Back to Journals » Drug Design, Development and Therapy » Volume 10
Anti-inflammatory drugs for Duchenne muscular dystrophy: focus on skeletal muscle-releasing factors
Authors Miyatake S, Shimizu-Motohashi Y, Takeda S, Aoki Y
Received 8 April 2016
Accepted for publication 26 May 2016
Published 30 August 2016 Volume 2016:10 Pages 2745—2758
DOI https://doi.org/10.2147/DDDT.S110163
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Shouta Miyatake,1 Yuko Shimizu-Motohashi,2 Shin’ichi Takeda,1 Yoshitsugu Aoki1
1Department of Molecular Therapy, National Institute of Neuroscience, National Center of Neurology and Psychiatry, Kodaira, Tokyo, Japan; 2Department of Child Neurology, National Center Hospital, National Center of Neurology and Psychiatry, Kodaira, Tokyo, Japan
Abstract: Duchenne muscular dystrophy (DMD), an incurable and a progressive muscle wasting disease, is caused by the absence of dystrophin protein, leading to recurrent muscle fiber damage during contraction. The inflammatory response to fiber damage is a compelling candidate mechanism for disease exacerbation. The only established pharmacological treatment for DMD is corticosteroids to suppress muscle inflammation, however this treatment is limited by its insufficient therapeutic efficacy and considerable side effects. Recent reports show the therapeutic potential of inhibiting or enhancing pro- or anti-inflammatory factors released from DMD skeletal muscles, resulting in significant recovery from muscle atrophy and dysfunction. We discuss and review the recent findings of DMD inflammation and opportunities for drug development targeting specific releasing factors from skeletal muscles. It has been speculated that nonsteroidal anti-inflammatory drugs targeting specific inflammatory factors are more effective and have less side effects for DMD compared with steroidal drugs. For example, calcium channels, reactive oxygen species, and nuclear factor-κB signaling factors are the most promising targets as master regulators of inflammatory response in DMD skeletal muscles. If they are combined with an oligonucleotide-based exon skipping therapy to restore dystrophin expression, the anti-inflammatory drug therapies may address the present therapeutic limitation of low efficiency for DMD.
Keywords: calcium channels, ryanodine receptor 1, exon skipping, NF-κB, myokine, ROS
Introduction
Duchenne muscular dystrophy (DMD), the most common form of muscular dystrophy, involves progressive deterioration of muscle function, affecting up to one in 3,800–6,000 live male births.1 DMD is caused mainly by a frameshift deletion, nonsense, or duplication mutations in the DMD gene on the X chromosome (Xp21.2), which encodes the protein dystrophin.2 Dystrophin is a member of the spectrin superfamily of cytoskeletal proteins. Its full-length mRNA is mainly expressed in skeletal and cardiac muscles, as well as in small amounts in brain. In healthy muscle, dystrophin is located on the intracellular surface of the sarcolemma, along with the sarcomeres.3 The structure is called the costamere, and it constitutes the structural cornerstone of muscular cells. At costameres, dystrophin assembles with the dystrophin-associated glycoprotein complex, which stabilizes the sarcolemma during muscular contractions. Dystrophin acts as a bridging and anchoring protein by binding to F-actin through its cytoplasmic N-terminal domain and to β-dystroglycan through its extracellular C-terminal domain.4,5 The loss of dystrophin disrupts the dystrophin-associated glycoprotein complex and causes membrane instability making it susceptible to damage and myofiber necrosis. Such circumstances provoke an abnormal persistence of inflammatory macrophages in the muscle, inducing chronic inflammation with impaired regeneration and ultimately fibrosis associated with the replacement of muscle by connective tissue, and consequently severe muscle dysfunction.6–8 Eventually, the loss of dystrophin results in severe muscle atrophy, respiratory and cardiac failure, and death before the age of 30 years.9 Despite these findings, the molecular mechanisms initiating and perpetuating inflammation in DMD are poorly understood.
The most promising therapeutic candidate to overcome the deletion of dystrophin in DMD is exon-skipping therapy using antisense oligonucleotides (ASOs). ASOs can switch splicing patterns by targeting specific sequences of pre-mRNA elements involved in exon recognition and/or consensus splice sites in a sequence-specific manner.10 Targeting splice sites or putative exon splicing enhancers with ASOs can induce the removal of exons from the mature DMD transcript so that a nonsense mutation is bypassed, or alternately removal of exons around a genomic deletion can restore the mRNA reading frame. ASOs have been extensively tested in disease models and are currently being evaluated in several clinical trials including ASO-based exon53 skipping therapy at our institute in Japan (http://www.ClinicalTrials.gov; NCT02081625).11,12
However, each ASO has a specific antisense sequence for single exon in the DMD gene and is considered a new drug; therefore, ASOs for separate exons have to undergo expensive and lengthy clinical trial stages. Furthermore, challenges involving exon skipping for duplications exist given the necessity to skip the only duplicated exon, in addition to the lack of an original exon for in-frame restoration and also the inability to recruit sufficient patients for clinical trials for exon skipping for rare mutations in DMD.13 Thus, in addition to the dystrophin restoration, an efficient treatment should consider the possibility of inhibiting the muscle inflammation associated with dystrophin deletion that is common to all DMD patients.
Regarding skeletal muscle inflammation, the functions of skeletal muscle as a secretory organ should be considered. The cytokines released by skeletal muscle are called “myokines”; they are considered as autocrine and paracrine factors and are important mediators of communication between skeletal muscle and other organs in the endocrine system. They can exert profound effects on glucose and lipid metabolism and can be important mediators in inflammatory processes. As such, they are involved in energy homeostasis and the pathogenesis of obesity, diabetes, and other diseases.14
In this review, the recent findings on the inflammatory mechanisms in DMD are discussed, especially focusing on inflammatory factors released from skeletal muscle, which are regulated by calcium influx, reactive oxygen species (ROS), and nuclear factor-kappa B (NF-κB) (Figure 1). This review emphasizes the future directions in DMD therapy targeting those master regulators of inflammation as well as each releasing factor regulated in DMD muscles.
 | Figure 1 The hypothetical mechanism of Ca2+ overflow-induced inflammation response in DMD muscles. |
Broad anti-inflammatory drug – corticosteroids and nonsteroidal anti-inflammatory drugs targeting cyclooxygenases
The only currently accepted pharmacological therapy for DMD is corticosteroid-based anti-inflammatory treatment. In short-term clinical trials, corticosteroids have been shown to improve muscle strength and function without clinically severe adverse effects.15,16 In addition, nonrandomized trials have been shown significant beneficial effects on ambulation and cardiac function, delayed onset of both scoliosis and respiratory dysfunction, and a general amelioration of quality of life in treatments with prednisone or deflazacort for >2 years.17
However, tolerance to chronic use and heterogeneous response to treatment are well-known drawbacks of corticosteroid therapy.18 Glucocorticoids act through multiple mechanisms of action, making it unclear and controversial which molecular pathways provide the efficacy in DMD treatment and which pathways are responsible for detrimental effects. For example, glucocorticoids have a side effect of impaired growth in children with asthma,19,20 but the related limited muscle workload and delayed muscle maturation have been proposed to contribute to efficacy in DMD.21
Like corticosteroids, treatment with commonly used nonsteroidal anti-inflammatory drugs (NSAIDs) broadly inhibiting cyclooxygenase (COX) enzymes had only partial therapeutic effects in an animal study. COXs generate prostaglandins and lipid autacoids from arachidonic acid, leading to pathogenic mechanisms, including the inflammatory response.22 Daily treatment with NSAIDs (COX inhibitors) such as aspirin and ibuprofen was effective in ameliorating muscle morphology and reducing macrophage infiltration and necrosis but did not modify the percentage of regenerating myofibers. In addition, isometric tension did not differ in treated and untreated muscles; however, resistance to fatigue decreased by treatment with aspirin and not with ibuprofen.23 In this study, parecoxib, a COX-2-selective inhibitor, was ineffective against DMD pathogenesis. COX-2 is known to be activated mainly in leukocytes, not muscles. In addition, steroids showed robust inhibition of COX-2 in leukocytes, which had only partial effects as mentioned earlier.24 Therefore, it is possible that in order to treat DMD efficiently, the inflammatory process needs to be inhibited not only in leukocytes but in muscles as well.
Considering how problematic and insufficient corticosteroids and NSAIDs appear to be as anti-inflammatory agents in the treatment of DMD, it is possible that more specific, safer, and more effective drugs need to be developed in this area.
Potential drugs targeting inflammatory mechanisms in DMD
Store-operated Ca2+ entry and transient receptor potential calcium channels
Based on the mechanism of inflammation in DMD, initial targets for anti-inflammatory drugs are calcium channels. Loss of dystrophin leads to a structurally weaker plasma membrane that is easily damaged during muscle contraction and allows both extracellular calcium influx and release of endogenous ligands. The upregulated cytoplasmic Ca2+ leads to calpain overactivation in DMD, which can activate the NF-κB inflammatory pathways mediated by degradation of IkBα (an NF-κB inhibitor)25 and stimulate inflammatory cytokine release, which further impairs muscle regeneration (Figure 1). This pathway induces abnormal persistence of inflammatory cells such as macrophages.26,27 In fact, elevated Ca2+ influx through calcium channels was sufficient to induce a dystrophic phenotype, including increased central nucleated myofibers, fibrosis, and infiltration of inflammatory cells28 (Figure 1). However, the mechanism of calcium over-influx in DMD muscle is not clearly understood. One of the proposed models suggest that extracellular calcium leaks into dystrophin-deficient myofibers during muscle contraction, inducing local hypercontraction after repeated contraction, which magnifies membrane damage on adjacent lengthened regions in the same or adjacent muscle fibers.29 In contrast, dystrophin deficiency increases calcium concentration mediated by 1) store-operated Ca2+ entry (SOCE) machinery as a result of by reduction of intracellular Ca2+ stores, 2) concentrated stretch-activated channels on plasma membrane such as the transient receptor potential (TRP) cation channel, and 3) the oxidized, overactivated “leaky” ryanodine receptor 1 (RyR1) described in “ROS and ryanodine receptor 1” section (Figure 1).
The depletion of endoplasmic/sarcoplasmic reticulum (ER/SR) Ca2+ stores promotes the translocation of stromal interaction molecule 1 (STIM1), a calcium sensor in the ER/SR membrane, to regions close to the plasma membrane, where STIM1 activates Orai, a pore-forming unit that allows Ca2+ influx through the plasma membrane into the cytosol.30 The expression of Orai1 was elevated in the dystrophic muscles, whereas STIM1 levels remained largely unchanged, together with increased SOCE activity in adult muscles of mdx mice. When Orai1 was inhibited with BTP-2, a specific SOCE inhibitor injected for 2 weeks in mdx mice, the cytosolic calpain1 activity in myofibers was significantly reduced, indicating that upregulation of Orai1-mediated SOCE pathway contributed to the disrupted Ca2+ homeostasis in mdx muscle.30
TRP channels promote calcium overload from extracellular calcium as a result of their expression in plasma membrane. In skeletal muscle, several isoforms of the TRPC (canonical), TRPV (vanilloid), and TRPM (melastatin) subfamilies are expressed. In particular, TRPC1, C3, and C6; TRPV1, V2, and V4; and TRPM4 and M7 have been consistently found in cultured myoblasts or in adult muscles; however, only some of the TRPC and TRPV isoforms have been studied in skeletal muscle.31
In the TRPC subfamily, expression of TRPC1, C3, and C6 was increased in dystrophic muscle.30,32 The abnormal activation of these channels in mdx fibers might be mediated by ROS production and Src kinase activation.32 TRPC1 may contribute through binding with STIM1–Orai1 complex as a result of Ca2+ store depletion.31 Additionally, Ca2+ influx through TRPC3 and C6 channels has been shown to be sufficient to induce muscle dystrophy.28
In the TRPV subfamily, Iwata et al33 reported that TRPV2 was abundantly expressed in the sarcolemma of dystrophic myocytes and that cyclic cellular stretch increased TRPV2 translocation to the membrane. It was found that dominant-negative inhibition of endogenous TRPV2 in mdx mice suppressed the calcium increase in muscle fibers and eased dystrophic pathology, that is, the increased number of central nucleus and fiber size variability/fibrosis/apoptosis, elevated serum creatine kinase levels, and reduced muscle performance.33 Tranilast, a clinically used antiallergic drug, is also known to be a TRPV2 blocker experimentally,34 and its administration was found to be efficacious in reducing serum creatine kinase levels in mdx mice35 and fibrosis in mdx skeletal muscles, leading to improved resistance to muscle fatigue.36
Thus far, the possible involvement of TRPV4 in DMD has not been investigated; however, it is notable that TRPV4 was activated by phospholipase A2,37 an enzyme involved in SOCE in dystophic myofibers,38 and that its expression is increased by 10- to 40-fold in dystrophinopathies39 (Table 1).
 | Table 1 Regulation of Ca2+ transport-related proteins in DMD muscles |
ROS and ryanodine receptor 1
In addition to calcium influx, elevated ROS production in DMD is a potential target for anti-inflammatory therapy, as suggested by the reports showing the inhibitory effect of the antioxidant N-acetylcysteine on muscle damage in mdx mice accompanied with reduced NF-κB activity40 and also the contribution of ROS to RyR1 or TRPV1-induced intracellular Ca2+ increase.41,42
The mechanical distension of the sarcolemma induced by contraction causes the formation of superoxide anion radicals by the action of nicotinamide adenine dinucleotide phosphate oxidases (NOX), including NOX type 2 and type 4 in skeletal muscle. This radical cannot be scavenged by nitric oxide to form peroxynitrite because of its low presence in DMD muscles, followed by the oxidization of RyRs and indirect activation of stretch-activated channels41 (Figure 2). Notably, NOX inhibition with diapocynin, a dimer of the commonly used NOX inhibitor apocynin, was shown to inhibit the loss of strength of skeletal muscles induced by eccentric contractions in mdx mice, a model of DMD, to levels similar to those in wild-type mice.43 In a potential therapy targeting downstream of ROS production, stabilization of the closed state of RyRs by Rycals, a RyR channel inhibitor, restored cardiac and skeletal muscle function by normalizing oxidized and overactivated RyRs.44,45
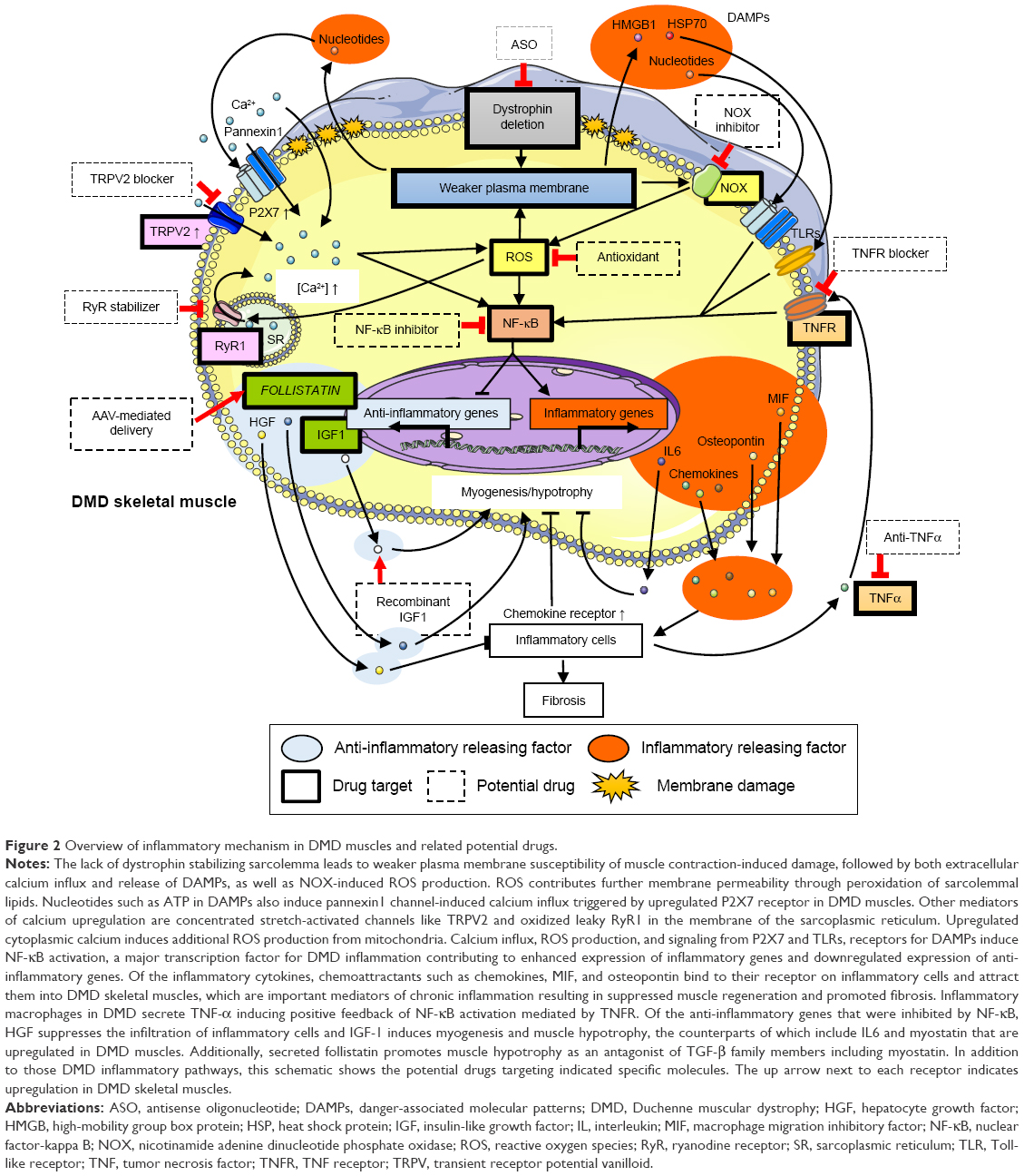 | Figure 2 Overview of inflammatory mechanism in DMD muscles and related potential drugs. |
In contrast, Ca2+ overload in mitochondria directly increases ROS levels,46 which further contributes to membrane permeability through sarcolemmal lipid peroxidation.47 The antioxidant idebenone (Catena®; Santhera Pharmaceuticals, Liestal, Switzerland), a synthetic analog of ubiquinone (coenzyme Q10), is currently under clinical trials to test its efficacy in the prevention of skeletal muscle function loss and heart pump function reduction. Santhera Pharmaceuticals has confirmed the safety of idebenone in DMD patients (https://ClinicalTrials.gov/ct2/show/NCT00654784). There is an ongoing Phase II extension trial to test its safety and efficacy in the long-term. A Phase III trial was also carried out to assess the effect of idebenone on pulmonary function, motor function, muscle strength, and quality of life in patients not treated with corticosteroids. The results show that idebenone was well tolerated, and a slower decline in respiratory function was observed for treated patients compared to that observed for the placebo group (https://ClinicalTrials.gov/ct2/show/NCT01027884). These beneficial effects of idebenone can be explained by its ability to protect against mitochondrial respiratory chain dysfunction and reduce ROS production.48
Santhera plans to file for marketing authorization in 2016. Furthermore, it is planning a placebo-controlled Phase III study in corticosteroids-treated patients. Additionally, preclinical studies with green tea extract and its active ingredient epigallocatechin gallate revealed histological and functional improvement in mdx mice.49,50 Furthermore, early treatment with green tea extract reduced dystrophic muscle pathology, potentially by regulating NF-κB activity in regenerating fibers.51 Epigallocatechin gallate for DMD patients is now under Phase II/III trials at Charité University Hospital Berlin, Germany (https://www.ClinicalTrials.gov/ct2/show/NCT01183767). Flavocoxid is another antioxidant that has been tested in a Phase I trial in DMD patients to assess its safety (https://www.ClinicalTrials.gov/ct2/show/CAT-1004) (Table 2; Figure 2).
 | Table 2 The potential drugs for DMD anti-inflammatory therapy |
NF-κB, tumor necrosis factor-α, and chemokines
NF-κB, one of the major transcription factor regulating inflammatory pathways, is also an important target for anti-inflammatory therapy. CAT-1004, an inhibitor of NF-κB has completed Phase I clinical trial (https://ClinicalTrials.gov/ct2/show/VBP15) (Table 2).
NF-κB is activated in dystrophic muscles, possibly following calcium influx and/or ROS production,25,40 and targets a wide range of genes, including inflammatory cytokines and chemokines.52 Interestingly, NF-κB is activated among the earliest histological features of DMD neonates,53–55 years before symptoms appear. It suggests that very early treatment of DMD patients with NF-κB suppression therapy may prevent or delay the onset of muscle dysfunction as a result of improved muscle regeneration and reduction of fibrosis.
Ablation of one allele of the p65 subunit of NF-κB was sufficient to improve pathology in mdx mice. In addition, a study explored conditional deletion of IκB kinase (IKK) β in mdx mice, a component of the IKK complex with IKKα as catalytic subunits that induce NF-κB activation by IκB degradation. It was found that NF-κB in activated macrophages promotes inflammation and muscle necrosis in skeletal muscle fibers, resulting in limited regeneration through the inhibition of muscle progenitor cells. In addition, specific pharmacological inhibition of IKK resulted in improved pathology and muscle function in mdx mice.26
Recently, Heier et al identified VBP15, a compound structurally related to glucocorticoids with similar anti-inflammatory properties but without steroidal side effects.56 VBP15 inhibited NF-κB-induced tumor necrosis factor (TNF)-α release mediated by the glucocorticoid receptor, independently of the glucocorticoid response elements (eg, classical steroid receptor transactivation or hormonal properties), activation or upregulation that is implicated in a number of glucocorticoid side effects. It also had protective physicochemical effects on the cell membrane. The translation of these drug mechanisms into mdx mice improved muscle strength, live imaging, and pathology through both preventive and post-onset intervention regimens. VBP15 is now under a Phase I clinical trial (Table 2).
Furthermore, the inhibitors of TNF-α, a chemokine that induces NF-κB activation, have shown some potential in improving DMD pathology in animal studies. Remicade, a human anti-TNF-α antibody and etanercept, a blocker of soluble TNF-α receptor, are two candidates clinically used to treat inflammatory disorders (Table 2). Remicade delayed and reduced the inflammation and disruption of dystrophic muscle without adverse effects in mdx mice.57 Etanercept inhibited exercise-induced force reduction in skeletal muscle58 and protected against exercise-induced myofiber necrosis in mdx mice.59 Furthermore, a murine-specific TNF-α antibody significantly reduced contractile dysfunction and myofiber necrosis.60
Chemokines, whose expression is regulated by NF-κB, are major contributors to DMD pathogenesis promoting persistent inflammatory cells such as CD4+ and CD8+ T-cells, neutrophils, eosinophils, and inflammatory macrophages.27 Besides immune cells, the main source of chemokines in skeletal muscle, muscle fibers can also chemoattract myeloid cells secreting chemokines. We recently found that NF-κB is activated in C2C12 myotubes upon muscle contraction and upregulate C-C motif chemokine ligand (CCL) 2 secretion, inducing THP-1 monocyte chemoattraction.61 In addition, several CC chemokines including CCL2, which are the components of the largest family of chemokines, and chemoattractive for diverse inflammatory cells, showed increased expression in mdx muscle.62 These findings suggest that increased CCL2 secretion from DMD muscles may also contribute to monocyte chemoattraction into skeletal muscles. Furthermore, CCL receptor (CCR) 2 is the only CCL2 receptor whose expression was upregulated in muscles of mdx mice.63 CCR2 is expressed highly in Ly6Chigh inflammatory monocytes, which polarize CD11bhigh inflammatory macrophages. CCR2 deficiency in mdx mice preferentially reduced the CD11bhigh population of macrophages and promoted the recovery of normal macrophage polarization characteristics. Consequently, these mice improved characteristic histopathological features of the disease and increased the force-generating capacity of the diaphragm.6
Also, C-X-C motif chemokine ligands (CXCL) such as CXCL1, CXCL2, CXCL3, CXCL8, and CXCL11 for neutrophils contributing to the inflammatory phase during regeneration and absent from normal muscle fibers were induced in DMD myofibers.62 In addition, we found the secretion of macrophage migration inhibitory factor (MIF), an inflammatory cytokine and chemoattractant, was regulated by contraction in C2C12 myotubes.64 MIF is known to be expressed mainly in leukocytes. However, in an immunohistochemical study, MIF was detected not only in immune cells but also in muscle fiber membrane areas of infiltration, necrosis, myophagocytosis, degeneration, and regeneration in muscular dystrophy samples.65 MIF binds to the CXC chemokine receptors CXCR2 and CXCR4. CXCR2 is the receptor for CXCL1, 2, 3, and 8, which are the CXCLs upregulated in DMD muscles, and CXCR4 is the receptor for CXCL12, which is also upregulated in regenerating myofibers from DMD patients62 and thus can contribute to the inflammation. However, CXCL12 and CXCR4 may be difficult to use as candidates for anti-inflammation therapy because of their major role in muscle regeneration.66 Table 3 describes the potential of chemoattractants and these receptors as future therapeutic targets for DMD.
 | Table 3 The upregulated inflammatory factors and their receptors in DMD muscles as the future targets for anti-inflammatory drugs |
A second mechanism by which IKK/NF-κB signaling regulates the dystrophic process is inhibition of muscle regeneration. Muscle regeneration after injury represents a coordinated sequence of events from the activation of quiescent satellite cells into myoblasts to their subsequent fusion to newly formed myotubes.67 NF-κB has been previously shown to inhibit the differentiation sequence by MyoD repression68 and by preventing myoblast fusion in vitro.69 Additionally, an in vivo study showed that TNF-α, a potent inducer of NF-κB signaling, inhibited myogenesis through repression of MyoD.68 Genetically or pharmacological blocking of NF-κB function promoted the formation of new myofibers in response to degeneration.26 IKK deletion in muscles in response to acute injury could also lead to improved regeneration70 (Figure 2).
In addition to upregulating inflammatory cytokines, NF-κB suppresses hepatocyte growth factor (HGF), an anti-inflammatory cytokine. The mdx; NF-κB p65+/− muscle at 4 weeks of age showed significantly higher HGF expression, correlating with reduced leukocyte infiltration and increased muscle fiber formation. The phenotypic improvements of muscle in these mice were reversed by silencing HGF preferentially in myogenic cells, resulting in significant degeneration of the diaphragm.71
Danger-associated molecular patterns
The disruption of plasma membrane or necrosis following calcium influx in DMD leads to the release of cytoplasmic molecules called danger-associated molecular patterns (DAMPs) that include nucleotides, high-mobility group box protein 1 (HMGB1), hyaluronic acid, biglycan, and heat shock proteins (HSPs). DAMPs can induce chronic inflammation as the ligands of Toll-like receptors (TLRs)72 (Figure 2). Recent proteomic studies of DMD subjects and mdx mice revealed an increased presence of several potential TLR2 ligands in the circulation, including serum amyloid A, HSP70, and fibrinogen.73,74 Other studies detected HMGB1, a shared TLR2/4 ligand, as a potential early inflammatory target in mdx mice,7 in addition to increased HSP70 expression in DMD muscles,75 and fibrinogen as a driver of fibrosis in dystrophic muscle76 (Table 3).
These reports suggest that endogenous TLR ligands released from injured skeletal muscle may be important signals for stimulating chemokine expression in both muscle fibers and hematopoietic progenitor cells mediated by TLR-dependent inflammasome and/or NF-κB activation, thereby helping to attract monocytes/macrophages to the site of damaged skeletal muscle. These TLR2 ligands also affected the function of other inflammatory cell types in DMD muscles such as T lymphocytes, which play an important role in regulating macrophage function and are also implicated in the pathogenesis.77,78 In fact, TLR2 deletion in mdx mice led to reduced macrophage numbers within DMD muscle during the acute inflammatory phase of the disease. The reduced macrophage infiltration within diaphragms of mdx-TLR2−/− mice was associated with a significant amelioration of DMD pathogenesis, that is, reduced necrotic injury, larger regenerated myofibers, and decreased fibrosis, all of which led to a higher force-generating capacity of the muscle. In detail, TLR2 deletion in this context not only reduced macrophage infiltration but also significantly modified macrophage polarization into iNOS−CD206+, anti-inflammatory macrophages.79 Furthermore, mdx mice lacking the TLR signaling adaptor protein MyD88 showed less myofiber necrosis in the diaphragm and improved limb muscle strength at 12 months of age.80 Similarly, TLR4 ablation in mdx mice resulted in significant reduction of the amount of inflammatory macrophages within the diaphragm early in the pathogenesis (6–12 weeks of age), together with improved muscle histology and strength.7
In DMD, ATP release, another major pattern in DAMPs, due to the fragility of myofibers can activate plasma membrane receptors for extracellular nucleotides termed “P2 receptors”. P2X7 was substantially upregulated in skeletal muscle from mdx mice and in myoblasts isolated from DMD patients.81–85 Additionally, exposure of mdx myoblasts to extracellular ATP induced a significant increase in P2X7/pannexin1 channel-dependent Ca2+ influx and release of interleukin (IL)-1β, suggesting that nucleotides released from dystrofic muscle can trigger inflammatory response in DMD through purinergic signaling.86 It was recently reported that in vivo blocking of the extracellular ATP/P2X purinergic signaling pathway by periodate-oxidized ATP delayed the progression of DMD pathogenesis and ameliorated the local inflammatory response in mdx mice, including reduced leukocyte infiltration and IL-6 expression87 (Table 3; Figure 2).
IL-6
IL-6 is one of the most extensively investigated myokines, and its secretion is regulated by exercise. Additionally, it can regulate glucose and lipid homeostasis affecting liver, adipose tissue, and skeletal muscle itself.88 IL-6 expression in skeletal muscle was found to be also upregulated in obesity and to contribute to increased white adipose tissue mass.89
IL-6 is present at high levels in the circulation and muscles from DMD patients and from young mdx mice. It also follows the disease time-course in DMD patients.90–92 IL-6 contributes to satellite cell proliferation93 and muscle growth,94,95 in addition to its major role of inducing the transition from acute neutrophil infiltration to chronic mononuclear cell infiltration.96 Therefore, repeated cycles of degeneration/regeneration, induced by chronic IL-6 upregulation in DMD, might induce the exhaustion of satellite cells, leading to enhancement of the dystrophic phenotype. In fact, IL-6-overexpressing mdx mice showed an exacerbation of the dystrophic phenotype wherein increased circulating levels of IL-6 promoted muscle degeneration, inflammation, exhaustion of muscle stem cells, and accumulation of fibro/adipogenic progenitors.92 Importantly, the neutralization of IL-6 activity in mdx mice using an anti-IL-6 receptor antibody resulted in increased robustness for the dystrophic muscle, impeded the chronic inflammatory response, reduced muscle necrosis, and promoted muscle differentiation and maturation. These events led to the reduction of exercise-induced fiber damage and mitigation of diminished muscle strength.91 The Food and Drug Administration has approved a number of novel compounds blocking IL-6 signaling for the treatment of other inflammatory disorders in the last decade; the application of these drugs to DMD patients can be due to their anti-inflammatory properties (Table 3; Figure 2).
Osteopontin
Osteopontin (OPN) is a secreted and chemotactic phosphoprotein that plays important roles in tissue remodeling following injury.97,98 Recently, elevated serum OPN levels were found in the dystrophic dogs compared with those in the wild type just before and an hour after a cesarean section birth and at the age of 3 months. In addition, the serum OPN level was significantly correlated with the phenotypic severity of dystrophic dogs at the onset of muscle weakness. Immunohistologically, OPN was upregulated in infiltrated macrophages and developmental myosin heavy chain-positive regenerating muscle fibers in the dystrophic dogs.99 It is reported that a promoter polymorphism on the OPN gene was associated with severity of DMD. The G allele in the locus (dominant model; 35% of subjects) inhibiting the binding of transcription factors, specificity protein-1 was associated with more rapid progression and less grip strength, suggesting OPN is a genetic modifier of DMD.100 In addition, the promoter structure of the OPN gene has multiple predicted steroid hormone enhancers and a NF-κB promoter element.101
Genetic ablation of OPN in mdx mice caused significant reduction in the amount of intramuscular neutrophils and natural killer T-like cells and an increased amount of regulatory T-cells. This anti-inflammatory state resulted in a net decrease in the amount of transforming growth factor-beta (TGF-β) in the later stages of DMD pathogenesis. Diminished TGF-β levels were correlated with a marked decrease in fibrosis of both diaphragmatic and cardiac muscles. These studies identified OPN as an immunomodulator and pro-fibrotic cytokine in DMD muscle78 (Table 3; Figure 2).
Myostatin and follistatin
Myostatin, a member of TGF-β family, is a negative regulator of skeletal muscle growth102,103 and is produced predominantly in muscle and blood.102,104 Its expression was significantly higher in mdx mice than in wild-type mice.105 The inhibitory effect of myostatin on postnatal growth is mediated by its negative regulation of satellite cell activation, proliferation, and self-renewal106 as well as myoblast proliferation and differentiation.103,107
Blockade of endogenous myostatin using blocking antibodies for 3 months resulted in increased body weight, muscle mass, muscle size, and absolute muscle strength in mdx muscle, along with decreased muscle degeneration and concentrations of serum creatine kinase.108 Myostatin propeptide also ameliorated the symptoms in mdx mice.109
However, clinical studies targeting myostain have not shown sufficient therapeutic effects. MYO-029, a human myostatin antibody,110 increased the muscle mass in immunodeficient mice by ~30% in 3 months.108 In contrast, a double-blind randomized clinical trial in Becker muscular dystrophy indicated that MYO-029 was safe but not efficacious.110 Another myostatin inhibitor, ACE-031, a soluble activin type IIB receptor was found promising in increasing muscle mass and whole body pulling tension in mdx mice.111 However, a safety–tolerability study of ACE-031 in DMD patients was prematurely terminated because of minor nosebleeds and/or gum bleeding.
Follistatin, a secreted glycoprotein and functional antagonist of the TGF-β family, which includes myostatin, is likely to be more effective as a therapeutic target to promote muscle growth than inhibition of only myostatin, as indicated by a report that heterozygous loss of follistatin resulted in retention of reduced muscle mass in a myostatin-null background.112 This implies that follistatin inhibits other TGF family members in addition to myostatin to regulate muscle size. Delivery of the FOLLISTATIN gene by adeno-associated virus (AAV1. CMV.FS344) in Becker muscular dystrophy patients in a Phase I/IIa clinical study showed safety and efficacy as determined by the distance walked in a 6-minute walk test, along with improved muscle histopathology.113 Based on this positive result, the same strategy for DMD is currently under a Phase I/II clinical study (https://ClinicalTrials.gov/ct2/show/NCT02354781) (Table 2; Figure 2).
Insulin-like growth factor-1
Contrary to the inhibition of inflammatory response, there is also the strategy of enhancement of anti-inflammatory response and hypotrophic factor targeting by insulin-like growth factor-1 (IGF-1) in skeletal muscles. The transgenic mdx mice expressing muscle-restricted IGF-1 (mIGF-1) (mdx:mIgf+/+) exhibited increase in muscle mass by at least 40% leading to similar increases in force generation in extensor digitorum longus muscles compared with mdx mice. The diaphragms from transgenic mdx:mIgf+/+ mice exhibited significant hypertrophy and hyperplasia at all ages. In addition, the enhanced IGF-1 expression significantly diminished the amount of fibrosis normally observed in diaphragms of aged mdx mice. Decreased myonecrosis was also observed in diaphragms and quadriceps from the transgenic mice compared with age-matched mdx animals. Furthermore, signaling pathways associated with muscle regeneration and protection against apoptosis were significantly elevated in the transgenic mice.114 Interestingly, mIGF-1 repressed the expression and activity of MIF, HMGB1, and NF-κB.115
IGF-1 is currently approved for severe primary IGF deficiency by the Food and Drug Administration. A Phase II clinical trial of recombinant IGF-1 (INCRELEX™) has been initiated in glucocorticoid-treated DMD patients to test its ability to preserve muscle function over 6 months (https://ClinicalTrials.gov/ct2/show/NCT01207908) (Table 2).
Conclusion
From the point of view of inflammatory mechanisms in DMD skeletal muscles, calcium channel, ROS production, and NF-κB pathway are potential targets for treatment as the master regulators. In fact, RyR channel blockers, antioxidants, and NF-κB inhibitors are under clinical trial. Those potential drugs targeting specific molecules are promising as more effective and less toxic compared with the current corticosteroid therapy used as a broad anti-inflammatory treatment. Furthermore, those master regulators of DMD inflammation eventually induce the release of inflammatory factors such as DAMPs and inflammatory cytokines and suppress anti-inflammatory cytokines such as HGF and IGF-1 that mediate DMD pathogenesis (Figure 1). Thus, we emphasize the need to understand the underlying mechanisms of DMD pathogenesis and the secretory functions of skeletal muscle as both contributor and healer of DMD pathogenesis in order to develop the next generation of DMD drugs.
Future perspectives
Dystrophin restoration by exon skipping in DMD is the most promising therapy, because it directly addresses the underlying pathogenic cause. Combined with this therapy, the anti-inflammatory treatments might show improved therapeutic potential. Interestingly, there are already trials using dual exon skipping of dystrophin and myostatin pre-mRNAs using ASOs.116 Although the additional or synergistic effect of this combination of dystrophin restoration and muscle growth promotion is not yet clear, this ASO-using epochal idea can be applied to the treatments targeting other inflammatory factors discussed in this review with dystrophin restoration at the same time.
Additionally, clustered regularly interspaced short palindromic repeat/Cas9-mediated genome editing was recently applied to correct the DMD gene mutation itself in the germ line of mdx mice, suggesting the potential to restore DMD protein levels.117 Future advances of this technique may enable genome editing of postnatal somatic cells and correction of DMD gene mutations in the muscle tissue. The combination of this strategy with anti-inflammation drugs may cooperate to attenuate DMD pathogenesis in the future.
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science Grant-in-Aid for Research Activity Start-up (grant to YA) (number 15H06883) and the Japan Agency for Medical Research and Development (AMED)(16ek0109154h0002 and 16am0301021h0002). We are grateful to Dr Urs T Ruegg and Dr Takashi Saito for their insightful comments and suggestions.
Disclosure
The authors report no conflicts of interest in this work.
References
Mendell JR, Shilling C, Leslie ND, et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. 2012;71(3):304–313. | ||
Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. | ||
Ljubicic V, Burt M, Jasmin BJ. The therapeutic potential of skeletal muscle plasticity in Duchenne muscular dystrophy: phenotypic modifiers as pharmacologic targets. FASEB J. 2014;28(2):548–568. | ||
Ogura Y, Tajrishi MM, Sato S, Hindi SM, Kumar A. Therapeutic potential of matrix metalloproteinases in Duchenne muscular dystrophy. Front Cell Dev Biol. 2014;2:11. | ||
Cirak S, Arechavala-Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378(9791):595–605. | ||
Mojumdar K, Liang F, Giordano C, et al. Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol Med. 2014;6(11):1476–1492. | ||
Giordano C, Mojumdar K, Liang F, et al. Toll-like receptor 4 ablation in mdx mice reveals innate immunity as a therapeutic target in Duchenne muscular dystrophy. Hum Mol Genet. 2015;24(8):2147–2162. | ||
Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidball JG. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum Mol Genet. 2009;18(3):482–496. | ||
Findlay AR, Wein N, Kaminoh Y, et al. Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann Neurol. 2015;77(4):668–674. | ||
Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev. 2012;11(2):125–140. | ||
Wood MJ, Gait MJ, Yin H. RNA-targeted splice-correction therapy for neuromuscular disease. Brain. 2010;133(Pt 4):957–972. | ||
Koo T, Wood MJ. Clinical trials using antisense oligonucleotides in Duchenne muscular dystrophy. Hum Gene Ther. 2013;24(5):479–488. | ||
Aartsma-Rus A, Fokkema I, Verschuuren J, et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. 2009;30(3):293–299. | ||
Ahima RS, Park HK. Connecting myokines and metabolism. Endocrinol Metab. 2015;30(3):235–245. | ||
Bonifati MD, Ruzza G, Bonometto P, et al. A multicenter, double-blind, randomized trial of deflazacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve. 2000;23(9):1344–1347. | ||
Escolar DM, Hache LP, Clemens PR, et al. Randomized, blinded trial of weekend vs daily prednisone in Duchenne muscular dystrophy. Neurology. 2011;77(5):444–452. | ||
Falzarano MS, Scotton C, Passarelli C, Ferlini A. Duchenne muscular dystrophy: from diagnosis to therapy. Molecules. 2015;20(10):18168–18184. | ||
Huizenga NA, Koper JW, De Lange P, et al. A polymorphism in the glucocorticoid receptor gene may be associated with and increased sensitivity to glucocorticoids in vivo. J Clin Endocrinol Metab. 1998;83(1):144–151. | ||
Wolthers OD, Pedersen S. Short term linear growth in asthmatic children during treatment with prednisolone. BMJ. 1990;301(6744):145–148. | ||
Avioli LV. Glucocorticoid effects on statural growth. Br J Rheumatol. 1993;32(Suppl 2):27–30. | ||
Grounds MD, Shavlakadze T. Growing muscle has different sarcolemmal properties from adult muscle: a proposal with scientific and clinical implications: reasons to reassess skeletal muscle molecular dynamics, cellular responses and suitability of experimental models of muscle disorders. BioEssays. 2011;33(6):458–468. | ||
Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler ThrombVasc Biol. 2011;31(5):986–1000. | ||
Serra F, Quarta M, Canato M, et al. Inflammation in muscular dystrophy and the beneficial effects of non-steroidal anti-inflammatory drugs. Muscle Nerve. 2012;46(5):773–784. | ||
Martinez L, Ermolova NV, Ishikawa TO, Stout DB, Herschman HR, Spencer MJ. A reporter mouse for optical imaging of inflammation in mdx muscles. Skelet Muscle. 2015;5:15. | ||
Schaecher K, Goust JM, Banik NL. The effects of calpain inhibition on IkB alpha degradation after activation of PBMCs: identification of the calpain cleavage sites. Neurochem Res. 2004;29(7):1443–1451. | ||
Acharyya S, Villalta SA, Bakkar N, et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007;117(4):889–901. | ||
Villalta SA, Rosenberg AS, Bluestone JA. The immune system in Duchenne muscular dystrophy: friend or foe. Rare Dis. 2015;3(1):e1010966. | ||
Millay DP, Goonasekera SA, Sargent MA, Maillet M, Aronow BJ, Molkentin JD. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc Natl Acad Sci U S A. 2009;106(45):19023–19028. | ||
Cooper ST, McNeil PL. Membrane repair: mechanisms and pathophysiology. Physiol Rev. 2015;95(4):1205–1240. | ||
Zhao X, Moloughney JG, Zhang S, Komazaki S, Weisleder N. Orai1 mediates exacerbated Ca(2+) entry in dystrophic skeletal muscle. PloS One. 2012;7(11):e49862. | ||
Gailly P. TRP channels in normal and dystrophic skeletal muscle. Curr Opin Pharmacol. 2012;12(3):326–334. | ||
Gervasio OL, Whitehead NP, Yeung EW, Phillips WD, Allen DG. TRPC1 binds to caveolin-3 and is regulated by Src kinase – role in Duchenne muscular dystrophy. J Cell Sci. 2008;121(Pt 13):2246–2255. | ||
Iwata Y, Katanosaka Y, Arai Y, Shigekawa M, Wakabayashi S. Dominant-negative inhibition of Ca2+ influx via TRPV2 ameliorates muscular dystrophy in animal models. Hum Mol Genet. 2009;18(5):824–834. | ||
Aoyagi K, Ohara-Imaizumi M, Nishiwaki C, Nakamichi Y, Nagamatsu S. Insulin/phosphoinositide 3-kinase pathway accelerates the glucose-induced first-phase insulin secretion through TrpV2 recruitment in pancreatic beta-cells. Biochem J. 2010;432(2):375–386. | ||
Iwata Y, Katanosaka Y, Shijun Z, et al. Protective effects of Ca2+ handling drugs against abnormal Ca2+ homeostasis and cell damage in myopathic skeletal muscle cells. Biochem Pharmacol. 2005;70(5):740–751. | ||
Swiderski K, Todorov M, Gehrig SM, et al. Tranilast administration reduces fibrosis and improves fatigue resistance in muscles of mdx dystrophic mice. Fibrogenesis Tissue Repair. 2014;7(1):1. | ||
Vriens J, Appendino G, Nilius B. Pharmacology of vanilloid transient receptor potential cation channels. Mol Pharmacol. 2009;75(6):1262–1279. | ||
Boittin FX, Petermann O, Hirn C, et al. Ca2+-independent phospholipase A2 enhances store-operated Ca2+ entry in dystrophic skeletal muscle fibers. J Cell Sci. 2006;119(Pt 18):3733–3742. | ||
Lindahl M, Backman E, Henriksson KG, Gorospe JR, Hoffman EP. Phospholipase A2 activity in dystrophinopathies. Neuromuscul Disord. 1995;5(3):193–199. | ||
Whitehead NP, Pham C, Gervasio OL, Allen DG. N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J Physiol. 2008;586(7):2003–2014. | ||
Ruegg UT. Pharmacological prospects in the treatment of Duchenne muscular dystrophy. Curr Opin Neurol. 2013;26(5):577–584. | ||
Ito N, Ruegg UT, Kudo A, Miyagoe-Suzuki Y, Takeda S. Activation of calcium signaling through Trpv1 by nNOS and peroxynitrite as a key trigger of skeletal muscle hypertrophy. Nat Med. 2013;19(1):101–106. | ||
Ismail HM, Scapozza L, Ruegg UT, Dorchies OM. Diapocynin, a dimer of the NADPH oxidase inhibitor apocynin, reduces ROS production and prevents force loss in eccentrically contracting dystrophic muscle. PloS One. 2014;9(10):e110708. | ||
Marx SO, Marks AR. Dysfunctional ryanodine receptors in the heart: new insights into complex cardiovascular diseases. J Mol Cell Cardiol. 2013;58:225–231. | ||
Andersson DC, Meli AC, Reiken S, et al. Leaky ryanodine receptors in beta-sarcoglycan deficient mice: a potential common defect in muscular dystrophy. Skelet Muscle. 2012;2(1):9. | ||
Nethery D, Callahan LA, Stofan D, Mattera R, DiMarco A, Supinski G. PLA(2) dependence of diaphragm mitochondrial formation of reactive oxygen species. J Appl Physiol. 2000;89(1):72–80. | ||
Hauser E, Hoger H, Bittner R, Widhalm K, Herkner K, Lubec G. Oxyradical damage and mitochondrial enzyme activities in the mdx mouse. Neuropediatrics. 1995;26(5):260–262. | ||
Buyse GM, Van der Mieren G, Erb M, et al. Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: cardiac protection and improved exercise performance. Eur Heart J. 2009;30(1):116–124. | ||
Nakae Y, Hirasaka K, Goto J, et al. Subcutaneous injection, from birth, of epigallocatechin-3-gallate, a component of green tea, limits the onset of muscular dystrophy in mdx mice: a quantitative histological, immunohistochemical and electrophysiological study. Histochem Cell Biol. 2008;129(4):489–501. | ||
Dorchies OM, Wagner S, Vuadens O, et al. Green tea extract and its major polyphenol (-)-epigallocatechin gallate improve muscle function in a mouse model for Duchenne muscular dystrophy. Am J Physiol. 2006;290(2):C616–C625. | ||
Evans NP, Call JA, Bassaganya-Riera J, Robertson JL, Grange RW. Green tea extract decreases muscle pathology and NF-kappaB immunostaining in regenerating muscle fibers of mdx mice. Clin Nutr. 2010;29(3):391–398. | ||
Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18(18):2195–2224. | ||
Chen YW, Nagaraju K, Bakay M, et al. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology. 2005;65(6):826–834. | ||
Porter JD, Khanna S, Kaminski HJ, et al. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum Mol Genet. 2002;11(3):263–272. | ||
Porter JD, Merriam AP, Leahy P, Gong B, Khanna S. Dissection of temporal gene expression signatures of affected and spared muscle groups in dystrophin-deficient (mdx) mice. Hum Mol Genet. 2003;12(15):1813–1821. | ||
Heier CR, Damsker JM, Yu Q, et al. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol Med. 2013;5(10):1569–1585. | ||
Grounds MD, Torrisi J. Anti-TNFalpha (Remicade) therapy protects dystrophic skeletal muscle from necrosis. FASEB J. 2004;18(6):676–682. | ||
Pierno S, Nico B, Burdi R, et al. Role of tumour necrosis factor alpha, but not of cyclo-oxygenase-2-derived eicosanoids, on functional and morphological indices of dystrophic progression in mdx mice: a pharmacological approach. Neuropathol Appl Neurobiol. 2007;33(3):344–359. | ||
Hodgetts S, Radley H, Davies M, Grounds MD. Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNFalpha function with Etanercept in mdx mice. Neuromuscul Disord. 2006;16(9–10):591–602. | ||
Piers AT, Lavin T, Radley-Crabb HG, Bakker AJ, Grounds MD, Pinniger GJ. Blockade of TNF in vivo using cV1q antibody reduces contractile dysfunction of skeletal muscle in response to eccentric exercise in dystrophic mdx and normal mice. Neuromuscul Disord. 2011;21(2):132–141. | ||
Miyatake S, Bilan PJ, Pillon NJ, Klip A. Contracting C2C12 myotubes release CCL2 in an NF-kappaB-dependent manner to induce monocyte chemoattraction. Am J Physiol. 2016;310(2):E160–E170. | ||
De Paepe B, Creus KK, Martin JJ, De Bleecker JL. Upregulation of chemokines and their receptors in Duchenne muscular dystrophy: potential for attenuation of myofiber necrosis. Muscle Nerve. 2012;46(6):917–925. | ||
Porter JD, Guo W, Merriam AP, et al. Persistent over-expression of specific CC class chemokines correlates with macrophage and T-cell recruitment in mdx skeletal muscle. Neuromuscul Disord. 2003;13(3):223–235. | ||
Miyatake S, Manabe Y, Inagaki A, et al. Macrophage migration inhibitory factor diminishes muscle glucose transport induced by insulin and AICAR in a muscle type-dependent manner. Biochem Biophys Res Commun. 2014;444(4):496–501. | ||
Reimann J, Schnell S, Schwartz S, Kappes-Horn K, Dodel R, Bacher M. Macrophage migration inhibitory factor in normal human skeletal muscle and inflammatory myopathies. J Neuropathol Exp Neurol. 2010;69(6):654–662. | ||
Brzoska E, Kowalski K, Markowska-Zagrajek A, et al. Sdf-1 (CXCL12) induces CD9 expression in stem cells engaged in muscle regeneration. Stem Cell Res Ther. 2015;6:46. | ||
Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84(1):209–238. | ||
Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS Jr. NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science. 2000;289(5488):2363–2366. | ||
Thaloor D, Miller KJ, Gephart J, Mitchell PO, Pavlath GK. Systemic administration of the NF-kappaB inhibitor curcumin stimulates muscle regeneration after traumatic injury. Am J Physiol. 1999;277(2 Pt 1):C320–C329. | ||
Mourkioti F, Kratsios P, Luedde T, et al. Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J Clin Invest. 2006;116(11):2945–2954. | ||
Proto JD, Tang Y, Lu A, et al. NF-kappaB inhibition reveals a novel role for HGF during skeletal muscle repair. Cell Death Dis. 2015;6:e1730. | ||
Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev. 2010;10(12):826–837. | ||
Hathout Y, Marathi RL, Rayavarapu S, et al. Discovery of serum protein biomarkers in the mdx mouse model and cross-species comparison to Duchenne muscular dystrophy patients. Hum Mol Genet. 2014;23(24):6458–6469. | ||
Hathout Y, Brody E, Clemens PR, et al. Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2015;112(23):7153–7158. | ||
Paepe BD, Creus KK, Weis J, Bleecker JL. Heat shock protein families 70 and 90 in Duchenne muscular dystrophy and inflammatory myopathy: balancing muscle protection and destruction. Neuromuscul Disord. 2012;22(1):26–33. | ||
Vidal B, Serrano AL, Tjwa M, et al. Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev. 2008;22(13):1747–1752. | ||
Villalta SA, Rosenthal W, Martinez L, et al. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci transl Med. 2014;6(258):258ra142. | ||
Vetrone SA, Montecino-Rodriguez E, Kudryashova E, et al. Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-beta. J Clin Invest. 2009;119(6):1583–1594. | ||
Mojumdar K, Giordano C, Lemaire C, et al. Divergent impact of Toll-like receptor 2 deficiency on repair mechanisms in healthy muscle versus Duchenne muscular dystrophy. J Pathol. 2016;239(1):10–22. | ||
Henriques-Pons A, Yu Q, Rayavarapu S, et al. Role of Toll-like receptors in the pathogenesis of dystrophin-deficient skeletal and heart muscle. Hum Mol Genet. 2014;23(10):2604–2617. | ||
Young CN, Brutkowski W, Lien CF, et al. P2X7 purinoceptor alterations in dystrophic mdx mouse muscles: relationship to pathology and potential target for treatment. J Cell Mol Med. 2012;16(5):1026–1037. | ||
Jiang T, Yeung D, Lien CF, Gorecki DC. Localized expression of specific P2X receptors in dystrophin-deficient DMD and mdx muscle. Neuromuscul Disord. 2005;15(3):225–236. | ||
Buvinic S, Almarza G, Bustamante M, et al. ATP released by electrical stimuli elicits calcium transients and gene expression in skeletal muscle. J Biol Chem. 2009;284(50):34490–34505. | ||
Yeung D, Zablocki K, Lien CF, et al. Increased susceptibility to ATP via alteration of P2X receptor function in dystrophic mdx mouse muscle cells. FASEB J. 2006;20(6):610–620. | ||
Young CN, Sinadinos A, Lefebvre A, et al. A novel mechanism of autophagic cell death in dystrophic muscle regulated by P2RX7 receptor large-pore formation and HSP90. Autophagy. 2015;11(1): 113–130. | ||
Rawat R, Cohen TV, Ampong B, et al. Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. Am J Pathol. 2010;176(6):2891–2900. | ||
Gazzerro E, Baldassari S, Assereto S, et al. Enhancement of muscle T regulatory cells and improvement of muscular dystrophic process in mdx mice by blockade of extracellular ATP/P2X axis. Am J Pathol. 2015;185(12):3349–3360. | ||
Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev. 2012;8(8):457–465. | ||
Knudsen JG, Bertholdt L, Joensen E, Lassen SB, Hidalgo J, Pilegaard H. Skeletal muscle interleukin-6 regulates metabolic factors in iWAT during HFD and exercise training. Obesity. 2015;23(8):1616–1624. | ||
Rufo A, Del Fattore A, Capulli M, et al. Mechanisms inducing low bone density in Duchenne muscular dystrophy in mice and humans. J Bone Miner Res. 2011;26(8):1891–1903. | ||
Pelosi L, Berardinelli MG, De Pasquale L, et al. Functional and morphological improvement of dystrophic muscle by interleukin 6 receptor blockade. EBioMedicine. 2015;2(4):285–293. | ||
Pelosi L, Berardinelli MG, Forcina L, et al. Increased levels of interleukin-6 exacerbate the dystrophic phenotype in mdx mice. Hum Mol Genet. 2015;24(21):6041–6053. | ||
Kurosaka M, Machida S. Interleukin-6-induced satellite cell proliferation is regulated by induction of the JAK2/STAT3 signalling pathway through cyclin D1 targeting. Cell Proliferat. 2013;46(4):365–373. | ||
Kurek JB, Nouri S, Kannourakis G, Murphy M, Austin L. Leukemia inhibitory factor and interleukin-6 are produced by diseased and regenerating skeletal muscle. Muscle Nerve. 1996;19(10): 1291–1301. | ||
Serrano AL, Baeza-Raja B, Perdiguero E, Jardi M, Munoz-Canoves P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metabol. 2008;7(1):33–44. | ||
Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. 2006;8(Suppl 2):S3. | ||
Pagel CN, Wasgewatte Wijesinghe DK, Taghavi Esfandouni N, Mackie EJ. Osteopontin, inflammation and myogenesis: influencing regeneration, fibrosis and size of skeletal muscle. J Cell Commun Signal. 2014;8(2):95–103. | ||
Uaesoontrachoon K, Wasgewatte Wijesinghe DK, Mackie EJ, Pagel CN. Osteopontin deficiency delays inflammatory infiltration and the onset of muscle regeneration in a mouse model of muscle injury. Dis Model Mech. 2013;6(1):197–205. | ||
Kuraoka M, Kimura E, Nagata T, et al. Serum osteopontin as a novel biomarker for muscle regeneration in Duchenne muscular dystrophy. Am J Pathol. 2016;186(5):1302–1312. | ||
Pegoraro E, Hoffman EP, Piva L, et al. SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy. Neurology. 2011;76(3):219–226. | ||
Barfield WL, Uaesoontrachoon K, Wu CS, et al. Eccentric muscle challenge shows osteopontin polymorphism modulation of muscle damage. Hum Mol Genet. 2014;23(15):4043–4050. | ||
McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387(6628):83–90. | ||
Thomas M, Langley B, Berry C, et al. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem. 2000;275(51):40235–40243. | ||
Zimmers TA, Davies MV, Koniaris LG, et al. Induction of cachexia in mice by systemically administered myostatin. Science. 2002;296(5572):1486–1488. | ||
Abe S, Soejima M, Iwanuma O, et al. Expression of myostatin and follistatin in Mdx mice, an animal model for muscular dystrophy. Zoolog Sci. 2009;26(5):315–320. | ||
McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R. Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol. 2003;162(6):1135–1147. | ||
Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002;277(51):49831–49840. | ||
Bogdanovich S, Krag TO, Barton ER, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420(6914):418–421. | ||
Bogdanovich S, Perkins KJ, Krag TO, Whittemore LA, Khurana TS. Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J. 2005;19(6):543–549. | ||
Wagner KR, Fleckenstein JL, Amato AA, et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63(5):561–571. | ||
Cadena SM, Tomkinson KN, Monnell TE, et al. Administration of a soluble activin type IIB receptor promotes skeletal muscle growth independent of fiber type. J Appl Physiol. 2010;109(3):635–642. | ||
Lee SJ, Lee YS, Zimmers TA, et al. Regulation of muscle mass by follistatin and activins. Mol Endocrinol. 2010;24(10):1998–2008. | ||
Mendell JR, Sahenk Z, Malik V, et al. A phase 1/2a follistatin gene therapy trial for Becker muscular dystrophy. Mol Ther. 2015;23(1):192–201. | ||
Barton ER, Morris L, Musaro A, Rosenthal N, Sweeney HL. Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J Cell Biol. 2002;157(1):137–148. | ||
Pelosi L, Giacinti C, Nardis C, et al. Local expression of IGF-1 accelerates muscle regeneration by rapidly modulating inflammatory cytokines and chemokines. FASEB J. 2007;21(7):1393–1402. | ||
Lu-Nguyen NB, Jarmin SA, Saleh AF, Popplewell L, Gait MJ, Dickson G. Combination antisense treatment for destructive exon skipping of myostatin and open reading frame rescue of dystrophin in neonatal mdx mice. Mol Ther. 2015;23(8):1341–1348. | ||
Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014;345(6201):1184–1188. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.