")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 14
Angiopoietin-Like Protein 3 (ANGPTL3) Inhibitors in the Management of Refractory Hypercholesterolemia
Authors Kosmas CE, Bousvarou MD, Sourlas A, Papakonstantinou EJ, Peña Genao E, Echavarria Uceta R, Guzman E
Received 24 March 2022
Accepted for publication 8 July 2022
Published 16 July 2022 Volume 2022:14 Pages 49—59
DOI https://doi.org/10.2147/CPAA.S345072
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Constantine E Kosmas,1,2 Maria D Bousvarou,3 Andreas Sourlas,3 Evangelia J Papakonstantinou,4 Edilberto Peña Genao,2 Rogers Echavarria Uceta,2 Eliscer Guzman1,2
1Division of Cardiology, Department of Medicine, Montefiore Medical Center, Bronx, NY, USA; 2Cardiology Clinic, Cardiology Unlimited, PC, New York, NY, USA; 3School of Medicine, University of Crete, Heraklion, Greece; 4General Directorate of Public Health and Social Welfare, Athens, Attica Region, Greece
Correspondence: Constantine E Kosmas, Email [email protected]
Abstract: Cardiovascular disease (CVD) is the most common cause of death in a global scale and significantly depends on the elevated plasma levels of low-density lipoprotein cholesterol (LDL-C) and the subsequent formation of atherosclerotic plaques. While physicians have several LDL-C-lowering agents with diverse mechanisms of action, including statins, ezetimibe, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors and inclisiran, angiopoietin-like protein 3 (ANGPTL3) inhibitors have recently emerged as a powerful addition in the armamentarium of lipid-lowering strategies, especially for patients with refractory hypercholesterolemia, as in the case of patients with homozygous familial hypercholesterolemia (HoFH). ANGPTL3 protein is a glycoprotein secreted by liver cells that is implicated in the metabolism of lipids along with other ANGPTL proteins. These proteins inhibit lipoprotein lipase (LPL) and endothelial lipase (EL) in tissues. Loss-of-function mutations affecting the gene encoding ANGPTL3 are linked with lower total cholesterol, LDL-C, and triglyceride (TG) levels. Evinacumab is a monoclonal antibody that targets, binds to, and pharmacologically inhibits ANGPTL3, which was recently approved by the United States Food and Drug Administration (FDA) as a complementary agent to other LDL-C lowering regimens for patients aged 12 or older with HoFH, based on clinical trial evidence that confirmed its safety and efficacy in those patients. Antisense oligonucleotides (ASOs) also represent an interesting class of agents that target and inhibit the mRNA derived from the transcription of ANGPTL3 gene. This review aims to present and discuss the current clinical and scientific data pertaining to the role of ANGPTL3 inhibitors, a novel lipid-modifying class of agents capable of reducing LDL-C levels via a mechanism independent of LDL receptors.
Keywords: cardiovascular disease, ANGPTL3 inhibitors, evinacumab, refractory hypercholesterolemia, familial hypercholesterolemia, LDL-cholesterol, LDL receptors
Introduction
The modern human society has been making an ongoing, undeniable progress in the course of the past decades, regarding optimization of the quality of life and ultimately longevity. Nonetheless, the leading cause of death in a global scale seems to be a rather common, plausibly preventable and manageable medical condition: cardiovascular disease (CVD). According to the WHO, about 17.9 million people departed this life due to cardiovascular events in 2019, representing 32% of global deaths, among which 85% were attributable to heart attack and stroke. CVD prevalence is high in the United States, Central Europe, North Africa, and the Middle East. CVD is expected to account for over 22.2 million deaths by 2030.1,2 Heart attack, stroke and peripheral vascular disease constitute the three main clinical manifestations of atherosclerotic cardiovascular disease (ASCVD). Atherosclerosis is an immunoinflammatory vascular condition in which lipid plaques form in the vessel walls; when unstable, these plaques may rupture with ensuing superimposed thrombosis, thus leading to CVD.3 The plasma levels of low-density lipoprotein cholesterol (LDL-C) are a well-established independent factor in the pathogenesis of atherosclerotic plaques. The lower the levels of LDL-C, preferably below 100 mg/dl from a young age, the lower the risk of CVD.4,5
Familial hypercholesterolemia (FH) is an inherited autosomal dominant disease, linked to remarkably high plasma levels of LDL-C due to its impaired catabolism. The condition is mainly attributed to gene mutations affecting the quality or quantity of LDL receptors (LDLR). More infrequent reported genetic determinants include Apolipoprotein B100 (ApoB100), proprotein convertase subtilisin/kexin type 9 (PCSK9) gain-of-function mutations, and LDL receptor adaptor protein 1 (LDLRAP1), as well as other gene mutations with very low prevalence that generate an FH phenotype. If not managed properly, the formation rate of lipid plaques increases and subsequently there is an excessive risk of premature CVD and death. Heterozygous FH (HeFH) has a prevalence of approximately 1:300, whereas the prevalence of homozygous FH (HoFH) is estimated to be 1:300.000.6 Levels of LDL-C in patients with HeFH tend to fluctuate between 190 and 500 mg/dl when untreated. Respectively, patients with HoFH have untreated LDL-C levels above 500 mg/dl.7
Besides FH, other clinical entities, namely polygenic hypercholesterolemia and familial combined hyperlipidemia, could lead to refractory hypercholesterolemia, defined as an LDL-C level of ≥70 mg/dl with ASCVD or ≥100 mg/dl without ASCVD, despite background lipid-lowering therapy at maximum tolerated doses.7 Familial combined hyperlipidemia (FCHL) is the most common form of dyslipidemia with an estimated prevalence of 1:100. Some of its features are hypercholesterolemia, hypertriglyceridemia or a combination of both high LDL-C and triglyceride levels. FCHL is a polygenic disorder with defects in multiple metabolic genetic loci, such as the lipoprotein lipase (LPL) gene and loci implicated in the clearance of LDL particles.8
Traditionally, statins are first-line agents for the management of hypercholesterolemia. Their mechanism of action involves competitive inhibition of 3-hydroxy-3 methylglutaryl-coenzyme A (HMG-CoA) reductase, an enzyme participating in the mevalonate pathway which converts HMG-CoA into mevalonic acid, a cholesterol precursor. Thence, the reduction of intrahepatic cholesterol synthesis leads to upregulation of LDLR, consequently leading to increased cholesterol uptake by the hepatocytes and lower plasma LDL-C levels.9 Ezetimibe is a second-line agent, reducing the absorption of cholesterol by the gastrointestinal tract via inhibition of the Niemann-Pick C1-like 1 (NPC1L1) protein.10 PCSK9 inhibitors is a new class of agents that is also a valuable addition to lipid-lowering therapy, blocking the lysosomal degradation of LDLR. When added to statin therapy, PCSK9 inhibitors cause LDL-C levels to plummet down by 60%.7,11 Furthermore, PCSK9 inhibitors have a favorable side effect profile and the adherence to treatment with PCSK9 inhibitors appears to be excellent.12,13 Many patients with severe hyperlipidemia have already benefited from this powerful LDL-C reduction, such as those with FH. In addition, inclisiran, a small interfering RNA (siRNA) molecule that increases the number of LDLR in the hepatocyte membranes by halting the transcription of PCSK9, is another promising LDL-C-lowering agent, which has been very recently approved by the European Medicines Agency (EMA) and the United States Food and Drug Administration (FDA) for the treatment of patients with HeFH or clinical ASCVD who require additional lowering of LDL-C. Inclisiran has been shown to cause significant LDL-C reductions, although its impact on clinical outcomes has not yet been definitely ascertained.14–16 However, given its very infrequent, 6-monthly dosing intervals, inclisiran is also expected to have an excellent patient adherence profile.12
Notwithstanding, standard high-intensity lipid-lowering therapies, such as statins, ezetimibe or PCSK9 inhibitors, might still fail to achieve target LDL-C plasma levels in patients with extremely high baseline LDL-C levels, such as those with FH, especially HoFH. Thus, extensive research is being conducted for the identification of novel agents that would be beneficial for patients who cannot respond desirably to maximally tolerated doses of the current lipid-lowering therapy.
This review aims to present and discuss the current clinical and scientific data pertaining to the role of angiopoietin-like protein 3 (ANGPTL3) inhibitors, a novel lipid-modifying class of agents capable of reducing LDL-C levels via a mechanism independent of LDLR.
Angiopoietin-Like Protein 3 (ANGPTL3)
Before shedding light on ANGPTL3 inhibitors, a brief reference to ANGPTL3 protein structure, function and clinical correlations ought to be made.
Angiopoietin-like proteins (ANGPTL) compose a family of secreted glycoproteins (ANGPTL1-8) with high homology to angiopoietins, which are implicated in the physiology of angiogenesis. In particular, the ANGPTL3-4-8 model is characterized by high sequence homology and is implicated in the inhibition of LPL and endothelial lipase (EL) in various tissues; ANGPTL3 requires to be bound with ANGPTL8 in order for its function to be enhanced, though not necessarily for the inhibition of EL. ANGPTL3 is a 70 kDA, 460-amino acid secreted glycoprotein expressed in the liver during embryonic and adult stage. A coiled-coil structure between N-terminal and C-terminal domain is required for the protein’s main function, which is cleavage and inhibition of LPL and EL activity. Genome-wide association studies (GWAS) have identified single-nucleotide polymorphisms (SNPs) near the locus of ANGPTL3 with strong effects on lipid metabolism, thus confirming the vital role of ANGPTL3 in lipid metabolism.17–20 The final structure and function of the glycoprotein is largely regulated by liver X receptors (LXR), as well as by a post-translational modulator named GALNT2.19,21 LPL is located on the luminal side of the vascular endothelium, whereas EL is a secreted glycoprotein that is active at the vascular endothelial surface. These enzymes catalyze the hydrolysis of triglycerides (TG) in chylomicrons (CM), very low-density lipoproteins (VLDL) and, in the case of EL, phospholipids.22
Mutations located in the gene encoding ANGPTL3 alter its function and, consequently, a large aspect of the lipid metabolism pathway. Dewey et al sequenced the exons of ANGPTL3 in 58,335 participants in the DiscovEHR human genetics study and identified 13 distinct loss-of-function variants in ANGPTL3. The condition caused by ANGPTL3 loss-of-function mutations, namely familial combined hypolipidemia (FCH), is associated with a decreased risk of coronary artery disease (CAD); in 13,102 patients with CAD and 40,430 controls, the presence of a loss-of-function variant was associated with a 41% lower risk of coronary artery disease (adjusted OR 0.59, 95% CI 0.41–0.85; P=0.004).23 Those findings are consistent with the findings of a meta-analysis of 19 studies involving 21,980 patients with CAD and 158,200 control participants, demonstrating that carriers of an ANGPTL3 loss-of-function mutation manifested a 34% lower risk of CAD,24 along with lower total plasma cholesterol, LDL-C and TG levels, compared with non-carriers.25 In particular, Dewey et al studied 45,226 individuals with documented serum lipid levels and determined that carriers of a loss-of-function ANGPTL3 variant had 27% lower TG levels, 9% lower LDL-C levels, and 4% lower HDL-cholesterol (HDL-C) levels than noncarriers.23 Ruhanen et al26 demonstrated that ANGPTL3 depletion precipitates alterations on the lipidome of lipoproteins in human subjects. Moreover, mice studies suggest that the low LDL-C levels in the presence of ANGPTL3 deficiency are attributed to reduced secretion and increased uptake of Apolipoprotein B (ApoB)-containing lipoproteins27 Interestingly though, a link between potential ANGPTL3 gain-of-function mutations and dyslipidemia profile, specifically in FCHL, is yet to be found.28 The aforementioned observations have already laid the foundations for the development of ANGPTL3 inhibitors.
ANGPTL3 Inhibitors – Evinacumab
As previously mentioned, ANGPTL3 inhibitors constitute novel medications, which inhibit ANGPTL3 activity or its production from hepatocytes in an attempt to imitate the phenotype of loss-of-function mutations.29
Evinacumab, available under the trade name EvkeezaTM, is a fully human monoclonal antibody invented by Regeneron Pharmaceuticals Inc., using VelocImmune technology. VelocImmune relies on genetically engineered, specifically modified mice with a humanized immune system, so as to produce fully humanized antibodies.30 Evinacumab is an IgG4-kappa antibody characterized by the standard Y morphology. It is composed of two gamma heavy chains, measuring 453 amino acids each in length, and a pair of kappa light chains with a length of 214 amino acids each. The heavy chains are interlinked with disulfide bonds at their hinge region.31 When administered subcutaneously (SC) or intravenously (IV), evinacumab binds to ANGPTL3, which, as we have mentioned above, prevents LPL and EL from hydrolyzing lipids. Consequently, pharmacological inhibition of ANGPTL3 by evinacumab preserves the function of LPL and EL with a subsequent decrement of TG, LDL-C and HDL-C plasma levels independently of LDLR function. The mechanism of action of evinacumab is shown in Figure 1.
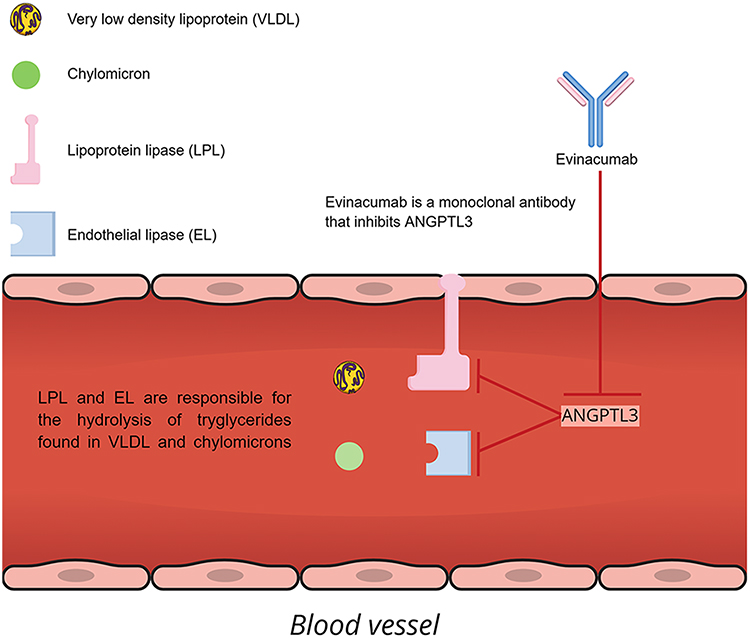 |
Figure 1 Mechanism of action of evinacumab. |
Safety, tolerability and pharmacokinetics of evinacumab were assessed in a randomized, double-blind, placebo-controlled, Phase 1 clinical trial in 96 healthy Caucasian and Japanese volunteers with LDL-C levels between 100 and 160 mg/dl. Participants were randomly assigned (3:1) in four different cohorts: evinacumab SC 300 mg single dose, SC 300 mg once weekly for eight doses, IV 5 mg/kg, or IV 15 mg/kg once every 4 weeks for two doses. Each cohort comprised 24 participants, 12 Japanese and 12 Caucasian, to receive evinacumab or placebo with a 24-week follow-up. Evinacumab was found to be well tolerated at all dose regimens, including the highest administered doses of 15 mg/kg IV every 4 weeks and 300 mg SC in a weekly basis. The safety profile of evinacumab in both ethnicities was similar to that of placebo with no serious treatment-emergent adverse events or adverse events leading to study discontinuation. In addition, the pharmacokinetic and pharmacodynamic profiles of evinacumab were similar in both ethnicities across all dose regimens. Pertaining to pharmacodynamics, evinacumab injections produced significant dose-dependent reductions in LDL-C, TG and ApoB levels both in Japanese and Caucasian subjects.32
Several trials have attempted to assess the efficacy of evinacumab in lowering LDL-C plasma levels in cases of severe and refractory hypercholesterolemia. A single-group, open-label, proof-of-concept, Phase 2 study was carried on nine adult patients with HoFH on maximum tolerated aggressive lipid-lowering regimens. Evinacumab was administered to the participants for a total of 4 weeks. At week 4, there was a mean reduction of LDL-C levels by 49±23% with an absolute decrease from baseline of 157±90 mg/dl. In addition, levels of ApoB, non-HDL-C, TG and HDL-C were reduced by 46±18%, 49±22%, 47% and 36±16%, respectively.33
A small, recently completed study, enrolled four subjects already participating in the above-mentioned study in order to assess the catabolic rate of ApoB lipoproteins before and after receiving evinacumab. A stable leucine isotope was administered and measured in VLDL, intermediate-density lipoprotein (IDL) and LDL before and after the IV administration of evinacumab 15 mg/kg. Evinacumab lowered LDL-C by 59±2% and increased IDL-ApoB and LDL-ApoB fractional catabolic rate in all four HoFH subjects, by 616±504% and 113±14%, respectively. VLDL-ApoB production rate decreased in two of the four subjects.34 Thus, increased clearance rate of ApoB may represent an interesting, worthy to be further studied mechanism of lowering LDL-C levels when ANGPTL3 is pharmacologically inhibited.
In a double-blind, placebo-controlled, Phase 3 trial, 65 patients with HoFH who were receiving stable maximum doses of lipid-lowering therapy were randomly assigned in a 2:1 ratio to receive an IV infusion of evinacumab (at a dose of 15 mg/kg) every 4 weeks or placebo. The mean baseline LDL-C plasma level in both groups was 255.1 mg/dl. At week 24, patients receiving evinacumab marked significantly lower LDL-C levels with a mean reduction of 47,1%, in comparison with an increase of 1.9% in the placebo group, resulting in a between-group least-squares mean difference of −49.0%. Lower LDL-C levels were observed in the evinacumab group compared to the placebo group, both in patients with absent or non-functional LDLR (null–null variants) and in those with partially impaired LDLR function (non-null variants); −43.4% vs +16.2% and –49.1% vs –3.8%, respectively. Similar incidence of adverse events was observed in both groups.30 From this pool of 65 people, two adolescents with null/null variants, aged 12 (patient A) and 16 years (patient B), were chosen in order to assess whether evinacumab is capable of causing regression of atherosclerotic plaques. Coronary computed tomography angiographies (CCTAs), before randomization and after 6 months of treatment with evinacumab, showed that total plaque volumes were lowered by 76% and 85% in patients A and B, respectively. Therefore, the study concluded that the formation of atherosclerotic plaques may be a potentially regressive process.35
Rosenson et al carried out a double-blind, placebo-controlled, phase 2 trial across 20 countries. This study enrolled 272 patients with hypercholesterolemia that was refractory to treatment with a PCSK9 inhibitor and a statin at a maximum tolerated dose, with or without ezetimibe. The screening LDL-C level was 70 mg/dl or higher for patients with clinical ASCVD and 100 mg/dl or higher for patients without clinical ASCVD. Patients were randomly assigned to receive SC or IV evinacumab or placebo for 16 weeks. At week 16, the differences in the least-squares mean change from baseline in the LDL-C level between the groups receiving SC evinacumab and placebo ranged from −38.5% to −56.0% depending on the received dose regimen. The respective differences between the groups receiving IV evinacumab and placebo ranged from −24.2% to −50.5% depending on the administered dose. Similar incidence of adverse events was observed in both groups receiving SC and IV evinacumab, with serious adverse events during the treatment period ranging from 3% to 16% across trial groups. Evinacumab significantly reduced LDL-C by more than 50% at the maximum dose.36
Furthermore, in a meta-analysis of five randomized controlled trials (RCTs) with a total of 568 subjects, treatment with evinacumab, as compared with placebo, significantly reduced LDL-C, TG, and HDL-C by 33.123%, 50.959%, and 12.773%, respectively (P < 0.0001 for all comparisons). The incidence of at least 1 treatment emergent adverse event was not significantly different between evinacumab and placebo groups. Thus, the findings of this meta-analysis indicate that evinacumab may be a valuable therapeutic choice in the management of hypercholesterolemia.37
As of ongoing trials, an open-label, single-group clinical trial, being conducted at 38 sites across 12 countries, will assess by January 2023 the long-term safety and efficacy of evinacumab in 116 adult and adolescent patients with HoFH.38 Last but not least, another currently conducted three-part, single-arm, open-label clinical trial with an estimated completion in April 2023 will evaluate the efficacy, safety, and pharmacokinetics of evinacumab in 20 pediatric patients with HoFH.39
Evinacumab is currently approved in the United States since February 11, 2021, as a complementary agent to other LDL-C lowering regimens for patients aged 12 or older with HoFH.40
Antisense Oligonucleotides (ASOs)
ANGPTL3-LRx constitutes an alternative, promising pharmacological agent to treat dyslipidemias. Otherwise known as vupanorsen, ANGPTL3-LRx is a second-generation ligand-conjugated antisense oligonucleotide (ASO) targeting the ANGPTL3 gene mRNA coding sequence in liver cells, ultimately inactivating its translation to ANGPTL3 protein. ASOs are relatively short molecules of 15–20 nucleotides in length, capable of binding precisely with complementary mRNA sequences, and act via RNA cleavage or blockage.41
Several experimental and clinical studies have assessed the role of ANGPTL3-LRx in dyslipidemia. In 2017, Graham et al demonstrated that use of ANGPTL3-LRx in mice effectively lowers levels of liver ANGPTL3 mRNA, ANGPTL3 protein, TG and LDL-C in a dose-dependent manner, along with reductions in hepatic TG content and atherosclerosis progression. Following this, they properly designed a randomized, double-blind, placebo-controlled, phase 1 clinical trial, enrolling 44 adult participants with an age ranging from 18 to 65, randomly assigned in a 3:1 ratio to receive ANGPTL3-LRx or placebo. The aim of the trial was to assess the safety, possible adverse events, pharmacokinetics and pharmacodynamics of single and multiple ascending doses of ANGPTL3-LRx. Findings were consistent with the preclinical mice trial, as at day 43, ANGPTL3-LRx reduced the levels of ANGPTL3 protein, TG, LDL-C, ApoB and non-HDL-C by up to 84.5%, 63.1%, 32.9%, 25.7%, and 36.6%, respectively, as compared with placebo. With the exception of headache and dizziness, reported by three participants who received the antisense oligonucleotide and three who received placebo, no serious adverse events were noted.42
Another randomized, double-blind, placebo-controlled, multicenter, dose-ranging, phase 2 study was conducted, enrolling 105 patients with elevated fasting plasma TG levels (>150 mg/dl), diabetes mellitus type 2, hepatic steatosis and a body mass index between 27 and 40 kg/m2. Subjects were randomized in a 3:1 ratio to receive SC vupanorsen or placebo for 6 months at doses of 40 or 80 mg every 4 weeks or 20 mg every week. The mean reduction of fasting TG levels from baseline in patients receiving vupanorsen reached a maximum of 53%, with 35–58% of the patients (depending on the administered dose regimen) achieving normal levels of plasma TGs. ANGPTL3 protein was markedly reduced by up to 62%. In addition, vupanorsen, as compared with placebo, reduced apolipoprotein C-III, remnant cholesterol, total cholesterol, non-HDL-C, HDL-C, LDL-C, and ApoB by 58%, 38%, 19%, 18%, 24%, 12%, and 9%, respectively. The above data indicate that vupanorsen, a second-generation N-acetyl galactosamine (GalNAc3)-modified ASO targeting hepatic ANGPTL3 mRNA, appears to be capable of decreasing the plasma levels of atherogenic particles and thus potentially reducing residual cardiovascular risk. Once again, no serious adverse events were reported in this study besides injection-site pruritus and injection-site erythema.43
Based on the above, and to further evaluate the role of vupanorsen in cardiovascular risk reduction and in severe hypertriglyceridemia, a phase 2b, multicenter, randomized, double-blind, placebo-controlled, dose-ranging, 8-arm parallel-group study of vupanorsen (TRANSLATE-TIMI 70), which enrolled 286 statin-treated participants with dyslipidemia, was started on September 28, 2020. Its primary endpoint was the percent change from baseline in non-HDL-C, with secondary endpoints being reduction of ANGPTL3, LDL-C, ApoB and TG levels at weeks 16 and 24.44 Although the study met its primary endpoint, achieving a statistically significant reduction in non-HDL-C, as well as statistically significant reductions in TG and ANGPTL3, the magnitude of the reduction of non-HDL-C and TG levels observed did not encourage the continuation of the study. In addition, a dose-dependent elevation in hepatic fat, as well as in liver enzymes alanine aminotransferase (ALT) and aspartic aminotransferase (AST) was observed. These events led Pfizer Inc. and Ionis Pharmaceuticals to discontinue the clinical development program of vupanorsen, as both parties announced on January 31, 2022, according to Pfizer’s official press release statement.45
Further clinical studies are indispensable to fully comprehend and determine the potential of ASOs as a therapeutic strategy for the management of dyslipidemias.
A summary of the results of the main trials discussed in this review is shown in Table 1.
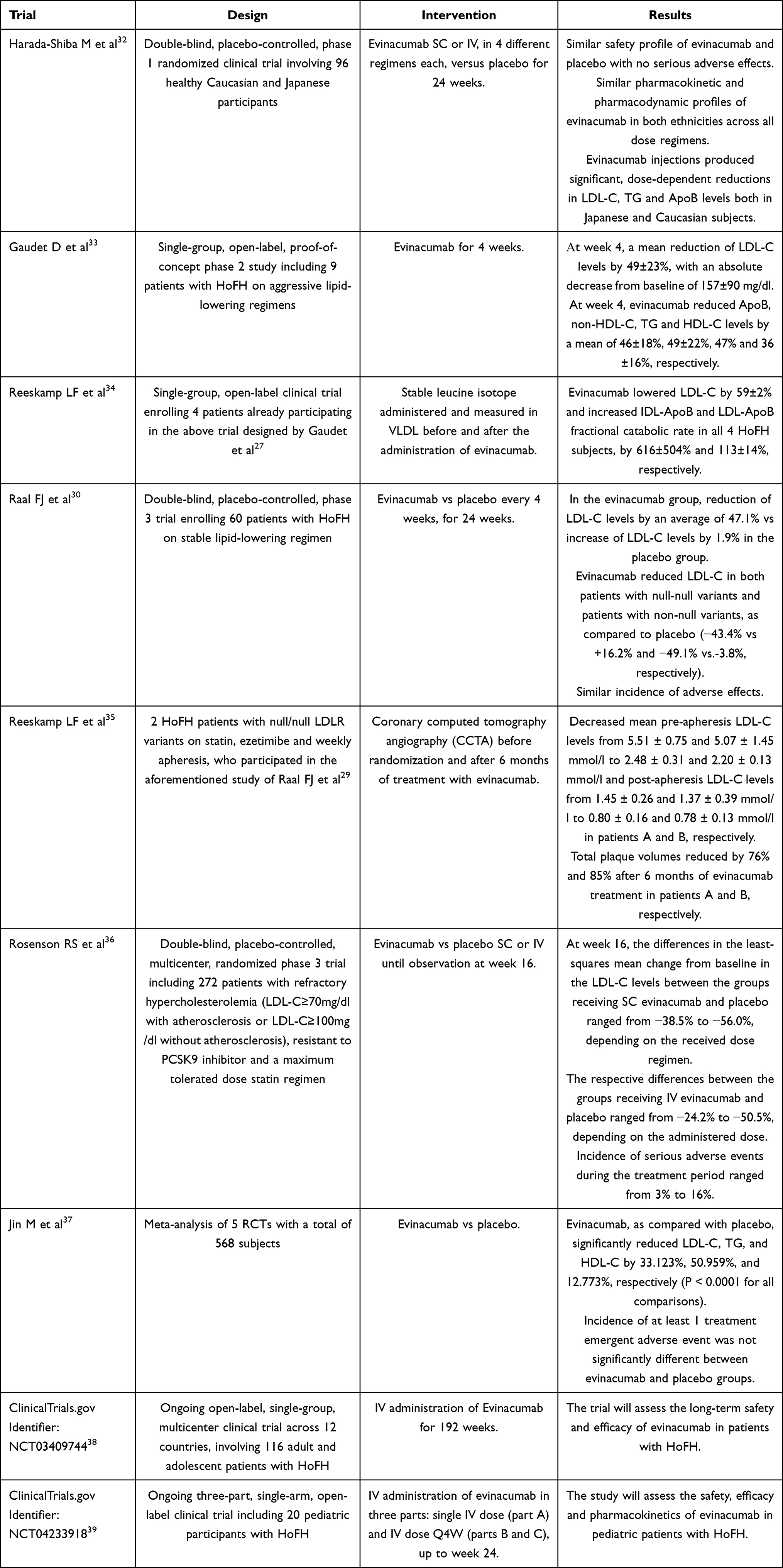 |
Table 1 Summary of the Results of the Clinical Studies Pertaining to ANGPTL3 Inhibitors |
Cost-Effectiveness of Evinacumab
Undeniably, the above discussed clinical and scientific data provide firm evidence that evinacumab would attain remarkable clinical results in terms of routine pharmacological management of HoFH, should it be widely available. However, the question arises if that would actually be feasible in the near future, given the nature of the drug pricing market.
Evinacumab belongs to a class of medications known as orphan drugs, which are defined as drugs used to treat rare diseases. The term “orphan” rises from prior abandoned or “orphaned” attempts to design such drugs due to inadequate funding or absence of interest in development. In an effort to incentivize the interest and initiative of the pharmaceutical companies in developing drugs for rare diseases, the United States Congress passed the Orphan Drug Law Act (ODA) in 1983, which offers several attractive tax credits, a waiver of the Prescription Drug User Fee, and extended market exclusivity options to participating drug developers. In the US, about 6000–8000 diseases, including HoFH, are classified as rare, affecting 25 million people. The ODA seems to achieve its purpose, as the number of secondary indication approvals has more than doubled in proportion since the 1980s, from 19% to 47% in the 2010s.46
The recent approval of evinacumab renders it the latest addition in the armamentarium of lipid-lowering agents; however, the expenditures from the patient’s perspective are by no means negligible. Regeneron stated that the annual wholesale cost of evinacumab will be $450,000 on average, raising serious concerns about its affordability. Given that HoFH is excessively difficult to be managed with the traditional drugs, this high price could set an obstacle in the treatment of patients who are most in need of evinacumab (especially those with null–null variants). However, this does not necessarily imply that the drug is overpriced in terms of its actual cost-effectiveness.47 Notwithstanding, one could assume that evinacumab might possibly eventually follow the example of PSCK9 inhibitors, the price of which was ultimately significantly reduced by the manufacturing companies from $14,100 per annum to $5850 per annum (a 60% price drop) in October 2018 and February 2019.13
Conclusions and Future Directions
Taking into consideration the clinical evidence provided above, ANGPTL3 targeting seems a tempting, efficient strategy to manage cases of severe and refractory hypercholesterolemia when other available treatment options appear to be, more or less, inadequate. By inhibiting either ANGPTL3 mRNA or its derivative protein, a significant reduction in LDL-C, non-HDL-C and TG levels is feasible, independently of LDLR function. This claim is supported by yet another proof-of-concept study, focusing on the effects of evinacumab in the LDLR activity of lymphocytes obtained by patients with HoFH, before and after treatment with evinacumab, and versus lymphocytes carrying wild-type LDLR, and also in an LDLR-defective Chinese hamster ovary cell line (CHO-ldlA7) transfected with plasmids encoding the LDLR variant. In that study, the researchers again clearly demonstrated that the inhibition of ANGPTL3 by evinacumab in humans lowers LDL-C in a mechanism independent of the LDLR.48 As discussed earlier, there is even proof that atherosclerotic plaques may regress with the administration of evinacumab.35 Thus, with its capability of reduction of LDL-C plasma levels and the potential regression of the atherosclerotic process being combined, evinacumab emerges as a very promising therapeutic strategy to lower the exceptionally high risk of CVD in patients with HoFH. Moreover, evinacumab appears to have a benign side-effect profile, as the same mild adverse events were reported in patients receiving evinacumab and placebo in most of the trials mentioned above with no reported serious adverse events leading to early termination of studies, thus validating the safety of the drug.
One ought to bear in mind that several issues are yet to be fully elucidated. The independent of LDLR function mechanism of action of ANGPTL3 inhibitors is very promising and may represent an important field of research. The improvement of CVD risk with the administration of evinacumab could be better defined with the conduction of carefully designed clinical trials in the near future. Efforts should be also made to study the effects of evinacumab on clinical outcomes in patients with HoFH, as well as in patients with refractory hypercholesterolemia attributed to different multifactorial causes, so as to broaden its indications. Results of two ongoing studies are eagerly awaited, concerning the effect and long-term safety and tolerability of evinacumab in adults and adolescents, as well as the outcomes of its administration in pediatric patients with HoFH.38,39 Administration of evinacumab in pregnant and lactating women is another interesting field of research. Lastly, the potential role of ASOs as a therapeutic strategy for the management of refractory dyslipidemias may still require further investigation.
A summary of the main scientific and clinical attributes of ANGPTL3 inhibitors is shown in Table 2.
 |
Table 2 Summary of the Main Scientific and Clinical Attributes of ANGPTL3 Inhibitors |
Disclosure
Dr Kosmas reports personal fees from Amgen, Inc. and Amarin Pharma, Inc., outside the submitted work. Dr Kosmas and Dr Guzman have served on the Dyslipidemia Speaker Bureau of Amgen, Inc. The authors report no other conflicts of interest in this work.
References
1. World Health Organization. Cardiovascular diseases (CVDs). Available from: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds).
2. Virani SS, Alonso A, Benjamin EJ, et al. Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation. 2020;141(9):e139–e596. doi:10.1161/CIR.0000000000000757
3. Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol. 2006;47(8 Suppl):C7–12. doi:10.1016/j.jacc.2005.09.068
4. Sharrett AR, Ballantyne CM, Coady SA, et al.; Atherosclerosis Risk in Communities Study Group. Coronary heart disease prediction from lipoprotein cholesterol levels, triglycerides, lipoprotein(a), apolipoproteins A-I and B, and HDL density subfractions: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2001;104(10):1108–1113. doi:10.1161/hc3501.095214
5. Ference BA, Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol. 2012;60(25):2631–2639. doi:10.1016/j.jacc.2012.09.017
6. Benito-Vicente A, Uribe KB, Jebari S, Galicia-Garcia U, Ostolaza H, Martin C. Familial hypercholesterolemia: the most frequent cholesterol metabolism disorder caused disease. Int J Mol Sci. 2018;19(11):3426. doi:10.3390/ijms19113426
7. Martin SS. Factoring in ANGPTL3 When LDL Is refractory. N Engl J Med. 2020;383(24):2385–2386. doi:10.1056/NEJMe2032798
8. Taghizadeh E, Esfehani RJ, Sahebkar A, et al. Familial combined hyperlipidemia: an overview of the underlying molecular mechanisms and therapeutic strategies. IUBMB Life. 2019;71(9):1221–1229. doi:10.1002/iub.2073
9. Kosmas CE, Pantou D, Sourlas A, Papakonstantinou EJ, Echavarria Uceta R, Guzman E. New and emerging lipid-modifying drugs to lower LDL cholesterol. Drugs Context. 2021;10:8–13. doi:10.7573/dic.2021-8-3
10. Musunuru K. Treating coronary artery disease: beyond statins, ezetimibe, and PCSK9 inhibition. Annu Rev Med. 2021;72(1):447–458. doi:10.1146/annurev-med-080819-044918
11. Michos ED, McEvoy JW, Blumenthal RS. Lipid management for the prevention of atherosclerotic cardiovascular disease. N Engl J Med. 2019;381(16):1557–1567. doi:10.1056/NEJMra1806939
12. Kosmas CE, Silverio D, Ovalle J, Montan PD, Guzman E. Patient adherence, compliance, and perspectives on evolocumab for the management of resistant hypercholesterolemia. Patient Prefer Adherence. 2018;12:2263–2266. doi:10.2147/PPA.S149423
13. Kosmas CE, Skavdis A, Sourlas A, et al. Safety and tolerability of PCSK9 inhibitors: current insights. Clin Pharmacol. 2020;12:191–202. doi:10.2147/CPAA.S288831
14. Kosmas CE, Muñoz Estrella A, Sourlas A, et al. Inclisiran: a new promising agent in the management of hypercholesterolemia. Diseases. 2018;6(3):63. doi:10.3390/diseases6030063
15. Kosmas CE, Muñoz Estrella A, Skavdis A, Peña Genao E, Martinez I, Guzman E. Inclisiran for the treatment of cardiovascular disease: a short review on the emerging data and therapeutic potential. Ther Clin Risk Manag. 2020;16:1031–1037. doi:10.2147/TCRM.S230592
16. Kosmas CE, Muñoz Estrella A, Sourlas A, Pantou D. Inclisiran in dyslipidemia. Drugs Today. 2021;57(5):311–319. doi:10.1358/dot.2021.57.5.3277083
17. Sylvers-Davie KL, Segura-Roman A, Salvi AM, Schache KJ, Davies BSJ. Angiopoietin-like 3 inhibition of endothelial lipase is not modulated by angiopoietin-like 8. J Lipid Res. 2021;62:100112. doi:10.1016/j.jlr.2021.100112
18. Chen PY, Gao WY, Liou JW, Lin CY, Wu MJ, Yen JH. Angiopoietin-like protein 3 (ANGPTL3) modulates lipoprotein metabolism and dyslipidemia. Int J Mol Sci. 2021;22(14):7310. doi:10.3390/ijms22147310
19. Lupo MG, Ferri N. Angiopoietin-Like 3 (ANGPTL3) and atherosclerosis: lipid and non-lipid related effects. J Cardiovasc Dev Dis. 2018;5(3):39. doi:10.3390/jcdd5030039
20. Chi X, Britt EC, Shows HW, et al. ANGPTL8 promotes the ability of ANGPTL3 to bind and inhibit lipoprotein lipase. Mol Metab. 2017;6(10):1137–1149. doi:10.1016/j.molmet.2017.06.014
21. Li X, Zhang Y, Zhang M, Wang Y. GALNT2 regulates ANGPTL3 cleavage in cells and in vivo of mice. Sci Rep. 2020;10(1):16168. doi:10.1038/s41598-020-73388-3
22. Tikka A, Jauhiainen M. The role of ANGPTL3 in controlling lipoprotein metabolism. Endocrine. 2016;52(2):187–193. doi:10.1007/s12020-015-0838-9
23. Dewey FE, Gusarova V, Dunbar RL, et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N Engl J Med. 2017;377(3):211–221. doi:10.1056/NEJMoa1612790
24. Stitziel NO, Khera AV, Wang X, et al. PROMIS and myocardial infarction genetics consortium investigators. ANGPTL3 deficiency and protection against coronary artery disease. J Am Coll Cardiol. 2017;69(16):2054–2063. doi:10.1016/j.jacc.2017.02.030
25. Kersten S. Angiopoietin-like 3 in lipoprotein metabolism. Nat Rev Endocrinol. 2017;13(12):731–739. doi:10.1038/nrendo.2017.119
26. Ruhanen H, Haridas PAN, Minicocci I, et al. ANGPTL3 deficiency alters the lipid profile and metabolism of cultured hepatocytes and human lipoproteins. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865(7):158679. doi:10.1016/j.bbalip.2020.158679
27. Xu YX, Redon V, Yu H, et al. Role of angiopoietin-like 3 (ANGPTL3) in regulating plasma level of low-density lipoprotein cholesterol. Atherosclerosis. 2018;268:196–206. doi:10.1016/j.atherosclerosis.2017.08.031
28. Bea AM, Franco-Marín E, Marco-Benedí V, et al. ANGPTL3 gene variants in subjects with familial combined hyperlipidemia. Sci Rep. 2021;11(1):7002. doi:10.1038/s41598-021-86384-y
29. Genest J. ANGPTL3: a gene, a protein, a new target? Aye, there’s the rub. J Am Coll Cardiol. 2017;69(16):2064–2066. doi:10.1016/j.jacc.2017.03.015
30. Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for homozygous familial hypercholesterolemia. N Engl J Med. 2020;383(8):711–720. doi:10.1056/NEJMoa2004215
31. Poiron C, Wu Y, Ginestoux C, Ehrenmann F, Duroux P, Lefranc M-P. IMGT/mAb-DB: the IMGT® database for therapeutic monoclonal antibodies. JOBIM. 2010;13.
32. Harada-Shiba M, Ali S, Gipe DA, et al. A randomized study investigating the safety, tolerability, and pharmacokinetics of evinacumab, an ANGPTL3 inhibitor, in healthy Japanese and Caucasian subjects. Atherosclerosis. 2020;314:33–40. doi:10.1016/j.atherosclerosis.2020.10.013
33. Gaudet D, Gipe DA, Pordy R, et al. ANGPTL3 inhibition in homozygous familial hypercholesterolemia. N Engl J Med. 2017;377(3):296–297. doi:10.1056/NEJMc1705994
34. Reeskamp LF, Millar JS, Wu L. et al. ANGPTL3 inhibition with evinacumab results in faster clearance of IDL and LDL apoB in patients with homozygous familial hypercholesterolemia-brief report. Arterioscler Thromb Vasc Biol. 2021;41(5):1753–1759. doi:10.1161/ATVBAHA.120.315204
35. Reeskamp LF, Nurmohamed NS, Bom MJ, et al. Marked plaque regression in homozygous familial hypercholesterolemia. Atherosclerosis. 2021;327:13–17. doi:10.1016/j.atherosclerosis.2021.04.014
36. Rosenson RS, Burgess LJ, Ebenbichler CF, et al. Evinacumab in patients with refractory hypercholesterolemia. N Engl J Med. 2020;383(24):2307–2319. doi:10.1056/NEJMoa2031049
37. Jin M, Meng F, Yang W, Liang L, Wang H, Fu Z. Efficacy and safety of evinacumab for the treatment of hypercholesterolemia: a meta-analysis. J Cardiovasc Pharmacol. 2021;78(3):394–402. doi:10.1097/FJC.0000000000001073
38. ClinicalTrials.gov Identifier. An open-label study to evaluate the long-term safety and efficacy of evinacumab in patients with homozygous familial hypercholesterolemia: NCT03409744. Available from. https://clinicaltrials.gov/ct2/show/NCT03409744.
39. ClinicalTrials.gov Identifier: NCT04233918. A three-part, single-arm, open-label study to evaluate the efficacy, safety, and pharmacokinetics of evinacumab in pediatric patients with homozygous familial hypercholesterolemia. Available from: https://clinicaltrials.gov/ct2/show/NCT04233918.
40. Markham A. Evinacumab: first approval. Drugs. 2021;81(9):1101–1105. doi:10.1007/s40265-021-01516-y
41. Dhuri K, Bechtold C, Quijano E, et al. Antisense oligonucleotides: an emerging area in drug discovery and development. J Clin Med. 2020;9(6):2004. doi:10.3390/jcm9062004
42. Graham MJ, Lee RG, Brandt TA, et al. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N Engl J Med. 2017;377(3):222–232. doi:10.1056/NEJMoa1701329
43. Gaudet D, Karwatowska-Prokopczuk E, Baum SJ, et al.; Vupanorsen Study Investigators. Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridaemia. Eur Heart J. 2020;41(40):3936–3945. doi:10.1093/eurheartj/ehaa689
44. ClinicalTrials.gov Identifier: NCT04516291. A phase 2b randomized, double-blind, placebo-controlled, parallel group, dose-ranging study to assess the efficacy, safety, and tolerability of vupanorsen (PF-07285557) in statin-treated participants with dyslipidemia. Available from: https://clinicaltrials.gov/ct2/show/NCT04516291.
45. Pfizer Inc. Pfizer and ionis announce discontinuation of vupanorsen clinical development program. Available from: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-ionis-announce-discontinuation-vupanorsen.
46. Roberts AD, Wadhwa R. Orphan drug approval laws. StatPearls. Treasure Island (FL):StatPearls Publishing;2022. https://www.ncbi.nlm.nih.gov/books/NBK572052/.
47. Kuehn BM. Evinacumab approval adds a new option for homozygous familial hypercholesterolemia with a hefty price tag. Circulation. 2021;143(25):2494–2496. doi:10.1161/CIRCULATIONAHA.121.055463
48. Banerjee P, Chan KC, Tarabocchia M, et al. Functional analysis of LDLR (Low-Density Lipoprotein Receptor) variants in patient lymphocytes to assess the effect of evinacumab in homozygous familial hypercholesterolemia patients with a spectrum of LDLR activity. Arterioscler Thromb Vasc Biol. 2019;39(11):2248–2260. doi:10.1161/ATVBAHA.119.313051
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.