Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
Angiotensin II: a new therapeutic option for vasodilatory shock
Authors Bussard RL ![]() , Busse LW
, Busse LW ![]()
Received 7 April 2018
Accepted for publication 10 June 2018
Published 26 July 2018 Volume 2018:14 Pages 1287—1298
DOI https://doi.org/10.2147/TCRM.S150434
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Rachel L Bussard,1 Laurence W Busse2,3
1Critical Care Pharmacy Specialist, Department of Pharmacy, Emory St Joseph’s Hospital, Atlanta, GA, USA; 2Department of Critical Care, Emory St Joseph’s Hospital, Atlanta, GA, USA; 3Division of Pulmonary, Critical Care, Allergy and Sleep Medicine, Department of Medicine, Emory University, Atlanta, GA, USA
Abstract: Angiotensin II (Ang II), part of the renin–angiotensin–aldosterone system (RAS), is a potent vasoconstrictor and has been recently approved for use by the US Food and Drug Administration in high-output shock. Though not a new drug, the recently published Angiotensin II for the Treatment of High Output Shock (ATHOS-3) trial, as well as a number of retrospective analyses have sparked renewed interest in the use of Ang II, which may have a role in treating refractory shock. We describe refractory shock, the unique mechanism of action of Ang II, RAS dysregulation in shock, and the evidence supporting the use of Ang II to restore blood pressure. Evidence suggests that Ang II may preferentially be of benefit in acute kidney injury and acute respiratory distress syndrome, where the RAS is known to be disrupted. Additionally, there may be a role for Ang II in cardiogenic shock, angiotensin converting enzyme inhibitor overdose, cardiac arrest, liver failure, and in settings of extracorporeal circulation.
Keywords: catecholamine resistance, refractory shock
Introduction
Circulatory shock is life-threatening, and is characterized by hypotension, tissue hypoperfusion, and inadequate cellular oxygen utilization.1 The most common form of circulatory shock is vasodilatory shock, which accounts for two-thirds of cases and is associated with the risk of multi-organ failure and death.2 A central tenet of the management of shock is the defense of the mean arterial pressure (MAP), which often requires vasopressor therapy. Traditionally, catecholamines and vasopressin have been used to achieve the MAP goal, sometimes at the risk of adverse events, including peripheral and splanchnic ischemia, dysrhythmias, and organ dysfunction.3,4 To date, no specific vasopressor has been shown to improve mortality. A 2016 Cochrane Review meta-analysis concluded that other than the potential arrhythmogenicity of dopamine, no vasopressor outperformed any other with respect to mortality.5 Angiotensin II (Ang II) has been shown to increase blood pressure in patients with hypotension6 and has recently been approved by the US Food and Drug Administration as a vasopressor (Giapreza™; La Jolla Pharmaceutical Company, La Jolla, CA, USA) in the treatment of distributive shock. With this approval and the renewed interest in Ang II, there exists a need for better understanding of its mechanisms of action, risks and opportunities for use, and future areas of research.
Refractory shock
The concept of inadequate hemodynamic response to high doses of vasopressors is often referred to as refractory shock.7 While no consensus definition exists, most efforts to delineate this level of illness highlight either failure of response to conventional therapy8–10 or increased levels of morbidity and mortality.4,11,12 Refractory shock is common, occurring in, by some estimates, 6%–7% of critically ill patients,13,14 and mortality can range from 37% to 66%, depending on the definition used.7 Catecholamines are traditionally used as first-line therapy in shock15 and at higher doses, are strongly associated with mortality.16–19 Additionally, high-dose catecholamines may have toxic effects, contributing substantially to morbidity20–22 and may be an independent predictor of mortality.23
The phenotype of refractory shock can be described as profound vasodilation and marked tissue hypoxia. Vasodilation is mediated by the presence of nitric oxide and the deficiency of important innate vasoactive peptides such as catecholamines, cortisol, vasopressin, and Ang II.24–28 The reduced catecholamine responsiveness seen in refractory shock may be a result of this dysfunction and deficiency at the subcellular level.7 However, the process is not uniform – considerable microcirculatory heterogeneity exists, which can contribute to regional and organ-specific hypoxia.29 Catecholamine therapy may also contribute to the maldistribution of blood flow and oxygenation.30,31 Defense of the MAP is the primary treatment goal, which is typically attempted with a myriad of therapeutic options, including catecholamine and non-catecholamine vasopressors, corticosteroids, and other rescue therapies.15
Non-catecholamine vasopressor support has emerged as an important idea in the treatment of refractory shock.32,33 Catecholamine sparing, or the reduction of catecholamine doses with non-adrenergic alternatives, may reduce the potential toxicities associated with high doses of catecholamine therapy via vasoconstriction using alternative mechanisms of action. Mounting evidence suggests that multimodal blood pressure support with catecholamine and non-catecholamine therapies may improve outcomes, such as reduction of tachyarrhythmia and resolution of renal failure.4,34 Data are conflicting as to whether this practice improves mortality.35–39 This approach makes teleological sense – cardiovascular homeostasis in humans is maintained through the complex, yet harmonious, interplay between sympathetic tone and hormonal systems, including the renin–angiotensin–aldosterone (RAS) system and the arginine–vasopressin system. A detailed discussion of the role of vasopressin in refractory shock is beyond the scope of this paper, but can be found throughout the literature.28,35 Future studies will be required to determine whether or not the mimicking of this innate response via exogenous multimodal therapy can translate into meaningful outcome-based endpoints.
The use of Ang II in shock
The randomized, controlled, double-blind, Phase 3 Angiotensin II for the Treatment of High-Output Shock (ATHOS-3) trial (ClinicalTrials.gov, NCT02338843) showed that Ang II significantly increased MAP vs placebo in patients with shock with a trend toward 28-day survival.36 Patients included in this study were required to 1) have vasodilatory features (cardiac index >2.3 L/min/m2 or central venous oxygen saturation >70% coupled with central venous pressure >8 mmHg) and 2) be on high doses of standard-of-care vasopressors (>0.2 μg/kg/min of norepinephrine equivalent dose). Additionally, the vasopressor effects of Ang II have been demonstrated throughout the historical literature in patients with distributive, cardiogenic, and other forms of shock, including hypotension associated with chronic dialysis, angiotensin converting enzyme (ACE) inhibitor overdose and hypotension in the perioperative setting.6 Multiple dose ranges have been studied including continuous infusions of 10 ng/kg/min to bolus dose therapy of 1,500 mcg. Appropriate doses, based on the currently available literature, appear to be in the range of 5 to 40 ng/kg/min.6,27,36,40
The use of Ang II in circulatory shock as part of a balanced approach, along with catecholamines and vasopressin, makes teleological sense. Ang II and the RAS system have evolved in conjunction with the arginine–vasopressin and sympathetic nervous systems over eons as part of human physiology to support blood pressure.41,42 Ang II does this through multiple mechanisms, including direct vasoconstriction of peripheral vessels, water reabsorption through potentiation of antidiuretic hormone, sodium retention via the synthesis of aldosterone, and through synergistic activity with catecholamines.43,44 During hypotension, the RAS is activated in response to decreased pressure and solute content in the kidney. The subsequent release of renin, Ang II, and aldosterone, as well as antidiuretic hormone and catecholamines acts to harmoniously restore blood pressure.45,46 There is some evidence to suggest that circulating levels of vasoactive peptides are decreased or dysfunctional in sepsis.24 Replacement of these peptides exogenously forms the basis of treatment in shock.
The RAS system and physiology of Ang II
The RAS plays a vital role in blood pressure regulation, fluid and electrolyte balance, and preservation of volume status and vascular tone, which together influence arterial pressure and cardiac output. Located ubiquitously throughout the body, the RAS participates in the regulation of cardiovascular homeostasis as well as other processes via autocrine, paracrine, and endocrine mechanisms and a network of intracellular signaling pathways.47 A schematic of the RAS is presented as Figure 1.
 | Figure 1 The renin–angiotensin–aldosterone system. |
Renin is a proteolytic enzyme secreted from the juxtaglomerular apparatus of the kidney in response to activation of the sympathetic nervous system, reduction in afferent arterial pressure, or a drop in solute concentrations in the distal tubules.48 Renin does not have any peripheral receptors and, therefore, does not directly affect hemodynamics, but does cleave angiotensinogen into angiotensin I (Ang I). Upregulation of renin synthesis occurs as a result of chronic RAS blockade (via ACE inhibition) and downregulation can occur in the presence of exogenous Ang II.49,50
Angiotensinogen is an α-glycoprotein produced by the liver and released into systemic circulation where it is transformed by renin converting it to Ang I. Ang I is then cleaved by ACE into Ang II. This occurs mostly by endothelial-bound ACE in the lungs, but can also occur in plasma as well as in the vascular bed of the kidneys, heart, and brain and, to a lesser extent, by chymases stored in granules in the mast cells.48 ACE also plays a role in the degradation of bradykinin, which causes vasodilation and natriuresis.51 A homologue of ACE named ACE2 is expressed in a more limited way throughout the body and efficiently hydrolyzes Ang II into angiotensin 1–7 (Ang 1–7), which opposes the actions of Ang II and causes vasodilation, natriuresis, and reduced blood pressure.52,53
Ang II, an octapeptide, has a myriad of physiologic effects, depending on the receptor type, which mediate cardiovascular, sodium, and water homeostasis. It is rapidly metabolized by aminopeptidase A and ACE2 to Ang 1–7 in plasma, erythrocytes, and many major organs.130 Because of its rapid degradation, its half-life is ~30 seconds in the circulation; however, in the tissues, the half-life is extended to 15–30 minutes. Ang II regulates fluid and electrolytes and maintains hemodynamic stability by binding to specific angiotensin (AT) receptors on the cell membrane. The major physiologic effects in humans are mediated by the AT-1 receptor, which is located in the kidneys, vascular smooth muscle, lung, heart, brain, adrenal gland, pituitary gland, and liver. These effects include direct vasoconstriction (via receptors located in the endothelium of peripheral vessels), inotropy and cardiac remodeling (via receptors located in the myocardium), potentiation of the sympathetic nervous system, vasopressin release, regulation of the thirst mechanism (via receptors in the brain, sympathetic ganglia, and pituitary), and regulation of aldosterone release (via receptors in the adrenal gland).54 AT-1 receptors have different subcellular signaling pathways, including G-coupled protein receptors (GPCRs) as well as a β-arrestins which, when activated by Ang II, carry out very different physiologic tasks55 including the physiologically opposed effects of hypertension (via a GPCR mechanism) and hypotension (via a β-arrestin mechanism).56 AT-2 receptors are expressed primarily in the vascular endothelium, brain, adrenals, and selected cutaneous, renal, and cardiac structures, and mediate antiproliferation, vasodilation, and anti-inflammation, and thereby antagonize the actions of the AT-1 receptor-mediated effects of Ang II. A discussion of some of the pharmacologic details pertaining to Ang II is provided in the Supplementary material.
Ang II is associated with myocardial ventricular remodeling in the setting of cardiac dysfunction or hypertension. A chronic increase in cardiac pressures produced by Ang II leads to hypertrophy and collagen deposition, and its blockade via ACE inhibition or AT receptor blocker therapy represents a cornerstone of cardiovascular protection against common disease states such as congestive heart failure (CHF) and hypertension.57,58 In addition to direct hypertrophic effects on smooth muscle cells, Ang II enhances sympathetic excitation, fibrogenesis, chemotaxis of fibroblasts, and inflammation, all of which contribute to remodeling.59–61 Patients with CHF can have elevated Ang II levels, despite commonly using ACE inhibition therapy, which is associated with increased morbidity and mortality.62,63 In contrast, patients with circulatory shock may exhibit a physiologic deficiency in Ang II.
The physiology of Ang II deficiency in shock
RAS dysregulation during sepsis is well described.64,65 The vasodilation seen in septic shock may be associated with activation of adenosine triphosphate (ATP)-sensitive potassium channels (KATP) in the vascular smooth muscle, which results from reduced cellular ATP or increased hydrogen and lactate concentrations.66 Activation of KATP channels is known to inhibit Ang II–induced vasoconstriction.67 RAS may be over- or understimulated, affecting both systemic and microcirculation, and varying levels of Ang I, Ang II, and renin have been reported in sepsis.68–70 Additionally, ACE levels in patients admitted with lung dysfunction are known to be reduced, compared to healthy controls.71 ACE dysfunction in sepsis is thought to be the result of both pulmonary and vascular endothelial damage from inflammatory molecules and blood-borne substrates.72 Additionally, endotoxins associated with gram-negative infections have the ability to inactivate ACE.70 Studies have also demonstrated a downregulation and dysfunction of both AT-1 and AT-2 receptors during septic shock.73,74 AT-1 downregulation may be a result of proinflammatory cytokines and nitric oxide production.75 The downregulation of these receptors ultimately leads to decreases in catecholamine and aldosterone levels and the risk of hypotension.47
Defects in ACE and AT receptors in sepsis suggest that a relative Ang II deficiency may be a root cause in the hemodynamic instability seen in sepsis. Low levels of Ang II and ACE in septic patients are associated with morbidity and mortality,68 and there is evidence that replacement of physiologic levels of Ang II in septic shock is effective in reversing hemodynamic instability.39 Based on early physiologic studies, a dose of 5 ng/kg/min of Ang II is thought to be analogous to replacement of physiologic serum levels,76 and it was found that among the ATHOS-3 Ang II recipients, almost half (48.5%) responded to doses ≤5 ng/kg/min. Dose-related efficacy and safety parameters were compared, post hoc, in patients enrolled in the Ang II arm of the ATHOS-3 study and dichotomized based on doses of ≤5 vs >5 ng/kg/min. More patients receiving the lower dose (≤5 ng/kg/min) exhibited a blood pressure response compared to patients requiring >5 ng/kg/min, and this variable was independently predictive of response. The lower dose of Ang II was also associated with a mortality and morbidity benefit. This phenomenon is further supported in an analysis of ACE capacity and functionality within the ATHOS-3 population. The precursor and product of ACE (Ang I and Ang II, respectively) and the ratio of Ang I/II are reflective of ACE functionality.77,78 While the normal Ang I/II ratio is 0.5, the median ratio was 1.63 in patients in the ATHOS-3 study,36 suggesting dysregulated ACE functionality in septic shock. In further post hoc analysis of ATHOS-3 patients with Ang I/II ratios of >1.63 (reflecting an Ang II deplete state and less ACE functionality), those receiving Ang II exhibited a mortality benefit compared to placebo, whereas patients with an Ang I/II ratio <1.63 (reflecting an Ang II replete state) receiving Ang II did not.79 As such, hyperresponsiveness to a low dose of Ang II or a high Ang I/II ratio identifies those patients with ACE deficiency who would most benefit from Ang II administration.
Historical uses and risks associated with the use of Ang II
Ang II was discovered in the 1930s, but use was not reported in the literature until the early 1960s. In total, use is reported in >31,000 subjects, including healthy humans, as well as a diagnostic tool in the identification of pre-eclampsia, for enhancing delivery of chemotherapy to solid tumors, in prevention of hypotension during anesthesia, and for treatment of hypotension following ACE Inhibitor overdose.40 Is has been given to patients suffering from numerous medical conditions, including pulmonary, renal, cardiovascular, endocrine, and hepatic failure, trauma, and malignancy.
In the obstetric population, Ang II was used to treat hypotension and compared favorably to ephedrine with regard to uterine oxygen delivery and pH levels.80–82 Similarly, the acid–base status in neonates of women given Ang II was better than with ephedrine infusions.83 Additionally, Ang II was used to risk-stratify pregnant women at risk for pre-eclampsia,84,85 who have high levels of circulating estrogen. Estrogen induces AGT gene transcription in the liver (which makes angiotensinogen), and the M235T variant is associated with pre-eclampsia.48 Moreover, the ratio of reduced to oxidized Ang II in pre-eclamptic women is different from healthy pregnant women, and RAS dysregulation was thought to elucidate the hypertension seen in pre-eclampsia.
Ang II is physiologically a rational treatment for hypotension following ACE inhibitor overdose. Exogenous infusion of Ang II restores the innate deficiency resulting from the inhibition of ACE. Several case studies have shown successful resolution of hypotension in ACE inhibitor overdose with Ang II.86–88 In these reports, patients were refractory to other treatment modalities, but experienced a profound increase in blood pressure upon receiving the drug.
Ang II has been used to enhance the delivery of chemo- and radiation therapy to solid tumors.89,90 By selectively increasing blood flow to tumor tissue with Ang II, investigators were able to simulate hyperbaric oxygenation radiation therapy, thus improving tumor response and minimizing healthy tissue damage.89 Additionally, chemotherapy delivery was found to be enhanced via selective Ang II-induced hypertension, resulting in reduction of tumor size and less toxicity.91 Mechanistically, the increase of tumor blood flow caused by Ang II was thought to demonstrate a loss of autoregulation and allow for increased delivery of therapy to the tumor.92 Despite the aforementioned investigations, there have been no recent reports of this use of Ang II.
Physiologic effects, side effects, and adverse events of Ang II were evaluated in a large systematic review of safety.40 Common findings included increased systemic and pulmonary blood pressure, reduced heart rate and cardiac output, and decreased renal blood flow and glomerular filtration rate. Additionally, investigators cited increased plasma aldosterone and other endocrine perturbations, alterations of electrolyte balance, and reduction in sodium and water excretion. Common side effects in the literature included headache, sensation of chest pressure, dyspepsia, and orthostatic hypotension upon cessation of the drug. Ang II was found to worsen bronchoconstriction during an asthma exacerbation93–95 and worsen ventricular function when administered to patients with acute CHF.96 Two deaths were found to be attributable to Ang II, including that of a 36-year-old healthy male who died of a hypertensive cerebral hemorrhage while receiving a 6-day infusion of Ang II,97 and that of a patient with pre-infusion symptoms of acute heart failure who failed to respond to Ang II during profound cardiogenic shock.96 While the adverse event rates were similar in the patients studied in ATHOS-3 (excluded from the aforementioned review), the incidence rates of thromboembolic events, delirium, and infection were higher in the Ang II cohort.36 While speculative at this time, it is plausible that immune dysregulation, alterations in microvascular blood flow, and the prothrombotic potential of Ang II may be causative. Further analyses are required to more clearly elucidate these potential areas of concern, and proper precaution is warranted in patients at risk for these conditions.
Ang II in acute kidney injury patients
Acute kidney injury (AKI) in septic shock is associated with poor outcomes.98 Mortality in patients with AKI who require renal replacement therapy can reach 50%.99,100 Though a common occurrence,101 the mechanisms involved in the development of sepsis-induced AKI are incompletely understood. It is thought that sepsis-induced AKI results in part not only from decreased renal perfusion in the setting of hypotension,102 but also from renal microvascular dysregulation and shunting, inflammatory and immune activation, and cell-cycle arrest.103–105 Systemic hypotension and intrarenal vasodilation are accompanied by a reduction in glomerular filtration rate due to reduced intra-glomerular perfusion pressure.106 Vasopressors, vasodilators, inotropes, and natriuretic peptides have failed to demonstrate improved outcomes in AKI.107,108 In the renal microcirculation, Ang II preferentially constricts efferent arteriolar tone, more so than the afferent tone, thereby restoring glomerular perfusion pressure109,110 and may have a unique role in sepsis-associated AKI. In an animal model, Ang II was found to restore systemic blood pressure, though with a concomitant decrease in renal blood flow.106 However, despite this decrease, animals receiving Ang II exhibited improved urinary output and creatinine clearance. The beneficial effects on renal function have been found in other pre-clinical work as well,111,112 though in clinical practice, results are equivocal.27,87,113–115
Ang II has recently been evaluated in patients with septic shock, as part of a post hoc analysis of ATHOS-3.34 Patients with shock and AKI requiring renal replacement were found to have improved 28-day survival, improved renal recovery (liberation from renal replacement therapy at day 7), fewer days on ventilation, and shorter intensive care unit and hospital length of stay. These results suggest that the AKI seen in septic shock is, at least in part, due to decreased glomerular perfusion, and that Ang II may mitigate this phenomenon. The microcirculatory properties of Ang II are not seen when using other vasopressors which do not reverse the phenotypical pattern of efferent vasodilation at the level of the glomerulus.103 Further supporting this is that pre-morbid RAS manipulation with ACE inhibition or AT receptor blocker therapy (which causes efferent arteriolar dilatation) increases the risk of sepsis-induced AKI.116 There remains continued interest in the manipulation of the RAS system to improve renal outcomes in septic shock.117
Ang II in acute respiratory distress syndrome patients
Some patients with septic shock also have acute respiratory distress syndrome (ARDS), which may in part be due to similar or linked pathophysiologic mechanisms between the two syndromes.118 The hallmark of ARDS is pulmonary endothelial injury and accumulation of protein-rich fluid inside the alveoli, which is due to increased pulmonary capillary permeability. Endothelium-bound ACE converts Ang I into Ang II,119 and in conditions of significant lung injury, reductions in this enzymatic process may result in decreased levels of Ang II.120 Supplementation with exogenous Ang II has been shown to be effective in supporting hemodynamics in patients with ARDS.33,36
In a post hoc analysis using data from the ATHOS-3 population, the efficacy of Ang II was evaluated in patients with septic shock and ARDS.121 Patients with ARDS based on the Berlin criteria122 at the time of study drug initiation were analyzed for blood pressure response and clinical outcomes. Almost three-quarters of the patients enrolled in ATHOS-3 had some degree of lung injury, and a greater proportion of lung-injured patients treated with Ang II experienced a blood pressure response compared to placebo. Moreover, the blood pressure response was most evident in patients with severe ARDS. While 28-day mortality increased in both the Ang II group and the placebo group with increasing ARDS severity, the progression in the rate of mortality by ARDS severity was lower in the Ang II group and the magnitude of the 28-day mortality benefit was most evident in severe ARDS (Figure 2).
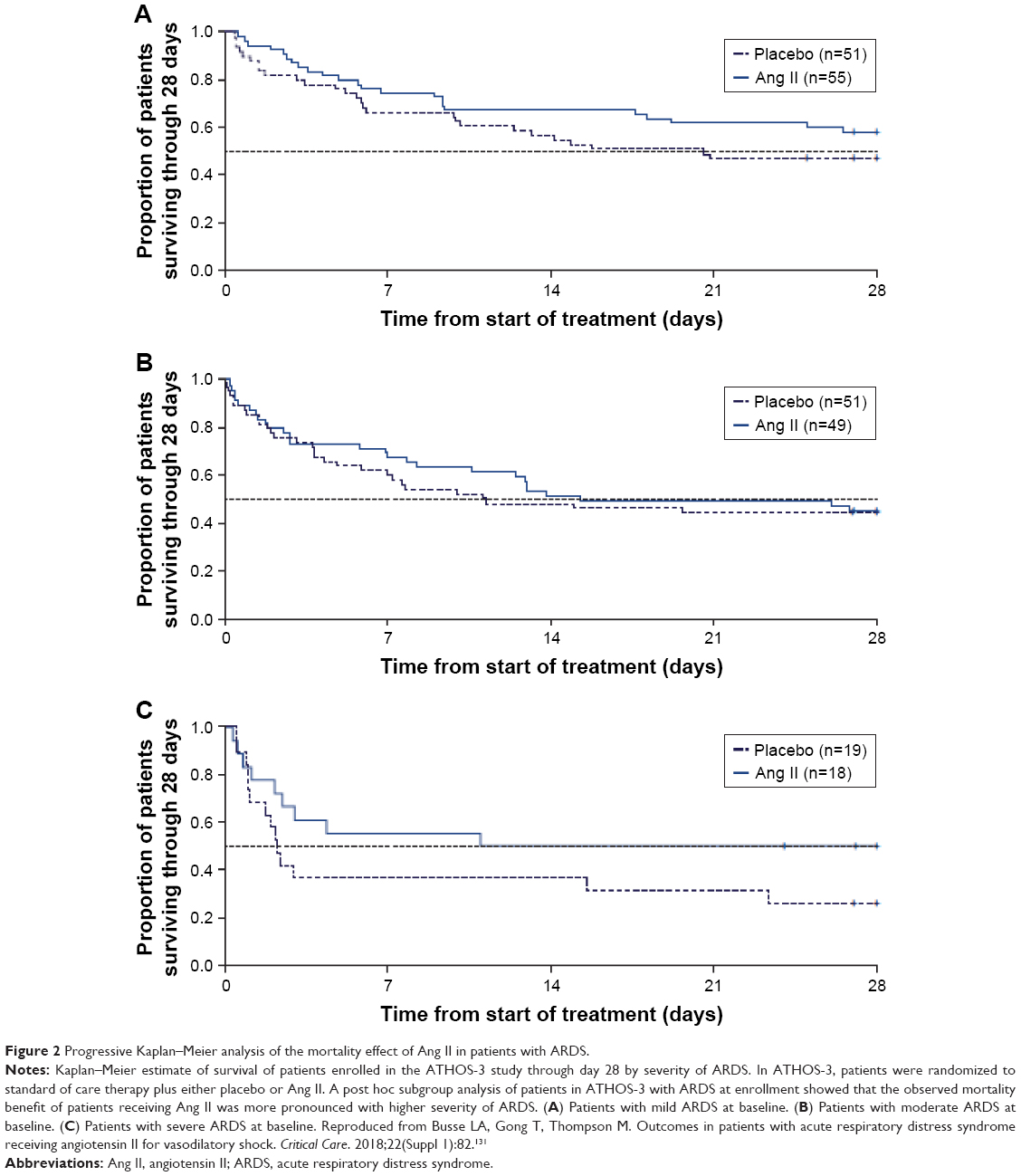 | Figure 2 Progressive Kaplan–Meier analysis of the mortality effect of Ang II in patients with ARDS. |
The findings in this analysis suggest that exogenous Ang II may be beneficial in reversing the hemodynamic compromise in patients with ARDS and septic shock. The pulmonary vascular endothelium is essential for the synthesis and degradation of Ang II, and it has been demonstrated that ACE activity is altered in severe lung injury, and this dysfunction correlates with the severity of the disease.72 While incompletely understood, other components of the RAS, including ACE homologues such as ACE2, may play a role in modulation of disease in ARDS.123,124 Further research is needed to more fully understand the unique role for Ang II in ARDS.
Future directions
Ang II has been well studied in distributive shock, but may play a role in other disease states as well. Some data have suggested that Ang II may be beneficial in both cardiogenic shock and cardiac arrest. Though data are limited, patients with cardiogenic shock who received Ang II infusion experienced robust improvement in blood pressure.6 Like patients with septic shock, patients with cardiogenic shock may exhibit RAS dysregulation via “ACE escape”, whereby the levels of Ang I, Ang II, renin, and ACE are altered in the setting of pre-morbid ACE inhibition.62,63 In a historical analysis of the use of Ang II in shock, five patients with heart failure who had pre-morbid ACE inhibitor exposure experienced a dramatic improvement in blood pressure when receiving Ang II for cardiogenic shock.6 The restoration of hemodynamic stability with intravenous Ang II in this patient population suggests both a role for Ang II in cardiogenic shock as well as ACE inhibitor overdose, the latter of which has been reported in the literature.86–88
In an analysis of the historical literature, 14 patients with presumed cardiac arrest were found to have a profound blood pressure effect upon return of spontaneous circulation after administration of Ang II.6 The authors hypothesized that successful resuscitation and normalization of blood pressure may have been from a number of potential mechanisms, including restoration of blood flow to vital organs, increased inotropy, potentiation of catecholamines, or improved coronary perfusion. To date, no human trials have sought to evaluate Ang II in cardiac arrest, but Ang II levels were evaluated in a porcine model of cardiac arrest and found to be elevated.125 The influence of Ang II on the RAS system during cardiac arrest is a potential target for future investigation.
Ang II may preferentially reverse shock in a number of disease states which, by their nature, can be associated with RAS dysregulation. Patients with liver failure or cirrhosis, for example, have impaired synthesis of angiotensinogen126,127 and may respond well to exogenous Ang II. Likewise, in patients receiving extracorporeal circulation support (cardiopulmonary bypass), circumvention of the pulmonary circulation is associated with altered ACE levels128,129 may be associated with increased Ang II sensitivity. Presumably, patients on extracorporeal membrane oxygenation circuits would exhibit similar ACE dysfunction, though there is no data describing this as of yet. Table 1 describes the current and contemplated uses of Ang II.
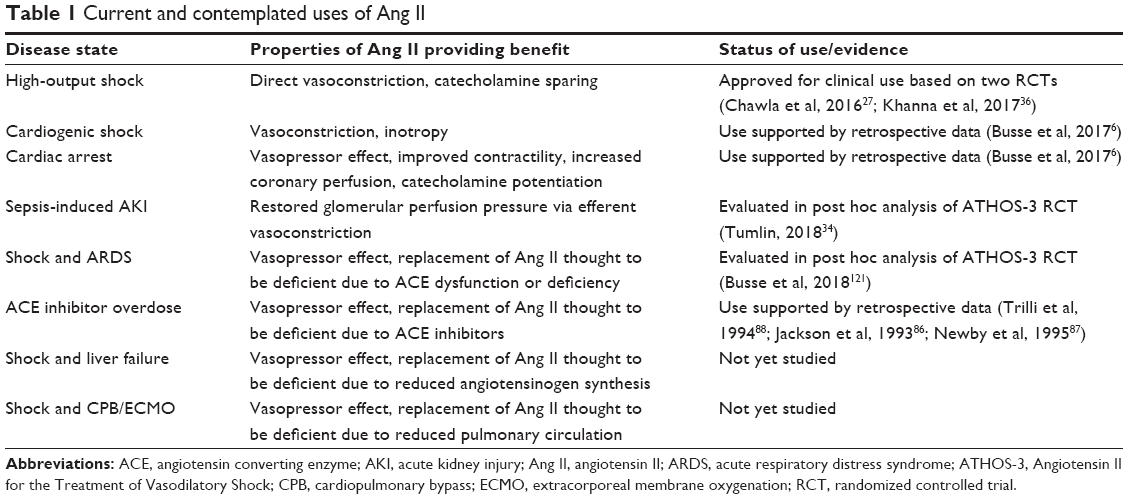 | Table 1 Current and contemplated uses of Ang II |
Conclusion
Ang II is a potent vasoconstrictor and part of the RAS, a highly conserved system involved in blood pressure control, fluid and electrolyte homeostasis, and hormonal activity in humans. Recently approved for use as a novel vasopressor in high-output shock, Ang II may prove to be a beneficial addition to the armamentarium against refractory shock by virtue of its unique mechanism of action, which is complementary to that of the currently available therapies. RAS dysregulation in shock is common, and evidence suggests that exogenous Ang II may reverse the deficiency seen in many patients with this syndrome. Multiple studies have evaluated Ang II, which has been shown to restore blood pressure in septic shock and may preferentially be of benefit in AKI and ARDS. Additionally, there may be a role for Ang II in cardiogenic shock, ACE inhibitor overdose, and cardiac arrest, which may be evaluated in future research efforts.
Disclosure
LWB reports receiving consulting fees from La Jolla Pharmaceutical Company. The authors report no other conflicts of interest in this work.
References
Vincent J-L, de Backer D. Circulatory Shock. N Engl J Med Overseas Ed. 2013;369(18):1726–1734. | ||
de Backer D, Biston P, Devriendt J, et al. Comparison of Dopamine and Norepinephrine in the Treatment of Shock. N Engl J Med Overseas Ed. 2010;362(9):779–789. | ||
Schmittinger CA, Torgersen C, Luckner G, Schröder DCH, Lorenz I, Dünser MW. Adverse cardiac events during catecholamine vasopressor therapy: a prospective observational study. Intensive Care Med. 2012;38(6):950–958. | ||
Dünser MW, Mayr AJ, Ulmer H, et al. Arginine Vasopressin in Advanced Vasodilatory Shock: A Prospective, Randomized, Controlled Study. Circulation. 2003;107(18):2313–2319. | ||
Gamper G, Havel C, Arrich J, et al. Vasopressors for hypotensive shock. Cochrane Database Syst Rev. 2016;33(9):Cd003709. | ||
Busse LW, Mccurdy MT, Ali O, Hall A, Chen H, Ostermann M. The effect of angiotensin II on blood pressure in patients with circulatory shock: a structured review of the literature. Critical Care. 2017;21(1):324. | ||
Jentzer JC, Vallabhajosyula S, Khanna AK, Chawla LS, Busse LW, Kashani KB. Management of Refractory Vasodilatory Shock. Chest. Epub 2018 Jan 9. | ||
Bassi E, Park M, Azevedo LCP. Therapeutic Strategies for High-Dose Vasopressor-Dependent Shock. Crit Care Res Pract. 2013;2013(2):654708–654710. | ||
Luckner G, Dünser MW, Jochberger S, et al. Arginine vasopressin in 316 patients with advanced vasodilatory shock. Crit Care Med. 2005;33(11):2659–2666. | ||
Brown SM, Lanspa MJ, Jones JP, et al. Survival After Shock Requiring High-Dose Vasopressor Therapy. Chest. 2013;143(3):664–671. | ||
Luckner G, Mayr VD, Jochberger S, et al. Comparison of two dose regimens of arginine vasopressin in advanced vasodilatory shock. Crit Care Med. 2007;35(10):2280–2285. | ||
Torgersen C, Dünser MW, Wenzel V, et al. Comparing two different arginine vasopressin doses in advanced vasodilatory shock: a randomized, controlled, open-label trial. Intensive Care Med. 2010;36(1):57–65. | ||
Benbenishty J, Weissman C, Sprung CL, Brodsky-Israeli M, Weiss Y. Characteristics of patients receiving vasopressors. Heart & Lung: The Journal of Acute and Critical Care. 2011;40(3):247–252. | ||
Jenkins CR, Gomersall CD, Leung P, Joynt GM. Outcome of patients receiving high dose vasopressor therapy: a retrospective cohort study. Anaesth Intensive Care. 2009;37(2):286–289. | ||
Rhodes A, Evans LE, Alhazzani W, et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med. 2017;43(3):304–377. | ||
Mayr VD, Dünser MW, Greil V, et al. Causes of death and determinants of outcome in critically ill patients. Critical Care. 2006;10(6):R154. | ||
Jentzer JC, Coons JC, Link CB, Schmidhofer M. Pharmacotherapy Update on the Use of Vasopressors and Inotropes in the Intensive Care Unit. J Cardiovasc Pharmacol Ther. 2015;20(3):249–260. | ||
Vincent JL, Moreno R, Takala J, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996;22(7):707–710. | ||
Dünser MW, Ruokonen E, Pettilä V, et al. Association of arterial blood pressure and vasopressor load with septic shock mortality: a post hoc analysis of a multicenter trial. Critical Care. 2009;13(6):R181. | ||
Dünser MW, Hasibeder WR. Sympathetic Overstimulation During Critical Illness: Adverse Effects of Adrenergic Stress. J Intensive Care Med. 2009;24(5):293–316. | ||
Andreis DT, Singer M. Catecholamines for inflammatory shock: a Jekyll-and-Hyde conundrum. Intensive Care Med. 2016;42(9):1387–1397. | ||
Asfar P, Meziani F, Hamel J-F, et al. High versus Low Blood-Pressure Target in Patients with Septic Shock. N Engl J Med Overseas Ed. 2014;370(17):1583–1593. | ||
Sviri S, Hashoul J, Stav I, van Heerden PV. Does high-dose vasopressor therapy in medical intensive care patients indicate what we already suspect? J Crit Care. 2014;29(1):157–160. | ||
Levy B, Collin S, Sennoun N, et al. Vascular hyporesponsiveness to vasopressors in septic shock: from bench to bedside. Intensive Care Med. 2010;36(12):2019–2029. | ||
Hosseinian L, Weiner M, Levin MA, Fischer GW. Methylene Blue: Magic Bullet for Vasoplegia? Anesth Analg. 2016;122(1):194–201. | ||
Marik PE, Pastores SM, Annane D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: Consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med. 2008;36(6):1937–1949. | ||
Chawla LS, Busse L, Brasha-Mitchell E, et al. Intravenous angiotensin II for the treatment of high-output shock (ATHOS trial): a pilot study. Critical Care. 2014;18(5):534. | ||
Russell JA. Bench-to-bedside review: Vasopressin in the management of septic shock. Critical Care. 2009;15(4):226. | ||
Walley KR. Heterogeneity of oxygen delivery impairs oxygen extraction by peripheral tissues: theory. J Appl Physiol. 1996;81(2):885–894. | ||
de Backer D, Creteur J, Dubois M-J, et al. The effects of dobutamine on microcirculatory alterations in patients with septic shock are independent of its systemic effects. Crit Care Med. 2006;34(2):403–408. | ||
de Backer D, Creteur J, Silva E, Vincent J-L. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: Which is best? Crit Care Med. 2003;31(6):1659–1667. | ||
Asfar P, Russell JA, Tuckermann J, Radermacher P. Selepressin in Septic Shock: A Step Toward Decatecholaminization? Crit Care Med. 2016;44(1):234–236. | ||
Chawla LS, Busse LW, Brasha-Mitchell E, Alotaibi Z. The use of angiotensin II in distributive shock. Critical Care. 2016;20(1):137. | ||
Tumlin JA, Murugan R, Deane AM, et al. Outcomes in Patients with Vasodilatory Shock and Renal Replacement Therapy Treated with Intravenous Angiotensin II. Crit Care Med. 2018;46(6):949–957. | ||
Russell JA, Walley KR, Singer J, et al. Vasopressin versus Norepinephrine Infusion in Patients with Septic Shock. N Engl J Med Overseas Ed. 2008;358(9):877–887. | ||
Khanna A, English SW, Wang XS, et al. Angiotensin II for the Treatment of Vasodilatory Shock. N Engl J Med Overseas Ed. 2017;377(5):419–430. | ||
Russell JA, Lee T, Singer J, Boyd JH, Walley KR. The Septic Shock 3.0 Definition and Trials: A Vasopressin and Septic Shock Trial Experience. Crit Care Med. 2017;45(6):940–948. | ||
Szerlip H, Bihorac A, Chang S, et al. Effect of disease severity on survival in patients receiving angiotensin II for vasodilatory shock. Crit Care Med. 2018;46(1):3–868. | ||
Mccurdy MB, Gong L, Boldt M, et al. Association of angiotensin II dose with all-cause mortality in patients with vasodilatory shock. Critical Care. 2018;22(Suppl 1):82. | ||
Busse LW, Wang XS, Chalikonda DM, et al. Clinical Experience With IV Angiotensin II Administration: A Systematic Review of Safety. Crit Care Med. 2017;45(8):1285–1294. | ||
Fournier D, Luft FC, Bader M, Ganten D, Andrade-Navarro MA. Emergence and evolution of the renin–angiotensin–aldosterone system. J Mol Med. 2012;90(5):495–508. | ||
Kuntz A. The evolution of the sympathetic nervous system in vertebrates. J Comp Neurol. 1911;21(3):215–236. | ||
Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. 2010;2(7):247–257. | ||
Zhuo JL, Li XC, Xc L. New insights and perspectives on intrarenal renin-angiotensin system: Focus on intracrine/intracellular angiotensin II. Peptides. 2011;32(7):1551–1565. | ||
Rolih CA, Ober KP. The endocrine response to critical illness. Med Clin North Am. 1995;79(1):211–224. | ||
du Cheyron D, Lesage A, Daubin C, Ramakers M, Charbonneau P. Hyperreninemic hypoaldosteronism: a possible etiological factor of septic shock-induced acute renal failure. Intensive Care Med. 2003;29(10):1703–1709. | ||
Hall A, Busse LW, Ostermann M. Angiotensin in Critical Care. Critical Care. 2018;22(1):69. | ||
Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM. Classical Renin-Angiotensin system in kidney physiology. Compr Physiol. 2014;4(3):1201–1228. | ||
Timmermans PB, Wong PC, Chiu AT, et al. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev. 1993;45(2):205–251. | ||
Kurtz A, Pfeilschifter J, Hutter A, et al. Role of protein kinase C in inhibition of renin release caused by vasoconstrictors. Am J Physiol Cell Physiol. 1986;250(4):C563–C571. | ||
Margolius HS. Kallikreins and Kinins: Molecular Characteristics and Cellular and Tissue Responses. Diabetes. 1996;45(Suppl 1):S14–S19. | ||
Donoghue M, Hsieh F, Baronas E, et al. A Novel Angiotensin-Converting Enzyme-Related Carboxypeptidase (ACE2) Converts Angiotensin I to Angiotensin 1-9. Circ Res. 2000;87(5):e1–e9. | ||
Ferrario CM, Brosnihan KB, Diz DI, et al. Angiotensin-(1-7): a new hormone of the angiotensin system. Hypertension. 1991;18(5_Suppl):III126–III133. | ||
Paul M, Poyan Mehr A, Kreutz R. Physiology of Local Renin-Angiotensin Systems. Physiol Rev. 2006;86(3):747–803. | ||
Shenoy SK, Lefkowitz RJ. Angiotensin II-stimulated signaling through G proteins and beta-arrestin. Sci STKE. 2005;2005(311):cm14. | ||
Dewire SM, Violin JD. Biased Ligands for Better Cardiovascular Drugs: Dissecting G-Protein-Coupled Receptor Pharmacology. Circ Res. 2011;109(2):205–216. | ||
Felker GM, Butler J, Collins SP, et al. Heart failure therapeutics on the basis of a biased ligand of the angiotensin-2 type 1 receptor. Rationale and design of the BLAST-AHF study (Biased Ligand of the Angiotensin Receptor Study in Acute Heart Failure). JACC Heart Failure. 2015;3(3):193–201. | ||
Ferrario CM, Schiffrin EL. Role of Mineralocorticoid Receptor Antagonists in Cardiovascular Disease. Circ Res. 2015;116(1):206–213. | ||
Davisson RL, Oliverio MI, Coffman TM, Sigmund CD. Divergent functions of angiotensin II receptor isoforms in the brain. J Clin Invest. 2000;106(1):103–106. | ||
Geisterfer AA, Peach MJ, Owens GK. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ Res. 1988;62(4):749–756. | ||
Intengan HD, Schiffrin EL. Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension. 2001;38(3 Pt 2):581–587. | ||
Roig E, Perez-Villa F, Morales M, et al. Clinical implications of increased plasma angiotensin II despite ACE inhibitor therapy in patients with congestive heart failure. Eur Heart J. 2000;21(1):53–57. | ||
van de Wal RMA, Plokker HWM, Lok DJA, et al. Determinants of increased angiotensin II levels in severe chronic heart failure patients despite ACE inhibition. Int J Cardiol. 2006;106(3):367–372. | ||
Doerschug KC, Delsing AS, Schmidt GA, Ashare A. Renin-angiotensin system activation correlates with microvascular dysfunction in a prospective cohort study of clinical sepsis. Critical Care. 2010;14(1):R24. | ||
Antonucci E, Gleeson PJ, Annoni F, et al. Angiotensin II in Refractory Septic Shock. Shock. 2017;47(5):560–566. | ||
Landry DW, Oliver JA. The Pathogenesis of Vasodilatory Shock. N Engl J Med Overseas Ed. 2001;345(8):588–595. | ||
Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77(4):1165–1232. | ||
Zhang WEI, Chen X, Huang L, et al. Severe sepsis: Low expression of the renin-angiotensin system is associated with poor prognosis. Exp Ther Med. 2014;7(5):1342–1348. | ||
Dong LW, Chang YZ, Tong LJ, Tang J, Jy S, Tang CS. Role of regulatory peptide in pathogenesis of shock. Science in China Series B, Chemistry, Life Sciences & Earth Sciences. 1994;37(2):162–169. | ||
Dunn CW, Horton JW. Role of angiotensin II in neonatal sepsis. Circ Shock. 1993;40(2):144–150. | ||
Casey L, Krieger B, Kohler J, Rice C, Oparil S, Szidon P. Decreased serum angiotensin converting enzyme in adult respiratory distress syndrome associated with sepsis: a preliminary report. Crit Care Med. 1981;9(9):651–654. | ||
Orfanos SE, Armaganidis A, Glynos C, et al. Pulmonary Capillary Endothelium-Bound Angiotensin-Converting Enzyme Activity in Acute Lung Injury. Circulation. 2000;102(16):2011–2018. | ||
Schmidt C, Höcherl K, Kurt B, Moritz S, Kurtz A, Bucher M. Blockade of multiple but not single cytokines abrogates downregulation of angiotensin II type-I receptors and anticipates septic shock. Cytokine. 2010;49(1):30–38. | ||
Bucher M, Hobbhahn J, Kurtz A. Nitric oxide-dependent down-regulation of angiotensin II type 2 receptors during experimental sepsis. Crit Care Med. 2001;29(9):1750–1755. | ||
Bucher M, Ittner K-P, Hobbhahn J, Taeger K, Kurtz A. Downregulation of Angiotensin II Type 1 Receptors During Sepsis. Hypertension. 2001;38(2):177–182. | ||
Wolf RL, Mendlowitz M, Gitlow SE, Naftchi N. Metabolism of Angiotensin II-I131 in Normotensive and Hypertensive Human Subjects. Circulation. 1961;23(5):754–758. | ||
Luque M, Martin P, Martell N, Fernandez C, Brosnihan KB, Ferrario CM. Effects of captopril related to increased levels of prostacyclin and angiotensin-(1-7) in essential hypertension. J Hypertens. 1996;14(6):799–805. | ||
Azizi M, Chatellier G, Guyene T-T, Murieta-Geoffroy D, Menard J. Additive Effects of Combined Angiotensin-Converting Enzyme Inhibition and Angiotensin II Antagonism on Blood Pressure and Renin Release in Sodium-Depleted Normotensives. Circulation. 1995;92(4):825–834. | ||
Wunderink RA, Busse T, Deane L, et al. Baseline angiotensin levels and ACE effects in patients with vasodilatory shock treated with angiotensin II. Intensive Care Medicine Experimental. 2017;5(Suppl 2):0703. | ||
Rosenfeld CR, Naden RP. Uterine and nonuterine vascular responses to angiotensin II in ovine pregnancy. Am J Physiol Heart Circ Physiol. 1989;257(1):H17–H24. | ||
Ramin SM, Ramin KD, Cox K, Magness RR, Shearer VE, Gant NF. Comparison of prophylactic angiotensin II versus ephedrine infusion for prevention of maternal hypotension during spinal anesthesia. Am J Obstet Gynecol. 1994;171(3):734–739. | ||
Naden RP, Rosenfeld CR. Effect of angiotensin II on uterine and systemic vasculature in pregnant sheep. J Clin Invest. 1981;68(2):468–474. | ||
Vincent RD, Werhan CF, Norman PF, et al. Prophylactic Angiotensin II Infusion during Spinal Anesthesia for Elective Cesarean Delivery. Anesthesiology. 1998;88(6):1475–1479. | ||
Conti C, Tranquilli AL, Garzetti GG, Romanini C. Modulation of vascular reactivity after acute calcium antagonist administration in pregnant women moderately sensitive to angiotensin infusion. Bollettino della Societa italiana di biologia sperimentale. 1994;70(10–11):243–248. | ||
Öney T, Kaulhausen H. The value of the angiotensin sensitivity test in the early diagnosis of hypertensive disorders in pregnancy. Am J Obstet Gynecol. 1982;142(1):17–20. | ||
Jackson T, Corke C, Agar J. Enalapril overdose treated with angiotensin infusion. The Lancet. 1993;341(8846):703. | ||
Newby DE, Lee MR, Gray AJ, Boon NA. Enalapril overdose and the corrective effect of intravenous angiotensin II [letter]. Br J Clin Pharmacol. 1995;40(1):103–104. | ||
Trilli LE, Johnson KA. Lisinopril Overdose and Management with Intravenous Angiotensin II. Ann Pharmacother. 1994;28(10):1165–1168. | ||
Kato T, Murakami Y, Saito Y, et al. New modality of radiation therapy under increased tumor oxygen tension with angiotensin II: a pilot study. Radiat Med. 1993;11(3):86–90. | ||
Sato H, Sugiyama K, Hoshi M, Urushiyama M, Ishizuka K. Angiotensin II (AII) induced hypertension chemotherapy (IHC) for unresectable gastric cancer: With reference to resection after down staging. World J Surg. 1995;19(6):836–842. | ||
Nagamitsu A, Greish K, Maeda H. Elevating blood pressure as a strategy to increase tumor-targeted delivery of macromolecular drug SMANCS: cases of advanced solid tumors. Jpn J Clin Oncol. 2009;39(11):756–766. | ||
Tomura N, Kato T, Kanno I, et al. Increased blood flow in human brain tumor after administration of angiotensin II: demonstration by PET. Comput Med Imaging Graph. 1993;17(6):443–449. | ||
Millar EA, Angus RM, Hulks G, Morton JJ, Connell JM, Thomson NC. Activity of the renin-angiotensin system in acute severe asthma and the effect of angiotensin II on lung function. Thorax. 1994;49(5):492–495. | ||
Millar EA, Nally JE, Thomson NC. Angiotensin II potentiates methacholine-induced bronchoconstriction in human airway both in vitro and in vivo. Eur Respir J. 1995;8(11):1838–1841. | ||
Ramsay SG, Clayton RA, Dagg KD, Thomson LJ, Nally JE, Thomson NC. Effect of angiotensin II on histamine-induced bronchoconstriction in the human airway both in vitro and in vivo. Respir Med. 1997;91(10):609–615. | ||
Cohn JN, Luria MH. Studies in clinical shock and hypotension. II. Hemodynamic effects of norepinephrine and angiotensin. J Clin Invest. 1965;44(9):1494–1504. | ||
Ames RP, Borkowski AJ, Sicinski AM, Laragh JH. Prolonged infusions of angiotensin II and norepinephrine and blood pressure, electrolyte balance, and aldosterone and cortisol secretion in normal man and in cirrhosis with ascites. J Clin Invest. 1965;44(7):1171–1186. | ||
Joannidis M, Druml W, Forni LG, et al. Prevention of acute kidney injury and protection of renal function in the intensive care unit: update 2017: Expert opinion of the Working Group on Prevention, AKI section, European Society of Intensive Care Medicine. Intensive Care Medicine. 2017;43(6):730–749. | ||
Gaudry S, Hajage D, Schortgen F, et al. Initiation strategies for renal-replacement therapy in the intensive care unit. N Engl J Med Overseas Ed. 2016;375(2):122–133. | ||
Zarbock A, Kellum JA, Schmidt C, et al. Effect of early vs delayed initiation of renal replacement therapy on mortality in critically Ill patients with acute kidney injury. JAMA. 2016;315(20):2190–2199. | ||
Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol. 2011;22(6):999–1006. | ||
Langenberg C, Bagshaw SM, May CN, Bellomo R. The histopathology of septic acute kidney injury: a systematic review. Crit Care. 2008;12(2):R38. | ||
Lankadeva YR, Kosaka J, Evans RG, Bailey SR, Bellomo R, May CN. Intrarenal and urinary oxygenation during norepinephrine resuscitation in ovine septic acute kidney injury. Kidney Int. 2016;90(1):100–108. | ||
Chen H, Busse LW. Novel therapies for acute kidney injury. Kidney Int Rep. 2017;2(5):785–799. | ||
Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest. 2004;114(1):5–14. | ||
Wan L, Langenberg C, Bellomo R, May CN. Angiotensin II in experimental hyperdynamic sepsis. Crit Care. 2009;13(6):R190. | ||
Herget-Rosenthal S, Saner F, Chawla LS. Approach to hemodynamic shock and vasopressors. Clin J Am Soc Nephrol. 2008;3(2):546–553. | ||
Gordon AC, Mason AJ, Thirunavukkarasu N, et al. Effect of early vasopressin vs norepinephrine on kidney failure in patients with septic shock: The VANISH randomized clinical trial. JAMA. 2016;316(5):509–518. | ||
Abuelo JG. Normotensive ischemic acute renal failure. N Engl J Med. 2007;357(8):797–805. | ||
Prowle JR, Bellomo R. Sepsis-associated acute kidney injury: macrohemodynamic and microhemodynamic alterations in the renal circulation. Semin Nephrol. 2015;35(1):64–74. | ||
Lankadeva YR, Kosaka J, Evans RG, Bellomo R, May CN. Urinary Oxygenation as a Surrogate Measure of Medullary Oxygenation During Angiotensin II Therapy in Septic Acute Kidney Injury. Crit Care Med. 2018;46(1):e41–e48. | ||
Brenner M, Schaer GL, Mallory DL, Suffredini AF, Parrillo JE. Detection of renal blood flow abnormalities in septic and critically ill patients using a newly designed indwelling thermodilution renal vein catheter. Chest. 1990;98(1):170–179. | ||
Thomas VL, Nielsen MS. Administration of angiotensin II in refractory septic shock. Crit Care Med. 1991;19(8):1084–1085. | ||
Tovar JL, Bujons I, Ruiz JC, Ibañez L, Salgado A. Treatment of severe combined overdose of calcium antagonists and converting enzyme inhibitors with angiotensin II. Nephron. 1997;77(2):239. | ||
Wray GM, Coakley JH. Severe septic shock unresponsive to noradrenaline. Lancet. 1995;346(8990):1604. | ||
Suberviola B, Rodrigo E, González-Castro A, Serrano M, Heras M, Castellanos-Ortega Á. Association between exposure to angiotensin-converting enzyme inhibitors and angiotensin receptor blockers prior to septic shock and acute kidney injury. Med Intensiva. 2017;41(1):21–27. | ||
Corrêa TD, Takala J, Jakob SM. Angiotensin II in septic shock. Crit Care. 2015;19:98. | ||
Umbrello M, Formenti P, Bolgiaghi L, Chiumello D. Current Concepts of ARDS: A Narrative Review. Int J Mol Sci. 2016;18(1):64. | ||
Ryan JW. Processing of endogenous polypeptides by the lungs. Annu Rev Physiol. 1982;44:241–255. | ||
Orfanos SE, Langleben D, Khoury J, et al. Pulmonary capillary endothelium-bound angiotensin-converting enzyme activity in humans. Circulation. 1999;99(12):1593–1599. | ||
Busse LA, Gong T, Thompson M. Outcomes in patients with acute respiratory distress syndrome receiving angiotensin II for vasodilatory shock. Critical Care. 2018;22(Suppl 1):82. | ||
ARDS Definition Task Force; Ranieri VM, Rubenfeld GD, Thompson BT, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526–2533. | ||
Imai Y, Kuba K, Rao S, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–116. | ||
Lambert DW, Hooper NM, Turner AJ. Angiotensin-converting enzyme 2 and new insights into the renin-angiotensin system. Biochem Pharmacol. 2008;75(4):781–786. | ||
Wang G, Zhang Q, Yuan W, Wu J, Li C. Enalapril protects against myocardial ischemia/reperfusion injury in a swine model of cardiac arrest and resuscitation. Int J Mol Med. 2016;38(5):1463–1473. | ||
Arnal JF, Cudek P, Plouin PF, Guyenne TT, Michel JB, Corvol P. Low angiotensinogen levels are related to the severity and liver dysfunction of congestive heart failure: implications for renin measurements. Am J Med. 1991;90(1):17–22. | ||
Imai M, Sokabe H. Plasma renin and angiotensinogen levels in pathological states associated with oedema. Arch Dis Child. 1968;43(230):475–479. | ||
Faymonville ME, Larbuisson R, Radermecker M, Limet R, Fourny J, Lamy M. Serum activity of angiotensin converting enzyme during extracorporeal circulation in man. C R Seances Soc Biol Fil. 1983;177(2):252–258. | ||
Panagiotopoulos I, Palatianos G, Michalopoulos A, Chatzigeorgiou A, Prapas S, Kamper EF. Alterations in biomarkers of endothelial function following on-pump coronary artery revascularization. J Clin Lab Anal. 2010;24(6):389–398. | ||
Giapreza. Lexi-Drugs. Lexicomp Online [database online]. Hudson, OH: Lexi-Comp, Inc; 2018. Available from: http://online.lexi.com/lco/action/doc/retrieve/docid/patch_f/6582570. Accessed March 26, 2018. | ||
Busse LA, Gong T, Thompson M. Outcomes in patients with acute respiratory distress syndrome receiving angiotensin II for vasodilatory shock. Critical Care. 2018;22(Suppl 1):82. |
Supplementary materials
Pharmacology of Ang II
Giapreza® (angiotensin II) was approved by the US Food and Drug Administration in December 2017 with a labeled indication for increasing blood pressure in adults with septic or other distributive shock. The medication is to be administered via continuous intravenous infusion with a starting rate, per the label, of 20 ng/kg/min and titrated every 5 minutes by increments of up to 15 ng/kg/minute as needed.1 Although off label, an initial starting rate of 10 ng/kg/min has been suggested, since some patients may respond with a rapid increase in blood pressure. The initial maximum infusion rate is 80 ng/kg/min within the first 3 hours, with a maximum maintenance infusion rate of 40 ng/kg/min.1 Because of the unique metabolism and very short half-life of <1 minute, Giapreza does not need to be dose adjusted for renal or hepatic impairment.2
Giapreza is available as a 2.5 mg/1 mL vial to be diluted in normal saline to a final concentration of 5,000 or 10,000 ng/mL for fluid-restricted patients.1 Vials may be stored at 2°C–8°C, and compounded drips may be stored diluted at room temperature or under refrigeration for no longer than 24 hours.2 Giapreza has intravenous compatibility with other vasopressors.2 Adverse reactions include thrombosis, tachycardia, deep vein thrombosis, peripheral ischemia, delirium, acidosis, hyperglycemia, thrombocytopenia, fungal infection.1 Of note, these adverse reactions are the result of combination therapy with other vasopressors. Drug interactions include angiotensin converting enzyme inhibitors and angiotensin receptor blockers.2 There are no contraindications listed in the manufacturer’s labeling.
References
Giapreza [angiotensin II] [prescribing information]. San Diego, CA; La Jolla Pharmaceutical Company: December 2017. | ||
Giapreza. Lexi-Drugs. Lexicomp Online [database online]. Hudson, OH: Lexi-Comp, Inc; 2018. Available from: http://online.lexi.com/lco/action/doc/retrieve/docid/patch_f/6582570. Accessed March 26, 2018. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.