")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Antibody-mediated targeting of iron oxide nanoparticles to the folate receptor alpha increases tumor cell association in vitro and in vivo
Authors Ndong C, Toraya-Brown S, Kekalo K, Baker I, Gerngross T, Fiering S, Griswold K
Received 16 December 2014
Accepted for publication 18 January 2015
Published 1 April 2015 Volume 2015:10(1) Pages 2595—2617
DOI https://doi.org/10.2147/IJN.S79367
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Christian Ndong,1 Seiko Toraya-Brown,2 Katsiaryna Kekalo,1 Ian Baker,1 Tillman U Gerngross,1,3,4 Steven N Fiering,2,5,6 Karl E Griswold1,3,6
1Thayer School of Engineering, Dartmouth, Hanover, NH, USA; 2Department of Microbiology and Immunology, Geisel School of Medicine at Dartmouth, Hanover, NH, USA; 3Department of Biological Sciences, Dartmouth, Hanover, NH, USA; 4Department of Chemistry, Dartmouth, Hanover, NH, USA; 5Department of Genetics, Geisel School of Medicine at Dartmouth, Hanover, NH, USA; 6Norris Cotton Cancer Center, Lebanon, NH, USA
Abstract: Active molecular targeting has become an important aspect of nanoparticle development for oncology indications. Here, we describe molecular targeting of iron oxide nanoparticles (IONPs) to the folate receptor alpha (FOLRα) using an engineered antibody fragment (Ffab). Compared to control nanoparticles targeting the non-relevant botulinum toxin, the Ffab-IONP constructs selectively accumulated on FOLRα-overexpressing cancer cells in vitro, where they exhibited the capacity to internalize into intracellular vesicles. Similarly, Ffab-IONPs homed to FOLRα-positive tumors upon intraperitoneal administration in an orthotopic murine xenograft model of ovarian cancer, whereas negative control particles showed no detectable tumor accumulation. Interestingly, Ffab-IONPs built with custom 120 nm nanoparticles exhibited lower in vitro targeting efficiency when compared to those built with commercially sourced 180 nm nanoparticles. In vivo, however, the two Ffab-IONP platforms achieved equivalent tumor homing, although the smaller 120 nm IONPs were more prone to liver sequestration. Overall, the results show that Ffab-mediated targeting of IONPs yields specific, high-level accumulation within cancer cells, and this fact suggests that Ffab-IONPs could have future utility in ovarian cancer diagnostics and therapy.
Keywords: nanoparticle targeting, antibody fragment, biodistribution, ovarian cancer
Introduction
Despite widespread advances in cancer diagnostics and treatment, ovarian cancers continue to have high mortality, with 5-year survival rates remaining near 45% since the mid-1990s.1 Hyperthermia represents one promising approach for peritoneal cancer therapy, as this modality has the capacity to kill cancer cells in a direct fashion and also indirectly stimulates an anticancer immune response.2–6 In seeking to apply hyperthermia therapy to dispersed peritoneal tumors, however, delivering thermal doses to malignant cells in a precise and controlled fashion represents a substantial technical barrier.
Iron oxide nanoparticles (IONPs) have a decades-long history as heating mediators in hyperthermia,7 and advances in nanoparticle fabrication and functionalization have fueled further interest in this research space.8–10 Preferential accumulation of IONPs in the tumor, however, remains a challenge in balancing efficacy and safety.8,11 One recent study co-opted tumor-associated peritoneal phagocytes to selectively deliver IONPs in an ovarian cancer model.6 In other work, luteinizing hormone–releasing hormone (LHRH) peptide was used as an IONP-targeting moiety for ovarian cancer cells overexpressing the LHRH receptor.12 Similarly, many ovarian cancers overexpress folate receptor alpha (FOLRα),13,14 and this fact has been leveraged to selectively target IONPs via functionalization with the cognate folic acid ligand.15 Monoclonal antibodies and antibody fragments have also been used to selectively target IONPs to ovarian cancer cells,16,17 but to date there is no report of antibody-mediated IONP targeting to the FOLRα surface protein. Antibody targeting of FOLRα might offer performance advantages over targeting with the folic acid ligand, as the former should be highly specific to FOLRα, while the latter is also bound with high affinity by folate receptors beta and gamma, and can interfere with uptake of circulating folate in patients.18,19
In the current research, we describe the development and characterization of IONPs functionalized with an engineered fab fragment of Farletuzumab, a humanized monoclonal antibody that has demonstrated tumor-inhibitory effects in pre-clinical models20–22 and in clinical trials.23 Tumor-specific homing of the antibody fragment Farletuzufab (Ffab)-targeted IONPs was assessed both in vitro and in vivo, and the results were compared to negative control particles targeting an irrelevant protein. In aggregate, these studies demonstrate the performance advantage of IONPs that actively target the FOLRα cancer marker.
Materials and methods
Cells lines and culture conditions
KB cells, derived from a human squamous cell carcinoma of the oral cavity, were obtained as a gift from Dr Philip S Low at Purdue University (West Lafayette, IN, USA). These KB cells were found to produce disseminated peritoneal tumors that are representative of advanced ovarian cancer in humans. The cells were maintained as a monolayer in folate-free Roswell Park Memorial Institute (RPMI) 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere consisting of 5% CO2 and 95% air. Cells were harvested with 0.25% trypsin, suspended, and spun down at 1,200 rpm prior to re-suspension and use in subsequent experiments.
Construction of Ffab and anti-botulinum toxin fab fragments
Ffab and negative control anti-botulinum toxin fab fragment (Bfab) were reformatted from their corresponding full length immunoglobulin G (IgG) monoclonal antibody sequences, which are available from the literature.24,25 Coding sequences for the variable and constant regions of the heavy and light chains from respective full length IgGs were reverse translated, codon optimized for expression in mammalian cells, and synthesized by DNA 2.0 (DNA 2.0 Inc., Menlo Park, CA, USA). Both Ffab and Bfab heavy chains were designed to carry an engineered cysteine tag at their respective C-termini for subsequent site-specific conjugation to maleimide-polyethylene glycol 2 (PEG2)-biotin (Thermo Fisher Scientific) or nanoparticles. Ffab and Bfab light- and heavy-chain constructs were individually sub-cloned into the CMVR VRC01 expression vector (National Institutes of Health [NIH] acquired immunodeficiency syndrome [AIDS] Reagent Program, Germantown, MD, USA).
Expression, extraction, and purification of Ffab protein
CMVR VRC01 expression vectors separately harboring Ffab light chain and heavy chain or Bfab light chain and heavy chain were co-transfected into suspension HEK 293 cells using polyethylenimine (Polysciences Inc, Warrington, PA, USA) as previously described.26 Secreted Ffab and Bfab were clarified through centrifugation at 8,000 rpm at 4°C for 15 minutes on an Avanti® J 25 centrifuge (Beckman Coulter Inc, Brea, CA, USA). The resulting supernatants were filtered through a 0.45 μm filter to remove any residual cell debris and other large particles before loading onto a fast protein liquid chromatography (FPLC) affinity column for purification.
Affinity purification was performed on a pre-packed 5 mL KappaSelect column from GE Healthcare Bio-Sciences Corporation (Piscataway, NJ, USA) as suggested by manufacturer instructions. Fabs were eluted with 100 mM glycine at pH 2.7 in 2 mL Eppendorf tubes prefilled with 50 μL of 1 M tris(hydroxymethyl)aminomethane (Tris), 5 mM ethylenediaminetetraacetic acid (EDTA). The purification process was automated on an AKTA™ FPLC system (GE Healthcare Bio-Sciences Corp). Purified proteins were subjected to a second step size exclusion chromatography column using Superdex® 75 (GE Healthcare Bio-Sciences Corp). Final products were eluted in phosphate-buffered saline (PBS) and stored at −20°C until used.
Reductive activation and chemical conjugation of the purified fabs to maleimide-PEG2-biotin (Thermo Fisher Scientific) were performed as described previously.26 Briefly, maleimide-PEG2-biotin was added to a final concentration of 2 mM (10 to 1 molar excess for maleimide-PEG2-biotin to proteins). The conjugation reaction was allowed to proceed for 2 hours at room temperature. The excess of maleimide-PEG2-biotin was removed by buffer exchange through a HiTrap™ desalting column using PBS pH 7.0, and the resultant Bfab and Ffab conjugates (Bfab-maleimide-PEG2-biotin and Ffab-maleimide-PEG2-biotin) were stored at −20°C until further use. Purified and conjugated proteins were analyzed using non-reduced and reduced sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) conditions and stained with Coomassie blue. Protein constructs were verified further using electrospray ionization time-of-flight (ESI-TOF) liquid chromatography mass spectrometry (Agilent Technologies, Santa Clara, CA, USA).
Affinity measurement of Ffab
The affinity of Ffab protein was analyzed using enzyme-linked immunosorbent assay (ELISA) and biolayer interferometry on the ForteBio Octet® Red instrument (ForteBio, Menlo Park, CA, USA). For ELISA, commercial rFOLR1-his protein (Sino Biological Inc, Beijing, People’s Republic of China) reconstituted at 50 μg/mL (1.9 μM) in water was diluted in coating buffer (100 mM sodium carbonate at pH 9.4) to 100 ng/mL (3 nM). Two 96-well Immulon™ 4HBX high protein binding plates (Thermo Fisher Scientific) were coated with 100 μL of rFOLR1-his at 4°C for 16 hours. Bindings with serial dilutions of Ffab and Bfab (0–200 nM) were performed as previously described.26 The monovalent affinity of Ffab was measured by biolayer interferometry. Briefly, rFOLR1-his was loaded in a 96-well plate at different concentrations (1.56–25 nM). Ffab was coupled to streptavidin biosensor tips (ForteBio) at 20 μg/mL and immersed into the rFOLR1-his dilutions. Ffab association and dissociation rates and equilibrium affinity were determined using software provided with the instrument.
Ffab cell-binding studies
For live cell binding, KB cells were harvested from T250 flasks (USA Scientific, Ocala, FL, USA) and seeded into a 96-well microplate at 50,000 cells per well using PBS containing 2% FBS. Different concentrations of Ffab or Bfab (0–200 nM) were added to cells and incubated at room temperature on a shaker (150 rpm) for 1 hour. Plates were centrifuged at 1,200 rpm for 5 minutes and washed twice with cold PBS containing 2% FBS using a vacuum unit. Cells were incubated with Phycolink® Streptavidin-R-Phycoerythrin (Prozyme, Hayward, CA, USA) at room temperature for 30 minutes, centrifuged, and washed as in previous step. Cells were resuspended in PBS, 2% FBS, and analyzed on a MacsQuant® instrument (Miltenyi Biotec Inc, Auburn, CA, USA).
Production of carboxymethyl dextran IONPs
Commercially available ferric chloride (FeCl3·6H2O), ferrous sulfate (FeSO4·7H2O), 25 wt% ammonium hydroxide solution, NaNO3, and NaOH were purchased from VWR International (Radnor, PA, USA). Carboxymethyl-dextran (CMD) 40 kDa was purchased from TdB Consultancy AB (Uppsala, Sweden). All reactants were used as received without further purification. Magnetic nanoparticles (MNPs) with CMD embedded in their structure, as described by Kekalo and Baker,27 were also obtained. Briefly, 10% solutions of salts of Fe and Fe(III) were precipitated by ammonia solution in the presence of excess of polysaccharide. The mixture was placed on a sand bath and heated up to 70°C. Then, NaOH and NaNO3 were added to oxidize Fe and maintain alkali media (pH >10). The temperature was raised up to 100°C at a speed of 10°C/hour. The resulting solution was spun at 5,000 rpm for 15 minutes to remove large aggregates. The remaining MNPs were purified using an LS magnetic column separator (Miltenyi Biotec).
Nanoparticle characterization
Transmission electron micrographs of the nanoparticles were taken using an FEI Technai F20ST field emission gun transmission electron microscope operated at 200 kV. Five hundred MNPs from three different locations on a grid were used to produce frequency vs particle size histograms.
Antibody-IONP conjugation and characterization
The 25 nm core size CMD-coated nanoparticles were obtained from the Dartmouth nanoparticle core facility, and the 100 nm core size aminodextran-coated bionized nanoferrite (BNF) nanoparticles were purchased from Micromod Partikeltechnologie GmbH (Rostock, Germany). Prior to use, CMD and BNF-IONPs were purified using MACS® separation LS columns (Miltenyi Biotec) and eluted with sterile water. CMD nanoparticles were maleimide functionalized by adding N-(2-Aminoethyl)maleimide and 1-ethyl-3-(3-dimethylaminopropyl)carboiimide (EDC) (both purchased from Sigma-Aldrich Co, St Louis, MO, USA) in 100-fold molar excess and incubating for 2 hours at room temperature in 100 mM 2-morpholinoethanesulfonic acid (MES) pH 6.3. BNF nanoparticles were maleimide functionalized by adding sulfo-GMBS ( N-[γ-maleimobutyryloxy] sulfosuccinimide ester) (Thermo Fisher Scientific) in 100-fold molar excess and incubating at room temperature for 2 hours in 100 mM sodium phosphate buffer pH 7.2. The excess N-(2-Aminoethyl)maleimide, EDC, and sulfo-GMBS were removed by buffer exchange with 30 mM MES pH 6.5 (for CMD particles) or 30 mM MES, 5 mM EDTA pH 6.5 (for BNF particles) using MACS separation LS columns.
Purified Ffab and Bfab were reduced with 20 mM cysteine as described previously,26 followed by buffer exchange on HiTrap desalting columns (GE Healthcare Bio-Sciences Corp) with 30 mM MES pH 6.5 (for CMD particles) or 30 mM MES, 5 mM EDTA pH 6.5 (for BNF particles). CMD-cysteine-reduced Ffab and Bfab were added to the maleimide-functionalized CMD and BNF-IONPs at a 1:10 (w/w) ratio and incubated at room temperature for 16 hours at 4°C on a shaker set at 125 rpm. This ratio was empirically determined to yield high-binding IONP conjugates and reproducible conjugation results. The unbound protein was then removed using MACS separation LS columns and magnetic field. All processes were performed in a sterile environment using sterile and endotoxin-free buffers. Micro BCA assay (Thermo Fisher Scientific) was used to determine the amount of Ffab and Bfab covalently bound to IONPs, as described previously.26
The hydrodynamic Z-average diameters (HDD) and zeta potentials (mV) of IONP-Fab conjugates were measured using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). For measuring the HDD, the IONPs were diluted to 0.05 mg/mL in PBS. For the determination of zeta potential, the IONPs were diluted to a concentration of 0.2 mg/mL in 10 mM NaCl.
In vitro IONP-fab binding studies
The commercial rFOLR1-his (Sino Biological Inc) was diluted in PBS to 500 ng/mL (20 nM), and a 96-well Immulon 4HBX high-protein binding plate (Thermo Fisher Scientific) was coated with 100 μL of rFOLR-his at 4°C for 16 hours. Coating buffer was removed by aspiration and replaced with 300 μL of blocking buffer (2% bovine serum albumin [BSA; w/v] in 1× PBS, pH 7.4). Serial dilutions of Ffab-CMD and Bfab-CMD (0–5 nM) or Ffab-BNF and Bfab-BNF (0–1 nM) in sample diluents (0.1% BSA [w/v] in PBS, pH 7.4) were added and incubated at room temperature for 1 hour. Nanoparticle uptake was measured using a ferrozine-based iron assay as described previously.26 Briefly, plates were washed three times using washing buffer (Tris buffer saline [TBS], 0.05% Tween20 [v/v]). After washing, 100 μL of 1.4 M HCl was added to each well, and plates were sealed and heated at 70°C for 2 hours. Plates were centrifuged at 200 rpm to settle liquids, 100 μL of ferrozine reagent (6.5 mM ferrozine, 13 mM neocuproine, and 2 M ascorbic acid diluted in 5 M ammonium acetate) were added to each well, and plates were shaken for 5 minutes. Plates were read at 562 nm (using a SpectraMax® 190; Molecular Devices LLC, Sunnyvale, CA, USA), and the amount of iron per well was determined from a standard curve of iron (FeCl3) in identically processed samples.
For cellular binding, KB cells were seeded at 100,000 cells per well on a 48-well plate (Corning Incorporated, Corning, NY, USA) and allowed to attach overnight. Old medium was removed and replaced with 300 μL of fresh medium containing 35 μg/mL of CMD-Ffab (0.6 nM), CMD-Bfab (0.6 nM), BNF-Ffab (0.035 nM), or BNF-Bfab (0.035 nM). Cells were incubated at 37°C for 8 hours, and unbound nanoparticles were removed by washing three times with PBS. Cells were treated with 100 μL of sodium hydroxide (NaOH), placed on a shaker in a 37°C room for 1 hour, and the ferrozine assay was performed as described above in this section, except 300 μL of ferrozine reagent was added.26
For transmission electron microscopy, KB cells treated with IONPs for 8 hours were washed with PBS and fixed with a fixative solution (3% glutaraldehyde and 1% paraformaldehyde in 0.1 M sodium cacodylate, pH 7.4) at room temperature for 15 minutes. After incubation, cells were briefly centrifuged and fresh fixative solution was added, followed by an additional 16 hours incubation at 4°C. Fixed cells were submitted to the Dartmouth Medical Electron Microscope Facility for imaging.
Peritoneal tumor model and IONP biodistribution
All mice were cared for according to approved Institutional Animal Care and Use Committee (IACUC) animal protocol. NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were originally obtained from Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA, and were bred at the Geisel School of Medicine at Dartmouth. At 5–6 weeks old, female mice were injected intraperitoneally (IP) on day 0 with 2×106 KB cells in 400 μL PBS. Within 2 weeks, tumors started growing on the peritoneal wall and in the peritoneal cavity, much like human ovarian cancer. For testing IONP biodistribution, Ffab-CMD, Bfab-CMD, Ffab-BNF, or Bfab-BNF were injected IP at 750 μg iron in 400 μL PBS on day 18. Approximately 18 hours after IONP injection, mice were euthanized using CO2 according to the IACUC approved protocol, followed by perfusion with 20 mL of PBS from the left ventricle. Fat, spleen, kidney, liver, and tumors growing on the peritoneal wall and in the peritoneal cavity were harvested in pre-weighed conical tubes. Each tissue was weighed and subjected to acid digestion using 3–10 mL of trace metal grade acid mixture (9:1 HNO3:HCl; Thermo Fisher Scientific) at 90°C for 1 hour. A 0.5 mL aliquot of 30% trace metal H2O2 (GFC Chemicals, Powell, OH, USA) was then added, and the samples were diluted to 15–50 mL total volume with double-distilled water. Dartmouth Trace Element Analysis Core Facility measured each digest tissue’s iron content using inductively coupled plasma mass spectrometry (ICP-MS).
In addition to quantitative iron analysis of tissues, qualitative histology was performed on representative tumor sections. Approximately 18 hours after IONP injection, mice were euthanized using CO2, and tumor cells on the peritoneal wall and cavity were harvested and embedded in paraffin. Paraffin sections (5 μm) on microscope slides were heated in an oven set at 79°C for 30 minutes and deparafinized. Samples were stained for iron using a Gomori Prussian blue iron stain kit (Newcomer Supply, Middleton, WI, USA) following the manufacturer’s protocol, and were then mounted with Permount™ (Thermo Fisher Scientific). Samples were immersed in hematoxylin (Thermo Fisher Scientific) for 5 minutes, washed with water, immersed in eosin (Fisher, Pittsburgh, PA) for 2 minutes, and washed with water again. Slides were dehydrated with increasing concentrations of ethanol and then xylene, and were mounted with Permount. Tissue histology was performed with the help of Dartmouth Pathology Translational Research Services.
Results
Fab design, production, and analysis
The workflow for IONP functionalization and subsequent conjugation with the engineered antibodies is shown in Figure 1. The two antibody fragments, Farletuzufab (Ffab), targeting the FOLRα cancer antigen, and Botulifab (Bfab), targeting the negative control protein botulinum toxin, were constructed such that each retained an unpaired C-terminal cysteine on the heavy chain. This design ultimately enabled site-specific conjugation to either maleimide-PEG2-biotin- or maleimide-functionalized IONPs. Ffab and Bfab were expressed in HEK 293 cells, and the desired fab monomers were isolated by Kappa select affinity purification followed by size exclusion chromatography (Figure S1A, B).
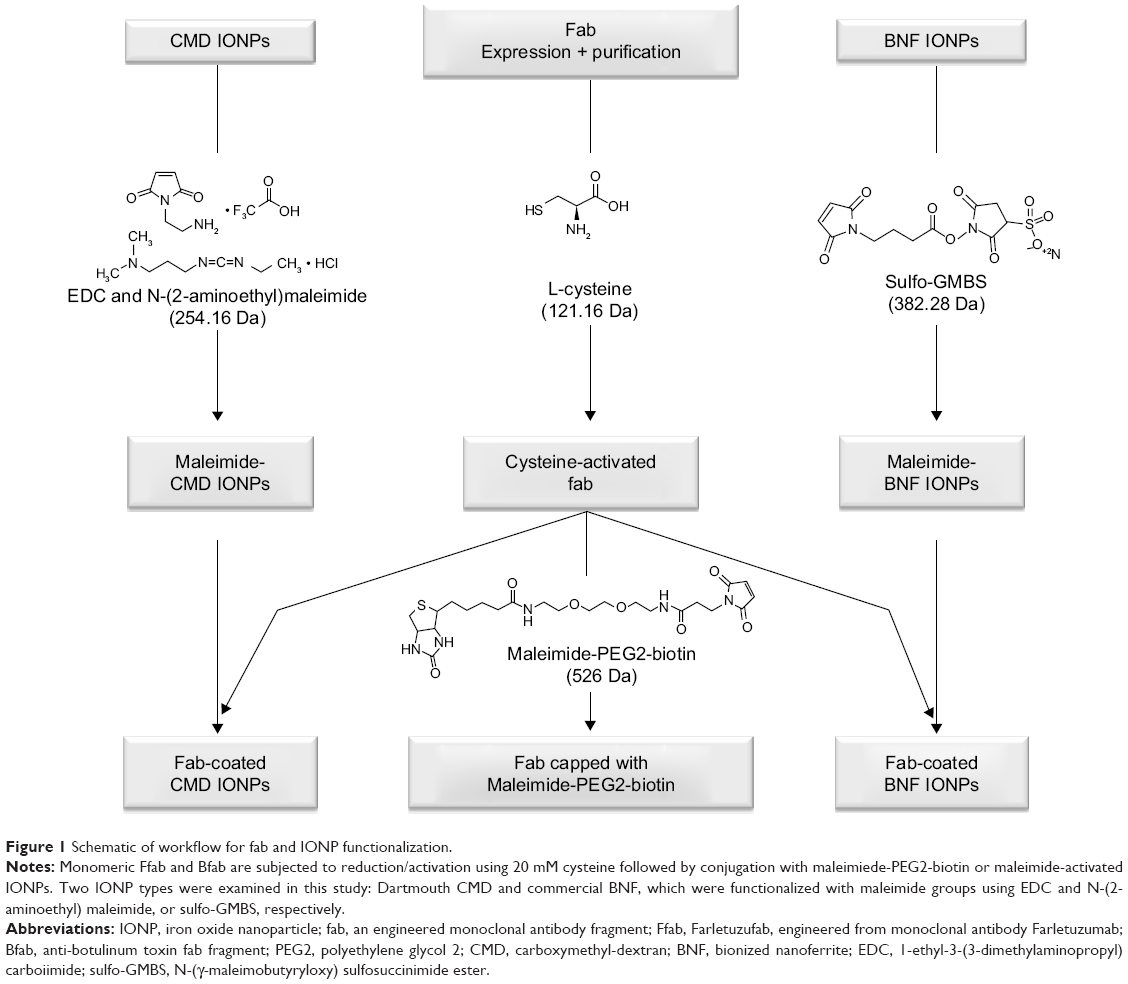 | Figure 1 Schematic of workflow for fab and IONP functionalization. |
Mass spectral analysis of Ffab and Bfab
The identities of monomeric Ffab and Bfab were verified by liquid chromatography-mass spectrometry (LC-MS). The base peak of Ffab (expected [exp] =47,989 Da, observed [obs] =48,109 Da) was 120 Da heavier than expected, suggesting the presence of a cysteine adduct (Figure S1C). Bfab, on the other hand, exhibited a +103 Da difference (exp =48,528 Da, obs =48,631 Da), the source of which was not immediately obvious (Figure S1D). Upon reductive activation with free cysteine, Ffab showed the expected peak at 47,989 Da as well as peaks corresponding to free heavy chain (exp =24,239 Da, obs =24,239 Da) and free light chain (exp =23,750 Da, obs =23,751 Da) (Figure S1E). Similar results were observed previously with a fab fragment of Trastuzumab, and it was determined then that the free heavy and light chains resulted from transient reduction of the intrachain disulfide bond, with no significant loss of binding activity or specificity.26 Cysteine-activated Bfab exhibited a −17 Da difference relative to the theoretical molecular mass (exp =48,528 Da, obs =48,511 Da). This difference is consistent with N-terminal deamination of glutamine,28 and it explains the observed mass of the pre-activation material (ie, a de-aminated cysteine adduct yields +103 Da). Similar to cysteine-activated Ffab, the activated Bfab exhibited two lower molecular mass peaks that corresponded with free heavy and light chains (Figure S1F).
To assess the chemical reactivity of the engineered antibody fragments, conjugation reactions were performed with maleimide-PEG2-biotin. As expected, the reactions resulted in addition of one biotin moiety (+526 Da) to each of the intact fab antibodies (Figure S1G, H), and this observation demonstrated that the engineered fab was amenable to site-specific conjugation as designed. The free heavy chain and light chain of both activated antibody preparations also reacted as expected, with the addition of one biotin moiety to the light chain and two to the heavy chain. These results are also consistent with previous work on the Trastuzumab fab antibody fragment.26
Ffab and Bfab purities were evaluated by SDS-PAGE. Under reducing conditions (presence of 50 mM dithiothreitol), the heavy and light chains of both Ffab and Bfab migrated as a single 25 kDa band (Figure 2A, B; lane 1). In non-reducing conditions (no dithiothreitol), Ffab and Bfab migrated predominantly as the expected 48 kDa band, although less intense low molecular weight bands corresponding to free heavy chain and light chain were also observed (Figure 2C, D; lane 1). Following reductive activation of both Ffab and Bfab, much of the intact fab band at 48 kDa was shifted to the lower molecular weight bands for free heavy chain and light chain (Figure 2C, D; lane 2), which was consistent with the appearance of these species during LC-MS analysis. Upon conjugation to maleimide-PEG2-biotin, substantial portions of the intact fabs were reconstituted, although bands for free heavy and light chain remained evident (Figure 2C, D; lane 3). The LC-MS analysis had identified heavy and light chain species in which the cysteines involved in interchain disulfide bond formation had been capped with maleimide-PEG2-biotin, and the corresponding free heavy and free light chain bands in the SDS-PAGE of biotinylated fabs corroborated that result (ie, maleimide-capped light and heavy chains are unable to reform intrachain disulfide bonds). We emphasize again that previous studies have shown that analogous, reduced yet intact fabs retain the binding activity and efficiency of their disulfide bonded counterparts.26,29
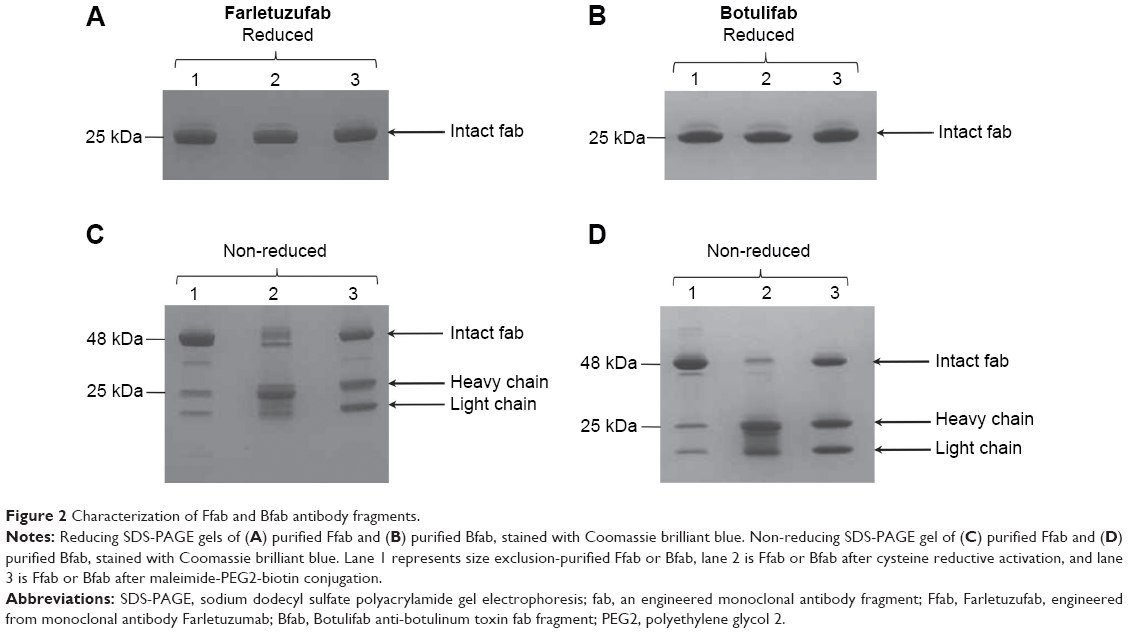 | Figure 2 Characterization of Ffab and Bfab antibody fragments. |
Ffab binds specifically to FOLRα
Binding of Ffab and Bfab to recombinant FOLRα (rFOLRα) was initially analyzed by ELISA. The biotin-labeled Ffab fragment exhibited a half maximal effective concentration (EC50) of 12 nM, whereas no binding was observed with the biotinylated Bfab control (Figure 3A). Additionally, more detailed rFOLRα binding kinetics were analyzed by biolayer interferometry (Figure S2). Ffab-maleimide-PEG2-biotin was immobilized on streptavidin biosensor tips and assayed with rFOLRα protein. The equilibrium dissociation constant for Ffab (KD =14 nM) was comparable to the EC50 obtained by ELISA (Figure 3A). Importantly, no detectable binding of Bfab-maleiemide-PEG2-biotin was observed with rFOLRα using the same streptavidin biosensors tips (data not shown). In aggregate, the quantitative binding studies with rFOLRα demonstrated that the engineered Ffab retained good binding affinity to rFOLRα, to which the parental Farletuzumab IgG was found to have a KD =2 nM.20
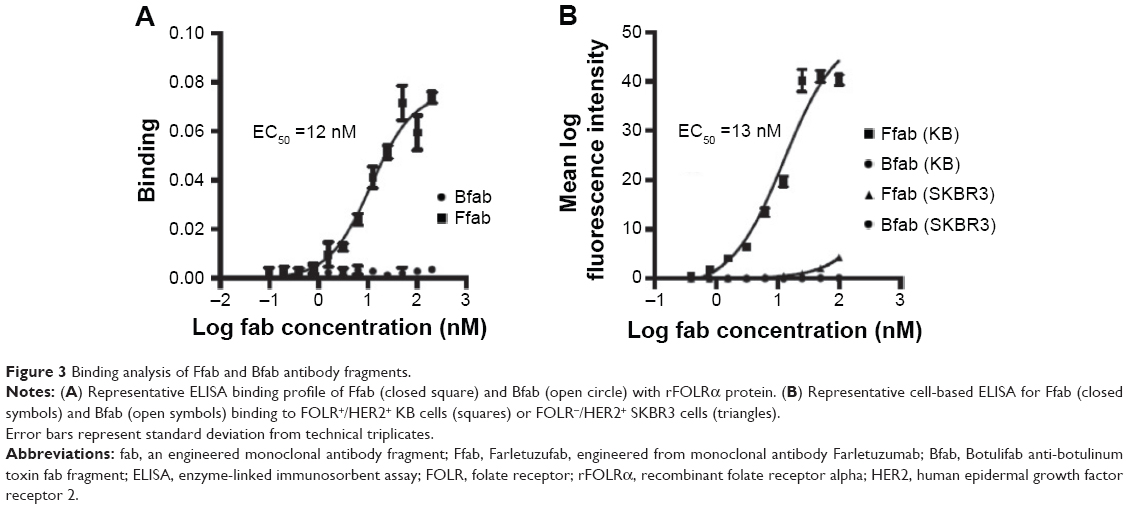 | Figure 3 Binding analysis of Ffab and Bfab antibody fragments. |
In addition to binding recombinant protein, it was critical that the engineered Ffab selectively bind the FOLRα surface protein in its native context. To assess cellular binding, the biotinylated Ffab and Bfab antibodies were assayed by flow cytometry using live human KB and SKBR3 cancer cells, where the former overexpress both FOLRα and human epidermal growth factor receptor 2 (HER2), and the latter overexpresses HER2 only. Ffab was found to exhibit high apparent binding affinity for KB cells (EC50 =13 nM), whereas no substantial binding was observed on SKBR3 cells (Figure 3B). As expected, the negative control Bfab failed to bind either KB or SKBR3 tumor cells (Figure 3B). Thus, the Ffab antibody efficiently targeted cancer cells in a FOLRα-dependent fashion.
Analysis of Ffab-functionalized IONPs
Having validated Ffab binding affinity and specificity for soluble and cell surface FOLRα, the antibody fragment was conjugated to both CMD and BNF-maleimide-IONPs that differ in nanoscale structure and carbohydrate coating (Figure S3). These conjugations yielded Ffab-CMD and Ffab-BNF constructs that differed in size (~120 nm vs ~190 nm hydrodynamic diameter, respectively) (Figure S4) and number of antibody-targeting moieties (~50 vs ~500 moieties, respectively; Table 1).
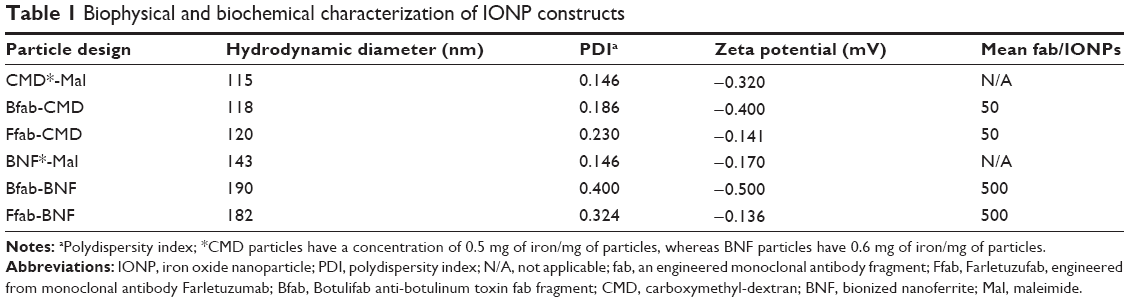 | Table 1 Biophysical and biochemical characterization of IONP constructs |
FOLRα binding of the Ffab-CMD and Ffab-BNF constructs, and their respective Bfab-IONP negative control counterparts was evaluated by ELISA (Figure 4A, B). Compared to the monomeric Ffab antibody, both Ffab-IONP constructs exhibited orders of magnitude higher apparent affinities (Ffab-BNF EC50 =0.013 nM; Ffab-CMD EC50 =0.16 nM), which reflects avidity effects derived from the nanoparticles’ polyvalent nature. Conversely, no substantial binding was observed with the Bfab-BNF or Bfab-CMD negative control nanoparticles. These results demonstrated that Ffab binding of rFOLRα was not compromised during IONP conjugation, and that the Bfab-IONP controls had no inherent affinity for the recombinant receptor. It is noteworthy that the larger Ffab-BNF particles exhibited 10-fold higher apparent affinity compared to the Ffab-CMD particles. This effect likely results from their 10-fold difference in number of conjugated antibodies (500 vs 50 antibodies for BNF and CMD, respectively; Table 1). The higher maximum signal of the Ffab-BNF particles is likely due, in part, to their higher apparent affinity, and in part, due to the fact that BNF nanoparticles possess greater iron content than CMD nanoparticles (0.6 mg iron/mg of particles vs 0.5 mg iron/mg of particles, respectively).
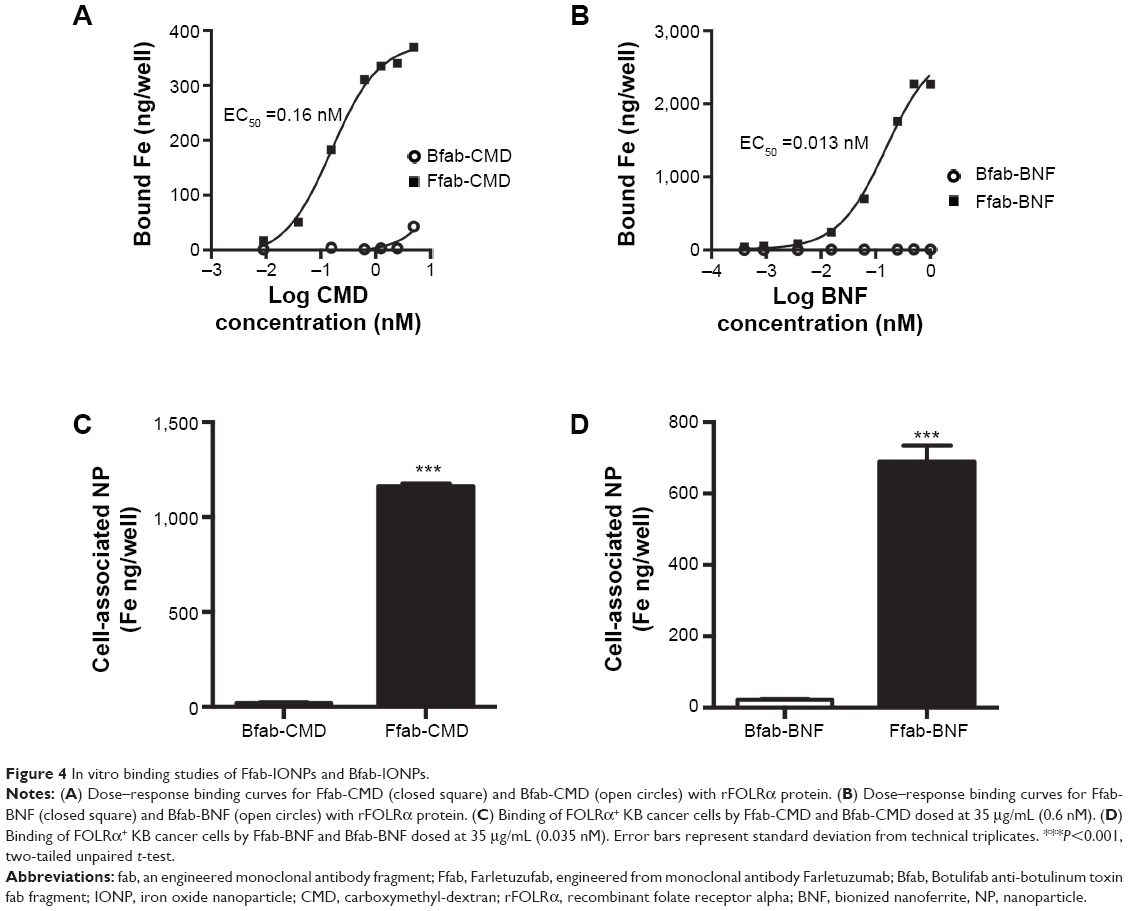 | Figure 4 In vitro binding studies of Ffab-IONPs and Bfab-IONPs. |
The targeting capacity of the nanoparticles was further assessed using FOLRα-positive cancer cells. Both Ffab-CMD and Ffab-BNF efficiently bound adherent KB tumor cells during an 8-hour incubation in complete medium, whereas the Bfab-targeted IONPs showed little to no cellular association (Figure 4C, D). Importantly, the greater accumulation of Ffab-CMD (0.6 nM) compared to Ffab-BNF (0.035 nM) is due to the experimental design, where both particle types were incubated at a mass concentration of 35 μg/mL, yielding a 17-fold greater molar concentration of CMD nanoparticles.
As part of the KB cellular binding studies, the subcellular localization of Ffab-CMD and Ffab-BNF conjugates was analyzed by transmission electron microscopy (TEM). TEM micrographs showed that, following an 8-hour cellular incubation in vitro, Ffab-CMD and Ffab-BNF can be found accumulated on both the cell membrane as well as within intracellular vesicles (Figure 5A, B, D, E). Consistent with the negligible binding of Bfab-CMD and Bfab-BNF observed above (Figure 4C, D), no Bfab-targeted particles could be found in corresponding TEM images (Figure 5C, F). In aggregate, these results demonstrate specific and efficient targeting of FOLRα-positive cancer cells by Ffab-CMD and Ffab-BNF, and additionally, they reveal that the Ffab-targeting moiety facilitates cellular internalization of both nanoparticle constructs.
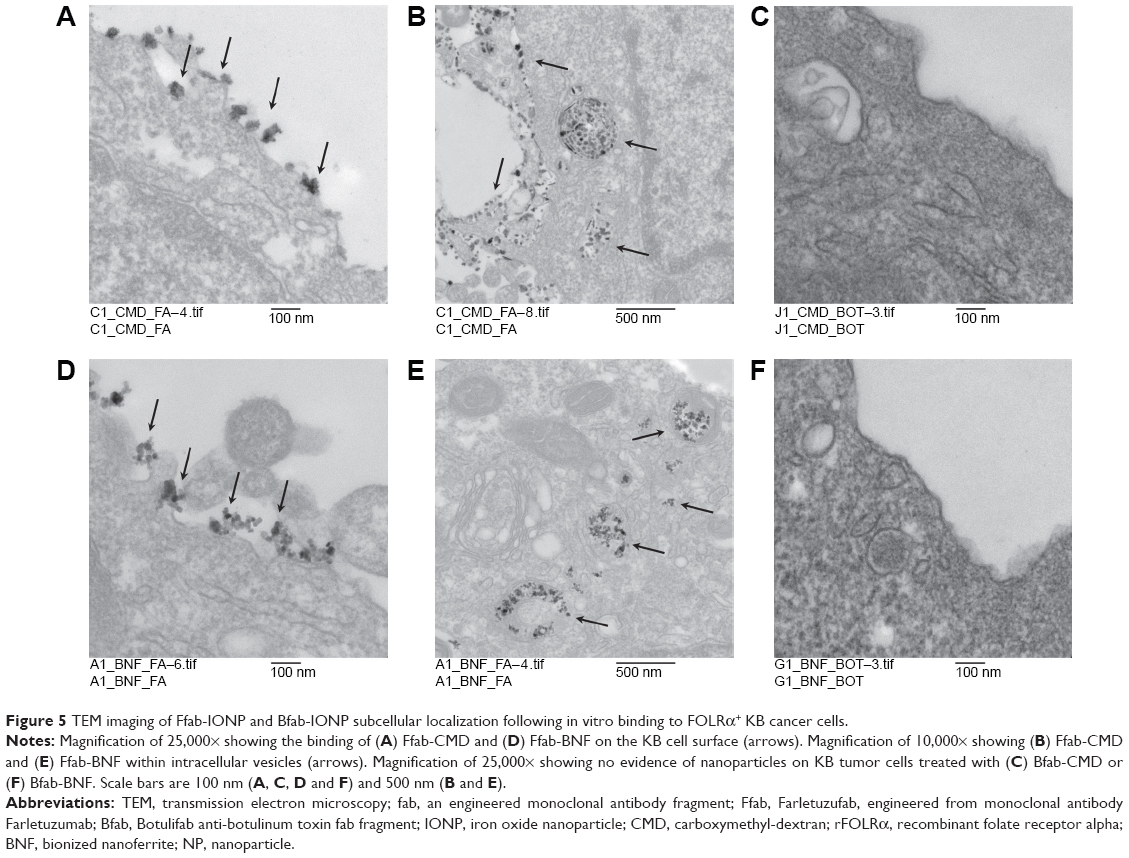 | Figure 5 TEM imaging of Ffab-IONP and Bfab-IONP subcellular localization following in vitro binding to FOLRα+ KB cancer cells. |
In vivo tumor targeting of IONPs
Based on the in vitro performance of targeted Ffab-IONPs, we anticipated that these nanoparticle constructs might also exhibit enhanced tumor localization in vivo. To test this hypothesis, a single dose (750 μg of iron) of each IONP was administered intraperitoneally (IP) to NSG mice bearing peritoneal tumors derived from human KB cells. Approximately 18 hours post-injection, tumors, surrounding fat, and other major organs in the abdominal cavity were harvested, and IONP content was quantified by ICP-MS (Figure 6 shows total tissue iron; Figure S5 shows tissue iron concentration). As seen in the in vitro cellular binding studies, the Bfab-CMD and Bfab-BNF negative controls failed to show significant iron accumulation within in vivo tumor masses (Figure 6A). In contrast, Ffab-BNF and Ffab-CMD yielded statistically significant increases in tumor-associated iron (Figure 6A), averaging ~5% or ~7%, respectively, of the initially injected dose. Histology of tissue sections showed the KB tumors to be largely localized on the peritoneal wall (Figure 7A–C), and Prussian blue staining for iron was qualitatively consistent with the ICP-MS analysis, revealing significant iron accumulation on the surface of and within tumors from animals treated with Ffab-BNF (compare Figure 7F to D and E).
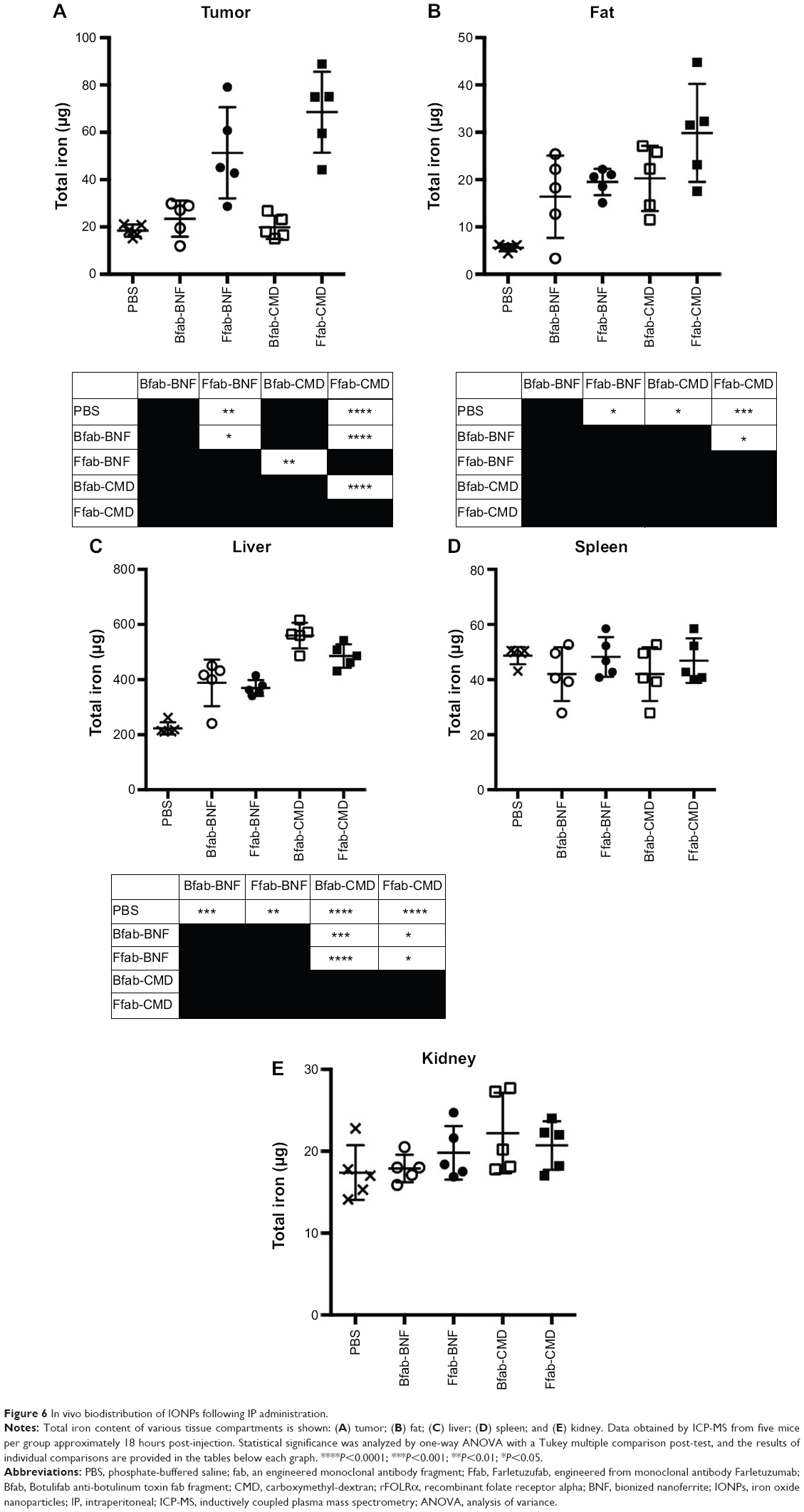 | Figure 6 In vivo biodistribution of IONPs following IP administration. |
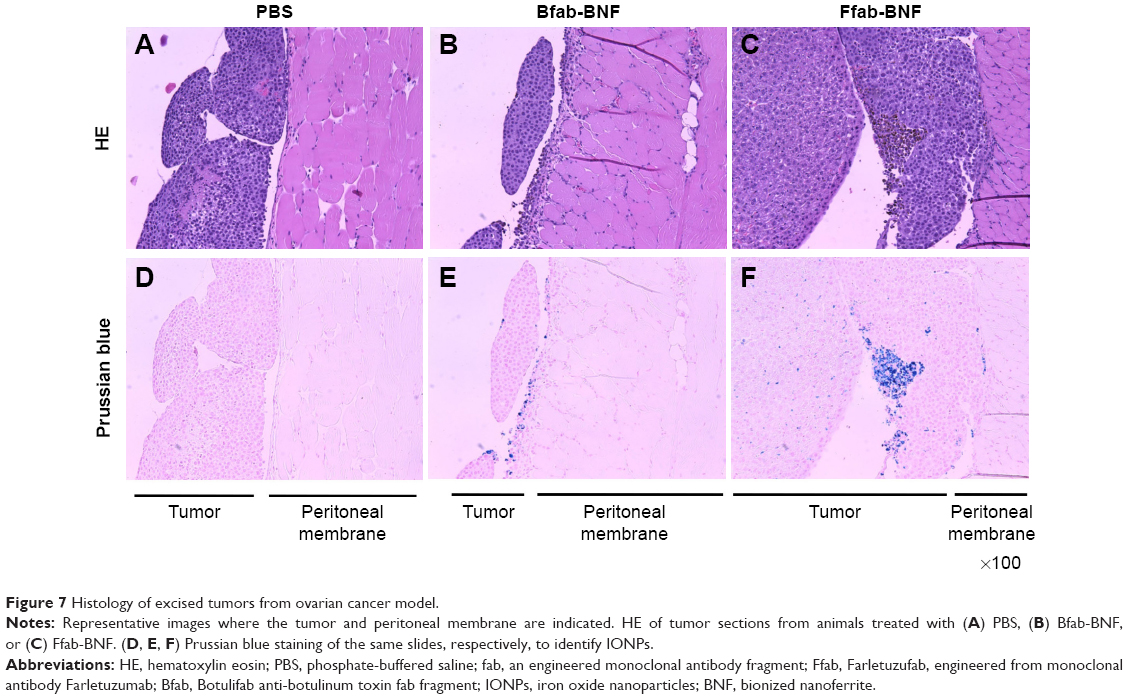 | Figure 7 Histology of excised tumors from ovarian cancer model. |
For both Ffab-IONPs and Bfab-IONPs, significant iron accumulation was also noted in the peritoneal fat tissue. There was a trend toward greater fat accumulation of Ffab-CMD vs the other constructs (~3% of the injected dose vs ~2% for other constructs), although the difference was only significant in comparison to Bfab-BNF (Figure 6B). For all four particle types, the majority of the recovered nanoparticles were found in the liver (Figure 6C). While the identity of the targeting antibody did not substantially influence liver accumulation, CMD-based nanoparticles showed a significantly higher liver concentration than did BNF nanoparticles (~40% injected dose vs ~20%, respectively). When compared to PBS-injected animals, animals treated with IONPs experienced no significant increase in splenic or kidney iron levels (Figure 6D and E, respectively).
Discussion
IONPs represent multi-functional nanomaterials with enormous value in the field of oncology. They have undergone extensive development as contrast agents for imaging, as cell capture reagents for diagnostics, as controlled release drug delivery platforms, and as energy converters for heat deposition in magnetic hyperthermia.7,9,10,30–54 In all of these applications, the ultimate utility of IONPs is critically dependent on differential association with malignant vs healthy cells; selective partitioning to the tumor environment and/or tumor cells is the key to enabling effective IONP diagnostics and therapeutics. In the current study, we have examined molecular targeting of IONPs to FOLRα-positive cancers using an engineered antibody fragment.
FOLRα is a glycosylphosphatidylinositol-anchored glycoprotein whose expression is generally restricted in normal tissues, while often exhibiting high expression levels in cancers of epithelial origin.13,14 IONPs have been targeted to ovarian cancers via functionalization with folic acid, the natural ligand of FOLRα.15 Importantly, however, folate is an essential nutrient, and healthy cells acquire folate from their environment via numerous high-affinity membrane receptor proteins and transporters, including FOLRα, FOLR beta, FOLR gamma,19 reduced folate carrier (solute carrier 19A1),55,56 and a proton-coupled folate transporter (solute carrier 46A1).57 Agents targeted by conjugation to folic acid can interfere with the uptake of naturally circulating folate, and they are known to bind both FOLRα and FOLRβ, which reduce their selectivity for cancer cells.19 On the other hand, the monoclonal IgG antibody Farletuzumab is a highly specific binder of FOLRα, and it has been shown to exhibit little to no cross-reactivity with healthy cells and tissues.20 Thus, Farletuzumab, or engineered fragments thereof, represent interesting candidates for tumor-specific targeting of IONPs.
In this study, we have demonstrated that 1) Ffab, a reformatted fragment of Farletuzumab, maintains the binding activity and specificity of the parental monoclonal antibody; and 2) functionalization of IONPs with Ffab substantially increases selective iron deposition in cultured cancer cells and in in vivo peritoneal tumors. Ffab-targeted IONPs were constructed from two different nanoparticle foundations: 1) commercially sourced BNF particles, and 2) custom synthesized CMD particles. The BNF particles had a larger hydrodynamic diameter (~190 nm vs ~120 nm), contained 1.2-fold more iron per particle, and bore a 10-fold greater number of Ffab-targeting moieties on their surface.
The distinct characteristics of the two IONP platforms resulted in differential targeting efficiency in vitro, where the larger Ffab-BNF particles had a 10-fold higher affinity for rFOLRα and accumulated 10-fold more iron when used at an equivalent molar concentration. Conversely, when used at equal mass concentrations, Ffab-CMD particles accumulated approximately 2-fold more iron during in vitro incubation with FOLRα-positive cancer cells. Importantly, however, both Ffab-BNF and Ffab-CMD outperformed controls targeted to an irrelevant protein, and both Ffab-targeted IONP types resulted in substantial cellular internalization. Internalization of antibody-targeted IONPs has been reported previously.26,58,59 and the capacity to internalize IONPs via FOLRα could have important implications for hyperthermia or cytotoxic drug delivery to ovarian cancers.3,60
In contrast to their differential in vitro binding activities, in vivo administration of the two IONP platforms at a fixed iron dose showed no significant difference with respect to tumor homing in a murine model of ovarian cancer. This observation underscores the fact that, with respect to cancer-specific targeting, the results of in vitro experiments cannot be readily extrapolated to predict in vivo performance. In a previous murine model study of breast cancer, we showed that IONP size was the dominant determinant of tumor localization following intravenous administration of either targeted or non-targeted constructs.26 Interestingly, in that study, smaller 30 nm IONPs localized to the tumor compartment, whereas larger 100 nm IONPs did not. Conversely, larger 100 nm IONPs localized almost exclusively to the liver, whereas smaller 30 nm IONPs manifested significantly lower liver accumulation. In the current study, IONP size also correlated with liver sequestration, but in an inverted fashion: smaller 120 nm IONPs showed greater liver accumulation than larger 180 nm IONPs (40% vs 20% of the injected dose, respectively).
Unfortunately, numerous confounding factors make it difficult to draw meaningful conclusions from comparison of the current ovarian cancer model and the previous breast cancer model,26 (eg, different nanoparticle types, nanoparticle sizes, cancer cell lines, the nature of the models, the route of IONP administration, etc). Nonetheless, the results reported here provide important insights into the biodistribution of IONPs following IP administration. In particular, molecular targeting of FOLRα resulted in a significant tumor accumulation of IONPs. It bears noting that, in larger tumor cross-sections, IONPs exhibited highly concentrated accumulation in limited areas, but smaller, punctate accumulations were also seen distributed throughout the cross-sectional fields of view (Figure 7F). This non-uniform but tumor-wide deposition is more analogous to results seen with intravenous IONP administration than with direct tumor injection, where the former has recently been shown to achieve better efficacy in thermal ablation treatments.49 Specifically, in their work, Huang and Hainfeld note that uniform IONP distribution throughout the tumor is not required to achieve therapeutic heating, but rather tumor-wide deposition and encasement is adequate.49
The 5%–7% of initially injected IONP dose that was found in the tumor in the current study equaled that of a previous study that leveraged tumor-associated peritoneal phagocytes to localize non-targeted IONPs to ovarian tumor masses.6 It is unclear whether the NSG mouse model of the current study possesses similar tumor-associated peritoneal phagocytes, but the lack of any negative control Bfab-targeted IONPs in the tumor mass might suggest that phagocyte-mediated trafficking of IONPs to the tumor is compromised in the NSG model. If true, we speculate that the use of FOLRα-targeted IONPs in a suitable immuno-competent model might result in additive tumor homing, and such a substantial IONP tumor accumulation would bode well for potential diagnostic and therapeutic applications.
In summary, in the present study, we have demonstrated molecular targeting of IONPs to the FOLRα cancer marker. Our targeting agent is based upon a clinically validated monoclonal antibody that has exquisite specificity for FOLRα vs other folate receptors and transporters, and this selectivity may enable enhanced tumor accumulation relative to alternative FOLRα targeting strategies. These FOLRα-specific iron oxide nanomaterials may ultimately prove useful in advancing diagnosis, imaging, and therapy of ovarian cancers.
Acknowledgments
The authors would like to thank Louisa Howard and Charles P Daghlian of the Dartmouth Electron Microscope Facility for their assistance with TEM data, and the Dartmouth Center for Cancer Nanotechnology Excellence Toxicology, Biodistribution and Pathology Core, and the Dartmouth Trace Element Core for ICP-MS data. We would also like to thank the Dartmouth Transgenic and Genetic Construct Shared Resources, and Jennifer Fields for maintaining the NSG mice. We also thank the Dartmouth CCOP Pathology Translational Research (especially Rebecca O’Meara and Eric York), for the assistance with tissue histology. Finally, we would like to thank Adimab LLC, in particular Yingda Xu and Felicia Reid, for their assistance with LC-MS data. This work was supported by NCI grant number 1 U54 CA151662-01.
Disclosure
The authors report no conflicts of interest in this work.
References
Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2011, National Cancer Institute. Bethesda, MD: National Cancer Institute; 2014. Available from: http://seer.cancer.gov/csr/1975_2011/. Accessed November 27, 2014. | ||
Zee Vd. Heating the patient: a promising approach. Annals of Oncology. 2002;13:1173–1184. | ||
Moroz P, Jones SK, Gray BN. Magnetically mediated hyperthermia: current status and future directions. Int J Hyperthermia. 2002;18:267–284. | ||
Ito A, Shinkai M, Honda H, Wakabayashi T, Yoshida J, Kobayashi T. Augmentation of MHC class I antigen presentation via heat shock protein expression by hyperthermia. Cancer Immunol Immunother. 2001;50:515–522. | ||
Ito A, Shinkai M, Honda H, et al. Heat shock protein 70 expression induces an antitumor immunity during intracellular hyperthermia using magnetite nanoparticles. Cancer Immunol Immunother. 2003;52:80–88. | ||
Toraya-Brown S, Sheen MR, Baird JR, et al. Phagocytes mediate targeting of iron oxide nanoparticles to tumors for cancer therapy. Integr Biol (Camb). 2013;5(1):159–171. | ||
Gilchrist RK, Medal R, Shorey WD, Hanselman RC, Parrott JC, Taylor CB. Selective inductive heating of lymph nodes. Ann Surg. 1957;146:596–606. | ||
Ivkov R, DeNardo SJ, Daum W, et al. Application of high amplitude alternating magnetic fields for heat induction of nanoparticles localized in cancer. Clin Cancer Res. 2005;11:7093S–7103S. | ||
Ito A, Shinkai M, Honda H, Kobayashi T. Medical application of functionalized magnetic nanoparticles. J Biosci Bioeng. 2005;100:1–11. | ||
Dennis CL, Jackson AJ, Borchers JA, et al. Nearly complete regression of tumors via collective behavior of magnetic nanoparticles in hyperthermia. Nanotechnology. 2009;20(39):395103. | ||
Fortin JP, Wilhem C, Servais J, Ménager C, Bacri JC, Gazeau F. Size-sorted anionic iron oxide nanomagnets as colloidal mediators for magnetic hyperthermia. J Am Chem Soc. 2007;129(9):2628–2635. | ||
Taratula O, Dani RK, Schumann C, et al. Multifunctional nanomedicine platform for concurrent delivery of chemotherapeutic drugs and mild hyperthermia to ovarian cancer cells. Int J Pharm. 2013;458:169–180. | ||
Weitman SD, Lark RH, Coney LR, et al. Distribution of the folate receptor GP38 in normal and malignant cells lines and tissues. Cancer Res. 1992;52:3396–3401. | ||
Garin-Chesa P, Campbell I, Saigo PE, Lewis JL Jr, Old LJ, Rettig WJ. Trophoblast and ovarian cancer antigen LK26. Sensitivity and specificity in immunopathology and molecular identification as a folate-binding protein. Am J Pathol. 1993;142:557–567. | ||
Krais A, Wortmann L, Hermanns L, et al. Targeted uptake of folic acid-functionalized iron oxide nanoparticles by ovarian cancer cells in the presence but not in the absence of serum. Nanomedicine. 2014;10(7):1421–1431. | ||
Quan G, Du X, Huo T, et al. Targeted molecular imaging of antigen OC183B2 in ovarian cancers using MR molecular probes. Acad Radiol. 2010;17:1468–1476. | ||
Prantner AM, Nguyen CV, Scholler N. Facile immunotargeting of nanoparticles against tumor antigens using site-specific biotinylated antibody fragments. J Biomed Nanotechnol. 2013;9(10):1686–1697. | ||
Kamen BA, Smith AK. Farletuzumab, an anti-folate receptor alpha antibody, does not block binding of folate or anti-folates to receptor nor does it alter the potency of anti-folates in vitro. Cancer Chemother Pharmacol. 2012;70(1):113–120. | ||
Marchetti C, Palaia I, Giorgini M, et al. Targeted drug delivery via folate receptors in recurrent ovarian cancer: a review. Onco Targets Ther. 2014;7:1223–1236. | ||
Ebel W, Routhier EL, Foley B, et al. Preclinical evaluation of MORAb-003, a humanized monoclonal antibody antagonizing folate receptor-alpha. Cancer Immun. 2007;7:6. | ||
Spannuth WA, Lyn YG, Merritt WM, et al. Therapeutic efficacy of folate receptor alpha blockade with MORAb-003 in ovarian cancer. Gynecol Oncol. 2008;108:S135. | ||
Spannuth WA, Sood AK, Coleman RL. Farletuzumab in epithelial ovarian carcinoma. Expert Opin Biol Ther. 2010;10(3):431–437. | ||
Armstrong DK, White AJ, Weil SC, Phillips M, Coleman RL. Farletuzumab (a monoclonal antibody against folate receptor alpha) in relapsed platinum-sensitive ovarian cancer. Gynecol Oncol. 2013;129:452–458. | ||
Wallace TP, Harris WJ, Carr FJ, Rettig WJ, Garin-Chesa P, Lloyd J, inventors. Sloan-Kettering Cancer Center, assignee. Recombinant human anti-LK26 antibodies. United States patent US 5952484. 1999 Sept 14. | ||
Marks JD, Amersdorfer P, Geren I, Lou J, Razai A, Consuelos MG, inventors. The Regents Of The University Of California, assignee. Therapeutic monoclonal antibodies that neutralize botulinum neurotoxins. United States patent US 20080124328A1. 2008 May 29. | ||
Ndong C, Tate JA, Kett WC, et al. Tumor cell targeting by iron oxide nanoparticles is dominated by different factors in vitro versus in vivo. PLoS One. 2015;10(2):e0115636. | ||
Kekalo KA, Baker I, inventors; Engineering DCTSo, assignee. Magnetic nanoparticles, composites, suspensions and colloids with high specific absorption rate (SAR). United States patent WO 2014089464 A1. 2014 Jun 12. | ||
Liu H, Gaza-Bulseco G, Faldu D, Chumsae C, Sun J. Heterogeneity of monoclonal antibodies. J Pharm Sci. 2008;97(7):2426–2447. | ||
Humphreys D, Heywood SP, Henry A, et al. Alternative antibody Fab’ fragment PEGylation strategies: combination of strong reducing agents, disruption of the interchain disulphide bond and disulphide engineering. Protein Eng Des Sel. 2007;20(5):227–234. | ||
Gupta AK, Gupta M. Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications. Biomaterials. 2005;26:3995–4021. | ||
Kohler N, Sun C, Wang J, Zhang M. Methotrexate-modified superparamagnetic nanoparticles and their intracellular uptake into human cancer cells. Langmuir. 2005;21:8858–8864. | ||
Lee H, Lee E, Kim DK, Jang NK, Jeong YY, Jon S. Antibiofouling polymer-coated superparamagnetic iron oxide nanoparticles as potential magnetic resonance contrast agents for in vivo cancer imaging. J Am Chem Soc. 2006;128:7383–7389. | ||
van Vlerken LE, Amiji MM. Multi-functionnal polymeric nanoparticles for tumor-targeted drug delivery. Expert Opin Drug Deliv. 2006;3(2):205–216. | ||
Lee JH, Huh YM, Jun YW, et al. Artificially engineered magnetic nanoparticles for ultra-sensitive molecular imaging. Nat Med. 2007;13(1):95–99. | ||
Peng X-H, Qian X, Mao H, et al. Targeted magnetic iron oxide nanoparticles for tumor imaging and therapy. Int J Nanomedicine. 2008;3(3):311–321. | ||
Cole AJ, Yang VC, David AE. Cancer theranostics: the rise of targted magnetic nanoparticles. Trends Biotechnol. 2011;29(7):323–332. | ||
Laurent S, Mahmoudi M. Superparamagnetic iron oxide nanoparticles: promises for diagnosis and treatment of cancer. Int J Mol Epidemiol Genet. 2011;2(4):367–390. | ||
Tassa C, Shaw SY, Weissleder R. Dextran-coated iron oxide nanoparticles: a versatile platform for targeted molecular imaging, molecular diagnostics, and therapy. Acc Chem Res. 2011;44(10):842–852. | ||
Kaaki K, Hervé-Aubert K, Chiper M, et al. Magnetic nanocarriers of doxorubicin coated with poly(ethylene glycol) and folic acid: Relation between coating strucutre, surface properties, colloidal stability and cancer cell targeting. Langmuir. 2012;28:1496–1505. | ||
Rosen JE, Chan L, Shieh DB, Gu FX. Iron oxide nanoparticles for targeted cancer imaging and diagnostics. Nanomedicine. 2012;8:275–290. | ||
Ittrich H, Peldschus K, Raabe N, Kaul M, Adam G. Superparmagnetic iron oxide nanoparticles in biomedicine: applications and developments in diagnostics and therapy. Rofo. 2013;185:1149–1166. | ||
Kirui DK, Khalidov I, Wang Y, Batt CA. Targeted near-IR hybrid magnetic nanoparticles for in vivo cancer therpay and imaging. Nanomedicine. 2013;9:702–711. | ||
Thomas R, Park I-K, Jeong YY. Magnetic iron oxide nanoparticles for multimodal imaging and therapy of cancer. Int J Mol Sci. 2013;14:15910–15930. | ||
Yen SK, Padmanabhan P, Selvan ST. Multifunctional iron oxide nanoparticles for diagnostics, therapy and macromolecule delivery. Theranostics. 2013;3(12):986–1003. | ||
Cortajarena AL, Ortega D, Ocampo SM, et al. Engineering Iron oxide nanoparticles for clinical settings. Nanobiomed. 2014;1(2):58841. | ||
Wabler M, Zhu W, Hedayati M, et al. Magnetic resonance imaging contrast of iron oxide nanoparticles developed for hyperthermia is dominated by iron content. Int J Hyperthermia. 2014;30(3):192–200. | ||
Silva AC, Oliveira TR, Mamami JB, et al. Application of hyperthermia induced by superparamagnetic iron oxide nanoparticles in glioma treatment. Int J Nanomedicine. 2011;6:591–603. | ||
Hilger I. In vivo applications of magnetic nanoparticles hyperthermia. Int J Hyperthermia. 2013;29(8):828–834. | ||
Huang HS, Hainfeld JF. Intravenous magnetic nanoparticles cancer hyperthermia. Int J Nanomedicine. 2013;8:2521–2532. | ||
Ivkov R. Magnetic nanoparticles hyperthermia: a new frontier in biology and medicine. Int J Hyperthermia. 2013;29(8):703–705. | ||
Petryk AA, Giustini AJ, Gottesman RE, Kaufman PA, Hoopes PJ. Magnetic nanoparticle hyperthermia enhancement of cisplatin chemotherapy cancer treatment. Int J Hyperthermia. 2013;29:845–851. | ||
Petryk AA, Giustini AJ, Gottesman RE, Trembly BS, Hoopes PJ. Comparison of magnetic nanoparticles and microwave hyperthermia cancer treatment methology and treatment effect in a rodent breast cancer model. Int J Hyperthermia. 2013;29:819–827. | ||
Toraya-Brown S, Sheen MR, Zhang P, et al. Local hyperthermia of tumors induces CD8(+) T cell-mediated resistance against distal and secondary tumors. Nanomedicine. 2014;10(6):1273–1285. | ||
Kossatz S, Ludwig R, Dähring H, et al. High therapeutic efficiency of magnetic hyperthermia in xenograft models achieved with moderate temperature dosages in tumor area. Pharm Res. 2014;31:3274–3288. | ||
Marthely LH, Hou Z, Deng Y. Human reduced folate carrier: translation of basic biology to cancer etiology and therapy. Cancer Metastasis Rev. 2007;26(1):111–128. | ||
Marthely LH, Hou Z. Structure and function of the reduced folate carrier: a paradigm of a major facilitator superfamily mammalian nutrient transporter. Vitam Horm. 2008;79:145–184. | ||
Qiu A, Jansen M, Sakaris A, et al. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell. 2006;127(5):917–928. | ||
Hilger I, Dietmar E, Linß W, Strec S, Kaiser WA. Developments for the minimally invasive treatment of tumours by targted magnetic heating. J Phys: Condens Mat. 2006;18:S2951–S2958. | ||
Zhang JP, Dewilde AH, Chinn P, et al. Herceptin-directed nanoparticles activated by an alternating magnetic field slectively kill HER-2 positive human breast cells in vitro via hyperthermia. Int J Hyperthermia. 2011;27:682–697. | ||
Wahajuddin, Arora S. Superparamagnetic iron oxide nanoparticles: magnetic nanoplatforms as drug carriers. Int J Nanomedicine. 2012;7:3445–3471. |
Supplementary materials
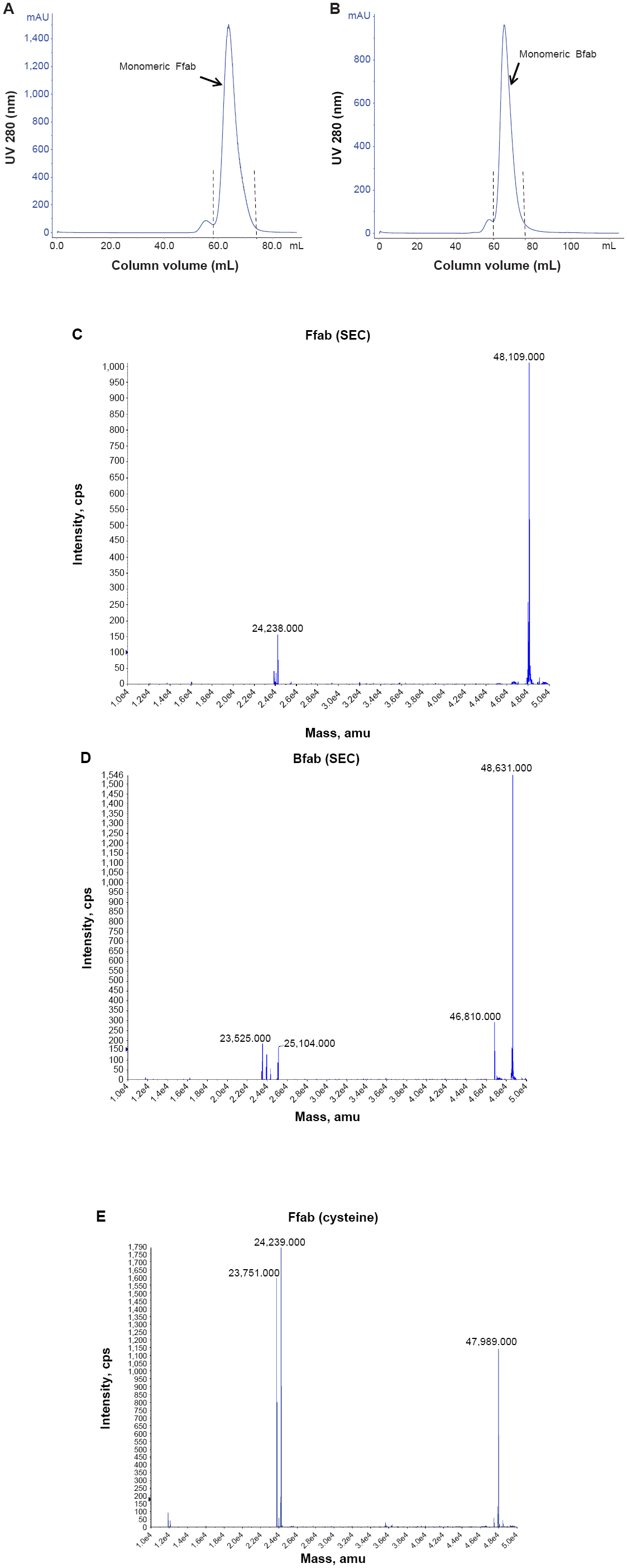 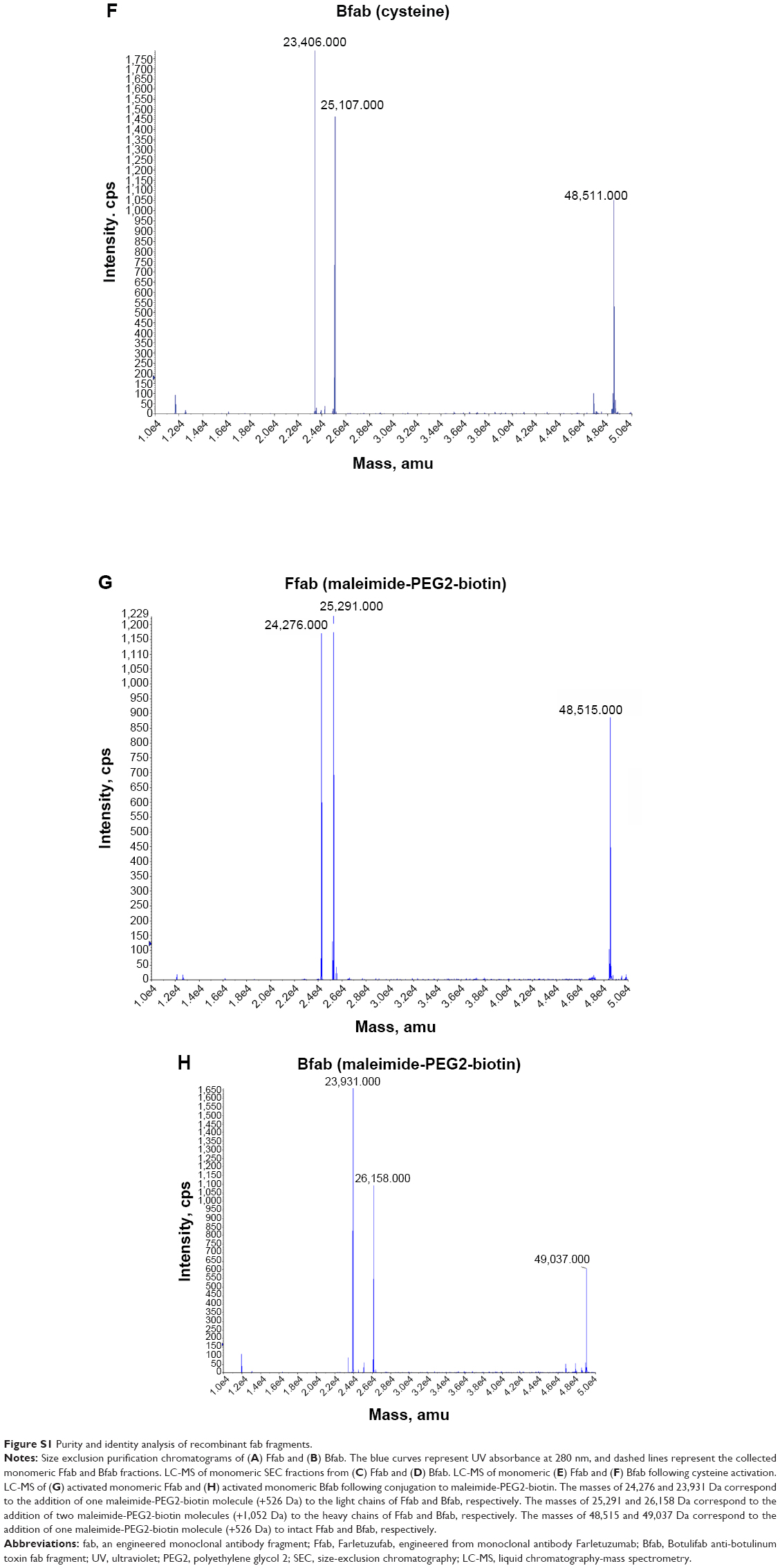 | Figure S1 Purity and identity analysis of recombinant fab fragments. |
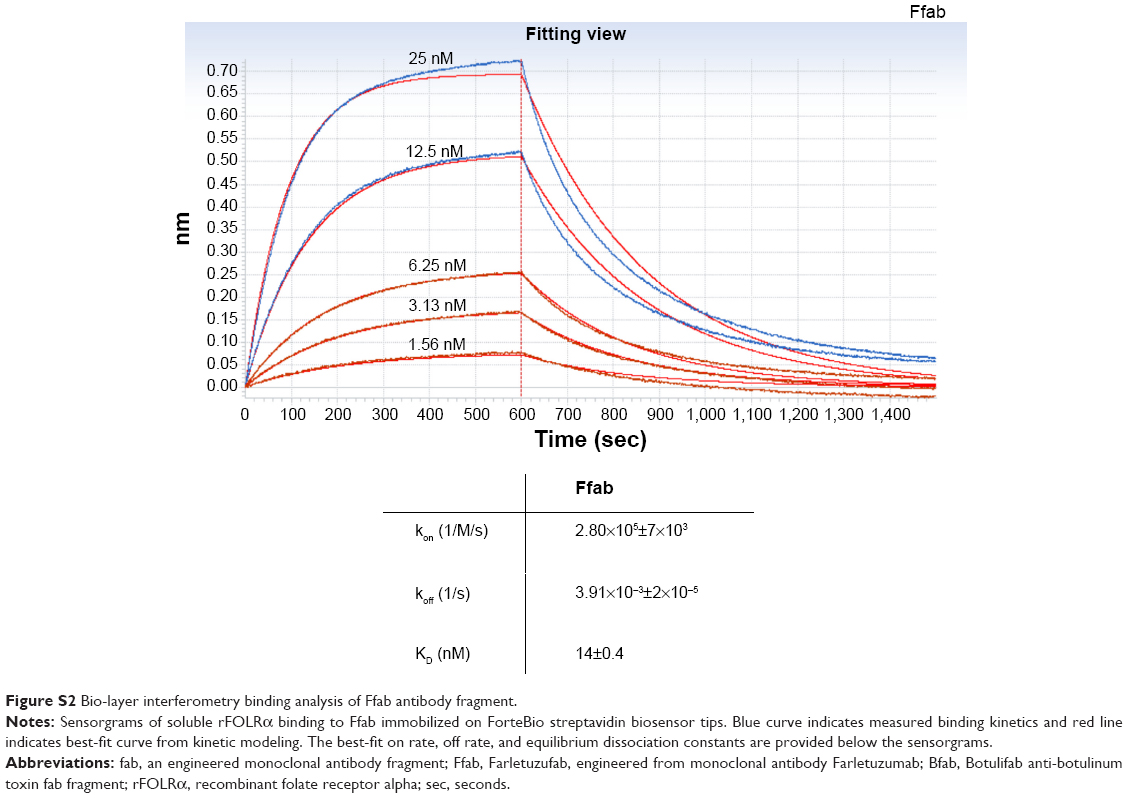 | Figure S2 Bio-layer interferometry binding analysis of Ffab antibody fragment. |
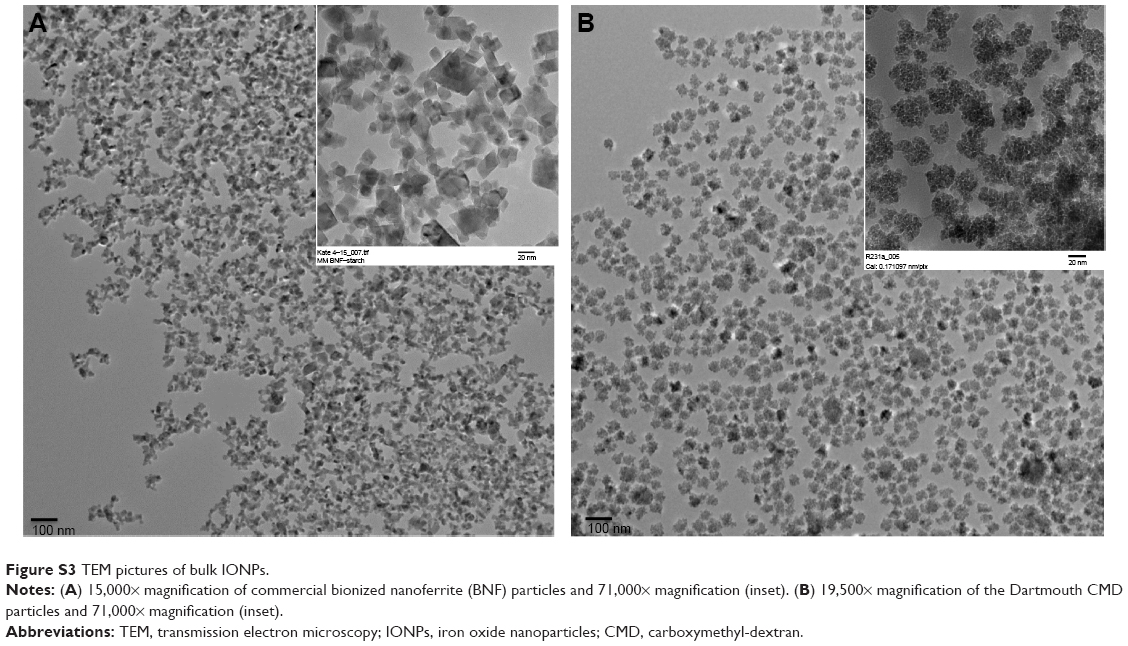 | Figure S3 TEM pictures of bulk IONPs. |
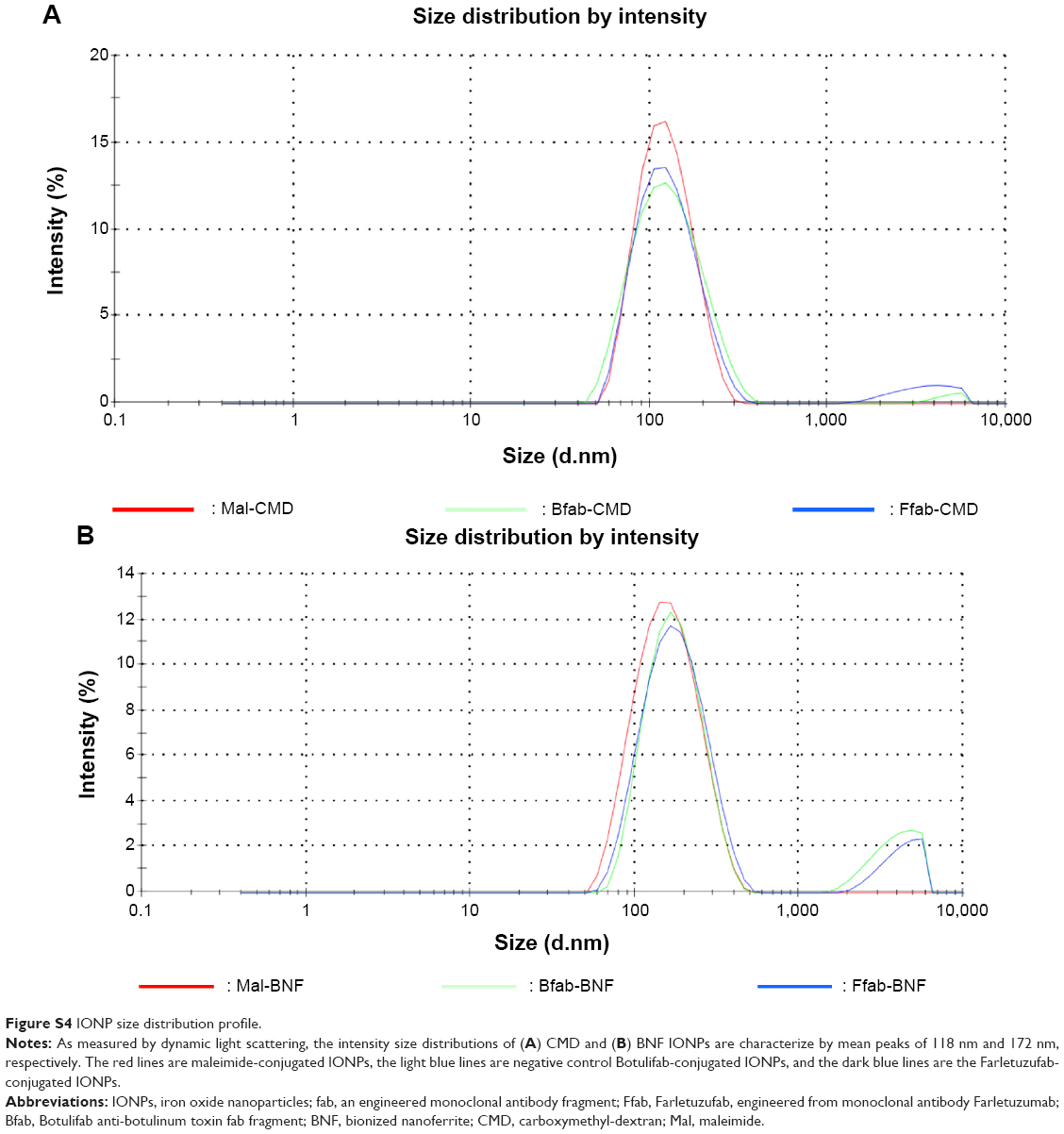 | Figure S4 IONP size distribution profile. |
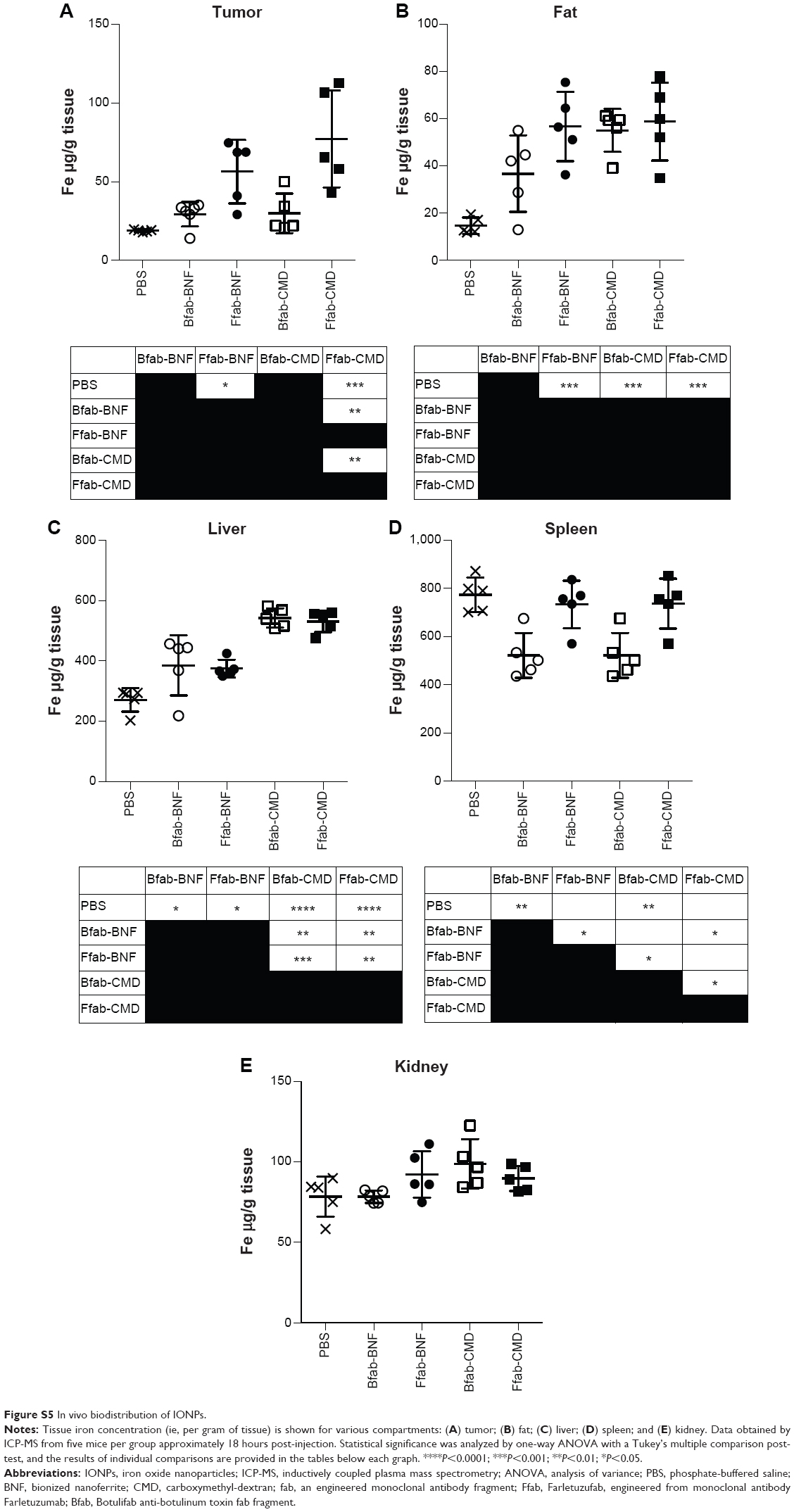 | Figure S5 In vivo biodistribution of IONPs. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.