Back to Journals » Drug Design, Development and Therapy » Volume 11
Analysis of pramipexole dose–response relationships in Parkinson's disease
Authors Wang Y, Sun SG, Zhu SQ, Liu CF, Liu YM, Di Q, Shang HF, Ren Y, Xiang W, Chen SD
Received 13 May 2016
Accepted for publication 15 October 2016
Published 23 December 2016 Volume 2017:11 Pages 83—89
DOI https://doi.org/10.2147/DDDT.S112723
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Georgios Panos
Ying Wang,1 Sheng-Gang Sun,2 Sui-Qiang Zhu,3 Chun-Feng Liu,4 Yi-Ming Liu,5 Qing Di,6 Hui-Fang Shang,7 Yan Ren,8 Wei Xiang,9 Sheng-Di Chen1
1Department of Neurology, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, 2Department of Neurology, Union Hospital Affiliated to Tongji Medical College of Huazhong University of Science and Technology, Wuhan, 3Department of Neurology, Tongji Hospital Affiliated to Tongji Medical College of Huazhong University of Science and Technology, Wuhan, 4Department of Neurology, The Second Affiliated Hospital of Soochow University, Suzhou, 5Department of Neurology, Qilu Hospital Affiliated to Shandong University, Jinan, 6Department of Neurology, Nanjing Brain Hospital, Nanjing, 7Department of Neurology, West China Hospital Affiliated to Sichuan University, Chengdu, 8Department of Neurology, First Affiliated Hospital of China Medical University, Shenyang, 9Medical Department, Boehringer Ingelheim (China) Investment Co., Ltd., Shanghai, People’s Republic of China
Background: Pramipexole (PPX), a non-ergot dopamine receptor agonist, is a first-line treatment for Parkinson’s disease (PD). A critical dose level above which a better benefit-to-harm ratio exists has not been examined.
Methods: Chinese PD patients (n=464) were retrospectively analyzed by PPX maintenance dose, PD stage, combined levodopa dose, and baseline tremor contribution. The sum score of Baseline Activities of Daily Living (part II) and Motor Examination (III) of the Unified Parkinson’s Disease Rating Scale (UPDRS II+III) was used as a covariate for final score adjustment.
Results: Sustained-release (SR) and immediate-release (IR) PPX showed similar efficacy based on score changes at 18 weeks, with comparable tolerability. Approximately two-third of patients received PPX at ≥1.5 mg/d, and one fourth of patients had ≥20% tremor contribution to UPDRS II+III. After treatment, patients receiving PPX ≥1.5 mg/d showed better improvement in UPDRS II+III scores (P=0.0025), with similar trends with the IR and SR formulations. Patients with ≥20% tremor contribution showed better improvement in UPDRS II+III scores (P=0.0017). No differences were seen based on PD stage or combined levodopa dose. The overall proportions of adverse events (AEs) were similar. More patients discontinued because of intolerable side effects, and more investigator-defined drug-related AEs were recorded in the <1.5 mg/d subgroup.
Conclusion: UPDRS II+III improvement was better with PPX ≥1.5 than with <1.5 mg/d in Chinese PD patients after 18 weeks of treatment, with similar trends seen with IR and SR formulations. The frequency of AEs in PPX ≥1.5 and <1.5 mg/d subgroups was similar.
Keywords: Parkinson’s disease, pramipexole, dose dependent, retrospective
Introduction
Pramipexole (PPX), a non-ergot dopamine receptor agonist (DA), is prescribed as initial monotherapy for early Parkinson’s disease (PD) and adjuvant treatment for advanced PD. PPX has neuroprotective effects in vitro and in vivo, which manifest, especially in early PD, as delayed development of levodopa-induced motor complications.1–3 Researchers postulated that DAs with a longer half-life than levodopa provide continuous activation of presynaptic dopaminergic receptors and/or intracellular kinase, which in turn reduces dopamine turnover and apoptosis and consequently the risk of motor complications.4 Initial PPX therapy reduced the risks of motor complications compared with levodopa5 and indicated a slower rate of dopamine neuron loss reflected by a surrogate biomarker.2 In addition, PPX can not only control motor symptoms and delay motor complications but also improve depressive symptoms in patients with PD.6,7
Currently, immediate-release (IR) PPX is administered orally three times daily. Sustained-release (SR) formulations of PPX, the same formulation with extended-release PPX, showed similar pharmacokinetics and tolerability as equivalent dose IR PPX.8,9 In clinical trials, PPX SR has demonstrated similar therapeutic efficacy and safety profile as PPX IR, both in early and advanced PD.9–16 In patients with previous PPX IR treatment, the success rates of switching from IR to SR and pseudo SR to SR were 86.2% and 83.8%, respectively.15 Moreover, 4 and 8 weeks after overnight switching from the IR to the SR formulation, patients’ adherence and motor symptoms (Unified Parkinson’s Disease Rating Scale [UPDRS] part III) improved without severe adverse effects; such improvement in efficacy might be attributable to a significantly higher adherence to the SR formulation than to the triple-dose formulation.17,18
While initiating PPX treatment, doses should be increased gradually from a starting dose of 0.375 mg/d and then increased every 5–7 days.19 Provided patients do not experience intolerable or undesirable side effects, the doses should be titrated to achieve a maximal therapeutic effect. Individual doses should range between 0.375 and 4.5 mg/d.19 Studies have shown that patients already taking PPX tablets may be switched to PPX SR tablets overnight at the same daily dose.20 During dose escalation in pivotal studies, both in early and advanced PD, efficacy was observed starting at a daily dose of 1.5 mg. As a preceding dose-escalation phase usually aids in achieving maximally tolerated doses in PD patients, the dose-dependent effects of PPX have not been fully explored. It is unclear whether the differences in different tolerated doses between patients indicate dose-dependent differences in benefit-to-risk ratios and/or risk profiles. In the present study, we retrospectively analyzed the raw data from a randomized clinical trial (ClinicalTrials.gov Identifier: NCT01191944) conducted in Chinese patients with early and advanced PD. In this study, patients received different dosages of PPX in maintenance period (0.375, 0.75, 1.5, 2.25, 3.0, 3.75, or 4.5 mg/d), and patient number is not enough to conduct statistical analysis between each dosage group. Patients received PPX at doses (mean dose in mg/d: SR, 1.5 and IR, 1.6) lower than those recommended by the Chinese PD consensus (usual PPX effective clinical dose: 1.5–2.25 mg/d, up to a maximum dosage of 4.5 mg/d).21 Accordingly, 1.5 mg/d was regarded as the critical dose level in this study to determine whether differences in the efficacy and tolerability of PPX in the Chinese population are dose dependent.
Methods
Patients and study design (heterogeneous participants)
Data from all patients included in the aforementioned Chinese study were analyzed in this retrospective analysis.16 Briefly, Chinese patients diagnosed with an idiopathic PD of >2 years’ duration were enrolled, including those with early- or advanced-stage PD and those with or without motor fluctuation. In addition, patients treated with stable doses of common anti-PD medications for >4 weeks prior to enrollment were included. After enrollment, the patients were randomized to receive PPX IR or PPX SR for 18 weeks, with no changes to the doses of the other combination anti-PD drugs. The optimal doses of PPX were up-titrated in the initial 7 weeks and maintained for 11 weeks thereafter. After completion of the study, the study drug was gradually withdrawn over 1 week. IR and SR PPX were administered at doses ranging between 0.375 and 4.5 mg/d. The study received ethical approval by the Ethics Committee of Ruijin Hospital, Shanghai, People’s Republic of China, and written informed consent was obtained for experimentation with human subjects.
Clinical assessment
Patients’ and treatment responses were assessed as previously described.16 Briefly, patients were assessed using the modified Hoehn and Yahr Scale. PD symptoms and primary treatment responses were assessed using UPDRS parts II and III. A treatment response was defined as a ≥20% decrease in UPDRS scores from the baseline. In addition, patients were evaluated using the Mini-Mental State Examination, and subjects’ self-reported likelihood of dosing was assessed using the Epworth Sleepiness Scale. The general status of patients was assessed using the Clinical Global Impressions of Improvement and the Patient Global Impression of Improvement scales.
Adverse events
Adverse events (AEs) were recorded as previously described.16 In brief, the occurrence, frequency, and severity of AEs were recorded throughout the trial. On the basis of severity, AEs were classified as mild if they were easily tolerated, moderate if they interfered with daily activities, and severe if these prevented patients from performing their daily activities or worse. AEs were considered serious if they resulted in death, were immediately life threatening, resulted in persistent or significant disability/incapacity, required or prolonged patient hospitalization, were a congenital anomaly/birth defect, or were deemed serious for any other reason.
Statistical analysis
Sample size estimation and different data sets have been described previously.16 The full analysis set (FAS) included patients who received at least 1 dose of the study drug and provided both a baseline and a post-baseline assessment of primary endpoints. Baseline refers to the last recorded measurements before administration of the study drug. Efficacy was analyzed using the FAS, and the last observation carried forward approach was used for missing data during follow-up. Because both PPX SR and IR improved symptoms in patients with early or advanced PD and showed similar efficacy and safety in this Chinese study, all patients were categorized into 2 subgroups based on the PPX maintenance dose (PPX <1.5 or ≥1.5 mg/d), PD stage (early or advanced), combined levodopa dose at baseline (low dose, 0–<400 mg/d or high dose, ≥400 mg/d), and the level of contribution of tremor to the sum score of Activities of Daily Living (part II) and Motor Examination (III) of the UPDRS score (UPDRS II+III score) at baseline (tremor scores/UPDRS II+III scores, <20% or ≥20%) for an exploratory analysis of the effects of these variables on efficacy. An analysis of covariance model was used to evaluate the improvement in UPDRS II+III scores in each of these subgroups based on the FAS. Formulation (IR or SR) and center were included as fixed effects, whereas baseline UPDRS II+III total scores formed a linear covariate. The incidence of AEs was presented for all treated patients who received PPX at a dose of ≥1.5 or <1.5 mg/d.
Results
Baseline characteristics
Patients who showed a comparable use of the 2 PPX formulations were regrouped by PPX dose, PD stage, levodopa dose, and contribution of tremor to UPDRS at baseline. The patients were almost equally distributed by disease stage (early and advanced) and levodopa dose (0–400 and ≥400 mg/d). Approximately two-third of patients were up-titrated to ≥1.5 mg/d in this trial. Approximately one-fourth of patients had a tremor contribution of ≥20% to the UPDRS II+III scores at baseline (Table 1).
 | Table 1 Patients’ distribution in subgroups (FAS) |
Dose-related efficacy
After 18 weeks of PPX treatment, patients receiving PPX ≥1.5 mg/d showed a greater reduction in UPDRS II+III scores compared with those receiving PPX <1.5 mg/d (14.83 vs 10.69, respectively). The adjusted difference between the PPX ≥1.5 and PPX <1.5 mg/d subgroups was 3.21 (95% CI [confidence interval], 1.14–5.28; P=0.0025; Figure 1 and Table 2).
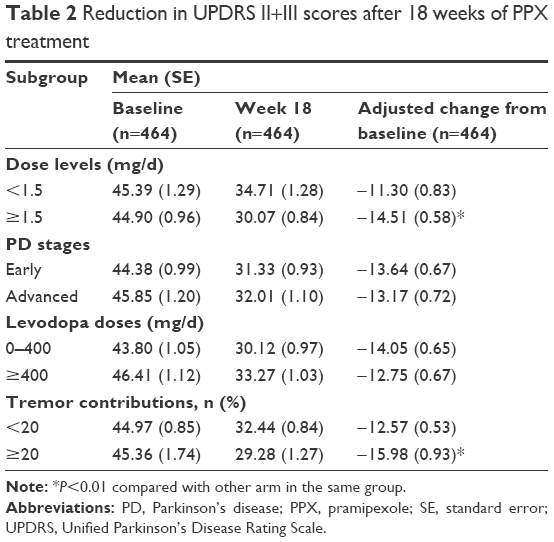 | Table 2 Reduction in UPDRS II+III scores after 18 weeks of PPX treatment |
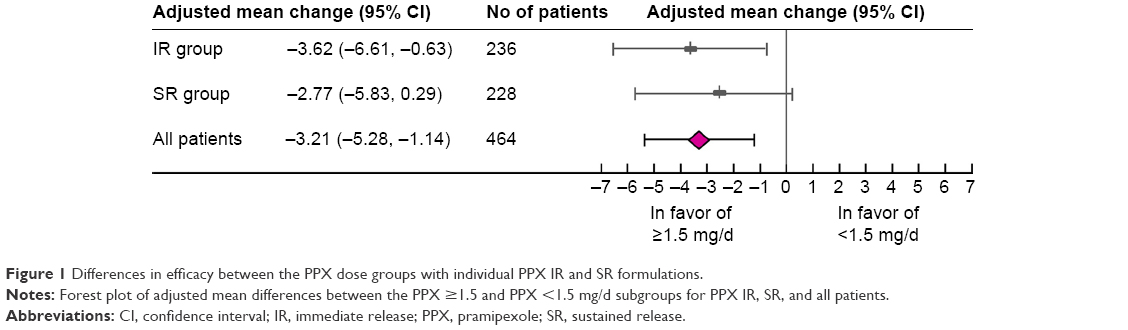 | Figure 1 Differences in efficacy between the PPX dose groups with individual PPX IR and SR formulations. |
In the individual PPX IR and SR groups, efficacy differences between the PPX ≥1.5 and <1.5 mg/d subgroups were comparable (3.62 vs 2.77, P=0.2924; Figure 1). These differences were greater than the minimal clinically important change (MCIC) of 2.5, thus indicating their clinical significance.22 The more serious a symptom was at baseline, the greater the improvement observed after 18 weeks of PPX treatment. Note the more prominent slope in the PPX ≥1.5 mg/d subgroup than in the PPX <1.5 mg/d subgroup in Figure 2 (slope: −0.3223 vs −0.3021). Therefore, patients in the PPX ≥1.5 mg/d group would get more improvement in UPDRS II+III scores than that in the PPX <1.5 mg/d group consistently across different baseline UPDRS II+III scores.
 | Figure 2 Differences in efficacy slopes between the PPX dose groups. |
In patients with a tremor contribution of ≥20% (tremor contribution = tremor scores [sum of 16th, 20th, and 21st items of UPDRS]/UPDRS II+III scores), PPX treatment resulted in greater improvements in UPDRS II+III scores; after adjustment for baseline UPDRS II+III scores, the average difference was 3.42 (95% CI, 1.29–5.55; P=0.0017; Table 2). However, patients with early and advanced PD responded similar to PPX (P=0.6580) as did those who had received levodopa at doses of 0–400 and ≥400 mg/d at baseline (P=0.1786).
Table 3 shows a subgroup analysis of improvements in major motor function characteristics, including bradykinesia (sum of 23rd, 24th, 25th, 26th, and 31st UPDRS items), rigidity (22nd item), postural instability gait difficulty (PIGD; sum of 13th, 14th, 15th, 29th, and 30th items), and tremors (sum of 16th, 20th, and 21st items). Score reduction for these 4 core motor symptoms and the corresponding proportions of patients who showed ≥20% improvement was greater in the PPX ≥1.5 mg/d subgroup than in the PPX <1.5 mg/d subgroup (Table 3). However, differences of <1.5 points were observed between the early and advanced PD subgroups and between the 0–400 and ≥400 mg/d levodopa dose groups, which were far below the MCIC of 2.5. Patients with dominant tremors (tremor contribution ≥20%) tended to achieve greater score reductions compared with those with nondominant tremors; however, score reductions for bradykinesia, rigidity, and PIGD were comparable between these patient types.
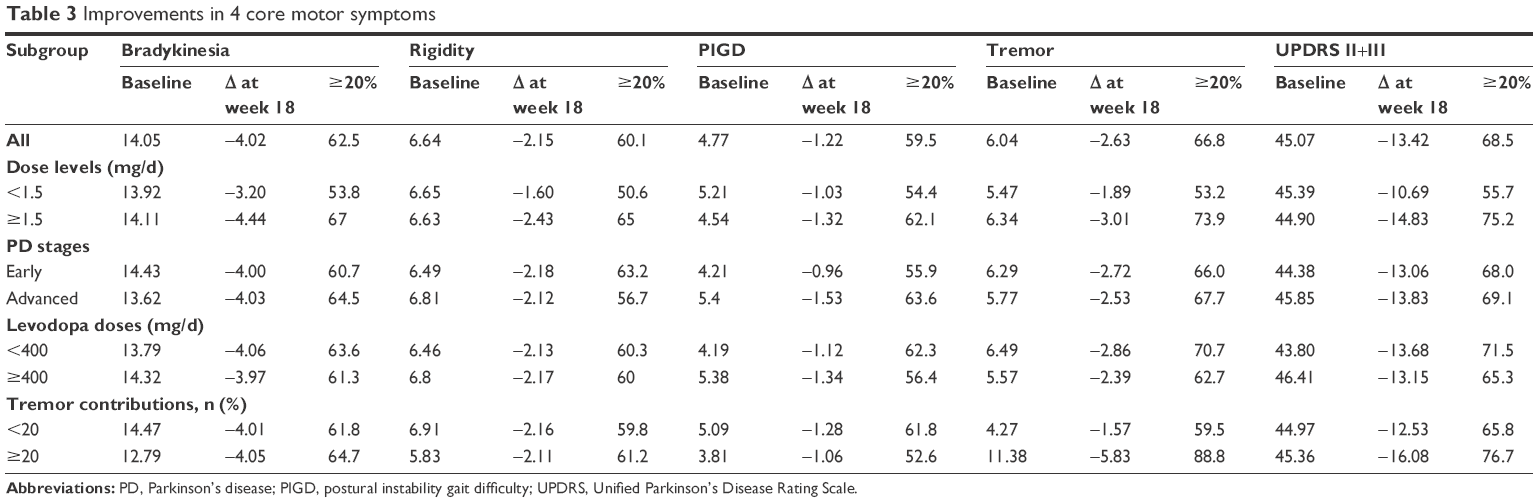 | Table 3 Improvements in 4 core motor symptoms |
Dose-related AEs
The incidence of AEs was lower in the PPX ≥1.5 mg/d subgroup (68.6%) than in the PPX <1.5 mg/d subgroup (75.4%). The 3 most common AEs were somnolence (18.6% vs 18.0%), dizziness (16.2% vs 11.1%), and nausea (10.8% vs 6.2%). Withdrawal due to AEs (12.6% vs 0.7%) and the incidence of investigator-defined drug-related AEs (61.1% vs 46.1%) were higher in the PPX <1.5 mg/d subgroup than in the PPX ≥1.5 mg/d subgroup (Table 4).
 | Table 4 Incidence of AEs for different PPX dose groups |
Discussion
The optimal dose of PPX for individual PD patients should be titrated to achieve a balance between efficacy and tolerability. A Japanese study reported that the increase in plasma concentration of PPX was proportional to the gradual increase in dose; however, current clinical data are insufficient to confirm an improvement in efficacy and AEs with increasing PPX doses.9 Previous studies have shown that the efficacy and safety profiles of PPX SR and IR were comparable.9,16 Therefore, we retrospectively analyzed the pooled raw data of PPX IR and SR groups from the Chinese study. Results of this analysis provide evidence for the existence of a critical dose level of PPX for PD patients, which in this case was 1.5 mg/d. Patients whose PPX doses could be titrated to ≥1.5 mg/d showed greater improvements in UPDRS II+III scores than those who received PPX at <1.5 mg/d at the end of the 18-week treatment period. In addition, patients in the PPX ≥1.5 mg/d subgroup reported fewer AEs, except for gastrointestinal disorders. Moreover, patients receiving PPX ≥1.5 mg/d showed greater improvements in motor function, particularly bradykinesia, rigidity, PIGD, and tremors. Both PPX IR and SR showed comparable efficacy improvement in terms of UPDRS II+III score reduction with increasing doses without any statistically significant differences. In the individual PPX IR and SR groups, and the IR + SR pooled groups, the differences between the ≥1.5 and <1.5 mg/d subgroups reached the MCIC of 2.5, which indicated superior efficacy of PPX at ≥1.5 mg/d.
In this study, tremor control contributed largely to the total UPDRS II+III score reduction. A large series of 100 patients with pathologically proven PD revealed tremor in 69% of patients at disease onset and in 75% during the disease course; in 9% of patients, tremors were lost late during the disease course.23 Previous data have demonstrated that PPX demonstrates favorable efficacy in tremor control, even for patients with refractory tremors.24 In this analysis, we observed that PPX improved the 4 core motor symptoms of PD, and this effect was more evident with increasing doses. Moreover, for patients with dominant tremor symptoms, improvements in UPDRS II+III and tremor scores were even more evident, which is consistent with previous data showing the superiority of PPX in improving tremor.
It remains unclear whether PD patients receiving PPX at different dose levels manifest different AE profiles. We observed a trend toward a marginally higher incidence of AEs in the <1.5 mg/d subgroup than in the ≥1.5 mg/d subgroup. However, these data are insufficient to draw concrete conclusions. Moreover, patients in the <1.5 mg/d subgroup had received low PPX doses, which might be related to poor tolerability; therefore, the incidence of AEs was higher in this subgroup than in the ≥1.5 mg/d subgroup. Accordingly, patients with better tolerability showed greater improvements in motor symptoms with PPX ≥1.5 mg/d without any significant increase in the incidence of AEs.
The retrospective analysis is a study limitation, and it is exploratory in nature. Furthermore, the comparisons were not based on a randomized sample although the analysis was adjusted for several important factors.
Conclusion
For PD patients receiving PPX treatment for 18 weeks, PPX at both dose levels can improve motor function and daily activities with comparable AE rates. However, compared with PPX <1.5 mg/d, administration of PPX ≥1.5 mg/d can result in further clinically significant efficacy improvements. Both IR and SR formulations displayed similar trends. Patients with dominant tremors tended to achieve greater improvements after PPX administration. PPX treatment can effectively improve patients’ symptoms regardless of the PD stage or dose of combined levodopa at baseline.
Acknowledgments
We thank Fei Liu and Paul Liu from Elsevier for linguistic assistance during the preparation of this manuscript, which was contracted and compensated by Boehringer Ingelheim (China) Investment Co., Ltd. Editorial assistance was provided by Cactus Communications and was funded by Boehringer Ingelheim. Boehringer Ingelheim (China) Investment Co., Ltd. was given the opportunity to review the manuscript content for medical and scientific accuracy and intellectual property considerations. In addition, we thank the Pramipexole SR Study Team for their help, and we thank Gang Cheng’s help.
Disclosure
Ying Wang has participated in clinical trials sponsored by GSK, Eisai, Lundbeck, and Novartis. Sheng-Gang Sun has participated in clinical trials sponsored by Novartis, Servier, Eisai, GSK, and Lundbeck. Sui-Qiang Zhu has participated in a clinical trial sponsored by UCB. Chun-Feng Liu has participated in clinical trials sponsored by Pfizer, UCB, and GSK. Yi-Ming Liu has participated in a clinical trial sponsored by UCB. Qing Di has participated in a clinical trial sponsored by Pfizer. Hui-Fang Shang has participated in clinical trials sponsored by GSK and UCB. Yan Ren reports no conflicts of interest in this work. Sheng-Di Chen has participated in clinical trials sponsored by Novartis, Lundbeck, Eisai, and Xian Janssen. All aforementioned authors served as investigators in this retrospective analysis sponsored by Boehringer Ingelheim (China) Investment Co., Ltd. Wei Xiang is employee of Boehringer Ingelheim (China) Investment Co., Ltd. The authors report no other conflicts of interest in this work.
References
Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA. 2000;284(15):1931–1938. | ||
Parkinson Study Group. Dopamine transporter brain imaging to assess the effects of pramipexole vs levodopa on Parkinson disease progression. JAMA. 2002;287(13):1653–1661. | ||
Albrecht S, Buerger E. Potential neuroprotection mechanisms in PD: focus on dopamine agonist pramipexole. Curr Med Res Opin. 2009;25(12):2977–2987. | ||
Antonini A, Tolosa E, Mizuno Y, Yamamoto M, Poewe WH. A reassessment of risks and benefits of dopamine agonists in Parkinson’s disease. Lancet Neurol. 2009;8(10):929–937. | ||
Holloway RG, Shoulson I, Fahn S, et al. Pramipexole vs levodopa as initial treatment for Parkinson disease: a 4-year randomized controlled trial. Arch Neurol. 2004;61(7):1044–1053. | ||
Costa FH, Rosso AL, Maultasch H, Nicaretta DH, Vincent MB. Depression in Parkinson’s disease: diagnosis and treatment. Arq Neuropsiquiatr. 2012;70(8):617–620. | ||
Cusin C, Iovieno N, Iosifescu DV, et al. A randomized, double-blind, placebo-controlled trial of pramipexole augmentation in treatment-resistant major depressive disorder. J Clin Psychiatry. 2013;74(7):e636–e641. | ||
Jenner P, Konen-Bergmann M, Schepers C, Haertter S. Pharmacokinetics of a once-daily extended-release formulation of pramipexole in healthy male volunteers: three studies. Clin Ther. 2009;31(11):2698–2711. | ||
Mizuno Y, Yamamoto M, Kuno S, et al. Efficacy and safety of extended- versus immediate-release pramipexole in Japanese patients with advanced and L-dopa-undertreated Parkinson disease: a double-blind, randomized trial. Clin Neuropharmacol. 2012;35(4):174–181. | ||
Safety and efficacy of pramipexole in early Parkinson disease. A randomized dose-ranging study. Parkinson Study Group. JAMA. 1997;278(2):125–130. | ||
Stocchi F, Hersh BP, Scott BL, Nausieda PA, Giorgi L; Ease-PD Monotherapy Study Investigators. Ropinirole 24-hour prolonged release and ropinirole immediate release in early Parkinson’s disease: a randomized, double-blind, non-inferiority crossover study. Curr Med Res Opin. 2008;24(10):2883–2895. | ||
Hauser RA, Schapira AH, Rascol O, et al. Randomized, double-blind, multicenter evaluation of pramipexole extended release once daily in early Parkinson’s disease. Mov Disord. 2010;25(15):2542–2549. | ||
Poewe W, Rascol O, Barone P, et al. Extended-release pramipexole in early Parkinson disease: a 33-week randomized controlled trial. Neurology. 2011;77(8):759–766. | ||
Schapira AH, Barone P, Hauser RA, et al; Pramipexole ER Studies Group. Extended-release pramipexole in advanced Parkinson disease: a randomized controlled trial. Neurology. 2011;77(8):767–774. | ||
Schapira AH, Barone P, Hauser RA, et al. Success rate, efficacy, and safety/tolerability of overnight switching from immediate- to extended-release pramipexole in advanced Parkinson’s disease. Eur J Neurol. 2013;20(1):180–187. | ||
Wang Y, Sun S, Zhu S, et al. The efficacy and safety of pramipexole ER versus IR in Chinese patients with Parkinson’s disease: a randomized, double-blind, double-dummy, parallel-group study. Transl Neurodegener. 2014;3:11. | ||
Takanashi M, Shimo Y, Hatano T, Oyama G, Hattori N. Efficacy and safety of a once-daily extended-release formulation of pramipexole switched from an immediate-release formulation in patients with advanced Parkinson’s disease: results from an open-label study. Drug Res (Stuttg). 2013;63(12):639–643. | ||
Grosset D, Antonini A, Canesi M, et al. Adherence to antiparkinson medication in a multicenter European study. Mov Disord. 2009;24(6):826–832. | ||
Antonini A, Barone P, Ceravolo R, Fabbrini G, Tinazzi M, Abbruzzese G. Role of pramipexole in the management of Parkinson’s disease. CNS Drugs. 2010;24(10):829–841. | ||
Rascol OBP, Debieuvre C, et al. Easy switching from immediate- to extended-release pramipexole in early Parkinson’s disease at the same daily dosage [abstract no Th-255]. Mov Disord. 2009;24 (Suppl 1):S362. | ||
Chen SD, Chan P, Sun SG, et al. The recommendations of Chinese Parkinson’s disease and movement disorder society consensus on therapeutic management of Parkinson’s disease. Transl Neurodegener. 2016;5:12. | ||
Hauser RA, Auinger P; Parkinson Study Group. Determination of minimal clinically important change in early and advanced Parkinson’s disease. Mov Disord. 2011;26(5):813–818. | ||
Ghika A, Kyrozis A, Potagas C, Louis ED. Motor and non-motor features: differences between patients with isolated essential tremor and patients with both essential tremor and Parkinson’s disease. Tremor Other Hyperkinet Mov (N Y). 2015;5:335. | ||
Navan P, Findley LJ, Jeffs JA, Pearce RK, Bain PG. Randomized, double-blind, 3-month parallel study of the effects of pramipexole, pergolide, and placebo on Parkinsonian tremor. Mov Disord. 2003;18(11):1324–1331. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.