buffer, and mixed with annexin V-FITC and PI at room temperature for 15 min. Finally, cell apoptosis induced by drug treatment was analyzed with BD FACSCalibur flow cytometer.
To visually observe the toxicity of TAIII and liposomes, HepG2 cells were seeded onto glass-bottomed culture dishes (7×104 cells/well) and incubated with TAIII, LP, and CD44-LP for 24 h. After 24 h incubation, cells were washed with PBS and then stained with annexin V-FITC/PI double staining kit, followed by observation under confocal laser scanning microscopy (CLSM).
The expression of apoptosis-related protein and cleaved forms of caspase-3 and caspase-9 was evaluated with Western blotting. Cells were treated with TAIII, LP, and CD44-LP at a dose of 10 μM for 24 h. After incubation, cells were collected and total protein was extracted using the cell lysis reagent. The total protein was quantified using the BCA protein assay kit (Promega Corporation, Fitchburg, WI, USA) and electrophoresed on SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were blotted with primary antibodies at recommended concentrations overnight at 4°C, followed by appropriate secondary antibodies. The immunoreactivities were visualized with an enhanced chemiluminescent system. Cells cultured as usual without treatment were used as a control. The band intensities were measured, and the protein signals were normalized to the GAPDH levels. As shown in Figure S6, the percentages of apoptosis in HepG2 cells incubated with TAIII alone, LP, and CD44-LP for 24 h were 18.0%±3.2%, 20.3%±1.9% and 23.5%±1.5%, respectively. Early- or late-stage apoptotic cells were stained green (annexin V-FITC), and dead or later apoptotic cells were stained red (PI). Western blotting revealed that TAIII and liposomes significantly increased the active cleaved forms of caspase-9 and caspase-3, suggesting that TAIII and liposomes initiate caspase-dependent apoptosis in HepG2 cells.
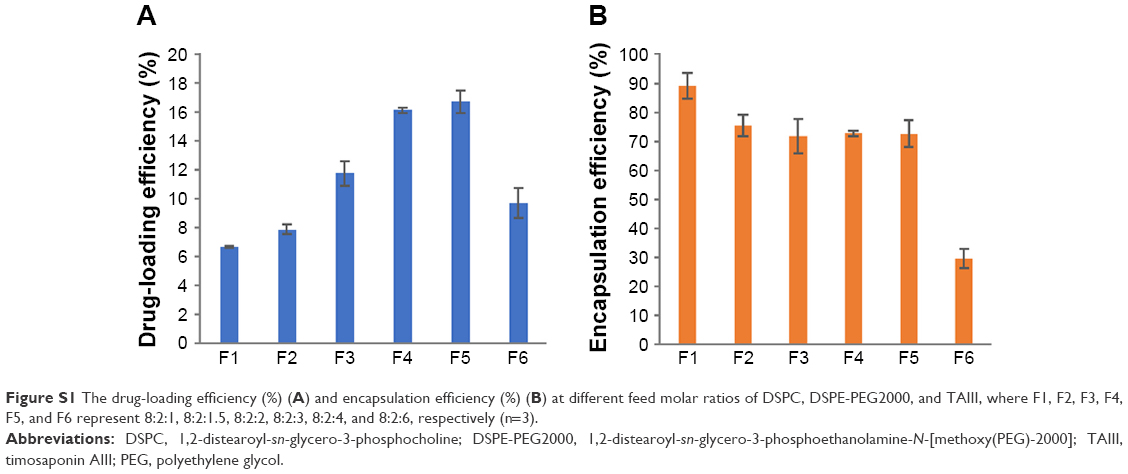 | Figure S1 The drug-loading efficiency (%) (A) and encapsulation efficiency (%) (B) at different feed molar ratios of DSPC, DSPE-PEG2000, and TAIII, where F1, F2, F3, F4, F5, and F6 represent 8:2:1, 8:2:1.5, 8:2:2, 8:2:3, 8:2:4, and 8:2:6, respectively (n=3). |
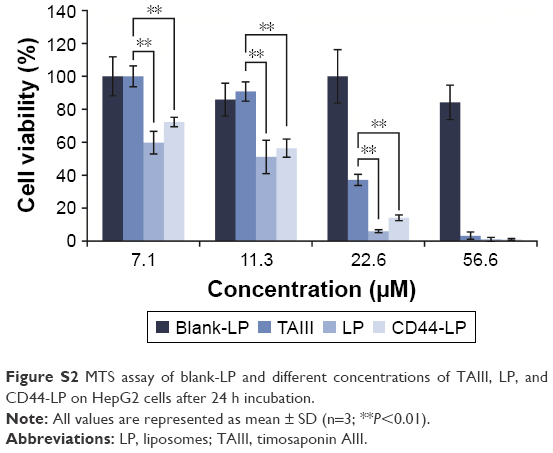 | Figure S2 MTS assay of blank-LP and different concentrations of TAIII, LP, and CD44-LP on HepG2 cells after 24 h incubation. |
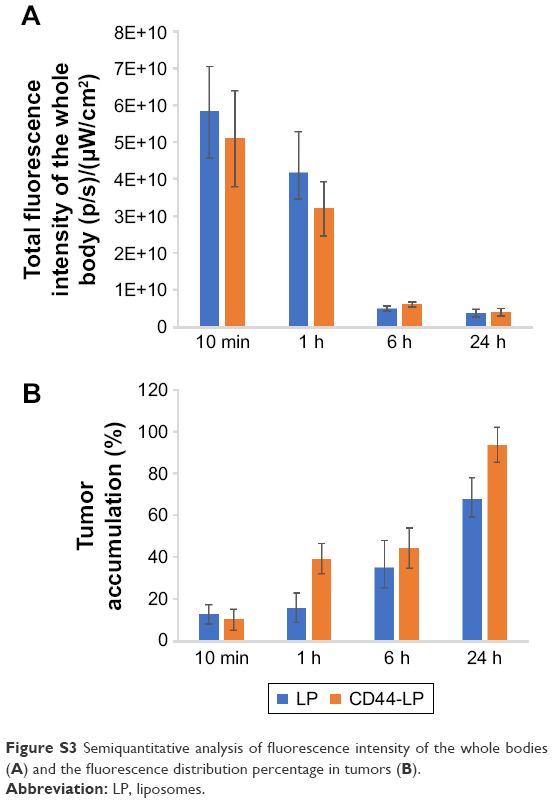 | Figure S3 Semiquantitative analysis of fluorescence intensity of the whole bodies (A) and the fluorescence distribution percentage in tumors (B). |
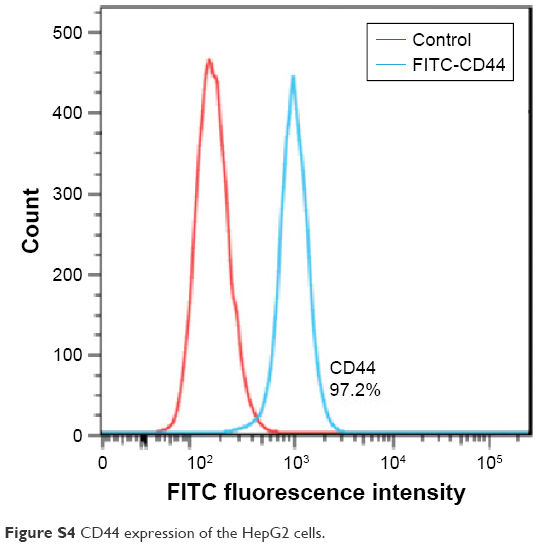 | Figure S4 CD44 expression of the HepG2 cells. |
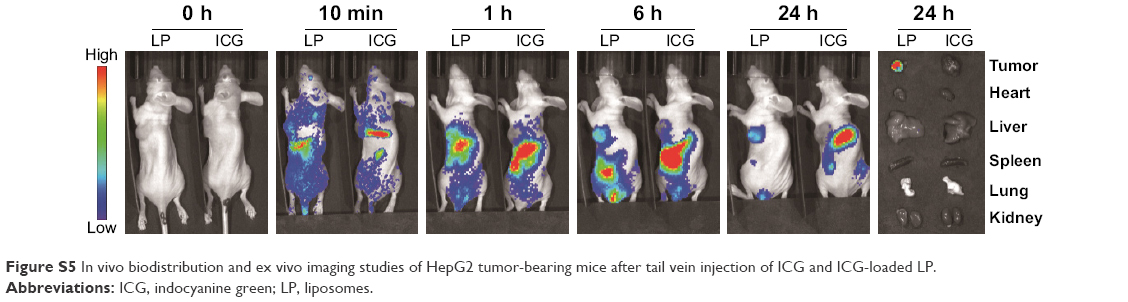 | Figure S5 In vivo biodistribution and ex vivo imaging studies of HepG2 tumor-bearing mice after tail vein injection of ICG and ICG-loaded LP. |
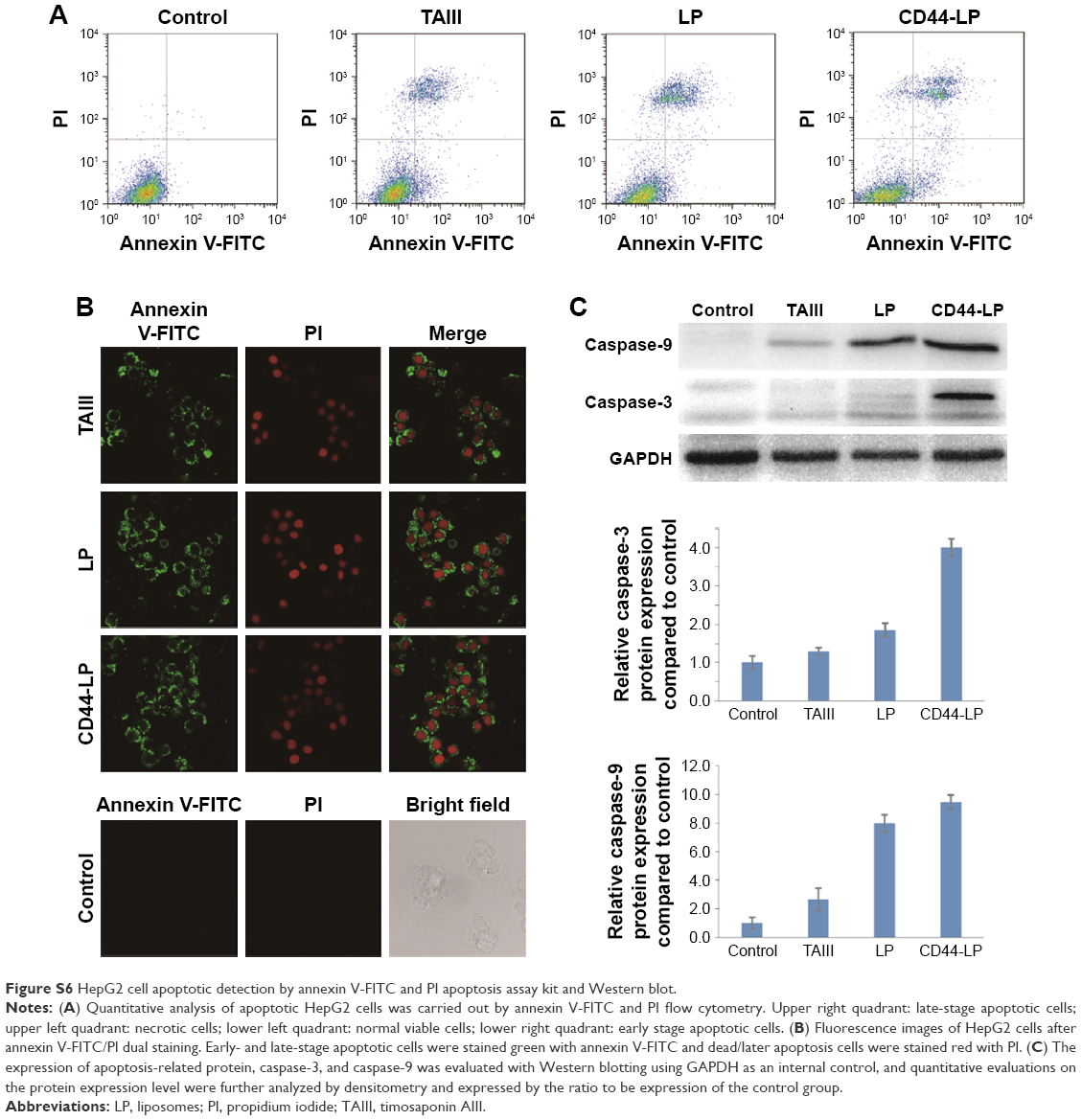 | Figure S6 HepG2 cell apoptotic detection by annexin V-FITC and PI apoptosis assay kit and Western blot. |
 | Table S1 In vitro cytotoxicity of TAIII, LP, and CD44-LP against HepG2 cells (n=3) |
References
Wang L, Su W, Liu Z, et al. CD44 antibody-targeted liposomal nanoparticles for molecular imaging and therapy of hepatocellular carcinoma. Biomaterials. 2012;33(20):5107–5114. | ||
Zheng X, Zhou F, Wu B, Chen WR, Xing D. Enhanced tumor treatment using biofunctional indocyanine green-containing nanostructure by intratumoral or intravenous injection. Mol Pharm. 2012;9(3):514–522. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.