Back to Journals » Drug Design, Development and Therapy » Volume 8
Anti-inflammatory effects of cordycepin in lipopolysaccharide-stimulated RAW 264.7 macrophages through Toll-like receptor 4-mediated suppression of mitogen-activated protein kinases and NF-κB signaling pathways
Received 30 July 2014
Accepted for publication 20 August 2014
Published 16 October 2014 Volume 2014:8 Pages 1941—1953
DOI https://doi.org/10.2147/DDDT.S71957
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Yung Hyun Choi,1,2 Gi-Young Kim,3 Hye Hyeon Lee4
1Department of Biochemistry, Dongeui University College of Korean Medicine, Busan, 2Anti-Aging Research Center and Blue-Bio Industry RIC, Dongeui University, Busan, 3Laboratory of Immunobiology, Department of Marine Life Sciences, Jeju National University, Jeju, 4Daegu Gyeongbuk Institute of Science and Technology, Daegu, Republic of Korea
Abstract: Cordycepin is the main functional component of the Cordyceps species, which has been widely used in traditional Oriental medicine. This compound possesses many pharmacological properties, such as an ability to enhance immune function, as well as antioxidant, antiaging, and anticancer effects. In the present study, we investigated the anti-inflammatory effects of cordycepin using a murine macrophage RAW 264.7 cell model. Our data demonstrated that cordycepin suppressed production of proinflammatory mediators such as nitric oxide (NO) and prostaglandin E2 by inhibiting inducible NO synthase and cyclooxygenase-2 gene expression. Cordycepin also inhibited the release of proinflammatory cytokines, including tumor necrosis factor-alpha and interleukin-1-beta, through downregulation of respective mRNA expression. In addition, pretreatment with cordycepin significantly inhibited lipopolysaccharide (LPS)-induced phosphorylation of mitogen-activating protein kinases and attenuated nuclear translocation of NF-κB by LPS, which was associated with abrogation of inhibitor kappa B-alpha degradation. Furthermore, cordycepin potently inhibited the binding of LPS to macrophages and LPS-induced Toll-like receptor 4 and myeloid differentiation factor 88 expression. Taken together, the results suggest that the inhibitory effects of cordycepin on LPS-stimulated inflammatory responses in RAW 264.7 macrophages are associated with suppression of mitogen-activating protein kinases and activation of NF-κB by inhibition of the Toll-like receptor 4 signaling pathway.
Keywords: cordycepin, anti-inflammation, mitogen-activated protein kinases, NF-κB, Toll-like receptor 4
Introduction
Inflammation is part of a complex biological response of the body to harmful stimuli, such as pathogens, damaged cells, or irritants; however, chronic inflammatory processes are involved in the pathogenesis of common inflammation-associated diseases, such as rheumatoid arthritis, atherosclerosis, chronic hepatitis, and pulmonary fibrosis.1,2 Macrophages, which are a type of differentiated tissue cells that originate as blood monocytes, play a critical role in the initiation and propagation of inflammatory responses by releasing proinflammatory mediators, ie, nitric oxide (NO) and prostaglandin (PG)E2, and cytokines to promote inflammatory responses.3,4 Therefore, the amount of proinflammatory mediators and cytokines may reflect the degree of inflammation and provide information to investigate the effect of pharmacological agents on the inflammatory process. In particular, due to their highly reproducible response to lipopolysaccharide (LPS), the main component of endotoxin, which is derived from Gram-negative bacterial cell walls,5 the RAW 264.7 mouse macrophage cell line is widely used for inflammation studies. LPS activates macrophages by binding to Toll-like receptor 4 (TLR4), and increases secretion of proinflammatory mediators and cytokines to promote the inflammatory response.6,7 The LPS-initiated signaling cascade leads to activation of mitogen-activating protein kinase (MAPK) and NF-κB signaling pathways in a myeloid differentiation factor 88 (MyD88)-dependent or MyD88-independent manner.8,9
Cordyceps is a genus of the family Clavicipitaceae that has been used in traditional Oriental medicine for centuries. Recent studies have demonstrated that the bioactive components isolated from this genus have various pharmacological actions.10–13 Among them, cordycepin (3′-deoxyadenosine), a derivative of the nucleoside adenosine, is a major functional component of the genus Cordyceps. Due to the absence of oxygen in the 3′-position of its ribose moiety, the incorporation of cordycepin during RNA synthesis results in premature termination of chain elongation.14,15 Recent studies indicate that cordycepin has various biological properties, including antifungal,16 antibacterial,17 antioxidant,18,19 immunomodulatory,20,21 anti-inflammatory,22–27 and antitumor effects.14,28,29 However, the molecular mechanisms underlying the anti-inflammatory effects of cordycepin are not yet completely understood.
In the current study, we investigated the inhibitory properties of cordycepin and established the possible mechanisms involved in its action on LPS-stimulated inflammatory response production in murine RAW 264.7 macrophages. The results demonstrated that cordycepin effectively suppressed LPS-induced inflammatory signaling through inactivation of the MAPK and NF-κB pathways by inhibiting TLR4-mediated signaling mechanisms.
Materials and methods
Materials
Cordycepin, LPS, Griess reagent, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Tween-20, bovine serum albumin, 4,6-diamidino-2-phenylindole (DAPI), and dimethyl sulfoxide were purchased from Sigma-Aldrich Chemical Co. (St Louis, MO, USA). Dulbecco’s modified Eagle’s minimum essential medium, fetal bovine serum, and other tissue culture reagents were purchased from Gibco-BRL (Grand Island, NY, USA). Enzyme-linked immunosorbent assay kits for PGE2, tumor necrosis factor-alpha (TNF-α), and interleukin-1 beta (IL-1β) were obtained from R&D Systems (Minneapolis, MN, USA). Antibodies against inducible NO synthase (iNOS), cyclooxygenase-2 (COX-2), IL-1β, TNF-α, NF-κB/p65, inhibitor kappa B (IκB), nucleolin, extracellular signal-regulated kinase (ERK) 1/2, phospho-ERK1/2, p38 MAPK, phospho-p38 MAPK, c-Jun N-terminal kinase (JNK), phospho-JNK, TLR4, Myd88, and actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The peroxidase-labeled donkey anti-rabbit immunoglobulin, peroxidase-labeled sheep anti-mouse immunoglobulin, and enhanced chemiluminescence detection kit were purchased from Amersham Corp (Arlington Heights, IL, USA). COX-2, iNOS, TNF-α, IL-1β, and glyceraldehyde-3-phosphate dehydrogenase oligonucleotide primers were purchased from Bioneer (Seoul, Republic of Korea). Fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit IgG and Fluoromount-G were obtained from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA, USA) and Southern Biotechnology Associates Inc. (Birmingham, AL, USA), respectively. Alexa Fluor 594-conjugated LPS (AF-LPS) was obtained from Invitrogen Corp (Carlsbad, CA, USA). All other chemicals were purchased from Sigma-Aldrich.
Cell culture, treatments, and MTT assay
The RAW 264.7 macrophage cell line was obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco’s modified Eagle’s minimum essential medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM L-glutamine at 37°C in a 5% CO2 humidified air environment. Cells were preincubated with and without various concentrations of cordycepin for 1 hour, then treated with LPS (100 ng/mL) for the indicated times. Cell viability was assessed by the MTT assay. Briefly, RAW 264.7 cells (5×105 cells/mL) were treated with the indicated concentrations of cordycepin or LPS (100 ng/mL) alone, or pretreated with different concentrations of cordycepin for 1 hour before LPS treatment. After 24 hours, the medium was removed and the cells were incubated with 0.5 mg/mL MTT solution for 2 hours. The supernatant was then discarded and the formazan blue that formed in the cells was dissolved with dimethyl sulfoxide. Optical density was measured at 540 nm with a microplate reader (Dynatech Laboratories, Chantilly, VA, USA).
Measurement of NO production
Nitrite accumulation in the culture supernatants were measured as an indicator of NO production based on the Griess reaction. Briefly, 100 μL of cell culture medium was collected at the end of culture, mixed with an equal volume of Griess reagent, and incubated at room temperature for 10 minutes. Absorbance at 540 nm was measured with a NaNO2 standard curve, and nitrite production was determined.30
Determination of PGE2, TNF-α, and IL-1β production
Proinflammatory cytokine (TNF-α and IL-1β) levels and PGE2 production in culture medium were determined using commercially available enzyme-linked immunosorbent assay kits according to the manufacturer’s instructions.31
RNA isolation and reverse transcriptase-polymerase chain reaction
Total RNA was isolated using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol, and 2 μg of RNA was used for complementary DNA synthesis using M-MLV reverse transcriptase (Promega, Madison, WI, USA). Reverse transcriptase-generated complementary DNA encoding iNOS, COX-2, TNF-α, and IL-1β genes was amplified by polymerase chain reaction using specific primers. The specific primers used were: mouse iNOS (forward 5′-ATGTCCGAA GCAAACATCAC-3′ and reverse 5′-TAATGTCCAGGA AGTAGGTG-3′), COX-2 (forward 5′-CAGCAAATCCTT GCTGTTCC-3′ and reverse 5′-TGGGCAAAGAATGCA AACATC-3′), IL-1β (forward 5′-ATGGCAACTGTTCCT GAACTCAACT-3′ and reverse 5′-TTTCCTTTCTTAGAT ATGGACAGGAC-3′), and TNF-α (forward 5′-ATGAGC ACAGAAAGCATGATC-3′ and reverse 5′-TACAGGCTT GTCACTCGAATT-3′). The amplified complementary DNA products were separated by 1% agarose gel electrophoresis and stained with ethidium bromide. In a parallel experiment, glyceraldehyde-3-phosphate dehydrogenase (forward 5′-CGGAGTCAACGGATTTGGTCGTAT-3′ and reverse 5′-AGCCTTCTCCATGGTGGTGAAGAC-3′) was used as an internal control.
Protein extraction and Western blotting
The cells were harvested and lysed with lysis buffer (20 mM sucrose, 1 mM ethylenediamine tetraacetic acid, 20 μM Tris-Cl, pH 7.2, 1 mM DTT, 10 mM KCl, 1.5 mM MgCl2, and 5 μg/mL aprotinin) for 1 hour. In a parallel experiment, nuclear and cytosolic proteins were prepared using nuclear extraction reagents (Pierce, Rockford, IL, USA) according to the manufacturer’s protocol. Protein concentration was measured using a Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer’s instructions. For Western blot analysis, an equal amount of protein was subjected to electrophoresis on sodium dodecyl sulfate-polyacrylamide gels and transferred by electroblotting to a nitrocellulose membrane (Schleicher and Schuell, Keene, NH, USA). The blots were probed with the desired antibodies for 1 hour, incubated with the diluted enzyme-linked secondary antibody, and visualized by enhanced chemiluminescence solution according to the recommended procedure.
Immunofluorescence staining
RAW 264.7 cells were cultured directly on glass coverslips in six-well plates for 24 hours to detect NF-κB/p65 localization by immunofluorescence assays using a fluorescence microscope. After stimulation with LPS in the presence or absence of cordycepin, the cells were fixed with 4% paraformaldehyde in phosphate-buffered saline for 10 minutes at room temperature and permeabilized with 100% MeOH for 10 minutes at 20°C. Polyclonal antibodies against anti-NF-κB/p65 were applied for 1 hour followed by a 1-hour incubation with FITC-conjugated donkey anti-rabbit IgG. After washing with phosphate-buffered saline, nuclei were stained with DAPI, and fluorescence was visualized using a fluorescence microscope (Carl Zeiss, Oberkochen, Germany). Cells were stimulated with AF-LPS for 30 minutes in the presence or absence of cordycepin for the LPS/TLR4 complex formation assay. The cells were fixed, stained with rabbit polyclonal anti-TLR4 antibody for 90 minutes at 4°C, and then incubated with secondary antibodies conjugated with Alexa Fluor 488 for 1 hour. The stained cells were observed under a fluorescence microscope.
Measurement of LPS binding on cell surface
RAW 264.7 cells were incubated with AF-LPS in the presence or absence of genistein for 1 hour. The cells were washed twice with phosphate-buffered saline, harvested with 0.005% ethylenediamine tetraacetic acid, and then analyzed by flow cytometry. Alexa 488 was excited using a 488 argon-ion laser line and detected on a channel FL1 using a 530 nm emission filter. The fluorescence emission of samples was recorded by a flow cytometer. The gate was not applied.
Statistical analyses
Data are presented as the mean ± standard deviation. Statistical significance was determined using analysis of variance followed by the Student’s t-test. A P-value <0.05 was considered to be statistically significant.
Results
Cordycepin inhibits LPS-induced NO and PGE2 production in RAW 264.7 macrophages
To determine the level of NO production, we measured nitrite released into the culture medium using Griess reagent. As a result, LPS alone markedly induced NO production compared with that generated by the control. However, pretreatment with cordycepin significantly repressed the levels of NO produced in LPS-stimulated RAW 264.7 cells in a concentration-dependent manner up to 30 μg/mL (Figure 1A). As shown in Figure 1B, treatment of RAW 264.7 cells with LPS also resulted in a marked increase in PGE2 release compared with the untreated control. However, cordycepin inhibited LPS-mediated PGE2 production in a concentration-dependent manner.
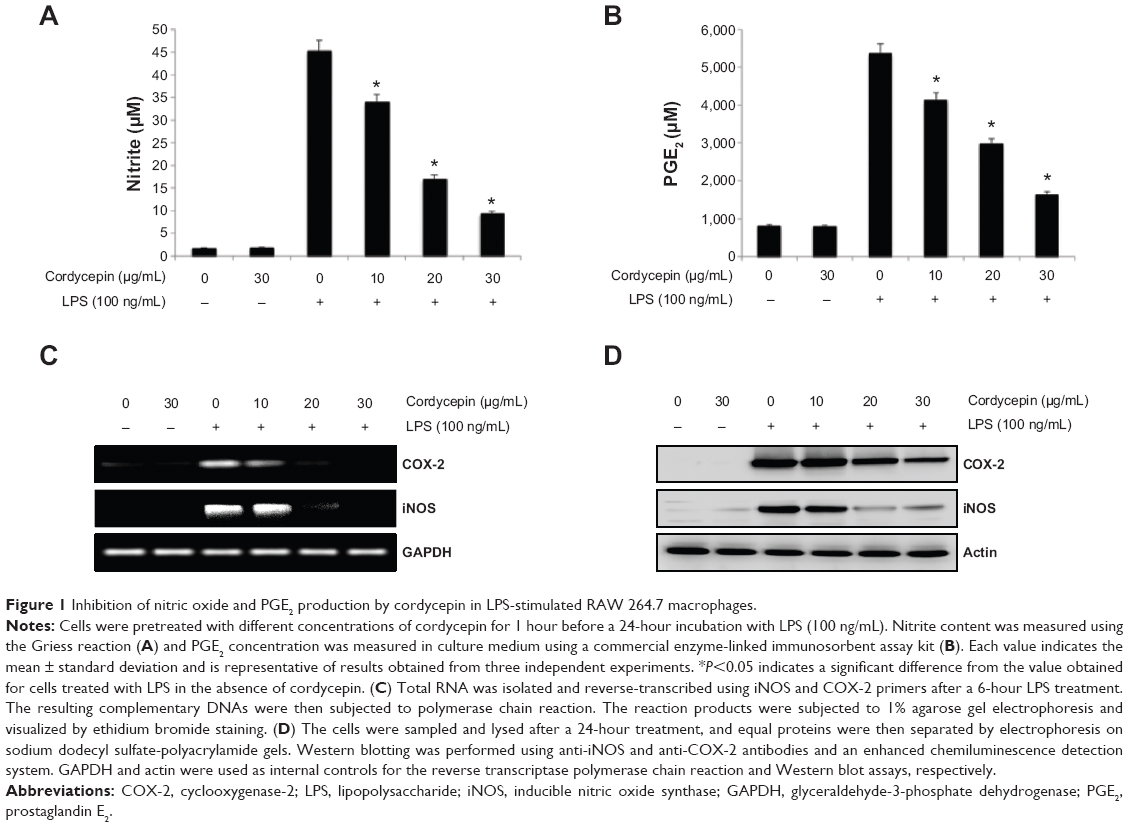 | Figure 1 Inhibition of nitric oxide and PGE2 production by cordycepin in LPS-stimulated RAW 264.7 macrophages. |
Cordycepin downregulates LPS-induced iNOS and COX-2 expression in RAW 264.7 macrophages
To elucidate the mechanism involved in the inhibition of NO and PGE2 generation by cordycepin in LPS-stimulated RAW 264.7 cells, we studied the effects of cordycepin on iNOS and COX-2 mRNA and protein expression by reverse transcriptase polymerase chain reaction and Western blot analyses. iNOS and COX-2 mRNA and protein expression was undetectable in unstimulated RAW 264.7 cells. However, COX-2 expression increased markedly in response to LPS and iNOS, which was significantly inhibited by pretreatment with cordycepin (Figure 2C and D). These results indicate that the reduced expression of iNOS and COX-2 at the transcriptional level contributed to the inhibitory effect of cordycepin on LPS-induced NO and PGE2 production.
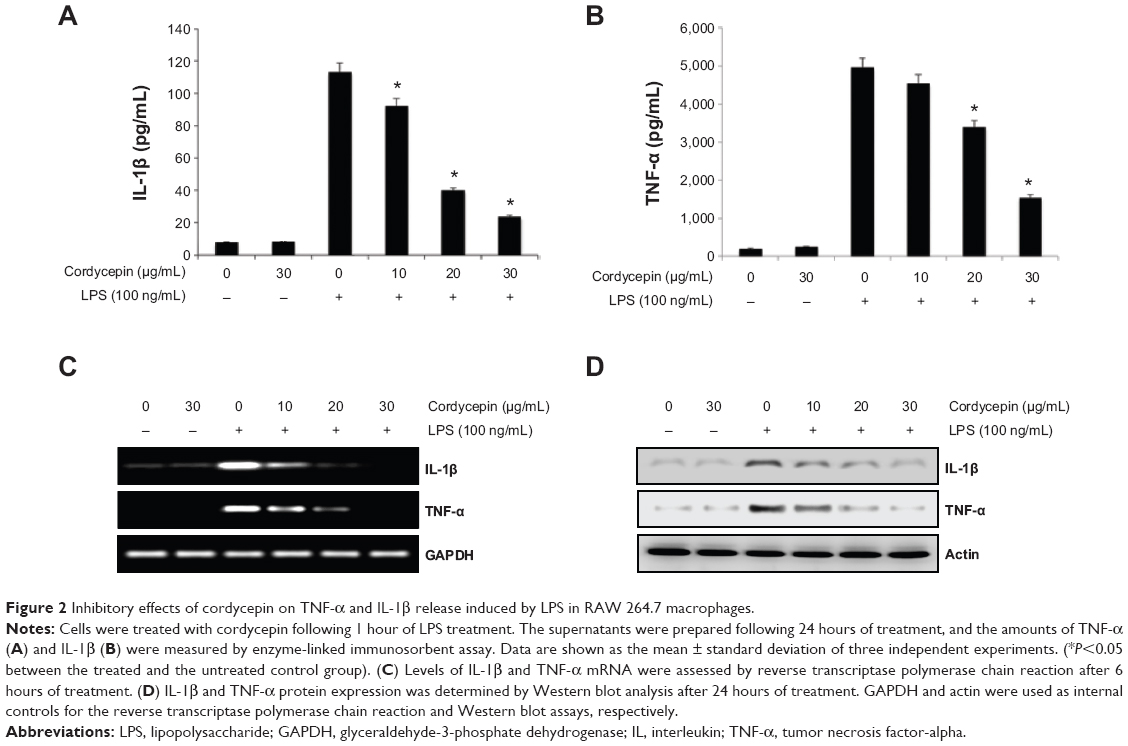 | Figure 2 Inhibitory effects of cordycepin on TNF-α and IL-1β release induced by LPS in RAW 264.7 macrophages. |
Cordycepin attenuates LPS-induced TNF-α and IL-1β production and expression in RAW 264.7 macrophages
In a parallel experiment, we investigated the effects of cordycepin on synthesis of the proinflammatory cytokines TNF-α and IL-1β by LPS treatment. As a result, TNF-α and IL-1β levels increased significantly in the culture medium of LPS-stimulated RAW 264.7 cells. However, pretreatment with cordycepin significantly decreased the release of both cytokines in a concentration-dependent manner (Figure 2A and B). Furthermore, the reverse transcriptase polymerase chain reaction results indicated that cordycepin markedly suppressed gene expression of these cytokines at the mRNA level in a similar fashion to the effects observed at the protein level (Figure 2C and D), suggesting that cordycepin-mediated inhibition of these cytokines may also be regulated at the transcriptional level.
Cordycepin blocks LPS-induced nuclear translocation of NF-κB/p65 in RAW 264.7 macrophages
Previous reports suggested that NF-κB is an important transcription factor regulating the expression of iNOS, COX-2, and inflammatory cytokines; thus, we explored whether cordycepin could block the NF-κB signaling pathway. Western blot analyses using cytosolic and nuclear fractions showed that the amount of NF-κB/p65 in the nucleus was markedly increased after 30 minutes of exposure to LPS alone, concomitant with degradation of Iκ B-alpha (IκBα) in the cytosol. However, LPS-induced NF-κB/p65 levels in the nuclear fraction were concentration-dependently reduced by cordycepin pretreatment, and LPS-induced IκBα degradation was obviously blocked by pretreatment with cordycepin (Figure 3A). Furthermore, the shift of NF-κB to the nucleus in RAW 264.7 cells was analyzed using immunofluorescence staining to clearly determine the influence of cordycepin on NF-κB/p65 nuclear translocation by LPS treatment (Figure 3B). The immunofluorescence images revealed that NF-κB/p65 was normally sequestered in the cytoplasm (Figure 3B, control panel), and that nuclear accumulation of NF-κB/p65 was strongly induced after stimulating RAW 264.7 cells with LPS (Figure 3B, LPS panel). However, nuclear translocation of NF-κB/p65 was not induced in the cells after pretreatment with cordycepin alone (Figure 3B, cordycepin panel), and the LPS-induced translocation of NF-κB/p65 was completely abolished after pretreating the cells with cordycepin (Figure 3B, LPS + cordycepin panel).
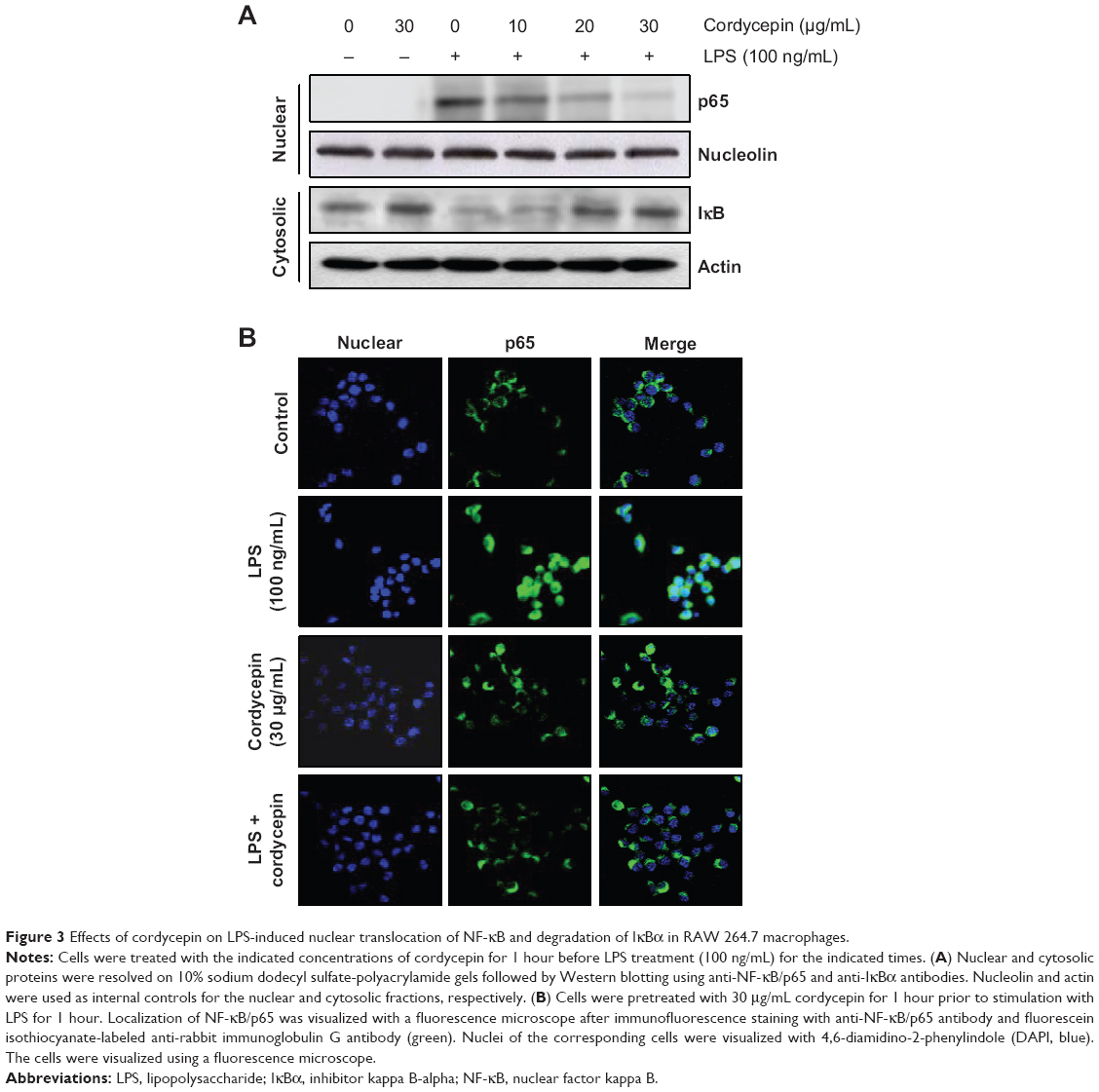 | Figure 3 Effects of cordycepin on LPS-induced nuclear translocation of NF-κB and degradation of IκBα in RAW 264.7 macrophages. |
Cordycepin suppresses LPS-induced phosphorylation of MAPKs in RAW 264.7 macrophages
Because MAPK signaling molecules also play a critical role in regulating the LPS-induced inflammatory process, we analyzed the phosphorylation levels of MAPKs in LPS-treated RAW 264.7 cells by Western blotting. As depicted in Figure 4, phosphorylation of p38 MAPK, ERK, and JNK by LPS stimulation occurred at 15 minutes and was sustained until 1–2 hours; however, pretreatment with cordycepin significantly suppressed phosphorylation in a concentration-dependent manner. In contrast, the levels of total MAPK proteins were unaffected by either LPS or cordycepin treatment, suggesting that inactivation of MAPK signaling may be involved in the inhibitory effect of cordycepin on the production of LPS-induced proinflammatory mediators and cytokines in RAW 264.7 cells.
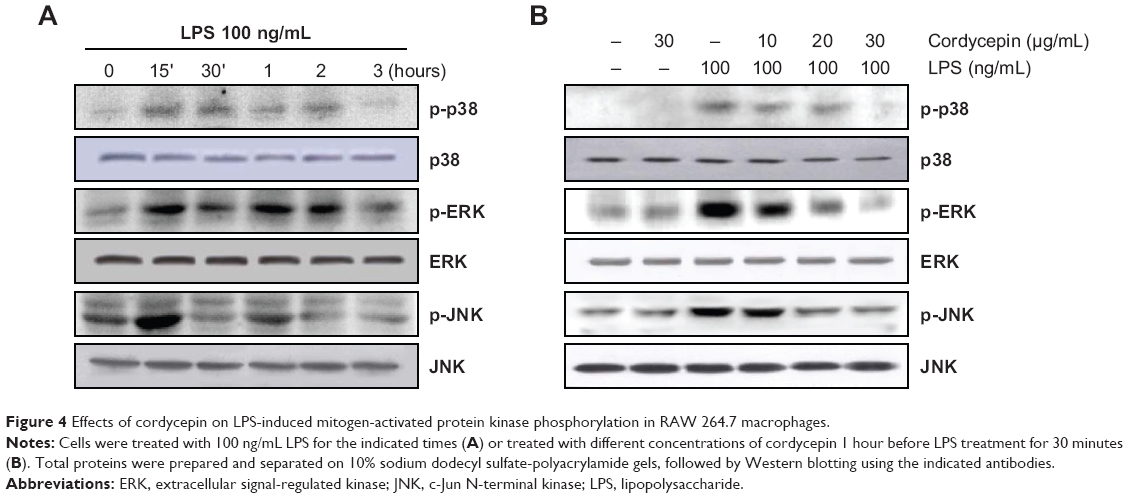 | Figure 4 Effects of cordycepin on LPS-induced mitogen-activated protein kinase phosphorylation in RAW 264.7 macrophages. |
Cordycepin inhibits LPS-induced TLR4 and MyD88 expression, and the interaction between LPS and TLR4 in RAW 264.7 macrophages
We next assessed the effects of cordycepin on the LPS-activated TLR4 signaling pathway to further determine the mechanisms underlying the anti-inflammatory effects of cordycepin. As indicated in Figure 5A, TLR4 protein expression increased significantly in RAW 264.7 cells treated with LPS compared with that in untreated cells, which was accompanied by up-regulation of MyD88 expression. However, treatment with cordycepin before LPS stimulation almost completely blocked the LPS-induced induction of TLR4 and MyD88. In addition, treatment with LPS in the presence of cordycepin significantly prevented the binding of LPS to the BV2 cell surface (Figure 5B). We further tested whether cordycepin could inhibit the interaction between LPS and TLR4 in RAW 264.7 cells using AF-LPS (Figure 5C). When cells were treated with AF-LPS alone, the fluorescence intensities of LPS and TLR4 were observed outside the cell membrane by immunofluorescence assay (Figure 5B, LPS panel). However, in the presence of cordycepin, the fluorescence intensity of TLR4 was markedly inhibited (Figure 5C, LPS + cordycepin panel), suggesting LPS-stimulated activation of TLR4 signaling pathway was potently blocked by cordycepin.
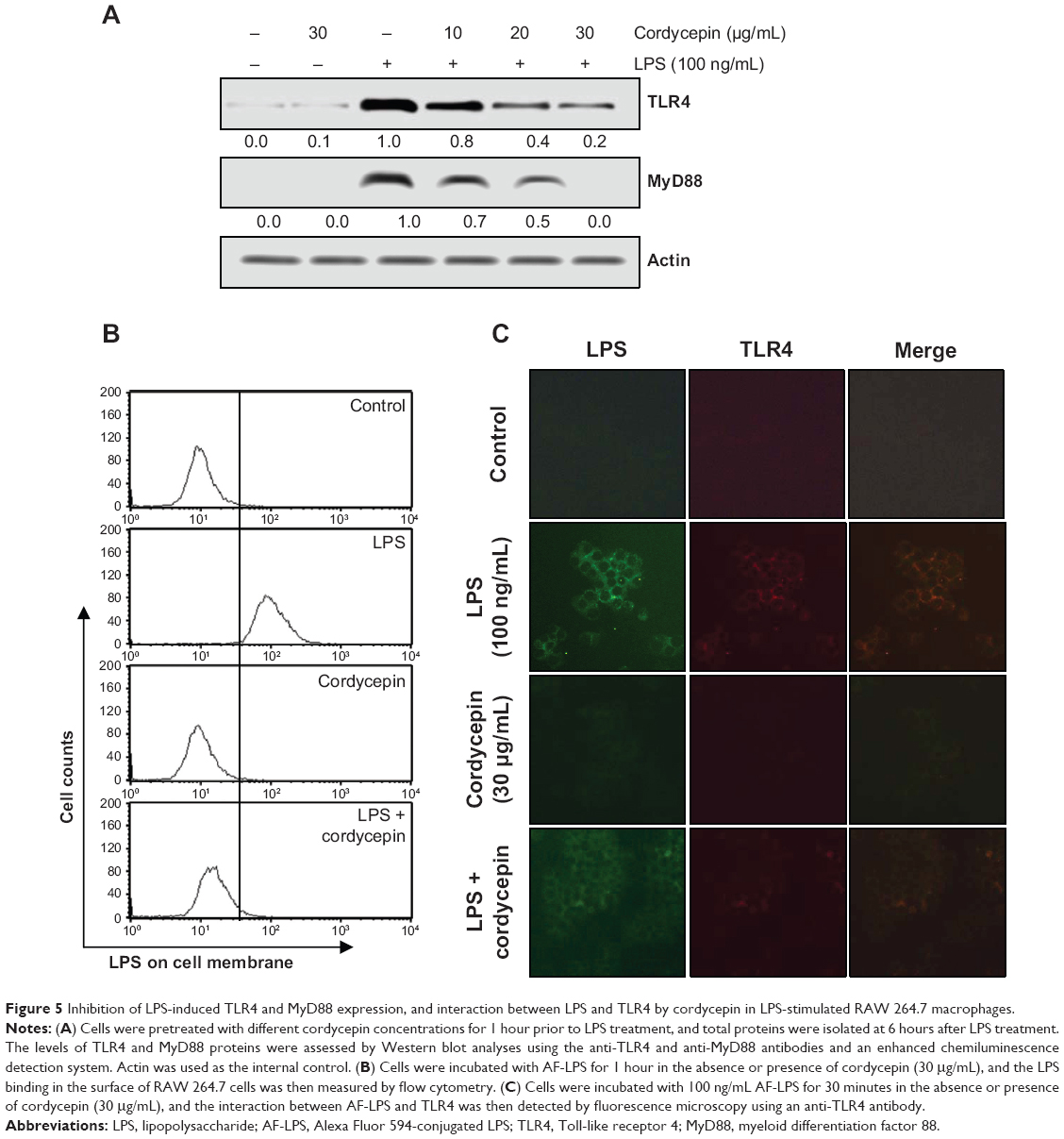 | Figure 5 Inhibition of LPS-induced TLR4 and MyD88 expression, and interaction between LPS and TLR4 by cordycepin in LPS-stimulated RAW 264.7 macrophages. |
Blocking TLR-4 with CLI-095 synergistically increases the anti-inflammatory potential of cordycepin in RAW 264.7 macrophages
To confirm involvement of the TLR4 signaling pathway in the cordycepin-mediated anti-inflammatory potential, we next evaluated the effects of the pharmacological agent CLI-095, which blocks TLR4-mediated signaling. As shown in Figure 6, we found that cordycepin and CLI-095 cotreatment synergistically inhibited LPS-induced production of NO and PGE2 as well as iNOS and COX-2 expression. Furthermore, the effects of LPS-enhanced TNF-α and IL-1β release were successfully inhibited by blocking TLR4 signaling with CLI-095, and cotreatment of cordycepin with CLI-095 almost completely inhibited the production of those proinflammatory cytokines to background levels similar to that in the untreated control (Figure 7). Taken together, these results verify that the inhibitory effects of cordycepin on the LPS-induced inflammatory response results, at least, from suppression of TLR4 signaling cascades in RAW 264.7 macrophages.
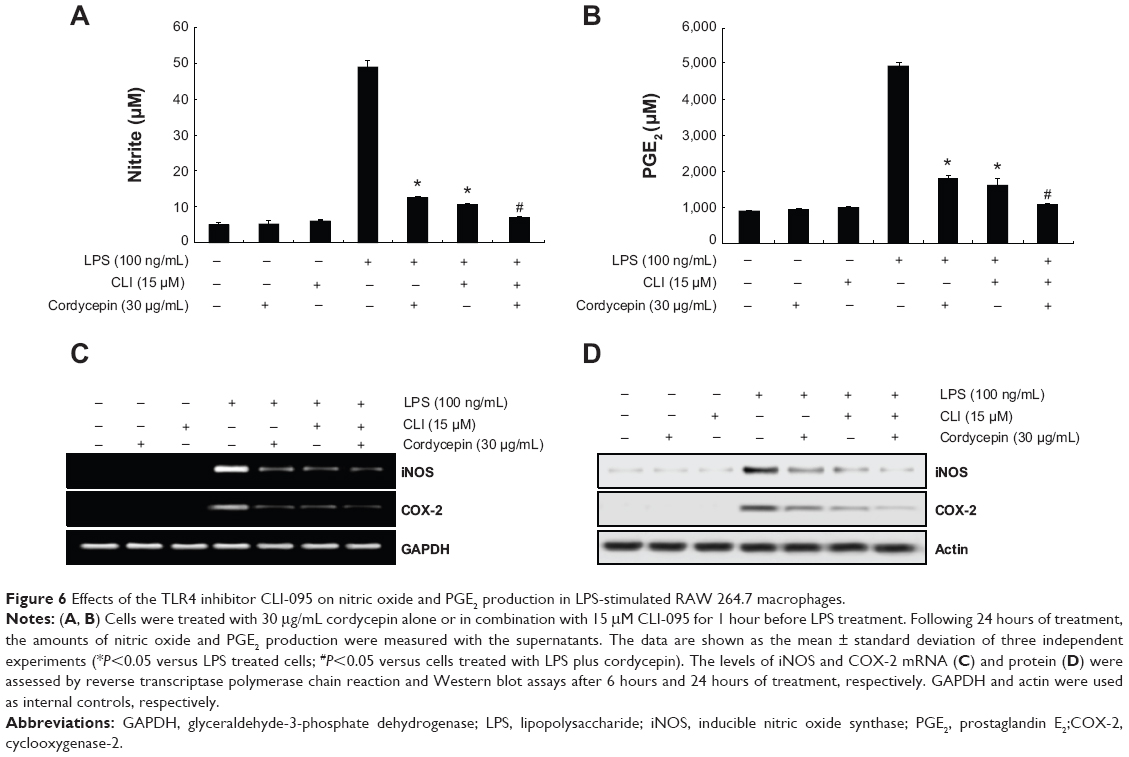 | Figure 6 Effects of the TLR4 inhibitor CLI-095 on nitric oxide and PGE2 production in LPS-stimulated RAW 264.7 macrophages. |
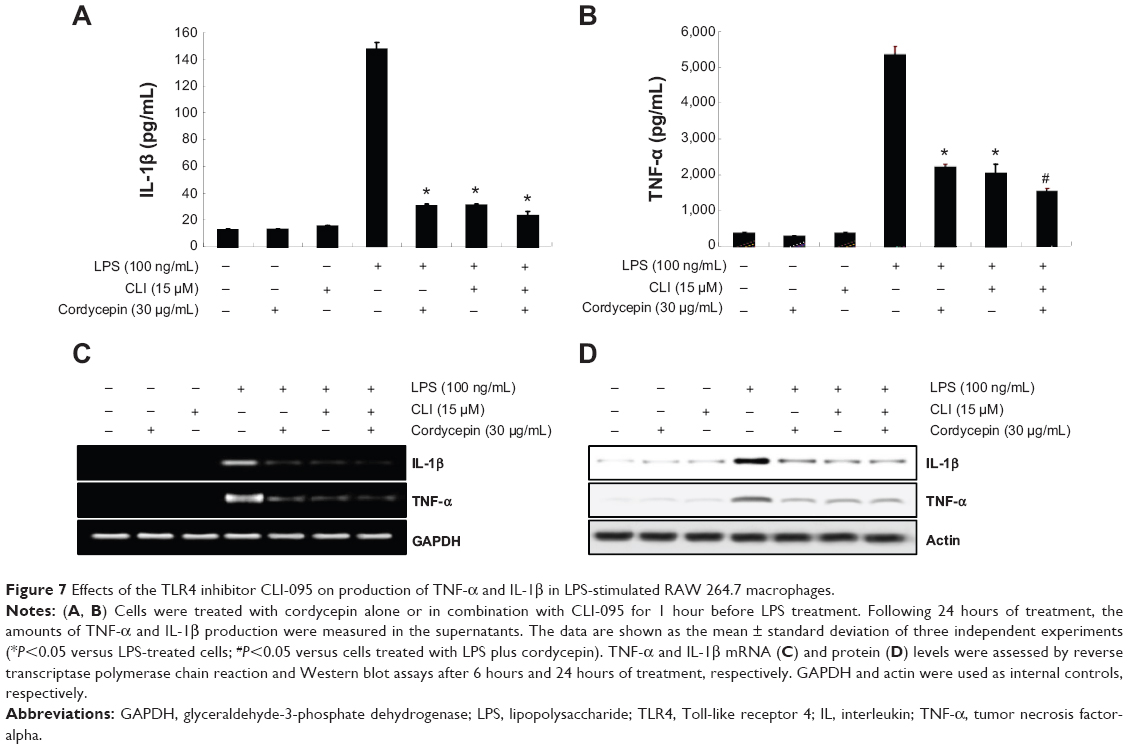 | Figure 7 Effects of the TLR4 inhibitor CLI-095 on production of TNF-α and IL-1β in LPS-stimulated RAW 264.7 macrophages. |
Finally, to examine whether cordycepin or CLI-095 is cytotoxic to RAW 264.7 cells, the cells were exposed to cordycepin alone and together with CLI-095 for 24 hours in the presence or absence of LPS, and cell viability was measured by the MTT assay. The results showed no cytotoxic effects within our tested concentrations (Figure 8). These results clearly indicate that the anti-inflammatory activity of cordycepin in LPS-stimulated RAW 264.7 macrophages was not due to cytotoxicity.
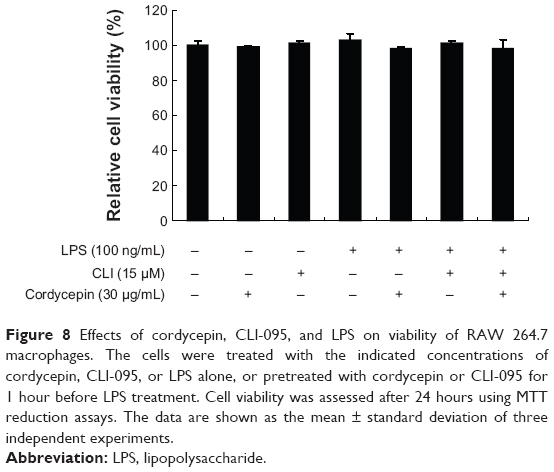 | Figure 8 Effects of cordycepin, CLI-095, and LPS on viability of RAW 264.7 macrophages. The cells were treated with the indicated concentrations of cordycepin, CLI-095, or LPS alone, or pretreated with cordycepin or CLI-095 for 1 hour before LPS treatment. Cell viability was assessed after 24 hours using MTT reduction assays. The data are shown as the mean ± standard deviation of three independent experiments. |
Discussion
Cordycepin is a specific polyadenylation inhibitor that was originally extracted from Cordyceps militaris and possesses many pharmacological activities, including immunological stimulation and antitumor activity. In the past few years, several investigations have indicated that cordycepin has an anti-inflammatory potential by suppressing the NF-κB signaling pathway, suggesting that cordycepin could be used as an anti-inflammatory agent in the treatment of inflammation-associated disorders. For example, cordycepin inhibits LPS-induced proinflammatory mediators and/or cytokines in RAW 264.7 macrophage26 and BV2 microglial cell models24 by blocking NF-κB activation. Cordycepin also prevents LPS-induced airway neutrophilia in mice and effectively blocks LPS-induced expression of vascular adhesion molecule-1 in human lung epithelial cells.27 Other studies have shown that this compound has anticancer effects by inhibiting the levels of some critical genes involved in cancer cell growth and metastasis by suppressing NF-κB activation.32–34 Although these observations suggest that cordycepin has anti-inflammatory and anticancer effects by modulating NF-κB signaling pathway, although the detailed anti-inflammatory signaling pathways remain to be explored.
Accumulating evidence indicates that NO and PGE2 are critical mediators of inflammation. NO plays a pivotal role in many body functions; however, its overproduction, particularly in macrophages, can lead to cytotoxicity, inflammation, and autoimmune disorders.35,36 iNOS is one of the key enzymes generating NO from arginine in response to various inflammatory stimuli. PGE2, which is produced by the inducible enzyme COX-2, has also been implicated as an important mediator in the development of many chronic inflammatory diseases. Therefore, production of endotoxin-induced NO and PGE2 can be used as a measure of the progression of inflammation, and inhibition of their production might have potential therapeutic value for preventing inflammatory reactions and disease. Consistent with previous results,25,26 we found that cordycepin significantly inhibited LPS-stimulated NO and PGE2 production in RAW 264.7 cells. This suppression was possibly due to inhibiting iNOS and COX-2 upregulation at the transcriptional level during RAW 264.7 cell activation by LPS (Figure 1).
Excessive production of proinflammatory cytokines such as TNF-α and IL-1β has also been linked to the development of chronic inflammatory diseases, including rheumatoid arthritis, septic shock, psoriasis, and cytotoxicity.37,38 This process is further increased by autocrine and paracrine routes, which markedly increased the severity of the immune response.39,40 Moreover, production of TNF-α and IL-1β is required for the synergistic induction of NO and PGE2 production in LPS-stimulated macrophages.37,41 Thus, overproduction of these cytokines is a histopathological hallmark of various inflammation-related diseases, and selective inhibition of their production and function may be effective therapeutically in the control of inflammatory disorders. As reported previously,25,26 our data also indicate that cordycepin significantly inhibits LPS-induced release of TNF-α and IL-1β in RAW 264.7 cells. This inhibitory effect may be attributable to the suppression of TNF-α and IL-1β transcription and subsequent decreased protein expression (Figure 2).
In particular, recent evidence had shown that LPS-mediated inflammation is highly associated with various intracellular signaling pathways, such as the NF-κB and MAPK cascades. Of these, NF-κB is important for LPS-stimulated inflammation, which regulates a number of inflammatory genes, including iNOS, COX-2, TNF-α, and IL-1β.42,43 It is well known that inactive NF-κB predominantly resides in the cytoplasm in a complex with IκBα, which is an IκB protein.44,45 However, IκB proteins are rapidly phosphorylated in response to proinflammatory stimuli and are subsequently degraded by the proteosomal pathway. The resulting free NF-κB then translocates to the nucleus where it binds to κB-binding sites in the promoter regions of target genes to promote their transcription, thereby reducing inflammation. NF-κB-targeted therapeutics could be effective for treating inflammatory diseases, as many anti-inflammatory agents exhibit their potency by suppressing NF-κB signaling. In agreement with previous observations,24,26,33 our data demonstrate that the anti-inflammatory effects of cordycepin appeared to involve inhibition of NF-κB activation by blocking LPS-stimulated IκBα degradation and translocation of the NF-κB/p65 protein to the nucleus (Figure 3).
The MAPK families, including p38 MPAK, ERK, and JNK, are a group of serine/threonine kinases that are activated in response to diverse extracellular stimuli and control cellular signal transduction from the cell surface to the nucleus.46,47 Moreover, phosphorylation and activation of MAPKs have previously been implicated in the signaling pathways relevant to LPS-induced inflammation, suggesting that MAPKs are important targets for anti-inflammatory molecules.40 Kim et al26 reported that cordycepin inhibits NO production and COX-2 gene expression by suppressing p38 MAPK phosphorylation, and a similar observation was made in this study. Moreover, pretreatment with cordycepin inhibited ERK and JNK phosphorylation (Figure 4). These results demonstrate that inactivation of MAPK signaling by cordycepin may contribute to suppressing proinflammatory responses in LPS-stimulated RAW 264.7 macrophages.
TLRs are pattern recognition receptors that play a central role in helping direct the innate immune system, which is generally involved in host defense mechanisms, either by identifying pathogens or as receptors for proinflammatory molecules.48–50 Among them, TLR4 is the most important LPS receptor.51 LPS-activated TLR4 induces activation of specific intracellular pathways through receptor dimerization and recruitment of different adapter molecules such as MyD88.52 Previous studies have shown that MAPKs become phosphorylated following TLR4 activation by LPS, and that the transcription factor NF-κB is directly activated.53,54 In the present study, we demonstrated that increased expression of TLR4 and Myd88 proteins was concentration-dependently reduced in the presence of cordycepin (Figure 5A). Our observations also show that pretreatment with genistein markedly attenuated LPS binding to the cell surface (Figure 5B), suggesting that genistein may interfere with the clustering of TLR4 with MyD88. In addition, pretreatment with cordycepin markedly inhibited LPS binding to the RAW 264.7 cell surface (Figure 5C), indicating the antagonistic effect of cordycepin against TLR4. Furthermore, cotreatment of cordycepin with CLI-095, a specific TLR4 signaling inhibitor, synergistically attenuated the release of NO and PGE2 as well as iNOS and COX-2 mRNA and protein expression (Figure 6). Similarly, we found that cotreatment inhibited the production and expression of TNF-α and IL-1β to a greater degree than that in the groups treated with cordycepin alone (Figure 7). In addition, under the same experimental conditions, no cytotoxic effect was observed within our tested concentrations of cordycepin, LPS, and CLI-095 in RAW 264.7 cells, indicating that the inhibitory effect did not result from cytotoxicity (Figure 8). These observations suggest that cordycepin inhibits the initiation of intracellular signaling cascades, which subsequently suppress activation of the MAPK and NF-κB signaling pathways. Several studies have reported that certain anti-inflammatory agents compete with LPS for binding to TLR4, resulting in suppression of downstream signaling pathways.55,56 Therefore, the antagonistic function of cordycepin against TLR4 may be responsible for the anti-inflammatory effects of cordycepin in LPS-stimulated RAW 264.7 macrophages.
Conclusion
In summary, we report herein the anti-inflammatory effects of cordycepin in activated macrophages, and analyze the molecular mechanisms. Our results demonstrate that cordycepin significantly reduced production of proinflammatory mediators and cytokines by inactivating their corresponding genes at the transcriptional levels, which was connected with inhibition of MAPK and NF-κB signaling cascades (Figure 9). Furthermore, we found that cordycepin inhibits TLR4 signaling by interfering with LPS and TLR4 interactions in RAW 264.7 cells. Based on these findings, we suggest that cordycepin blocks the initiation of intracellular signaling cascades through LPS and through inhibiting MAPK and NF-κB activation by regulating the binding of LPS to TLR4 on macrophages.
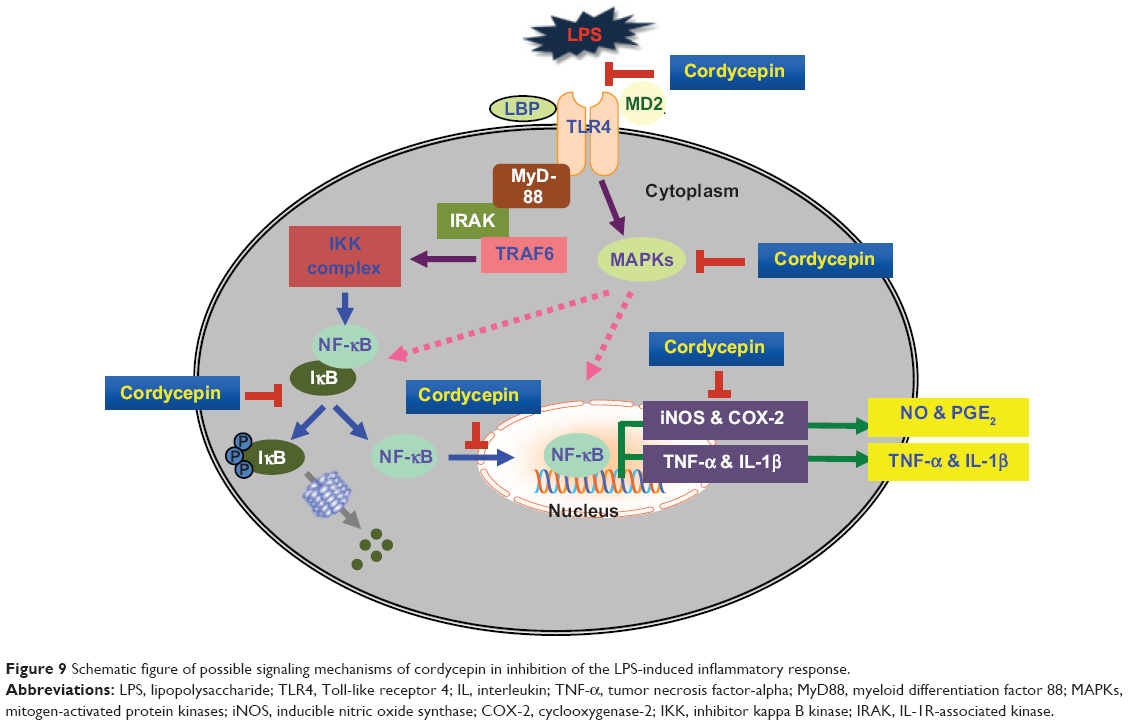 | Figure 9 Schematic figure of possible signaling mechanisms of cordycepin in inhibition of the LPS-induced inflammatory response. |
Acknowledgment
This study was supported by the Basic Science Research Program through a National Research Foundation of Korea grant (2012046358) funded by the government of the Republic of Korea.
Disclosure
The authors report no conflicts of interest in this work.
References
Lind L. Circulating markers of inflammation and atherosclerosis. Atherosclerosis. 2003;169(2):203–214. | ||
Bertolini A, Ottani A, Sandrini M. Dual acting anti-inflammatory drugs: a reappraisal. Pharmacol Res. 2001;44(6):437–450. | ||
Kanno S, Shouji A, Tomizawa A, et al. Inhibitory effect of naringin on lipopolysaccharide (LPS)-induced endotoxin shock in mice and nitric oxide production in RAW 264.7 macrophages. Life Sci. 2006;78(7):673–681. | ||
Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. | ||
Rietschel ET, Kirikae T, Schade FU, et al. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 1994;8(2):217–225. | ||
Anwar MA, Basith S, Choi S. Negative regulatory approaches to the attenuation of Toll-like receptor signaling. Exp Mol Med. 2013;45:e11. | ||
Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. | ||
Chan ED, Riches DW. IFN-gamma + LPS induction of iNOS is modulated by ERK, JNK/SAPK, and p38(mapk) in a mouse macrophage cell line. Am J Physiol Cell Physiol. 2001;280(3):C441–C450. | ||
Hattori Y, Hattori S, Kasai K. Lipopolysaccharide activates Akt in vascular smooth muscle cells resulting in induction of inducible nitric oxide synthase through nuclear factor-kappa B activation. Eur J Pharmacol. 2003;481(2/3):153–158. | ||
Yue K, Ye M, Zhou Z, Sun W, Lin X. The genus Cordyceps: a chemical and pharmacological review. J Pharm Pharmacol. 2013;65(4):474–493. | ||
Paterson RR. Cordyceps: a traditional Chinese medicine and another fungal therapeutic biofactory? Phytochemistry. 2008;69(7):1469–1495. | ||
Zhou X, Gong Z, Su Y, Lin J, Tang K. Cordyceps fungi: natural products, pharmacological functions and developmental products. J Pharm Pharmacol. 2009;61(3):279–291. | ||
Ng TB, Wang HX. Pharmacological actions of Cordyceps, a prized folk medicine. J Pharm Pharmacol. 2005;57(12):1509–1519. | ||
Müller WE, Seibert G, Beyer R, Breter HJ, Maidhof A, Zahn RK. Effect of cordycepin on nucleic acid metabolism in L5178Y cells and on nucleic acid-synthesizing enzyme systems. Cancer Res. 1977;37(10):3824–3833. | ||
Siev M, Weinberg R, Penman S. The selective interruption of nucleolar RNA synthesis in HeLa cells by cordycepin. J Cell Biol. 1969;41(2):510–520. | ||
Sugar AM, McCaffrey RP. Antifungal activity of 3′-deoxyadenosine (cordycepin). Antimicrob Agents Chemother. 1998;42(6):1424–1427. | ||
Ahn YJ, Park SJ, Lee SG, Shin SC, Choi DH. Cordycepin: selective growth inhibitor derived from liquid culture of Cordyceps militaris against Clostridium spp. J Agric Food Chem. 2000;48(7):2744–2748. | ||
Ramesh T, Yoo SK, Kim SW, et al. Cordycepin (3′-deoxyadenosine) attenuates age-related oxidative stress and ameliorates antioxidant capacity in rats. Exp Gerontol. 2012;47(12):979–987. | ||
Xiao L, Ge Y, Sun L, et al. Cordycepin inhibits albumin-induced epithelial-mesenchymal transition of renal tubular epithelial cells by reducing reactive oxygen species production. Free Radic Res. 2012;46(2):174–183. | ||
Zhou X, Luo L, Dressel W, et al. Cordycepin is an immunoregulatory active ingredient of Cordyceps sinensis. Am J Chin Med. 2008;36(5):967–980. | ||
Jeong MH, Seo MJ, Park JU, et al. Effect of cordycepin purified from Cordyceps militaris on Th1 and Th2 cytokines in mouse splenocytes. J Microbiol Biotechnol. 2012;22(8):1161–1164. | ||
Kondrashov A, Meijer HA, Barthet-Barateig A, et al. Inhibition of polyadenylation reduces inflammatory gene induction. RNA. 2012;18(12):2236–2250. | ||
Ren Z, Cui J, Huo Z, et al. Cordycepin suppresses TNF-α-induced NF-κB activation by reducing p65 transcriptional activity, inhibiting IκBα phosphorylation, and blocking IKKγ ubiquitination. Int Immunopharmacol. 2012;14(4):698–703. | ||
Jeong JW, Jin CY, Kim GY, et al. Anti-inflammatory effects of cordycepin via suppression of inflammatory mediators in BV2 microglial cells. Int Immunopharmacol. 2010;10(12):1580–1586. | ||
Shin S, Lee S, Kwon J, et al. Cordycepin suppresses expression of diabetes regulating genes by inhibition of lipopolysaccharide-induced inflammation in macrophages. Immune Netw. 2009;9(3):98–105. | ||
Kim HG, Shrestha B, Lim SY, et al. Cordycepin inhibits lipopolysaccharide-induced inflammation by the suppression of NF-kappaB through Akt and p38 inhibition in RAW 264.7 macrophage cells. Eur J Pharmacol. 2006;545(2/3):192–199. | ||
Kim H, Naura AS, Errami Y, Ju J, Boulares AH. Cordycepin blocks lung injury associated inflammation and promotes BRCA1-deficient breast cancer cell killing by effectively inhibiting PARP. Mol Med. 2011;17(9/10):893–900. | ||
Foss FM. Combination therapy with purine nucleoside analogs. Oncology (Williston Park). 2000;14(6 Suppl 2):S31–S35. | ||
Nakamura K, Yoshikawa N, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M. Antitumor effect of cordycepin (3′-deoxyadenosine) on mouse melanoma and lung carcinoma cells involves adenosine A3 receptor stimulation. Anticancer Res. 2006;26(1A):43–47. | ||
Kyung J, Kim D, Park D, et al. Synergistic anti-inflammatory effects of Laminaria japonica fucoidan and Cistanche tubulosa extract. Lab Anim Res. 2012;28(2):91–97. | ||
Lee SH, Kim DW, Eom SA, et al. Suppression of 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced skin inflammation in mice by transduced Tat-Annexin protein. BMB Rep. 2012;45(6):354–359. | ||
Lee EJ, Kim WJ, Moon SK. Cordycepin suppresses TNF-alpha-induced invasion, migration and matrix metalloproteinase-9 expression in human bladder cancer cells. Phytother Res. 2010;24(12):1755–1761. | ||
Lee YR, Noh EM, Jeong EY, et al. Cordycepin inhibits UVB-induced matrix metalloproteinase expression by suppressing the NFkappaB pathway in human dermal fibroblasts. Exp Mol Med. 2009;41(8):548–554. | ||
Noh EM, Kim JS, Hur H, et al. Cordycepin inhibits IL-1beta-induced MMP-1 and MMP-3 expression in rheumatoid arthritis synovial fibroblasts. Rheumatology (Oxford). 2009;48(1):45–48. | ||
Meda L, Cassatella MA, Szendrei GI, et al. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374(6523):647–650. | ||
Dandona P, Chaudhuri A, Dhindsa S. Proinflammatory and prothrombotic effects of hypoglycemia. Diabetes Care. 2010;33(7):1686–1687. | ||
Aggarwal BB, Natarajan K. Tumor necrosis factors: developments during the last decade. Eur Cytokine Netw. 1996;7(2):93–124. | ||
Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118(11):3537–3545. | ||
Pearlman DS. Pathophysiology of the inflammatory response. J Allergy Clin Immunol. 1999;104(4 Pt 1):S132–S137. | ||
Beutler B, Cerami A. The biology of cachectin/TNF – primary mediator of the host response. Annu Rev Immunol. 1989;7:625–655. | ||
Blatteis CM, Li S, Li Z, Perlik V, Feleder C. Signaling the brain in systemic inflammation: the role of complement. Front Biosci. 2004;9:915–931. | ||
Islam MN, Choi RJ, Jin SE, et al. Mechanism of anti-inflammatory activity of umbelliferone 6-carboxylic acid isolated from Angelica decursiva. J Ethnopharmacol. 2012;144(1):175–181. | ||
Chen JJ, Huang WC, Chen CC. Transcriptional regulation of cyclooxygenase-2 in response to proteasome inhibitors involves reactive oxygen species-mediated signaling pathway and recruitment of CCAAT/enhancer-binding protein delta and CREB-binding protein. Mol Biol Cell. 2005;16(12):5579–5591. | ||
Rajapakse N, Kim MM, Mendis E, Kim SK. Inhibition of inducible nitric oxide synthase and cyclooxygenase-2 in lipopolysaccharide-stimulated RAW264.7 cells by carboxybutyrylated glucosamine takes place via down-regulation of mitogen-activated protein kinase-mediated nuclear factor-kappaB signaling. Immunology. 2008;123(3):348–357. | ||
Denkers EY, Butcher BA, Del Rio L, Kim L. Manipulation of mitogen-activated protein kinase/nuclear factor-kappaB-signaling cascades during intracellular Toxoplasma gondii infection. Immunol Rev. 2004;201:191–205. | ||
Kirkwood KL, Rossa C Jr. The potential of p38 MAPK inhibitors to modulate periodontal infections. Curr Drug Metab. 2009;10(1):55–67. | ||
Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy-from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta. 2005;1754(1/2):253–262. | ||
Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13(2):85–94. | ||
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388(6640):394–397. | ||
Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Microbes Infect. 2004;6(15):1382–1387. | ||
Shuto T, Kato K, Mori Y, et al. Membrane-anchored CD14 is required for LPS-induced TLR4 endocytosis in TLR4/MD-2/CD14 overexpressing CHO cells. Biochem Biophys Res Commun. 2005;338(3):1402–1409. | ||
Fitzgerald KA, Palsson-McDermott EM, Bowie AG, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413(6851):78–83. | ||
Jawan B, Kao YH, Goto S, et al. Propofol pretreatment attenuates LPS-induced granulocyte-macrophage colony-stimulating factor production in cultured hepatocytes by suppressing MAPK/ERK activity and NF-kappaB translocation. Toxicol Appl Pharmacol. 2008;229(3):362–373. | ||
Kawai T, Takeuchi O, Fujita T, et al. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001;167(10):5887–5894. | ||
Jung JS, Shin KO, Lee YM, et al. Anti-inflammatory mechanism of exogenous C2 ceramide in lipopolysaccharide-stimulated microglia. Biochim Biophys Acta. 2013;1831(6):1016–1026. | ||
Lee JY, Ye J, Gao Z, et al. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. 2003;278(39):37041–37051. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.