")
Back to Journals » The Application of Clinical Genetics » Volume 7
Angelman syndrome: review of clinical and molecular aspects
Authors Bird L
Received 11 November 2013
Accepted for publication 16 February 2014
Published 16 May 2014 Volume 2014:7 Pages 93—104
DOI https://doi.org/10.2147/TACG.S57386
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Lynne M Bird
1Department of Pediatrics, University of California, Division of Genetics, Rady Children’s Hospital, San Diego, California, USA
Abstract: “Angelman syndrome” (AS) is a neurodevelopmental disorder whose main features are intellectual disability, lack of speech, seizures, and a characteristic behavioral profile. The behavioral features of AS include a happy demeanor, easily provoked laughter, short attention span, hypermotoric behavior, mouthing of objects, sleep disturbance, and an affinity for water. Microcephaly and subtle dysmorphic features, as well as ataxia and other movement disturbances, are additional features seen in most affected individuals. AS is due to deficient expression of the ubiquitin protein ligase E3A (UBE3A) gene, which displays paternal imprinting. There are four molecular classes of AS, and some genotype–phenotype correlations have emerged. Much remains to be understood regarding how insufficiency of E6-AP, the protein product of UBE3A, results in the observed neurodevelopmental deficits. Studies of mouse models of AS have implicated UBE3A in experience-dependent synaptic remodeling.
Keywords: Angelman syndrome, chromosome 15q11-13, UBE3A, imprinting
Introduction
Harry Angelman, an English pediatrician, first described this condition in 1965 when he reported three children that he referred to as “Puppet Children” because of their unusual arm position and jerky movements.1 In addition to the characteristic movements, Angelman noted severe intellectual disability, absent speech, and bouts of inappropriate laughter. In the nearly 50 years since that original report, the AS phenotype has been elaborated, and the etiology of the disorder identified as deficiency of UBE3A.2,3 The molecular pathogenesis of how UBE3A deficiency leads to this phenotype is beginning to be clarified. What follows is a description of our current understanding of the clinical and molecular aspects of AS.
Clinical review
“AS” is a neurodevelopmental disorder whose main features are intellectual disability, lack of speech, seizures, and a characteristic behavioral profile.4–9 It has a prevalence of between 1/10,000 and 1/20,000 individuals.10,11 See Table 1 for consistent, frequent, and occasional features of AS.
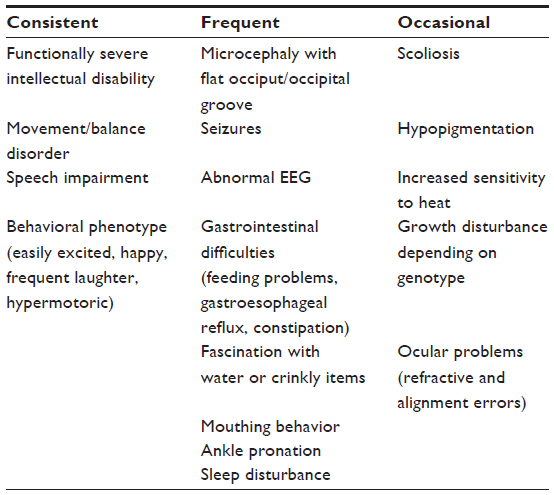 | Table 1 Clinical features of Angelman syndrome |
Performance
Development delays in AS are usually evident within the first year of life, with delayed attainment of gross motor, fine motor, receptive language, expressive language, and social skills. Reportedly, individuals with AS plateau at a developmental level of between 24 and 30 months,12 and cognitive performance is usually in the range of severe functional impairment.13 Language development in those with AS is significantly impaired. Most individuals lack speech entirely, a few individuals have small single-word vocabularies,13 and rare individuals are able to use phrases. The combination of deficits exhibited by individuals with AS make the commonly used developmental assessment tools difficult to apply, and these tests underestimate the abilities of AS children (author’s personal observations). Receptive language is superior to expressive language.14 Nonverbal communication using a variety of systems (picture exchange cards, communication devices, modified sign language) is possible in a substantial proportion of individuals with AS (author’s personal observations). Although all skills are delayed, there is variability in adaptive behavior functioning, with relative strength in socialization and relative weakness in motor skills.12,14 A substantial subset of children with AS qualify for a comorbid diagnosis of autism,15–21 independent of the severity of their cognitive and adaptive behavior functioning.21 AS adults are not able to live independently, but most are able to use feeding utensils and perform some household tasks with supervision.22–25 Dressing skills are dependent on the degree of fine-motor dexterity. Daytime continence is possible with prompted voiding and habit training.
Seizures
Seizures occur in 80%–95% of children with AS and usually start in childhood.26–29 The onset of seizures is before age 3 years in 75% of affected individuals.29 Seizure types include myoclonic, atypical absence, generalized tonic–clonic, and atonic (“drop”) seizures. Many individuals exhibit multiple seizures types.27,28 Seizures usually require broad-spectrum anticonvulsant medication and often combination therapy. Efficacy appears to be highest with valproate and clonazepam and lowest with phenobarbital and carbamazepine;28 vigabatrin and carbamazepine may exacerbate seizures.26,29 Some patients have responded to vagal nerve stimulation or ketogenic diet30 for seizures that were medically refractory.28,29 There can be attenuation of seizure activity in adolescents.27–29 There is a characteristic electroencephalogram (EEG) “signature” in AS,31,32 which can sometimes be useful in pointing toward the diagnosis.31 Various combinations of very high amplitude rhythmic (primarily anterior) delta activity, diffuse high amplitude rhythmic theta activity, and posterior-predominant spike and sharp waves are seen in >90% of individuals with AS.33–38
Behavior
The behavioral features of AS include a happy demeanor, easily provoked laughter, short attention span, hypermotoric behavior, mouthing of objects, sleep disturbance with reduced need for sleep, and an affinity for water.39–41 Though infancy can be difficult due to feeding problems and general irritability, happy disposition and increased smiling characterize most children.39 Rarely, unhappy or irritable affect persists, and gastrointestinal difficulties such as dysmotility and gastroesophageal reflux disease may play a role (author’s personal observations). Mouthing of objects becomes very prominent in the young child, along with drooling and tongue thrusting; these behaviors can be lessened or extinguished with behavioral modification. Individuals with AS have an apparently increased desire for social interaction.39 Children are described as easily excited. Though paroxysms of laughter are said to occur in AS, the laughter is not truly “unprovoked”, since an inciting event can usually be identified; however, the responding laughter is frequently excessive or inappropriate to the triggering stimulus. The majority of AS patients exhibit a short attention span, though this characteristic does not discriminate from other conditions with intellectual disability,42 and most children are hypermotoric/hyperactive, becoming calmer in adolescence and adulthood. Disruptive behaviors43,44 are displayed by the majority of patients, including biting, pinching, hair-pulling, and grabbing. Rarely are these behaviors intended to cause harm; they usually result from easy excitability, desire for attention, poor control over movements, reduced repertoire of need expression, and occasionally frustration over an inability to communicate effectively. Behavioral noncompliance, tantrums, and repetitive or stereotyped behaviors have also been described.43,44
Sleep
Most children have an apparently reduced need for sleep (sometimes as little as 5–6 hours per night) and abnormalities of the sleep–wake cycle, with long or frequent periods of wakefulness during the night.45–47 Sleep problems can involve the initiation and/or maintenance of sleep and early morning awakening.45–48 In a small study, melatonin levels were found to be low in those with AS,49 corroborating prior observations that melatonin improves the sleep of children with AS.50,51 Despite sleep disruption, most individuals with AS do not exhibit daytime somnolence. With behavior modification52 and/or pharmacologic treatment, sleep difficulties can be overcome in most patients, and sleep patterns improve with age.48 Epilepsy severity correlates with sleep problems, but whether more severe seizures create sleep disturbance or whether poor sleep patterns exacerbate epilepsy remains unclear.53
Other
Growth in AS varies with molecular diagnosis (see the “Genotype–phenotype correlations” section), and microcephaly is common (80%).54 Individuals with AS are generally non-dysmorphic as infants, but a subtle craniofacial phenotype develops with time, consisting of midface recession, prognathism, and broad mouth (the latter two are possibly consequences of tongue thrusting, mouthing behaviors and increased smiling). A subset of patients have hypopigmentation of the skin, hair, and eyes; this is more common in those with a deletion,55 who lack the maternal copy of OCA2 and presumably have a hypomorphic allele on the paternal chromosome.56 Patients with AS and oculocutaneous albinism type 2 (OCA2) have been reported, mostly commonly due to deletion of the maternal OCA2 and mutation in the paternal OCA2.57 A non-deletion patient with AS and OCA2 presumably has this constellation due to mutation in the paternal OCA2 and isodisomy for paternal chromosome 15 (author’s personal observations). AS patients with UBE3A mutations may have hypopigmentation on the basis of UBE3A’s regulation of the melanocortin-1 receptor, which is downregulated in Ube3a null mice.58
Ocular problems in AS include refractive errors (usually hyperopia and astigmatism), iris and choroidal hypopigmentation, and esotropia or exotropia.59–61 Nystagmus is reported but is not common.61 Ocular hypopigmentation is seen in all molecular classes but is more common in those with deletion.61 Patchy retinochoroidal atrophy was reported in two adults with AS.62 The author knows of one deletion patient with bilateral ocular pterygia requiring corneal transplantation to restore vision.
Truncal hypotonia and distal extremity hypertonia/hyperreflexia characterize the neurological examination of children with AS.63 Movement disturbances, abnormalities of tone, and impaired balance contribute to the delayed acquisition of motor skills (sitting after 12 months, walking between 2 and 6 years). Movement disorders include jerkiness, ataxic gait, and tremors.63,64 Many walk with arms held up and flexed at the elbows, true to the original description. The incidence of nonambulation is said to be 10%,65 but whether this will remain true in the modern era (of earlier diagnosis and prompt intervention and continued therapy) remains to be seen. The early institution and continuation of physical therapy may change the natural history of scoliosis, previously reported to occur in 10% of children and up to 70% of adults,66 by improving truncal tone.
Life expectancy appears to be normal;22–26 however, early death by accidental drowning has claimed the lives of some children, and the author is aware of premature deaths due to choking, pneumonia, suffocation, and seizures (personal communication).
Genetic basis
AS is caused by a lack of expression of the maternally inherited UBE3A gene in the brain. UBE3A is one of a small subset of human genes that are imprinted – that is, expressed depending on parent of origin, in a tissue-specific manner.65,66 While in most tissues, UBE3A appears to be expressed from both alleles (though perhaps unequally favoring the maternal allele67), in the brain, the paternally derived UBE3A gene is silenced, and only the maternally inherited copy is active.68–70 UBE3A was initially discarded as a potential candidate for the AS gene because it appeared not to be imprinted when studied in lymphocytes and fibroblasts,71 and its widespread expression ran counter to expectation since the phenotype is exclusively neurological. Establishing brain-only imprinting of UBE3A in mice resurrected UBE3A’s status as a candidate gene for AS.70 Analysis of Ube3a in AS patients with biparental contribution to 15q11-q13 and no imprinting abnormalities showed point mutations in several unrelated patients, identifying it as the gene responsible for the AS phenotype.2,3
AS is caused by deficient expression of the maternal copy of the UBE3A gene due to one of four molecular etiologies: deletion of the AS critical region on maternal chromosome 15q11-q13, paternal uniparental disomy (UPD) for chromosome 15, an imprinting defect causing lack of expression of the maternal copy of UBE3A, and mutations in the maternally inherited copy of UBE3A.55 There is a subgroup of patients with a clinical diagnosis of AS for whom no abnormality of UBE3A can be identified. Two potential explanations for these test-negative patients are 1) novel mechanisms for repression of UBE3A expression yet to be identified and 2) misdiagnoses of phenotypically similar conditions. Patients with AS and negative molecular analyses are now being recognized to have a variety of Angelman-like syndromes.72,73
In a cohort of AS patients participating in a natural history study conducted as part of the Rare Diseases Clinical Research Network, the distribution of molecular diagnoses among 286 patients was as follows: 31.1% class II deletion, 23.8% class I deletion, 10.8% unspecified deletion, 3.8% atypical deletion, 8.7% UPD, 7.7% imprinting defect, and 11.2% UBE3A mutation (unpublished data). Among individuals with a deletion, 40.5% have a common 5.9 megabases (Mb) (class I) deletion; 53% have a smaller 5.0 Mb (class II) deletion, differing only by the location of the proximal (centromeric) breakpoint; and 6.5% have atypical deletions. Deletions are mediated by homologous misalignment and meiotic recombination between low-copy-number repeats (duplicons) that have been identified in proximal and distal 15q11-q13. These duplicons arose with the amplification of an ancestral gene, homologous to the E6-AP carboxy terminus (HECT) and regulator of chromatin condensation 1-like domain (RLD)-containing E3 ubiquitin protein ligase 2 (HERC2). Duplicons having 90%–99% identity to the first 79 exons of HERC2 are found in at least ten copies in the 15q11-q13.74
Paternal UPD is isodisomic in almost all cases. The most likely origin of this event is maternal nondisjunction producing a monosomy 15 conception, with post-zygotic rescue by duplication of the paternal chromosome 15.75 As in the deletion cases, this class of AS represents de novo mutational events and has a very low risk for recurrence.
“Imprinting defect” occurs when a paternal imprint is erroneously assigned to the maternally inherited allele. Two types of imprinting defects are known: those due to a submicroscopic deletion of the imprinting center, and those with no detectable mutation.76–78 Most submicroscopic imprinting center deletions are familial and carry a 50% risk for recurrence. Thus far, all imprinting defects with undetectable mutations have been sporadic events. They are presumed to result from failure to establish or maintain the imprint during oogenesis, due to a stochastic event or perhaps (as yet unknown) environmental factors.78
Intragenic UBE3A mutations include insertion, deletion, nonsense, missense, and splice site mutations.79,80 A substantial portion of UBE3A mutations are inherited from the mother’s paternally acquired allele. In this circumstance, a 50% recurrence risk pertains.81
Mechanisms of imprinting and gene regulation
Genetic imprinting is the process of conferring functional differences onto specific genes such that their expression occurs from only one parent’s allele.65 There is incomplete understanding of the mechanism(s) of imprint establishment in the germ line, imprint maintenance during development and postnatal life, and imprint reversal in the germ line of the next generation. Further complexities, such as tissue-specific imprinting and age-related changes in imprinting are poorly understood.
Mechanisms of controlling gene expression include DNA insulators (DNA elements which prevent nearby chromatin domains from interacting); histone modifications (such as acetylation, phosphorylation, and methylation), which alter chromatin structure and influence transcriptional accessibility; DNA methylation; and transcriptional enhancer competition (promoters of linked imprinted genes competing for access to enhancers).82–84 Each of these epigenetic mechanisms overlays the information contained within the nucleotide sequence. Imprinted genes are found in clusters in specific areas of the genome, suggesting coordinated regulation by a regional element, which has been designated the “imprinting control region” (ICR).
DNA methylation is the most well understood of the recognized epigenetic mechanisms. The addition of a methyl group to the cytosine base of a CpG dinucleotide is found in most imprinted genes and in all ICRs.82 Loss and reacquisition of DNA methylation occurs during specific phases of germ cell development, and probably represents erasure of the imprint from the previous generation and re-establishment of the parent-of-origin specific epigenotype. DNA methylation may play a role in establishing and/or maintaining the imprint.82–84
Deficits in the imprinted gene cluster on chromosome 15q11-13 cause Prader–Willi syndrome (PWS) and AS.7,66,78,85,86 Loss of expression of maternally derived UBE3A causes AS, while loss of expression of paternally derived gene(s) causes PWS. The main features of PWS are neonatal hypotonia and failure to thrive, childhood onset hyperphagia and obesity, small hands and feet, short stature, hypogonadism, and cognitive impairment.87 Though many features of PWS can be reproduced by absence of a small nucleolar organizing RNA gene, SNORD116, the full PWS phenotype requires the loss of expression of several genes, indicating that PWS is truly a contiguous gene syndrome.87
Differential methylation of chromosome 15q11-q13 provides the basis for diagnostic testing for PWS and AS. The maternal chromosome is highly methylated while the paternal chromosome is mostly unmethylated in the 15q11-q13 region, and this can be demonstrated using methylation-sensitive restriction enzymes and Southern blot analysis or polymerase chain reaction assay of the promoter region of the small nuclear ribonucleoprotein polypeptide N (SNRPN) gene. A maternal-only contribution is diagnostic of PWS, while an exclusively paternal contribution indicates AS. A normal methylation profile having both a maternal and paternal contribution excludes PWS but does not rule out AS, because 10%–20% of cases of AS are due to maternally inherited UBE3A mutations.79,80
The ICR for the 15q11-q13 cluster has been designated the “IC”.76,85 It was defined by small submicroscopic deletions in a subgroup of PWS and AS multiplex families that demonstrated biparental inheritance of the 15q11-13 region yet uniparental imprint.76 The IC regulates in cis the establishment and maintenance of the imprint for the entire cluster. The IC has a bipartite structure, the Prader–Willi syndrome imprinting center (PWS-IC) and the Angelman syndrome imprinting center (AS-IC), separated by 35 kb (Figure 1). All AS patients with IC deletions are missing the AS-IC (880 bp centromeric of PWS-IC).88 The PWS-IC establishes and maintains paternal gene expression. During oogenesis, the AS-IC negatively regulates the PWS-IC and prevents the paternal imprint from being established. Deletion of the AS-IC leads to a paternal imprint of the entire 15q11-q13 region, and AS occurs when this is transmitted through the maternal germ line. The PWS-IC and AS-IC must be proximal and correctly related in order for the maternal imprint to be established.89
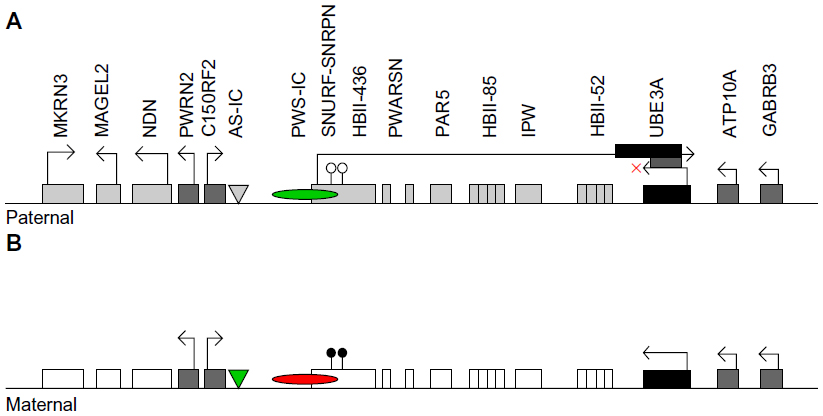 | Figure 1 Imprinting map of the human chromosome 15q11-13 region. Paternal and maternal chromosome 15q11-13 regions around the Angelman syndrome imprinting center (AS-IC) and Prader–Willi syndrome imprinting center (PWS-IC) are represented in (A and B), respectively. |
The genes makorin ring finger protein 3 (MKRN3), necdin (NDN), melanoma-associated antigen-like 2 (MAGEL2) and SNRPN upstream reading frame (SNURF-SNRPN) are expressed only from the paternal chromosome where their promoter regions are unmethylated.66,90,91 Between SNURF-SNRPN and UBE3A are located more than 70 small nucleolar RNA (snoRNA) genes. These noncoding genes produce snoRNAs, which modify ribosomal RNA.66 Though not differentially methylated, snoRNAs in this region are indirectly under the control of methylation, because they are processed from the differentially methylated and paternally expressed SNURF-SNRPN sense/UBE3A antisense transcript.
Studies of the orthologous mouse region (chromosome 7) form the basis for much of what is known about methylation status and imprinting of human chromosome 15q11-q13. However, important differences exist.92 The Frat3 gene, for which there is no human homologue, has joined the mouse PWS/AS region, acquiring the paternal pattern of methylation and expression. Transgenic studies, where human elements of the PWS/AS region are inserted in the mouse, have shown that the regulatory elements of the imprinting machinery have diverged between the two species. No mouse equivalent of the human AS-IC has been identified. Further study of the mouse PWS/AS region imprinting will probably yield important insights,93 but some may not apply to human UBE3A imprinting.
UBE3A itself is not differentially methylated;55 its imprinted expression is indirectly regulated by a long non-coding antisense RNA transcript (UBE3A-ATS) which is part of a larger SNURF-SNRPN transcript.66 UBE3A-ATS is active on the paternal chromosome and blocks UBE3A transcription in cis.94–97 Changes in DNA methylation and histone acetylation of the PWS-IC control production of the UBE3A-ATS from the paternal allele.98 The mechanism by which UBE3A-ATS blocks UBE3A transcription is unknown, but may involve histone-mediated repression, transcriptional interference, or repressive three-dimensional chromatin structure.66
Molecular pathogenesis
UBE3A spans 120 kb of genomic DNA.9 Mutations have been detected throughout all regions of the gene. Sixteen exons have been identified; since this region displays alternative splicing, additional exons at the 5′ end of the gene are possible. UBE3A is transcribed in the direction from telomere to centromere, producing RNA transcripts of 5–6 kb that include 2 kb of 3′ untranslated region sequence. There are three main transcripts producing three isoforms of UBE3A, and eight to ten additional transcripts of uncertain function. The function of the three different isoforms of UBE3A is unknown and the significance of tissue-specific variations in RNA splicing and isoform predominance is unclear.
UBE3A encodes E6-associated protein (E6-AP), an E3 ubiquitin ligase.9 E6-AP derives its name from its initial characterization, in which it was found to be associated with the E6 protein of papillomaviruses to promote degradation of p53. However, E6-AP does not maintain a stable association with p53, and its main function is believed to be participation in protein degradation in proteasomes via the ubiquitin pathway. The ubiquitin-proteasome system targets cellular proteins for destruction by covalently attaching ubiquitin to one or more lysine residues of proteins destined for degradation. Ubiquitination involves a three-step process: 1) activation of ubiquitin by an E1 enzyme, 2) transfer to an E2 conjugating enzyme, and 3) covalent ligation of ubiquitin to the protein substrate by an E3 ligase; E6-AP is one of many E3 ligases. E6-AP also interacts with proteins involved in such cellular functions as cell-cycle regulation and synaptic function and plasticity,99,100 and acts as a transcriptional co-activator of steroid hormone receptors.101,102 The C-terminus of E6-AP is a functionally important and highly conserved domain that is shared by a family of proteins (HECT domain), of which E6-AP is the founding member. The last six amino acids of the E6-AP C-terminus are essential for activity in vitro. It is unknown how substrate specificity is determined. E6-AP is found in all tissues that have been studied.
Presumably, defective UBE3A activity results in failure to degrade its substrates because of impaired ubiquitination. Several E6-AP targets have been identified, including activity-regulated cytoskeleton-associated protein (Arc) and Ephexin5.9 E6-AP regulates Arc levels either by regulating estradiol-induced transcription of Arc103 or by direct ubiquitination of Arc.104 Arc regulates surface expression of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionate receptors (AMPARs). When E6-AP is deficient, the excitatory postsynaptic AMPARs are internalized, which impairs synaptic transmission.104 Ephexin5 has a role in controlling synapse number.105 Recently proposed as a substrate for E6-AP is sacsin,104 defects of which cause a form of spastic ataxia;106 given the ataxic gait in AS, sacsin is an attractive target. There is also mounting evidence that UBE3A and methyl CpG binding protein 2 interact to regulate the expression of target genes.107 Recently, a role for E6-AP in Golgi acidification and protein sialylation was proposed.108
Mouse models have facilitated the progress in understanding the molecular pathogenesis of AS.109 The Ube3a knockout mouse, generated with target disruption of Ube3a on the maternal chromosome 7, nicely recapitulates the human disorder.110,111 Ube3am-p+ mice demonstrate reduced brain size, ataxia, motor impairment, abnormal EEG, sleep disturbance,112 learning and memory impairment, and deficits in hippocampal long-term potentiation (LTP).110,111 In the hippocampus of Ube3am-p+ mice, the calcium/calmodulin-dependent protein kinase II (CaMKII), which plays a role in induction of LTP that is critical for memory, has an increased level of inhibitory phosphorylation and reduced activity.113 Ube3am-p+ mice with a concomitant mutation in CaMKII that blocks inhibitory phosphorylation are indistinguishable from wild-type mice,114 indicating the defect of E6-AP can be overcome with excess CaMKII activity. E6-AP appears to play a role in activity-dependent synaptic plasticity; that is, the remodeling of synapses depending on experience.99,100 The observations of abnormal dendritic spine morphology115 and increase in synapses lacking AMPARs104 in Ube3am-p+ mice fit with this hypothesis. Ube3a acts as a transcriptional co-activator of many steroid hormone receptors, including the glucocorticoid receptor, and glucocorticoid receptor-mediated signaling is dysregulated in the brains of Ube3am-p+ mice, which show increased serum levels of corticosterone and increased anxiety-like behavior.116 Ube3am-p+ mice also have enhanced neuregulin-ErbB4 signaling117 that correlates with abnormal synaptic plasticity and memory impairment, not mediated through differences in AMPARs and N-methyl-D-aspartate receptors and not mediated through direct interaction of neuregulin or ErbB4 with UBE3A. The deficits in LTP can be rescued by ErbB inhibitors infused directly into the hippocampus of Ube3am-p+ mice.117
Diagnostic algorithm
The most sensitive test for AS is a methylation analysis of the chromosome 15q11-13 region, using either methylation-specific polymerase chain reaction or methylation-sensitive multiplex ligation-dependent probe amplification. Further testing is needed to parse those with abnormal methylation testing into deletion, UPD, and imprinting defect categories.117 Figure 2 depicts the diagnostic algorithm for testing for AS.
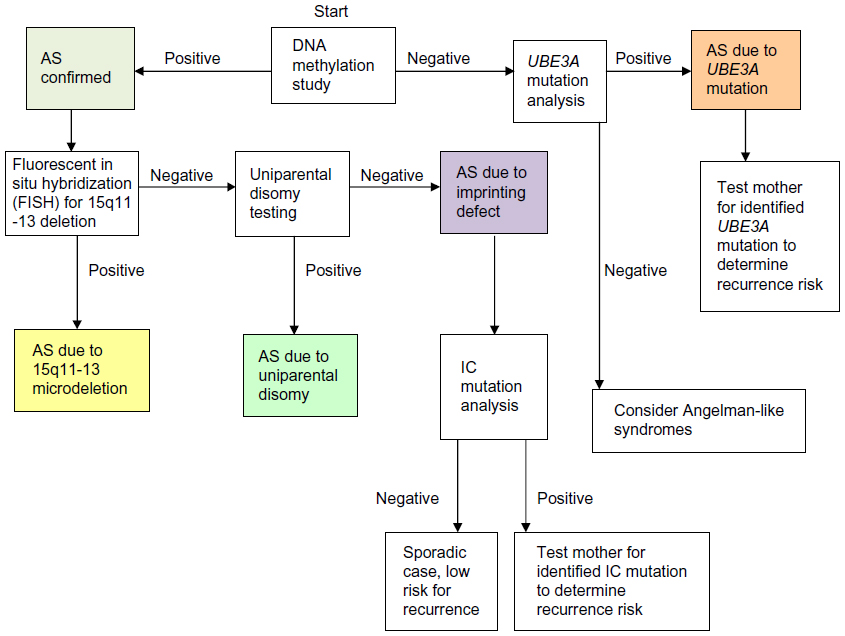 | Figure 2 Molecular diagnostic algorithm for AS. |
If DNA methylation analysis is negative, then UBE3A sequencing is appropriate for those with a convincing AS phenotype. When both DNA methylation analysis and UBE3A mutation testing are negative, the likelihood of AS is small.18 Though once thought to represent a substantial portion of cases, test-negative (“clinical diagnosis”) patients with AS are probably rare. Patients with an AS phenotype in whom testing returns normal should be considered for an alternative diagnosis, such as Pitt–Hopkins syndrome, Mowat–Wilson syndrome, Kleefstra syndrome, Phelan–McDermid syndrome, Koolen–de Vries syndrome, Christianson syndrome, and MBD5 haploinsufficiency.72,73
Recurrence risk for AS due to microdeletion or UPD is negligible, whereas UBE3A mutations and imprinting defects can have a 50% risk for recurrence if the mother is found to carry the mutation on her paternally inherited chromosome 15.81
Genotype–phenotype correlations
Clinical differences between the molecular classes of AS have been recognized.55,119–123 Those with deletion tend to be shorter and lighter than the general population, while those with UPD or imprinting defects tend to be taller and heavier; growth in those with UBE3A mutations is variable.54 There is a higher incidence of microcephaly in the deletion group compared with non-deletion subtypes.54,119 As a group, patients with deletions tend to have a more severe phenotype (later onset of independent walking, earlier onset and increased severity of seizures, complete absence of speech) than patients with UPD, imprinting defect, or UBE3A mutation (non-deletion patients).14,55 In younger patients with AS, those with UBE3A mutations scored higher in tests of cognition, gross motor and fine motor skills, and receptive language than deletion patients.14 Though speech is usually absent in AS patients with deletions, use of up to 20 words has been reported in AS patients of other molecular classes; however, Gentile et al found no differences in expressive language skills in patients of less than 5 years of age with AS with respect to molecular subtype.14 Compared with patients with a smaller class II deletion (~5 Mb), patients with a larger class I deletion (~6.0 Mb) are more likely to meet criteria for a comorbid diagnosis of autism, have lower cognitive scores, and require more seizure medications.122–124 It has been proposed that other deleted genes in the 15q11-13 region account for the increased phenotypic severity, but the precise contribution and mechanisms remain to be clarified. Three genes for gamma-Aminobutyric acid (GABA)-receptor subunits (GABRB3, GABRA5, GABRG3) are located telomeric of UBE3A and contained within the deleted region. Though these genes show biallelic expression, a role for them in the genesis of epilepsy in AS has been suggested.125 The GABA receptor subunit genes are deleted in both of the common deletion classes (I, II) so these genes do not account for differences seen among deletion-class groups but may contribute to the differences observed between deletion and non-deletion patients. No correlation between genotype and electroencephalographic pattern has emerged.31
Hypopigmentation occurs more frequently in AS deletion patients.55 The (un-imprinted) OCA2 gene located telomeric within the commonly deleted region is responsible for autosomal recessive OCA2. Individuals with AS and OCA2 have been reported, the mechanism being a maternal chromosome deletion and paternal OCA2 mutation.57,126 Semidominant behavior of the OCA2 product (expression of a hypomorphic allele) has been offered to explain the hypopigmentation seen in AS deletion patients. However, hypopigmentation has been reported in other classes of AS patients, including siblings with a maternally inherited intragenic deletion of exons 8–16 of UBE3A, suggesting that Ube3a can alter pigmentation.127 The melanocortin-1 receptor, which is downregulated in Ube3a-null mice, is a potential effector.58
Status of clinical research
Clinical trials conducted thus far have produced negative results. Attempts to increase transcription from the paternal allele through the use of pro-methylation vitamin supplements did not result in any noticeable improvement.128,129 There is an ongoing randomized, placebo-controlled trial using levodopa/carbidopa to treat AS.130 The rationale for this trial was based on the observations that levodopa was able to influence phosphorylation of CaMKII threonine residues in a rat model of Parkinson’s disease;131,132 the finding of dopaminergic neuronal loss in AS mouse models;133 and a report of two adults with AS and Parkinsonian symptoms who responded to levodopa.134 There was a short open-label trial of minocycline treatment,135 results of which have not yet been subjected to peer review. The rationale for minocycline was made based on the observation of elevated matrix metalloproteinase-9 activity in the hippocampi of AS mice (personal communication) and minocycline’s ability to reduce matrix metalloproteinase-9 activity.
Status of basic research
Interrupting the AS pathophysiology may be undertaken at the level of gene transcription or at the point of protein interactions.136 Mouse models for AS (the history of which was summarized recently137), have been invaluable in exploring both of these avenues. The Ube3am-p+ mouse has reduced brain size, ataxia, motor deficits, abnormal EEG, inducible seizures, and behavioral alterations.111,133 The AS mouse demonstrates impairments of context-dependent learning and memory and hippocampal LTP.110 Abnormalities of function in cerebellar Purkinje cells110,137,138 and nigrostriatal pathways133,139 have been reported. The behavioral phenotype of the Ube3am-p+ mouse was recently characterized in a comprehensive manner,140 which will assist greatly in the evaluation of future potential treatments.
Transcriptional upregulation has been approached by both gene therapy141 and pharmacologic intervention.142 Daily et al showed that direct injection of a recombinant adeno-associated viral vector carrying Ube3a into the hippocampi of adult AS mice could significantly improve associative learning (contextual fear conditioning).141 Huang and colleagues have demonstrated that the topoisomerase topotecan can un-silence the paternal Ube3a allele in cultured mouse primary cortical neurons, apparently by reducing transcription of Ube3a-ats.142 Recently the mechanism of inhibition of Ube3a-ats was shown to be mediated through RNA:DNA hybrid loop stabilization with the paternal snoRNA cluster.143
Identifying the pathways in which Ube3a participates may lead to targeted intervention.134 Studies have shown a role for CaMKII,113,114 neuregulin-ErbB4 signaling,117 EphB/Ephexin5 signaling,105 and Arc (possibly mediated through brain-derived neurotrophic factor-induced TrkB-PSD-95144) signaling.103,104 These observations point to a critical role for Ube3a in experience-dependent synaptic remodeling, possibly through more than one molecular pathway.
Conclusion
AS is a syndromic form of intellectual disability with a distinctive clinical presentation that can be best recognized by behavioral and performance characteristics. Molecular testing can diagnose most, if not all, cases. The causative protein, UBE3A, is critical for the processes of learning and memory through activity-dependent synaptic plasticity. Strides are being made in understanding the molecular pathogenesis, aided by mouse models that faithfully recapitulate the clinical syndrome.
Disclosure
The author declares no conflicts of interest in this work.
References
Angelman H. ‘Puppet’ children. A report on three cases. Dev Med Child Neurol. 1965;7(6):681–688. | |
Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15(1):70–73. Erratum in: Nat Genet. 1997;15(4):411. | |
Matsuura T, Sutcliffe JS, Fang P, et al. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15(1):74–77. | |
Clayton-Smith J, Laan L. Angelman syndrome: a review of the clinical and genetic aspects. J Med Genet. 2003;40(2):87–95. | |
Williams CA, Angelman H, Clayton-Smith J, et al. Angelman syndrome: consensus for diagnostic criteria. Angelman Syndrome Foundation. Am J Med Genet. 1995;56(2):237–238. | |
Williams CA, Beaudet AL, Clayton-Smith J, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet. 2006;140(5):413–418. | |
Chamberlain SJ, Lalande M. Angelman syndrome, a genomic imprinting disorder of the brain. J Neurosci. 2010;30(30):9958–9963. | |
Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med. 2010;12(7):385–395. | |
Dagli A, Buiting K, Williams CA. Molecular and clinical aspects of Angelman syndrome. Mol Syndromol. 2012;2(3–5):100–112. | |
Petersen MB, Brøndum-Nielsen K, Hansen LK, Wulff K. Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: estimated prevalence rate in a Danish county. Am J Med Genet. 1995;60(3):261–262. | |
Kyllerman M. On the prevalence of Angelman syndrome. Am J Med Genet. 1995;59(3):405. | |
Peters SU, Goddard-Finegold J, Beaudet AL, Madduri N, Turcich M, Bacino CA. Cognitive and adaptive behavior profiles of children with Angelman syndrome. Am J Med Genet A. 2004;128A(2):110–113. | |
Andersen WH, Rasmussen RK, Strømme P. Levels of cognitive and linguistic development in Angelman syndrome: a study of 20 children. Logoped Phoniatr Vocol. 2001;26(1):2–9. | |
Gentile JK, Tan WH, Horowitz LT, et al. A neurodevelopmental survey of Angelman syndrome with genotype-phenotype correlations. J Dev Behav Pediatr. 2010;31(7):592–601. Erratum in: J Dev Behav Pediatr. 2011;32(3):267. | |
Trillingsgaard A, øStergaard JR. Autism in Angelman syndrome: an exploration of comorbidity. Autism. 2004;8(2):163–174. | |
Peters SU, Beaudet AL, Madduri N, Bacino CA. Autism in Angelman syndrome: implications for autism research. Clin Genet. 2004;66(6):530–536. | |
Dykens EM, Sutcliffe JS, Levitt P. Autism and 15q11-q13 disorders: behavioral, genetic, and pathophysiological issues. Ment Retard Dev Disabil Res Rev. 2004;10(4):284–291. | |
Walz NC. Parent report of stereotyped behaviors, social interaction, and developmental disturbances in individuals with Angelman syndrome. J Autism Dev Disord. 2007;37(5):940–947. | |
Bonati MT, Russo S, Finelli P, et al. Evaluation of autism traits in Angelman syndrome: a resource to unfold autism genes. Neurogenetics. 2007;8(3):169–178. | |
Hogart A, Wu D, LaSalle JM, Schanen NC. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol Dis. 2010;38(2):181–191. | |
Peters SU, Horowitz L, Barbieri-Welge R, Taylor JL, Hundley RJ. Longitudinal follow-up of autism spectrum features and sensory behaviors in Angelman syndrome by deletion class. J Child Psychol Psychiatry. 2012;53(2):152–159. | |
Clayton-Smith J, Pembrey ME. Angelman syndrome. J Med Genet. 1992;29(6):412–415. | |
Laan LA, den Boer AT, Hennekam RC, Renier WO, Brouwer OF. Angelman syndrome in adulthood. Am J Med Genet. 1996;66(3):356–360. | |
Clayton-Smith J. Clinical research on Angelman syndrome in the United Kingdom: observations on 82 affected individuals. Am J Med Genet. 1993;46(1):12–15. | |
Buntinx IM, Hennekam RC, Brouwer OF, et al. Clinical profile of Angelman syndrome at different ages. Am J Med Genet. 1995;56(2):176–183. | |
Valente KD, Koiffmann CP, Fridman C, et al. Epilepsy in patients with angelman syndrome caused by deletion of the chromosome 15q11-13. Arch Neurol. 2006;63(1):122–128. | |
Pelc K, Boyd SG, Cheron G, Dan B. Epilepsy in Angelman syndrome. Seizure. 2008;17(3):211–217. | |
Thibert RL, Conant KD, Braun EK, et al. Epilepsy in Angelman syndrome: a questionnaire-based assessment of the natural history and current treatment options. Epilepsia. 2009;50(11):2369–2376. | |
Thibert RL, Larson AM, Hsieh DT, Raby AR, Thiele EA. Neurologic manifestations of Angelman syndrome. Pediatr Neurol. 2013;48(4):271–279. | |
Thibert RL, Pfeifer HH, Larson AM, et al. Low glycemic index treatment for seizures in Angelman syndrome. Epilepsia. 2012;53(9):1498–1502. | |
Boyd SG, Harden A, Patton MA. The EEG in early diagnosis of the Angelman (happy puppet) syndrome. Eur J Pediatr. 1988;147(5):508–513. | |
Dan B, Boyd SG. Angelman syndrome reviewed from a neurophysiological perspective. The UBE3A-GABRB3 hypothesis. Neuropediatrics. 2003;34(4):169–176. | |
Laan LA, Renier WO, Arts WF, et al. Evolution of epilepsy and EEG findings in Angelman syndrome. Epilepsia. 1997;38(2):195–199. | |
Minassian BA, DeLorey TM, Olsen RW, et al. Angelman syndrome: correlations between epilepsy phenotypes and genotypes. Ann Neurol. 1998;43(4):485–493. | |
Valente KD, Andrade JQ, Grossmann RM, et al. Angelman syndrome: difficulties in EEG pattern recognition and possible misinterpretations. Epilepsia. 2003;44(8):1051–1063. | |
Korff CM, Kelley KR, Nordli DR Jr. Notched delta, phenotype, and Angelman syndrome. J Clin Neurophysiol. 2005;22(4):238–243. | |
Laan LA, Vein AA. Angelman syndrome: is there a characteristic EEG? Brain Dev. 2005;27(2):80–87. | |
Vendrame M, Loddenkemper T, Zarowski M, et al. Analysis of EEG patterns and genotypes in patients with Angelman syndrome. Epilepsy Behav. 2012;23(3):261–265. | |
Williams CA. The behavioral phenotype of the Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010;154C(4):432–437. | |
Pelc K, Cheron G, Dan B. Behavior and neuropsychiatric manifestations in Angelman syndrome. Neuropsychiatr Dis Treat. 2008;4(3):577–584. | |
Horsler K, Oliver C. The behavioural phenotype of Angelman syndrome. J Intellect Disabil Res. 2006;50(Pt 1):33–53. | |
Barry RJ, Leitner RP, Clarke AR, Einfeld SL. Behavioral aspects of Angelman syndrome: a case control study. Am J Med Genet A. 2005;132A(1):8–12. Erratum in: Am J Med Genet A. 2005;136(1):111. | |
Summers JA, Allison DB, Lynch PS, Sandler L. Behaviour problems in Angelman syndrome. J Intellect Disabil Res. 1995;39(Pt 2):97–106. | |
Clarke DJ, Marston G. Problem behaviors associated with 15q- Angelman syndrome. Am J Ment Retard. 2000;105(1):25–31. | |
Bruni O, Ferri R, D’Agostino G, Miano S, Roccella M, Elia M. Sleep disturbances in Angelman syndrome: a questionnaire study. Brain Dev. 2004;26(4):233–240. | |
Didden R, Korzilius H, Smits MG, Curfs LM. Sleep problems in individuals with Angelman syndrome. Am J Ment Retard. 2004;109(4):275–284. | |
Walz NC, Beebe D, Byars K. Sleep in individuals with Angelman syndrome: parent perceptions of patterns and problems. Am J Ment Retard. 2005;110(4):243–252. | |
Pelc K, Cheron G, Boyd SG, Dan B. Are there distinctive sleep problems in Angelman syndrome? Sleep Med. 2008;9(4):434–441. | |
Takaesu Y, Komada Y, Inoue Y. Melatonin profile and its relation to circadian rhythm sleep disorders in Angelman syndrome patients. Sleep Med. 2012;13(9):1164–1170. | |
Zhandova IV, Wurtman RJ, Wagstaff J. Effects of a low dose of melatonin on sleep in children with Angelman syndrome. J Pediatr Endocrinol Metab. 1999;12(1):57–67. | |
Braam W, Didden R, Smits MG, Curfs LM. Melatonin for chronic insomnia in Angelman syndrome: a randomized placebo-controlled trial. J Child Neurol. 2008;23(6):649–654. | |
Allen KD, Kuhn BR, DeHaai KA, Wallace DP. Evaluation of a behavioral treatment package to reduce sleep problems in children with Angelman Syndrome. Res Dev Disabil. 2013;34(1):676–686. | |
Conant KD, Thibert RL, Thiele EA. Epilepsy and the sleep-wake patterns found in Angelman syndrome. Epilepsia. 2009;50(11):2497–2500. | |
Tan WH, Bacino CA, Skinner SA, et al. Angelman syndrome: Mutations influence features in early childhood. Am J Med Genet A. 2011;155A(1):81–90. | |
Lossie AC, Whitney MM, Amidon D, et al. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet. 2001;38(12):834–845. | |
Brilliant MH, King R, Francke U, et al. The mouse pink-eyed dilution gene: association with hypopigmentation in Prader-Willi and Angelman syndromes and with human OCA2. Pigment Cell Res. 1994;7(6):398–402. | |
Fridman C, Hosomi N, Varela MC, Souza AH, Fukai K, Koiffmann CP. Angelman syndrome associated with oculocutaneous albinism due to an intragenic deletion of the OCA2. Am J Med Genet A. 2003;119A(2):180–183. | |
Low D, Chen KS. UBE3A regulates MC1R expression: a link to hypopigmentation in Angelman syndrome. Pigment Cell Melanoma Res. 2011;24(5):944–952. | |
Mah ML, Wallace DK, Powell CM. Ophthalmic manifestations of Angelman syndrome. J AAPOS. 2000;4(4):248–249. | |
Schneider BB, Maino DM. Angelman syndrome. J Am Optom Assoc. 1993;64(7):502–506. | |
Michieletto P, Bonanni P, Pensiero S. Ophthalmic findings in Angelman syndrome. J AAPOS. 2011;15(2):158–161. | |
Rufa A, Dotti MT, Orrico A, Battisti C, Carletto F, Federico A. Retinochoroidal atrophy in two adult patients with Angelman syndrome. Am J Med Genet A. 2003;122A(2):155–158. | |
Williams CA. Neurological aspects of the Angelman syndrome. Brain Dev. 2005;27(2):88–94. | |
Laan LA, v Haeringen A, Brouwer OF. Angelman syndrome: a review of clinical and genetic aspects. Clin Neurol Neurosurg. 1999;101(3):161–170. | |
Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2(1):21–32. | |
Chamberlain SJ. RNAs of the human chromosome 15q11-q13 imprinted region. Wiley Interdiscip Rev RNA. 2013;4(2):155–166. | |
Gustin RM, Bichell TJ, Bubser M, et al. Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol Dis. 2010;39(3):283–291. | |
Rougeulle C, Glatt H, Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat Genet. 1997;17(1):14–15. | |
Vu TH, Hoffman AR. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet. 1997;17(1):12–13. | |
Albrecht U, Sutcliffe JS, Cattanach BM, et al. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet. 1997;17(1):75–78. | |
Nakao M, Sutcliffe JS, Durtschi B, Mutirangura A, Ledbetter DH, Beaudet AL. Imprinting analysis of three genes in the Prader-Willi/Angelman region: SNRPN, E6-associated protein, and PAR-2 (D15S225E). Hum Mol Genet. 1994;3(2):309–315. | |
Williams CA, Lossie A, Driscoll D; RC Phillips Unit. Angelman syndrome: mimicking conditions and phenotypes. Am J Med Genet. 2001;101(1):59–64. | |
Tan WH, Bird LM, Thibert RL, Williams CA. If not Angelman, what is it? A review of Angelman-like syndromes. Am J Med Genet. Epub January 29, 2014. | |
Ji Y, Rebert NA, Joslin JM, Higgins MJ, Schultz RA, Nicholls RD. Structure of the highly conserved HERC2 gene and of multiple partially duplicated paralogs in human. Genome Res. 2000;10(3):319–329. | |
Mutirangura A, Greenberg F, Butler MG, et al. Multiplex PCR of three dinucleotide repeats in the Prader-Willi/Angelman critical region (15q11-q13): molecular diagnosis and mechanism of uniparental disomy. Hum Mol Genet. 1993;2(2):143–151. | |
Buiting K, Saitoh S, Gross S, et al. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat Genet. 1995;9(4):395–400. | |
Buiting K, Gross S, Lich C, Gillessen-Kaesbach G, el-Maarri O, Horsthemke B. Epimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet. 2003;72(3):571–577. | |
Horsthemke B, Buiting K. Imprinting defects on human chromosome 15. Cytogenet Genome Res. 2006;113(1–4):292–299. | |
Malzac P, Webber H, Moncla A, et al. Mutation analysis of UBE3A in Angelman syndrome patients. Am J Hum Genet. 1998;62(6):1353–1360. | |
Fang P, Lev-Lehman E, Tsai TF, et al. The spectrum of mutations in UBE3A causing Angelman syndrome. Hum Mol Genet. 1999;8(1):129–135. | |
Van Buggenhout G, Fryns JP. Angelman syndrome (AS, MIM 105830). Eur J Hum Genet. 2009;17(11):1367–1373. | |
Abramowitz LK, Bartolomei MS. Genomic imprinting: recognition and marking of imprinted loci. Curr Opin Genet Dev. 2012;22(2):72–78. | |
Kelsey G, Feil R. New insights into establishment and maintenance of DNA methylation imprints in mammals. Philos Trans R Soc Lond B Biol Sci. 2013;368(1609):20110336. | |
Ishida M, Moore GE. The role of imprinted genes in humans. Mol Aspects Med. 2013;34(4):826–840. | |
Horsthemke B. Structure and function of the human chromosome 15 imprinting center. J Cell Physiol. 1997;173(2):237–241. | |
Lalande M, Calciano MA. Molecular epigenetics of Angelman syndrome. Cell Mol Life Sci. 2007;64(7–8):947–960. | |
Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14(1):10–26. | |
Buiting K, Lich C, Cottrell S, Barnicoat A, Horsthemke B. A 5-kb imprinting center deletion in a family with Angelman syndrome reduces the shortest region of deletion overlap to 880 bp. Hum Genet. 1999;105:665–666. | |
Buiting K, Barnicoat A, Lich C, Pembrey M, Malcolm S, Horsthemke B. Disruption of the bipartite imprinting center in a family with Angelman syndrome. Am J Hum Genet. 2001;68(5):1290–1294. | |
Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153–175. | |
Horsthemke B, Wagstaff J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am J Med Genet A. 2008;146A(16):2041–2052. | |
Johnstone KA, DuBose AJ, Futtner CR, Elmore MD, Brannan CI, Resnick JL. A human imprinting centre demonstrates conserved acquisition but diverged maintenance of imprinting in a mouse model for Angelman syndrome imprinting defects. Hum Mol Genet. 2006;15(3):393–404. | |
Wu MY, Jiang M, Zhai X, Beaudet AL, Wu RC. An unexpected function of the Prader-Willi syndrome imprinting center in maternal imprinting in mice. PLoS One. 2012;7(4):e34348. | |
Rougeulle C, Cardoso C, Fontés M, Colleaux L, Lalande M. An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat Genet. 1998;19(1):15–16. | |
Runte M, Hüttenhofer A, Gross S, Kiefmann M, Horsthemke B, Buiting K. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum Mol Genet. 2001;10(23):2687–2700. | |
Landers M, Bancescu DL, Le Meur E, et al. Regulation of the large (approximately 1000 kb) imprinted murine Ube3a antisense transcript by alternative exons upstream of Snurf/Snrpn. Nucleic Acids Res. 2004;32(11):3480–3492. | |
Meng L, Person RE, Beaudet AL. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum Mol Genet. 2012;21(13):3001–3012. | |
Chamberlain SJ, Brannan CI. The Prader-Willi syndrome imprinting center activates the paternally expressed murine Ube3a antisense transcript but represses paternal Ube3a. Genomics. 2001;73(3):316–322. | |
Yashiro K, Riday TT, Condon KH, et al. Ube3a is required for experience-dependent maturation of the neocortex. Nat Neurosci. 2009;12(6):777–783. | |
Sato M, Stryker MP. Genomic imprinting of experience-dependent cortical plasticity by the ubiquitin ligase gene Ube3a. Proc Natl Acad Sci U S A. 2010;107(12):5611–5616. | |
Nawaz Z, Lonard DM, Smith CL, et al. The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol Cell Biol. 1999;19(2):1182–1189. | |
El Hokayem J, Nawaz Z. E6AP in the Brain: One Protein, Dual Function, Multiple Diseases. Mol Neurobiol. Epub October 5, 2013. | |
Kühnle S, Mothes B, Matentzoglu K, Scheffner M. Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein Arc. Proc Natl Acad Sci U S A. 2013;110(22):8888–8893. | |
Greer PL, Hanayama R, Bloodgood BL, et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell. 2010;140(5):704–716. | |
Margolis SS, Salogiannis J, Lipton DM, et al. EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell. 2010;143(3):442–455. | |
Gregianin E, Vazza G, Scaramel E, et al. A novel SACS mutation results in non-ataxic spastic paraplegia and peripheral neuropathy. Eur J Neurol. 2013;20(11):1486–1491. | |
Kim S, Chahrour M, Ben-Shachar S, Lim J. Ube3a/E6AP is involved in a subset of MeCP2 functions. Biochem Biophys Res Commun. 2013;437(1):67–73. | |
Condon KH, Ho J, Robinson CG, Hanus C, Ehlers MD. The Angelman syndrome protein Ube3a/E6AP is required for Golgi acidification and surface protein sialylation. J Neurosci. 2013;33(9):3799–3814. | |
Mabb AM, Judson MC, Zylka MJ, Philpot BD. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011;34(6):293–303. | |
Jiang YH, Armstrong D, Albrecht U, et al. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21(4):799–811. | |
Miura K, Kishino T, Li E, et al. Neurobehavioral and electroencephalographic abnormalities in Ube3a maternal-deficient mice. Neurobiol Dis. 2002;9(2):149–159. | |
Colas D, Wagstaff J, Fort P, Salvert D, Sarda N. Sleep disturbances in Ube3a maternal-deficient mice modeling Angelman syndrome. Neurobiol Dis. 2005;20(2):471–478. | |
Weeber EJ, Jiang YH, Elgersma Y, et al. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J Neurosci. 2003;23(7):2634–2644. | |
van Woerden GM, Harris KD, Hojjati MR, et al. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci. 2007;10(3):280–282. | |
Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet. 2008;17(1):111–118. | |
Godavarthi SK, Dey P, Maheshwari M, Jana NR. Defective glucocorticoid hormone receptor signaling leads to increased stress and anxiety in a mouse model of Angelman syndrome. Hum Mol Genet. 2012;21(8):1824–1834. | |
Kaphzan H, Hernandez P, Jung JI, et al. Reversal of impaired hippocampal long-term potentiation and contextual fear memory deficits in Angelman syndrome model mice by ErbB inhibitors. Biol Psychiatry. 2012;72(3):182–190. | |
Ramsden SC, Clayton-Smith J, Birch R, Buiting K. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med Genet. 2010;11:70. | |
Bürger J, Kunze J, Sperling K, Reis A. Phenotypic differences in Angelman syndrome patients: imprinting mutations show less frequently microcephaly and hypopigmentation than deletions. Am J Med Genet. 1996;66(2):221–226. | |
Moncla A, Malzac P, Voelckel MA, et al. Phenotype-genotype correlation in 20 deletion and 20 non-deletion Angelman syndrome patients. Eur J Hum Genet. 1999;7(2):131–139. | |
Varela MC, Kok F, Otto PA, Koiffmann CP. Phenotypic variability in Angelman syndrome: comparison among different deletion classes and between deletion and UPD subjects. Eur J Hum Genet. 2004;12(12):987–992. | |
Sahoo T, Peters SU, Madduri NS, et al. Microarray based comparative genomic hybridization testing in deletion bearing patients with Angelman syndrome: genotype-phenotype correlations. J Med Genet. 2006;43(6):512–516. | |
Sahoo T, Bacino CA, German JR, et al. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. 2007;15(9):943–949. | |
Valente KD, Varela MC, Koiffmann CP, et al. Angelman syndrome caused by deletion: a genotype-phenotype correlation determined by breakpoint. Epilepsy Res. 2013;105(1–2):234–239. | |
Dan B, Boyd SG. Angelman syndrome reviewed from a neurophysiological perspective. The UBE3A-GABRB3 hypothesis. Neuropediatrics. 2003;34(4):169–176. | |
Saitoh S, Oiso N, Wada T, Narazaki O, Fukai K. Oculocutaneous albinism type 2 with a OCA2 missense mutation in a patient with Angelman syndrome. J Med Genet. 2000;37(5):392–394. | |
Boyes L, Wallace AJ, Krajewska-Walasek M, Chrzanowska KH, Clayton-Smith J, Ramsden S. Detection of a deletion of exons 8–16 of the UBE3A gene in familial Angelman syndrome using a semi-quantitative dosage PCR based assay. Eur J Med Genet. 2006;49(6):472–480. | |
Peters SU, Bird LM, Kimonis V, et al. Double-blind therapeutic trial in Angelman syndrome using betaine and folic acid. Am J Med Genet A. 2010;152A(8):1994–2001. | |
Bird LM, Tan WH, Bacino CA, et al. A therapeutic trial of pro-methylation dietary supplements in Angelman syndrome. Am J Med Genet A. 2011;155A(12):2956–2963. | |
Wen-Hann Tan. A trial of levodopa in Angelman syndrome. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2011 [updated November 18, 2013]. Available from: http://clinicaltrials.gov/show/NCT01281475. NLM identifier: NCT01281475. Accessed February 23, 2014. | |
Picconi B, Centonze D, Rossi S, Bernardi G, Calabresi P. Therapeutic doses of L-dopa reverse hypersensitivity of corticostriatal D2-dopamine receptors and glutamatergic overactivity in experimental parkinsonism. Brain. 2004;127(Pt 7):1661–1669. | |
Brown AM, Deutch AY, Colbran RJ. Dopamine depletion alters phosphorylation of striatal proteins in a model of Parkinsonism. Eur J Neurosci. 2005;22(1):247–256. | |
Mulherkar SA, Jana NR. Loss of dopaminergic neurons and resulting behavioural deficits in mouse model of Angelman syndrome. Neurobiol Dis. 2010;40(3):586–592. | |
Harbord M. Levodopa responsive Parkinsonism in adults with Angelman Syndrome. J Clin Neuroscience. 2001;8(5):421–422. | |
University of South Florida. Minocycline in the treatment of Angelman syndrome. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2012 [updated June 27, 2013]. Available from: http://clinicaltrials.gov/show/NCT01531582. NLM identifier: NCT01531582. Accessed February 23, 2014. | |
Dan B. Angelman syndrome: current understanding and research prospects. Epilepsia. 2009;50(11):2331–2339. | |
Jana NR. Understanding the pathogenesis of Angelman syndrome through animal models. Neural Plast. 2012;2012:710943. | |
Cheron G, Servais L, Wagstaff J, Dan B. Fast cerebellar oscillation associated with ataxia in a mouse model of Angelman syndrome. Neuroscience. 2005;130(3):631–637. | |
Riday TT, Dankoski EC, Krouse MC, et al. Pathway-specific dopaminergic deficits in a mouse model of Angelman syndrome. J Clin Invest. 2012;122(12):4544–4554. | |
Huang HS, Burns AJ, Nonneman RJ, et al. Behavioral deficits in an Angelman syndrome model: effects of genetic background and age. Behav Brain Res. 2013;243:79–90. | |
Daily JL, Nash K, Jinwal U, et al. Adeno-associated virus-mediated rescue of the cognitive defects in a mouse model for Angelman syndrome. PLoS One. 2011;6(12):e27221. | |
Huang HS, Allen JA, Mabb AM, et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2011;481(7380):185–189. | |
Powell WT, Coulson RL, Gonzales ML, et al. R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proc Natl Acad Sci U S A. 2013;110(34):13938–13943. | |
Cao C, Rioult-Pedotti MS, Migani P, et al. Impairment of TrkB-PSD-95 signaling in Angelman syndrome. PLoS Biol. 2013;11(2):e1001478. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.