Back to Journals » Journal of Pain Research » Volume 9
Anti-nerve growth factor in pain management: current evidence
Authors Chang D, Hsu E, Hottinger D, Cohen SP
Received 21 January 2016
Accepted for publication 9 March 2016
Published 8 June 2016 Volume 2016:9 Pages 373—383
DOI https://doi.org/10.2147/JPR.S89061
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Micheal Schatman
David S Chang,1 Eugene Hsu,2 Daniel G Hottinger,1 Steven P Cohen1,3–5
1Department of Anesthesiology and Critical Care Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, 2Clinical Excellence Research Center, Stanford University School of Medicine, Stanford, CA, 3Department of Anesthesiology, 4Department of Physical Medicine and Rehabilitation, Uniformed Services University of the Health Sciences, Bethesda 5Department of Physical Medicine and Rehabilitation, Johns Hopkins University School of Medicine, Baltimore, MD, USA
Abstract: There continues to be an unmet need for safe and effective pain medications. Opioids and nonsteroidal anti-inflammatory drugs (NSAIDs) dominate the clinical landscape despite limited effectiveness and considerable side-effect profiles. Although significant advancements have identified myriad potential pain targets over the past several decades, the majority of new pain pharmacotherapies have failed to come to market. The discovery of nerve growth factor (NGF) and its interaction with tropomyosin receptor kinase A (trkA) have been well characterized as important mediators of pain initiation and maintenance, and pharmacotherapies targeting this pathway have the potential to be considered promising methods in the treatment of a variety of nociceptive and neuropathic pain conditions. Several methodologic approaches, including sequestration of free NGF, prevention of NGF binding and trkA activation, and inhibition of trkA function, have been investigated in the development of new pharmacotherapies. Among these, NGF-sequestering antibodies have exhibited the most promise in clinical trials. However, in 2010, reports of rapid joint destruction leading to joint replacement prompted the Food and Drug Administration (FDA) to place a hold on all clinical trials involving anti-NGF antibodies. Although the FDA has since lifted this hold and a number of new trials are under way, the long-term efficacy and safety profile of anti-NGF antibodies are yet to be established.
Keywords: nociceptive pain, neuropathic pain, drug discovery, tanezumab, fulranumab, fasinumab
Introduction
Chronic pain is a disease unto itself, a state in which the protective role of pain transmission becomes deranged and pathologic. According to a 2010 analysis,1 chronic pain affects ~100 million Americans at an estimated annual cost of US$560–US$635 billion. The public health and economic burden of chronic pain is enormous and indicative of both the complexity of the disease as well as the limitations of current treatment modalities.
Despite their limited effectiveness for many chronic pain conditions and considerable side-effect profile, opioids and nonsteroidal anti-inflammatory drugs (NSAIDs) continue to dominate clinical practice.2 It is largely believed that mechanism-based treatments, rather than disease- or diagnosis-based treatments, hold the key to the development of new successful therapies.3 Shortcomings in the pharmacologic management of pain are thought to be attributed to a failure to target underlying mechanisms of chronic pain.3 Although tremendous strides have been made over the past several decades to better understand pain pathophysiology and identify potential drug therapies, the vast majority of new pharmacotherapies have failed in clinical trials.4–6 In fact, a 2015 scientometric analysis6 suggests that interest in analgesic drug discovery by the pharmaceutical industry is waning. It is within this context that nerve growth factor (NGF) and its related molecular targets represent a completely novel therapy mode, with potentially broad clinical applications for both nociceptive and neuropathic pain conditions.
NGF background
NGF was initially discovered in the 1950s as a tumor tissue-produced soluble factor that promotes the growth and differentiation of sensory and sympathetic ganglia.7,8 NGF was the first growth factor to be identified and its discovery represented a landmark achievement in developmental neurobiology. The illumination of NGF’s critical role in neuronal development eventually led to the creation of the “neurotrophic factor hypothesis” and the classical neurotrophic model in which NGF is synthesized and released by target tissues during embryonic development, promoting the growth, differentiation, and survival of neurons in a dose-dependent manner.7,9 Subsequent studies have broadened our understanding of this process and the role that neurotrophic factors play in the mammalian nervous system.7,8
NGF belongs to a family of neurotrophic factors or neurotrophins comprising brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4). Neurotrophins act by binding to two types of cell surface receptors: neurotrophin receptor (NGFR or p75) and a family of tyrosine kinase receptors, tropomyosin-related kinase A (trkA), trkB, and trkC. All neurotrophins bind NGFR/p75 with similar affinity, but each neurotrophin binds preferentially to a specific trk receptor; NGF preferentially binds trkA, BDNF binds trkB, and NT-3 binds trkC.10 TrkA is highly expressed by sensory neurons of the dorsal root ganglia (DRG) during embryogenesis; however, by the postnatal period, trkA expression and NGF sensitivity decline, and the role of NGF–trkA signaling shifts from promoting neuron growth and survival to regulating the sensitivity of the peripheral nervous system to noxious stimuli.11
Rationale: NGF and pain
NGF levels are elevated in preclinical models of both inflammation and peripheral nerve injury. Clinically, NGF concentration is increased in chronic pain conditions such as interstitial cystitis, prostatitis, arthritis, pancreatitis, chronic headaches, cancer pain, diabetic neuropathy, and noncancer pain, suggesting that NGF-mediated signaling is an ongoing and active process in chronic nociceptive and neuropathic pain states.10,12–14 A number of studies14–16 involving direct intradermal injection of NGF in rodents and humans have demonstrated a clear functional role for NGF in both activation and sensitization of nociceptors.14–16
Mechanism of action in nociceptive pain
Nociceptive pain occurs through the activation of nociceptors located in peripheral tissues in response to noxious stimuli. A noxious stimulus is any stimulus (eg, chemical, thermal, or mechanical) that either damages or threatens to cause damage to normal tissues.4 NGF is produced and released by peripheral tissues following noxious stimuli (eg, injury and inflammation) secondary to the production of inflammatory cytokines, such as interleukin-1 and tumor necrosis factor-alpha. NGF binds to trkA receptors on multiple targets, with multiple modulating effects on pain signaling.14
NGF binds to trkA that is selectively expressed on the peripheral terminals of A-delta and peptidergic unmyelinated C-fibers.10,14 The NGF–trkA complex is then internalized and transported retrogradely to DRG cell bodies, modulating and/or increasing the expression of a variety of cell surface receptors involved in nociception, including bradykinin receptors, acid-sensing ion channels (ASIC) 2/3, voltage-gated sodium channels, voltage-gated calcium channels, delayed rectifier potassium channels, putative mechanotransducers, as well as transient receptor potential cation channel subfamily V member 1 (TRPV1) receptor-mediated currents (Figure 1). There is some debate over whether the increase in TRPV1 signaling is due to a decrease in the TRPV1 activation threshold or an increase in receptor trafficking to the cell surface.13,17–19 Nevertheless, the increase in TRPV1 signaling and the increased activity of other channels result in peripheral sensitization and pain hypersensitivity. NGF–trkA signaling also leads to transcriptional changes that result in the increased expression of the pronociceptive neurotransmitters substance P (SP), calcitonin gene-related peptide (CGRP), and BDNF, thereby leading to central sensitization (Figure 1).13,14
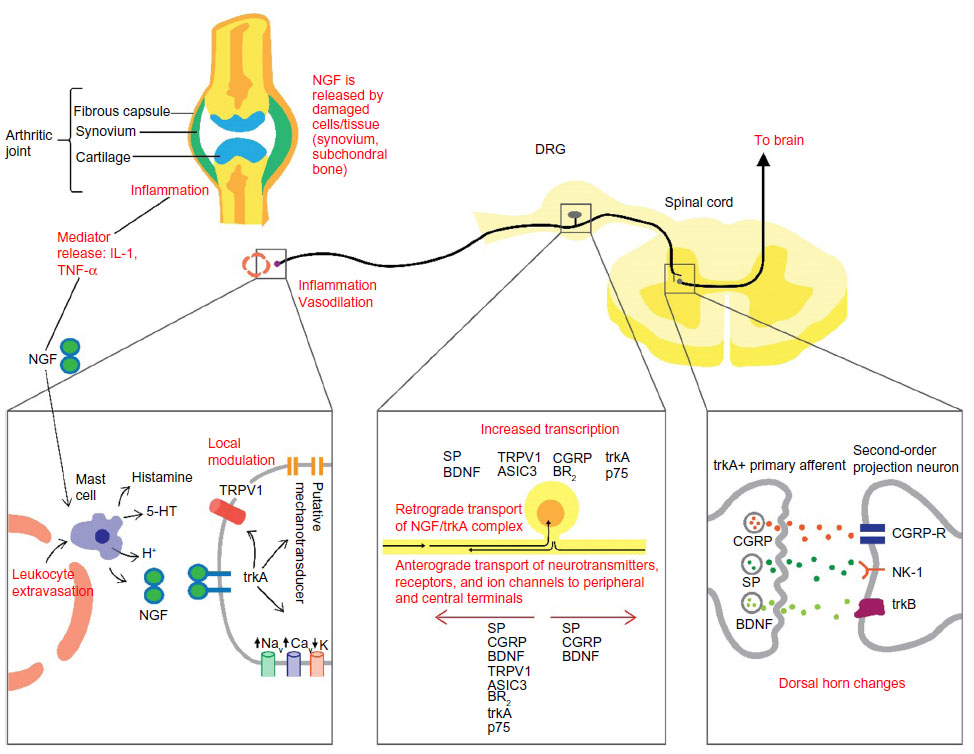 | Figure 1 Schematic diagram of the NGF mechanisms involved in the initiation and maintenance of pain. |
An additional effect of NGF on pain processing occurs through its binding of trkA receptors located on mast cells. This process is proinflammatory and elicits the release of inflammatory mediators such as histamine, serotonin or 5-hydroxytryptamine (5-HT), protons, as well as NGF itself, resulting in a positive feedback loop (Figure 1).13,14 Thus, not only does NGF signaling increase the expression of peripheral nociceptive receptors and centrally located pronociceptive neurotransmitters, but it also sensitizes adjacent nociceptive neurons in response to inflammation.
Mechanism of action in neuropathic pain
Neuropathic pain results from damage to the neurons of the somatosensory system, secondary to either direct injury or disease-related dysfunction, and results in the generation of ectopic discharges that occur independently of somatic stimuli.4 As mentioned previously, NGF levels are generally increased in chronic neuropathic conditions such as diabetic neuropathy and cancer pain, in particular, invasive nerve cancers.20,21 But the relationship between NGF signaling and neuropathic pain states is complex, and in some patients with diabetic neuropathy, NGF levels are actually decreased.21 In patients with chemotherapy-induced peripheral neuropathy, a decrease in circulating NGF is correlated with an increased severity of neuropathy.8 In preclinical studies, NGF has demonstrated a trophic and neuroprotective action on peptidergic small-diameter DRG cells after nerve injury,22 and a number of clinical studies have been conducted investigating the negative correlation between NGF levels and peripheral neuropathy by examining the administration of subcutaneous injections of recombinant NGF.8 In a Phase II trial23 involving patients with diabetic neuropathy, endogenous NGF administration resulted in relief of neuropathic pain, whereas a subsequent Phase III trial24 found no difference in neuropathic symptoms compared to placebo. In patients with HIV-associated peripheral neuropathy, two completed Phase II studies exhibited mixed results.25,26 It is noteworthy to mention that all clinical studies23–26 have reported significant dose-dependent hyperalgesia at the site of NGF injection. NGF administration has also shown the ability to induce nerve sprouting of trk-A-positive nociceptive as well as sympathetic nerve fibers, while NGF blockade by systemic injection of neutralizing antibodies in models of neuropathic pain appears to prevent allodynia and hyperalgesia.14,21 Mechanistically, NGF sequestration has demonstrated inhibition of neuroma formation and a decrease in ectopic discharges. In models of bone cancer, where neuroma formation and reorganization of sensory and sympathetic fibers is prominent, the administration of an NGF-sequestering antibody prevents this pathologic reorganization and inhibits the development of cancer pain.27
Pharmacotherapy
A number of approaches have been developed to target the NGF pathway and its effect on pain initiation and maintenance. The majority of these efforts have centered on the NGF–trkA pathway and focus on three methodologic approaches: 1) sequestration of free NGF; 2) prevention of NGF binding and activation of trkA; and 3) inhibition of trkA function.10,14
NGF-sequestering agents
Preclinical studies
Antibodies possess significant advantages compared to small molecules due to generally higher specificity and reduced off-target effects, culminating in quicker clinical development and faster US Food and Drug Administration (FDA) approval.12 Preclinical studies14,28 involving pretreatment with systemic anti-NGF antibody have shown successfully reduced acute thermal and mechanical hypersensitivity in response to Freund’s adjuvant in models of inflammatory pain. This preventative effect is also observed in models of visceral inflammation such as acetic acid-induced gastric inflammation and acrolein-induced cystitis.29,30 In models of established pain, such as visceral hyperalgesia involving trinitrobenzene sulfonic acid-induced colonic hypersensitivity, anti-NGF antibody administration has demonstrated the ability to reverse colonic hypersensitivity.31 An additional study32 involving the use of NGF antibodies in models of autoimmune arthritis have demonstrated analgesia equivalent to indomethacin despite continued joint destruction and inflammation. Anti-NGF administration has also resulted in significant analgesic effects in the treatment of hypersensitivity associated with chronic injury models of bone cancer and closed femur fracture, effectively reducing the neurochemical changes associated with peripheral and central sensitization.33,34 To highlight the specificity of anti-NGF antibody therapy in each of these models, despite significant reductions in pain after fracture and tumor growth, the density and number of sensory and sympathetic fibers were not affected.33,34 Furthermore, in models of inflammatory pain, NGF blockade shows reduced inflammation-mediated hypersensitivity but does so without altering other inflammatory processes such as erythema and edema.14,28 Additional preclinical work has also revealed no effect of anti-NGF therapy on bone healing after fracture.35
Clinical trials
Osteoarthritis
Phase I studies investigating tanezumab were initially reported in 2005, and a subsequent proof-of-concept Phase II trial in patients with moderate-to-severe knee osteoarthritis (OA) who had an unsatisfactory response to nonopioid pharmacotherapy, demonstrated dose-dependent efficacy and treatment-related adverse effects compared to placebo, with hypoesthesia and paresthesias being the most prominent.36–38 Dosing ranges in these Phase II studies included 10 μg/kg, 25 μg/kg, 50 μg/kg, 100 μg/kg, and 200 μg/kg administered intravenously (IV) at an 8-week interval, with follow-up conducted at a minimum of 8 weeks after the last dose.36,39,40 Subsequently, five placebo-controlled Phase III studies36,41–44 involving tanezumab in patients with hip and/or knee OA found statistically significant benefit across dosing ranges of 2.5 mg, 5 mg, and 10 mg, as assessed by the Western Ontario and McMaster Universities Osteoarthritis Index subscales. Fulranumab and fasinumab have also been studied in patients with chronic hip/knee OA and knee OA, respectively, and both medications demonstrated significant analgesia compared to placebo, with a low incidence of adverse effects.45,46 When assessed in a 2015 systematic review and meta-analysis that included 13 randomized controlled trials (ten tanezumab, two fulranumab, and one fasinumab), all anti-NGF agents across a variety of dosages were superior to placebo, and the three studies comparing tanezumab monotherapy to an active control (NSAID or opioid) also demonstrated superior efficacy (Table 1).36,39–50
 | Table 1 Summary of evidence: monoclonal anti-NGF antibodies |
Low back pain
In a 2011 proof-of-concept study,51 220 patients with chronic nonradicular low back pain (LBP) were administered a single dose of IV tanezumab 200 μg/kg plus oral placebo, IV placebo plus naproxen twice daily, or IV placebo plus oral placebo. At 6 weeks, compared to both naproxen and placebo, patients in the tanezumab treatment arm exhibited significantly greater reductions in pain intensity and corresponding improvements in physical function.51 A subsequent Phase II study52 also performed in patients with chronic mechanical LBP extended IV tanezumab therapy to two doses administered at an 8-week interval. Study arms included three fixed doses of 5 mg, 10 mg, and 20 mg, as well as IV and oral placebos and naproxen, in a double-blind, double-dummy design, in which all patients received two doses of IV treatment and daily oral administrations. The investigators found that both 10 mg and 20 mg dosages exhibited efficacy in pain and physical function at 16 weeks, which was statistically superior to both placebo and naproxen; the 5 mg dose was significant vs placebo at 4 weeks, 8 weeks, and 12 weeks, but not at 16 weeks, and was not superior to naproxen at any time point.52 A successive open-label extension study,53 whose objective was to further evaluate the long-term safety and efficacy of tanezumab, took patients from the aforementioned 16-week parent study52 and rerandomized them to receive either three IV injections of 10 mg or 20 mg of tanezumab, followed by four subcutaneous injections at the same dose every 8 weeks. Patients who had received 10 mg or 20 mg in the parent study52 were maintained on the same dose for the extension study.53 At 4 weeks, 8 weeks, 16 weeks, and 24 weeks of the extension study,53 all patients reported improvements in pain from baseline, with slightly greater efficacy in the 20 mg group vs the 10 mg treatment group. Additional Phase II studies54,55 in patients with chronic LBP have been undertaken with the NGF-neutralizing antibodies fulranumab and fasinumab. Subcutaneous injections of fulranumab in doses ranging from 1 mg to 10 mg at 4-week intervals failed to achieve a significant reduction in average pain at Week 12.54 Fasinumab administered to patients with radicular pain as a single subcutaneous injection at doses of 0.1 mg/kg (n=54) and 0.3 mg/kg (n=54) demonstrated no benefit for average daily back or leg pain at 4 weeks compared to placebo (n=51).55
Diabetic neuropathy and postherpetic neuralgia
A number of anti-NGF proof-of-concept clinical studies have been conducted in patients with diabetic neuropathy.56,57 Tanezumab administered as a single IV dose of 20 mg resulted in significant reductions in average pain scores at 4 weeks and 8 weeks after treatment, with 39.6%, 31.6%, 21.1%, and 13.2% experiencing ≥30%, ≥50%, ≥70%, and ≥90% pain reduction, respectively.56 In a Phase II double-blind placebo-controlled trial,57 fulranumab given as a subcutaneous injection of 1 mg, 3 mg, or 10 mg at 4-week intervals demonstrated dose-dependent efficacy, with the 10 mg dose resulting in superior relief compared to placebo at 12 weeks. At the 10 mg dose, 60.9% and 30.4% of patients reported ≥30% and ≥50% relief, respectively. A post hoc analysis57 revealed that Neuropathic Pain Symptom Inventory scores in burning (superficial) spontaneous pain >5/10 or pressing (deep) spontaneous pain >3.5/10 were more likely to respond to treatment, suggesting that phenotypic differences within this population may predict response to anti-NGF therapy, and that perhaps the variable increase or decrease in NGF levels in patients may also play a predictive role. In contrast, a parallel group study by Bramson et al56 examining tanezumab in patients with postherpetic neuralgia failed to yield significant reductions in average pain score.
Visceral pain
Tanezumab has been studied in two small-scale proof-of-concept trials58,59 involving chronic visceral pain. Patients with interstitial cystitis were administered a single dose of IV tanezumab 200 μg/kg (n=34) or placebo (n=30), with a primary end point of change in average daily pain score from baseline to 6 weeks. Patients in the tanezumab arm not only experienced a statistically significant reduction in pain scores vs placebo but also displayed reduced urgency episode frequency.58 In a study59 evaluating patients with moderate-to-severe chronic prostatitis/chronic pelvic pain syndrome, 30 patients received a single IV dose of tanezumab (20 mg) and 32 received a placebo. Although average pain score and urgency episode frequency trended downward at 6-week follow-up, neither outcome achieved significance.59
Cancer pain
A single study in patients with metastatic bone cancer involved an initial 16-week placebo-controlled parent study, followed by a 40-week uncontrolled open-label extension period.60 In the parent study, 59 patients were randomized and treated with an initial 10 mg IV tanezumab infusion (n=29) or placebo (n=30). Whereas no significant change was observed in daily average pain scores, a post hoc analysis suggested greater efficacy, with lower baseline opioid use and/or greater baseline pain. It should be noted that by Week 8, 48.3% of tanezumab-treated patients reported a ≥30% reduction in pain compared to only 20% in the placebo group, though no significant difference was observed in patients reporting ≥50%, ≥70%, and ≥90% improvement in average pain scores. In the 40-week extension, patients received 10 mg IV tanezumab infusions at 8-week intervals. These individuals experienced a significant decrease in pain scores compared to the baselines in the parent and extension studies60 at Week 8, with 43.3% and 36.7% of patients reporting ≥30% and ≥50% reductions in average daily pain, respectively, and, through Week 40, had improvements of –1.27 (0.68) and –1.40 (0.60) for average daily pain and daily worst pain, respectively.
Safety
In one recent review, Bannwarth and Kostine61 found that the most common treatment-related side effects of anti-NGF therapy were peripheral edema, arthralgia, extremity pain, and neurosensory symptoms (paresthesia and hypoesthesia). Moreover, treatment-related adverse events were similar for all anti-NGF therapies, suggestive of a class-specific effect. In the largest randomized controlled trial50 to date, 2,700 subjects with knee or hip OA receiving inadequate relief on a stable oral NSAID regimen were randomized into five treatment groups receiving IV tanezumab 5 mg (plus NSAID or oral placebo), tanezumab 10 mg (plus NSAID or oral placebo), or IV placebo plus NSAID; the study did not include an IV placebo plus oral placebo group. At 16 weeks, after all study patients received at least one IV treatment, the frequencies of the majority of adverse events were similar between the 5 mg and 10 mg tanezumab doses, with the exception of paresthesias and pain in the extremity, which were slightly more frequent at 10 mg in both the NSAID-combined and placebo-combined treatment arms (paresthesias: 11.1% vs 9.0% and pain in extremity: 5.5% vs 3.9% in the NSAID-combined group; paresthesias: 9.0% vs 7.2% and pain in extremity: 6.6% vs 3.5% in the placebo-combined group). A number of adverse events were more common in the tanezumab–plus-NSAID group compared to the group on mono-NGF therapy, such as peripheral edema (at 5 mg, 7.1% vs 6.1%; at 10 mg, 9.2% vs 5.0%), paresthesias (at 5 mg, 9.0% vs 6.1%; at 10 mg, 11.1% vs 7.2%), and hypoesthesias (at 5 mg, 6.5% vs 4.6%; at 10 mg, 6.5% vs 5.7%), whereas joint swelling (at 5 mg, 3.4% vs 4.1%; at 10 mg, 3.1% vs 5.0%) was less common in the combination treatment group compared to that in monotherapy group.50 In a meta-analysis of anti-NGF treatment in OA patients, overall odds ratio (OR) for withdrawals due to adverse events in all patients receiving tanzumab monotherapy was 1.50 (95% confidence interval [CI]: 0.94–2.38) compared to placebo.36 However, no statistical difference was observed in rates of withdrawal with the 2.5 mg (OR: 1.23; CI: 0.5–3.02) and 5.0 mg (OR: 1.09; CI: 0.55–2.16) doses relative to the placebo. The 10 mg dose had an OR of 1.92 (CI: 1.20–3.09), suggesting that some adverse effects are dose dependent.36 In a similar population and study design, the rates of withdrawal due to adverse effects with fulranumab (OR: 1.77; CI: 0.74–4.22) or fasinumab (OR: 1.53; CI: 0.50–4.73) failed to reach statistical significance.36 In the meta-analysis36 of the tanezumab-plus-NSAID combination therapy vs NSAID alone in OA patients, withdrawals due to adverse events reached statistical significance (OR: 1.90; CI: 1.39–2.61). Moreover, the incidence of serious adverse events was also higher in the combination group compared to the NSAID-alone group (OR: 1.39; CI: 1.00–1.94). Nevertheless, the overall incidence of adverse events was small, and anti-NGF therapy was generally well-tolerated. However, in 2010, all clinical trials for anti-NGF antibodies were put on hold by the FDA due to reports of rapidly progressive OA and osteonecrosis leading to joint replacement. Reported cases occurred in OA subjects receiving tanezumab, tanezumab-plus-NSAID, or fulranumab and involved extensive bone damage and joint destruction. Several cases occurred in multiple joints and nonindex joints, including shoulders. Pfizer (New York, NY, USA) and Janssen (Beerse, Belgium), the manufacturers of tanezumab and fulranumab, respectively, each assembled separate multidisciplinary adjudication committees comprising bone pathologists, orthopedic surgeons, and rheumatologists, charged with reviewing all reports of osteonecrosis leading to joint replacement.62 The tanezumab adjudication included a total of 87 subjects, 81 of whom were enrolled in Phase III OA studies and six who were enrolled in Phase II chronic LBP studies.62 Review of all available case reports diagnosed osteonecrosis and rapid OA progression in two (2.3%) and 34 (66.7%) subjects, respectively. The remaining cases were distinguished as normal OA progression (n=17, 33%), other diseases (n=21, 24.1%), lacking a consensus (n=5, 5.8%), and lacking sufficient information for a determination (n=8, 9.2%). In the fulranumab adjudication, among 1,353 subjects, 88 reported at least one joint replacement and, in total, there were 101 joint replacements (97 initial replacements and four revisions).63 Moreover, 65 (65%) of these cases were determined to be due to normal OA progression, 18 (18%) were classified as rapidly progressive OA, 14 (14%) cases lacked sufficient information for a determination, and the four (4%) revisions were considered not applicable. In 2012, the FDA commissioned an independent arthritis advisory committee to further investigate these claims and concluded that joint failures were probably related to anti-NGF treatment and represented a unique clinical form of rapidly progressive OA, citing rapid and considerable joint destruction, typically within 6–12 months of exposure. These cases were characterized by particular pathological features, including femoral head flattening and medial femoral condyle involvement with subchondral fractures, as well as associated edema, joint effusions, and marked pain.64 The committee also determined that events were more likely to occur with longer exposures and concurrent NSAID use, possibly through the inhibition of bone healing. Although the precise etiology was not clear, several plausible mechanisms were discussed, including higher susceptibility in patients with atrophic and neuropathic forms of OA, subchondral bone pathology, overuse of joints, and possible drug toxicity with concurrent NSAID use. Nevertheless, the committee conceded that more study was needed to fully understand the risk of anti-NGF therapy to bones and joints, in addition to voting in favor of continued development of the drug class due to the potential benefit of anti-NGF therapy for a multitude of pain conditions and the absence of any direct link between the administration of anti-NGF antibodies and joint destruction.61,65 Whereas a number of measures to mitigate the risks were discussed, such as limiting trials to the use of the lowest effective doses, restriction of NSAID use, and the development of a screening radiologic protocol, no consensus criteria for study continuation has been reached to date.61,66 In 2013, after negotiations between pharmaceutical companies and the FDA, the FDA issued a notification to Pfizer that the “clinical hold” for tanezumab would be lifted pending submission and review of nonclinical data.61 A recent search of the National Institutes of Health Web site “clinicaltrials.gov” revealed two ongoing long-term safety and efficacy studies67,68 evaluating tanezumab monotherapy in patients with OA of the hip or knee, as well as in patients with chronic LBP. There are four fulranumab studies69–72 in active recruitment for patients with OA, two of which will evaluate its adjunctive use with other medications, including NSAIDs. There is also ongoing recruitment for a Phase I trial evaluating fasinumab.73
NGF–trkA binding inhibitors
Initially developed for the characterization of the NGF-trkA signaling pathway, mouse monoclonal anti-trkA, MNAC13, is capable of inducing analgesia in models of inflammatory (formalin injection) and neuropathic pain (sciatic nerve ligation).20 When used in combination with low-dose opioids, the class demonstrates a synergistic effect. However, the inability to develop an equivalent humanized antibody has prevented the introduction of an analogous species into clinical trials. The nonpeptide small molecule ALE0540 is an NGF inhibitor that prevents NGF binding to both trkA and p75.74 Despite its demonstrated antinociceptive effect in animal models of neuropathic pain following intrathecal administration, ALE0540 appears to suffer from a lack of specificity when tested with other receptors in vitro and thus has not been advanced into clinical trials (Table 2).
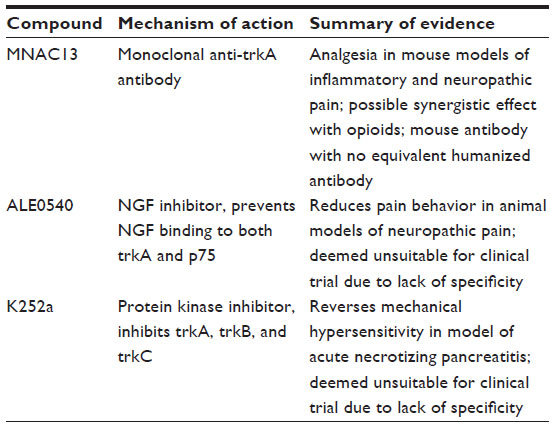 | Table 2 Summary of evidence: NGF–trkA binding inhibitors and trkA inhibitors |
Inhibitors of trkA
k252a is a small-molecule protein kinase inhibitor that inhibits the activation of the entire tropomyosin receptor kinase family (trkA, trkB, and trkC). In a rat model of acute necrotizing pancreatitis, k252a administration reversed DRG CGRP and SP upregulation and alleviated mechanical hypersensitivity.75 However, due to lack of specificity, no human trials have ever been initiated and no additional compounds targeting trkA activation have been developed to date, underscoring the challenges in targeting a single component of a pervasive receptor class.
Other potential targets
As efforts continue to be made to further characterize the NGF–trkA pathway, additional molecular targets are likely to emerge. One such target is the trkA-specific Q-SNARE protein, Syntaxin 8 (STX8).76 STX8 facilitates trkA receptor transport from the Golgi apparatus to the plasma membrane. Furthermore, knockdown of STX8 in rat DRG resulted in analgesia in models of inflammatory pain and could eventually lead to the generation of additional pain therapeutics.76
Conclusion and future directions
In nociceptive and inflammatory pain, NGF activity and its interaction with trkA have been well characterized as important mediators of pain initiation and maintenance. In preclinical models of inflammatory and visceral pain, NGF sequestration and inhibition of trkA signaling have demonstrated a consistent analgesic effect. In contrast, the role played by NGF in the pathophysiology of neuropathic pain is less clear. Preclinical studies evaluating NGF and trkA antagonism exhibit a consistent benefit in the prevention of hyperalgesia and allodynia,14,28–34 yet pharmacotherapies targeting this pathway are yet to make it to market. Monoclonal antibodies have produced among the most promising new therapies for the treatment of cancers and immunological disorders, but their use in the treatment of pain has been limited. Protein kinases, CGRP, and Nav1.7 have been investigated as potential targets; however, only cytokine- and NGF-directed monoclonal antibodies have reached clinical trials.6,77,78 To date, the systematic study of anti-NGF monoclonal antibodies in humans has yielded a mixed efficacy and safety record, and long-term follow-up studies are lacking, particularly in chronic disease indications. Even if anti-NGF monoclonal antibody-based treatments gain FDA clearance, the high costs of the therapy may outweigh its potential clinical value over existing treatment options. In rheumatoid arthritis, for instance, the price of therapies with monoclonal antibodies in the chronic setting can reach up to US$24,000 per patient-year and the cost of tanezumab and fulranumab is more than an order of magnitude greater than other existing pain treatments.6,79 Furthermore, the authors believe that while several studies in OA patients have exhibited superior efficacy compared to active controls (oxycodone, naproxen, and celecoxib),43,45 and one study in patients with chronic mechanical LBP demonstrated superior efficacy to naproxen at 10 mg and 20 mg doses,52 additional large, pragmatic, comparative-effectiveness studies with long-term follow-up periods are needed across a variety of pain conditions and patient populations to fully assess the merits of anti-NGF therapy for chronic pain conditions.43,45,52,80 Due to the occurrence of rapidly progressive OA and joint destruction in clinical trials, it is also incumbent upon investigators to further delineate the risks of anti-NGF antibody therapeutics and improve their safety profile if anti-NGF therapy is to someday become a mainstay treatment for chronic pain. Nevertheless, in spite of its high cost, the relatively short-term follow-up periods in currently published trials, its uncertain adverse-effect profile, and its high cost, anti-NGF therapy may find a role as a short-term treatment in properly screened patients with refractory pain conditions.
Disclosure
The authors report no conflicts of interest in this work.
References
Gaskin DJ, Richard P. The economic costs of pain in the United States. J Pain. 2012;13(8):715–724. | |
Kissin I. The development of new analgesics over the past 50 years: a lack of real breakthrough drugs. Anesth Analg. 2010;110(3):780–789. | |
Woolf CJ, Max MB. Mechanism-based pain diagnosis: issues for analgesic drug development. Anesthesiology. 2001;95(1):241–249. | |
Cohen SP, Mao J. Neuropathic pain: mechanisms and their clinical implications. BMJ. 2014;348:f7656. | |
Hsu E, Murphy S, Chang D, Cohen SP. Expert opinion on emerging drugs: chronic low back pain. Expert Opin Emerg Drugs. 2015;20(1):103–127. | |
Kissin I. Scientometrics of drug discovery efforts: pain-related molecular targets. Drug Des Devel Ther. 2015;9:3393–3404. | |
Aloe L. Rita Levi-Montalcini: the discovery of nerve growth factor and modern neurobiology. Trends Cell Biol. 2004;14(7):395–399. | |
Aloe L, Rocco ML, Bianchi P, Manni L. Nerve growth factor: from the early discoveries to potential clinical use. J Transl Med. 2012;10:239. | |
Yuen EC, Howe CL, Li Y, Holtzman DM, Mobley WC. Nerve growth factor and the neurotrophic factor hypothesis. Brain Dev. 1996;18(5):362–368. | |
Hefti FF, Rosenthal A, Walicke PA, et al. Novel class of pain drugs based on antagonism of NGF. Trends Pharmacol Sci. 2006;27(2):85–91. | |
Bennett DLH. Neurotrophic factors: important regulators of nociceptive function. Neuroscientist. 2001;7(1):13–17. | |
Watson JJ, Allen SJ, Dawbarn D. Targeting nerve growth factor in pain: what is the therapeutic potential? BioDrugs. 2008;22(6):349–359. | |
McKelvey L, Shorten GD, O’Keeffe GW. Nerve growth factor-mediated regulation of pain signaling and proposed new intervention strategies in clinical pain management. J Neurochem. 2013;124(3):276–289. | |
Mantyh PW, Koltzenburg M, Mendell LM, Tive L, Shelton DL. Antagonism of nerve growth factor-trkA signaling and the relief of pain. Anesthesiology. 2011;115(1):189–204. | |
Andreev NY, Dimitrieva N, Kolzenburg M, McMahon SB. Peripheral administration of nerve growth factor in the adult rat produces a thermal hyperalgesia that requires the presence of sympathetic post-ganglionic neurons. Pain. 1995;63(1):109–115. | |
Dyck PJ, Peroutka S, Rask C, et al. Intradermal recombinant human nerve growth factor induces pressure allodynia and lowered heat-pain threshold in humans. Neurology. 1997;48(2):501–505. | |
Chuang H, Prescott ED, Kong H, et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411(6840):957–962. | |
Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. P38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36(1):57–68. | |
Stein AT, Ufret-Vincenty CA, Hua L, Santana LF, Gordon SE. Phosphoinositide 3-kinase binds to TRPV1 and mediates NGF-stimulated TRPV1 trafficking to the plasma membrane. J Gen Physiol. 2006;128(5):509–522. | |
Ugolini G, Marinelli S, Covaceuszach S, Cattaneo A, Pavone F. The function neutralizing anti-TrkA antibody MNAC13 reduces inflammatory and neuropathic pain. Proc Natl Acad Sci U S A. 2007;104(8):2985–2990. | |
Wild KD, Bian D, Zhu D, et al. Antibodies to nerve growth factor reverse established tactile allodynia in rodent models of neuropathic pain without tolerance. J Pharmacol Exp Ther. 2007;322(1):282–287. | |
Gold BG, Mobley WC, Matheson SF. Regulation of axonal caliber, neurofilament content, and nuclear localization in mature sensory neurons by nerve growth factor. J Neurosci. 1991;11(4):943–945. | |
Apfel SC, Kessler JA, Adomato BT, Litchy WJ, Sanders C, Rask CA. Recombinant human nerve growth factor in the treatment of diabetic polyneuropathy. NGF study group. Neurology. 1998;51:695–702. | |
Apfel SC, Schwartz S, Adomato BT, Freeman R, Biton V, et al. Efficacy and safety of recombinant human nerve growth factor in patients with diabetic polyneuropathy: a randomized controlled trial. rhNGF clinical investigative group. JAMA. 2000;284:2215–2221. | |
McArthur JC, Yiannoutsos C, Simpson DM, et al. A phase II trial of nerve growth factor for sensory neuropathy associated with HIV infection. AIDS Clinical Trials Group Team 291. Neurology. 2000;54(5):1080–1088. | |
Schifitto G, Yiannoutsos C, Simpson DM, et al. Long-term treatment with recombinant nerve growth factor for HIV-associated sensory neuropathy. Neurology. 2001;57(7):1313–1316. | |
Mantyh WG, Jimenez-Andrade JM, Stake JI, et al. Blockade of nerve sprouting and neuroma formation markedly attenuates the development of late stage cancer pain. Neuroscience. 2010;171(2):588–598. | |
Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience. 1994;62(2):327–331. | |
Lamb K, Kang YM, Gebhart GF, Bielefeldt K. Nerve growth factor and gastric hyperalgesia in the rat. Neurogastroenterol Motil. 2003; | |
Guerios SD, Wang ZY, Boldon K, Bushman W, Bjorling DE. Blockade of NGF and trk receptors inhibits increased peripheral mechanical sensitivity accompanying cystitis in rats. Am J Physiol Regul Integr Comp Physiol. 2008;295(1):R111–R122. | |
Delafoy L, Raymond F, Doherty AM, Eschalier A, Diop L. Role of nerve growth factor in the trinitrobenzene sulfonic acid-induced colonic hypersensitivity. Pain. 2003;105(3):489–497. | |
Shelton DL, Zeller J, Ho WH, Pons J, Rosenthal A. Nerve growth factor mediates hyperalgesia and cachexia in auto-immune arthritis. Pain. 2005;116(1–2):8–16. | |
Sevcik MA, Ghilardi JR, Peters CM, et al. Anti-NGF therapy profoundly reduces bone cancer pain and the accompanying increase in markers of peripheral and central sensitization. Pain. 2005;115(1–2):128–141. | |
Jiminez-Andrade JM, Martin CD, Koewler NJ, et al. Nerve growth factor sequestering therapy attenuates non-malignant skeletal pain following fracture. Pain. 2007;133(1–3):183–196. | |
Koewler NJ, Freeman KT, Buus RJ, et al. Effects of monoclonal antibody raised against nerve growth factor on skeletal pain and bone healing after fracture of the C57BL/6J mouse femur. J Bone Miner Res. 2007;22(11):1732–1742. | |
Schnitzer TJ, Marks JA. A systematic review of the efficacy and general safety of antibodies to NGF in the treatment of OA of the hip or knee. Osteoarthritis Cartilage. 2015;23(Suppl 1):S8–S17. | |
Lane NE, Webster L, Shiao-ping L, Gray M, Hefti F, Walicke P. RN624 (anti-NGF) improves pain and function in subjects with moderate knee osteoarthritis: a phase I study. Arthritis Rheum. 2005;52:S461. | |
Hefti F, Mokhtarani M, Gray M, Zhao C, Chan C. RN624 (anti-NGF) reduces pain and improves function in subjects with moderate to severe pain from osteoarthritis of the knee. J Pain. 2006;7(4):S45. | |
Lane NE, Schnitzer TJ, Birbara CA, et al. Tanezumab for the treatment of pain from osteoarthritis of the knee. N Engl J Med. 2010; | |
Schnitzer TJ, Lane NE, Birbara C, Smith MD, Simpson SL, Brown MT. Long-term open-label study of tanezumab for moderate to severe osteoarthritic knee pain. Osteoarthritis Cartilage. 2011;19(6):639–646. | |
Brown MT, Murphy FT, Radin DM, Davignon I, Smith MD, West CR. Tanezumab reduces osteoarthritic knee pain: results of a randomized, double-blind, placebo-controlled phase III trial. J Pain. 2012;13(8): | |
Brown MT, Murphy FT, Radin DM, Davignon I, Smith MD, West CR. Tanezumab reduces osteoarthritic hip pain: results of a randomized, double-blind, placebo-controlled phase III trial. Arthritis Rheum. 2013; | |
Spierings EL, Fidelholtz J, Wolfram G, Smith MD, Brown MT, West CR. A phase III placebo- and oxycodone-controlled study of tanezumab in adults with osteoarthritis pain of the hip or knee. Pain. 2013;154(9):1603–1612. | |
Ekman EF, Gimbel JS, Bello AE, et al. Efficacy and safety of intravenous tanezumab for the symptomatic treatment of osteoarthritis: 2 randomized controlled trials versus naproxen. J Rheumatol. 2014;41(11):2249–2259. | |
Sanga P, Katz N, Polverejan E, et al. Efficacy, safety, and tolerability of fulranumab, an anti-nerve growth factor antibody, in the treatment of patients with moderate to severe osteoarthritis pain. Pain. 2013;154(10):1910–1919. | |
Tiseo PJ, Kivitz AJ, Ervin JE, et al. Fasinumab (REGN475), an antibody against nerve growth factor for the treatment of pain: results from a double blind, placebo-controlled exploratory study in osteoarthritis of the knee. Pain. 2014;155(7):1245–1252. | |
Balanescu AR, Feist E, Wolfram G, et al. Efficacy and safety of tanezumab added on to diclofenac sustained release in patients with knee or hip osteoarthritis: a double-blind, placebo-controlled, parallel-group, multicentre phase III randomised clinical trial. Ann Rheum Dis. 2014;73(9):1665–1672. | |
Mayorga A, Wang S, Wang S, Kelley K, Thipphawong J. Double-blind, randomized study to evaluate efficacy, and safety of fulranumab in patients with moderate to severe, chronic knee pain from osteoarthritis: interim analysis results. J Pain. 2013;14(4):S69. | |
Nagashima H, Suzuki M, Araki S, Yamabe T, Muto C. Preliminary assessment of the safety and efficacy of tanezumab in Japanese patients with moderate to severe osteoarthritis of the knee: a randomized, double-blind, dose-escalation, placebocontrolled study. Osteoarthritis Cartilage. 2011;19(12):1405–1412. | |
Schnitzer TJ, Ekman EF, Spierings EL, et al. Efficacy and safety of tanezumab monotherapy or combined with non-steroidal anti-inflammatory drugs in the treatment of knee or hip osteoarthritis pain. Ann Rheum Dis. 2015;74(6):1202–1211. | |
Katz N, Borenstein DG, Birbara C, et al. Efficacy and safety of tanezumab in the treatment of chronic low back pain. Pain. 2011;152(10):2248–2258. | |
Kivitz AJ, Gimbel JS, Bramson C, et al. Efficacy and safety of tanezumab versus naproxen in the treatment of chronic low back pain. Pain. 2013;154(7):1009–1021. | |
Gimbel JS, Kivitz AJ, Bramson C, et al. Long-term safety and effectiveness of tanezumab as treatment for chronic low back pain. Pain. 2014;155(9):1793–1801. | |
Sanga P, Karcher K, Wang S, et al. Efficacy, safety, and tolerability of fulranumab in treatment of patients with moderate-to-severe, chronic low back pain [abstract]. J Pain. 2011;12(Suppl4):53. | |
Tiseo PJ, Ren H, Mellis S. Fasinumab (REGN475), an antinerve growth factor monoclonal antibody, for the treatment of acute sciatic pain: results of a proof-of-concept study. J Pain Res. 2014;7:523–530. | |
Bramson C, Hermann DN, Carey W, et al. Exploring the role of tanezumab as a novel treatment for the relief of neuropathic pain. Pain Med. 2015;16(6):1163–1176. | |
Wang H, Romano G, Frutasci ME, et al. Fulranumab for treatment of diabetic peripheral neuropathic pain: A randomized controlled trial. Neurology. 2014;83:628–637. | |
Evans RJ, Moldwin RM, Cossons N, Darekar A, Mills IW, Scholfield D. Proof of concept trial of tanezumab for the treatment of symptoms associated with interstitial cystitis. J Urol. 2011;185(5):1716–1721. | |
Nickel JC, Atkinson G, Krieger JN, et al. Preliminary assessment of safety and efficacy in proof-of-concept, randomized clinical trial of tanezumab for chronic prostatitis/chronic pelvic pain syndrome. Urology. 2012;80(5):1105–1110. | |
Sopata M, Katz N, Carey W, et al. Efficacy and safety of tanezumab in the treatment of pain from bone metastases. Pain. 2015; | |
Bannwarth B, Kostine M. Targeting nerve growth factor (NGF) for pain management: what does the future hold for NGF antagonists? Drugs. 2014;74(6):619–626. | |
Hochberg MC. Serious joint-related adverse events in randomized controlled trials of anti-nerve growth factor monoclonal antibodies. Osteoarthritis Cartilage. 2015;23(Suppl 1):S18–S21. | |
Janssen Research & Development, LLC [webpage on the Internet]. Advisory Committee Briefing Document, JNJ-42160443 (Fulranumab). 2012. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM295204.pdf. Accessed November 14, 2015. | |
Food and Drug Administration Center for Drug Evaluation and Research [webpage on the Internet]. Arthritis Advisory Committee Meeting. 2012. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisDrugsAdvisoryCommittee/UCM301302.pdf. Accessed November 14, 2015. | |
Food and Drug Administration Center for Drug Evaluation and Research [webpage on the Internet]. Summary Minutes of the Arthritis Advisory Committee Meeting. 2012. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM307879.pdf. Accessed November 14, 2015. | |
Roemer FW, Hayes CW, Miller CG, et al. Imaging atlas for eligibility and on-study safety of potential knee adverse events in anti0NGF studies (Part 1). Osteoarthritis Cartilage. 2015;23(Suppl 1):S22–S42. | |
Pfizer [webpage on the Internet]. Long Term Safety and Efficacy Study of Tanezumab in Subjects with Osteoarthritis of the Hip or Knee. Available from: https://clinicaltrials.gov/ct2/show/NCT02528188. Accessed November 14, 2015. | |
Pfizer [webpage on the Internet]. A Phase 3 Study of Tanezumab for Chronic Low Back Pain (TANGO). Available from: https://clinicaltrials.gov/ct2/show/NCT02528253. Accessed November 14, 2015. | |
Janssen Research & Development, LLC [webpage on the Internet]. Study of Efficacy, Safety of Fulranumab Monotherapy for OA of Hip or Knee, PAI3003. Available from: https://clinicaltrials.gov/ct2/show/NCT02289716. Accessed November 14, 2015. | |
Janssen Research & Development, LLC [webpage on the Internet]. Study of Efficacy, Safety of Fulranumab Monotherapy for OA of Hip or Knee, PAI3002. Available from: https://clinicaltrials.gov/ct2/show/NCT02336698. Accessed November 14, 2015. | |
Janssen Research & Development, LLC [webpage on the Internet]. Study of Efficacy, Safety of Fulranumab Adjunctive Use in OA of Hip or Knee, PAI3001. Available from: https://clinicaltrials.gov/ct2/show/NCT02336685. Accessed November 14, 2015. | |
Janssen Research & Development, LLC [webpage on the Internet]. Study of Safety, Efficacy of Fulranumab Adjunctive Use in OA of Hip or Knee, PAI3007. Available from: https://clinicaltrials.gov/ct2/show/NCT02301234. Accessed November 14, 2015. | |
Regeneron Pharmaceuticals [webpage on the Internet]. Study of Fasinumab (REGN475) in Healthy Japanese and Caucasian Subjects. Available from: https://clinicaltrials.gov/ct2/show/NCT02516618. Accessed November 14, 2015. | |
Owolabi JB, Rizkalla G, Tehim A, et al. Characterization of antiiallodynic actions of ALE-0540, a novel nerve growth factor receptor antagonist, in rat. J Pharmacol Exp Ther. 1999;289(3):1271–1276. | |
Winston JH, Toma H, Shenoy M, et al. Acute pancreatitis results in referred mechanical hypersensitivity and neuropeptide up-regulation that can be suppressed by the protein kinase inhibitor k252a. J Pain. 2003;4(6):329–337. | |
Chen B, Zhao L, Li X, et al. Syntaxin 8 modulates the post-synthetic trafficking of the TrkA receptor and inflammatory pain transmission. J Biol Chem. 2014;289(28):19556–19569. | |
Bowler KE, Worsley MA, Broad L, et al. The effect of a monoclonal antibody to calcitonin-gene related peptide (CGRP) on injury-induced ectopic discharge following lingual nerve injury. Neurosci Lett. 2011;505(2):146–149. | |
Lee JH, Park CK, Chen G, et al. A monoclonal antibody that targets a NaV1.7 channel voltage sensor for pain and itch relief. Cell. 2014;157(6):1393–1404. | |
Bachmann MF, Whitehead P. Active immunotherapy for chronic disease. Vaccine. 2013;31(14):1777–1784. | |
Bach PB. New math on drug cost-effectiveness. N Engl J Med. 2015;373(19):1797–1799. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.