Back to Journals » Clinical Ophthalmology » Volume 16
Analysis of Molecular Genetic Testing Referrals for Inherited Retinal Dystrophies in a Quebec Tertiary Care Center Over a Decade
Authors Lachance A ![]() , Hébert M
, Hébert M ![]() , Hébert M, Salesse C, Bourgault S, Dirani A
, Hébert M, Salesse C, Bourgault S, Dirani A ![]()
Received 2 November 2021
Accepted for publication 14 January 2022
Published 2 February 2022 Volume 2022:16 Pages 239—244
DOI https://doi.org/10.2147/OPTH.S346103
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Alexandre Lachance,1,2 Mélanie Hébert,1,2 Marc Hébert,1– 3 Christian Salesse,1,2 Serge Bourgault,1,2 Ali Dirani1,2
1Faculté de médecine, Université Laval, Québec, QC, Canada; 2Département d’ophtalmologie et d’oto-rhino-laryngologie – chirurgie cervico-faciale, Centre Universitaire d’Ophtalmologie, Hôpital du Saint-Sacrement, CHU de Québec - Université Laval, Québec, QC, Canada; 3CERVO Brain Research Centre, Quebec, QC, Canada
Correspondence: Ali Dirani
Département d’ophtalmologie et d’oto-rhino-laryngologie – chirurgie cervico-faciale, Centre Universitaire d’Ophtalmologie, Hôpital du Saint-Sacrement, CHU de Québec - Université Laval, 1050 Chemin Ste-Foy, Québec, G1S 4L8, QC, Canada, Email [email protected]
Introduction
Inherited retinal dystrophy (IRD) is a subset of degenerative diseases of the retina which have marked clinical, molecular, and genetic heterogeneity. Clinical presentation can be variable and include night or color blindness, tunnel vision, visual field defects, and subsequent progression to complete blindness. Pathogenic variants in more than 250 genes can result in IRD.1
On October 15, 2020, Health Canada approved the first gene therapy (Luxturna (Voretigene Neparvovec-rzyl) Spark Therapeutics, Inc., Philadelphia, USA) for patients with biallelic RPE65 mutations. This approval marked the dawn of a new era of treatments for IRD previously thought to be untreatable.
Despite few available treatments for IRD, molecular genetic testing (MGT) remains crucial. Genetic diagnosis is not only important to improve genetic counselling to families and to advise on prognosis, but it also allows to identify patients that might be candidates for new treatments such as gene therapy. Moreover, gene therapy has grown rapidly in recent years: more than 36 clinical trials related to gene therapy to treat IRD are underway.2 The process for obtaining MGT must be efficient enough to facilitate access to these new innovative treatments for patients.
Methods
To evaluate this, we analyzed MGT referrals for patients with a suspected diagnosis of IRD in a tertiary care center in Quebec between 2011 and 2020. Given an average prevalence of 1 IRD case in 2000 people, we estimate an average 1000 cases of IRD in the Quebec regions served by our hospital (ie, Bas-St-Laurent, Saguenay-Lac-St-Jean, Capitale-Nationale, Côte-Nord, Nord-du-Quebec, Gaspésie, and Chaudière-Appalaches).
We reviewed the electronic records of all eyes scheduled for an electroretinogram (ERG) during the study period and all suspected cases of IRD were identified. All clinically suspected IRD patients had ERG, and only patients with abnormal ERG were included in the analysis. MGT was considered conclusive when a minimum of one pathogenic or likely pathogenic variant was identified, and inconclusive when no mutations or only variants of uncertain significance were found. The study followed the tenets of the Declaration of Helsinki, and ethics approval was obtained from the Institutional Review Board of the Centre Hospitalier Universitaire (CHU) de Québec-Université Laval (2022-6022).
Results
We identified 324 patients with suspected IRD diagnosis based on available clinical records (clinical diagnosis, clinical history, fundus changes, optical coherence tomography imaging, ERG, visual acuity, visual fields, and family history). The types of IRD diagnosis are shown in Figure 1.
 |
Figure 1 (A) Types of suspected IRD diagnoses by clinical presentation and ERG results; (B) number of MGT referrals to genetics and number of patients seen by genetics with or without MGT performed per year. Abbreviations: IRD, inherited retinal dystrophy; MGT, molecular genetic testing. |
A total of 67 patients (mean age 39 ± 16 (range: 10–83) years) (21%) were referred to a genetic specialist by an ophthalmologist; only 19 out of 67 patients (28%) received a MGT result by the end of 2020. No referrals had been made by primary health-care physicians. The number of patients referred per year increased steadily during the study period (from 0 to 9 referrals per year between 2011 and 2020), but the number of patients seen by geneticist for MGT has been stable over the years (Figure 1). The average time between MGT referral and MGT results (or the end of 2020 if the patient was not seen by genetics yet) was 35 ± 23 (range: 1–87) months. This delay is also likely underestimated because most patients (n = 48, 72%) still did not obtain MGT results by the end of the study period. However, for those who got MGT results, 14/19 patients (74%) had a conclusive result with pathogenic or likely pathogenic gene mutations (Figure 2). This is similar to another large worldwide study that showed conclusive results in 76% (n = 760) patients.3 The most common mutated gene in our cohort was USH2A (Figure 2). The pathogenic or likely pathogenic mutations found in this gene were: heterozygous variants c.5329C>T, p.Arg1777Trp and c.7311+1G>T, splice site; heterozygous variant c.148083C>T, p.Arg4935*.
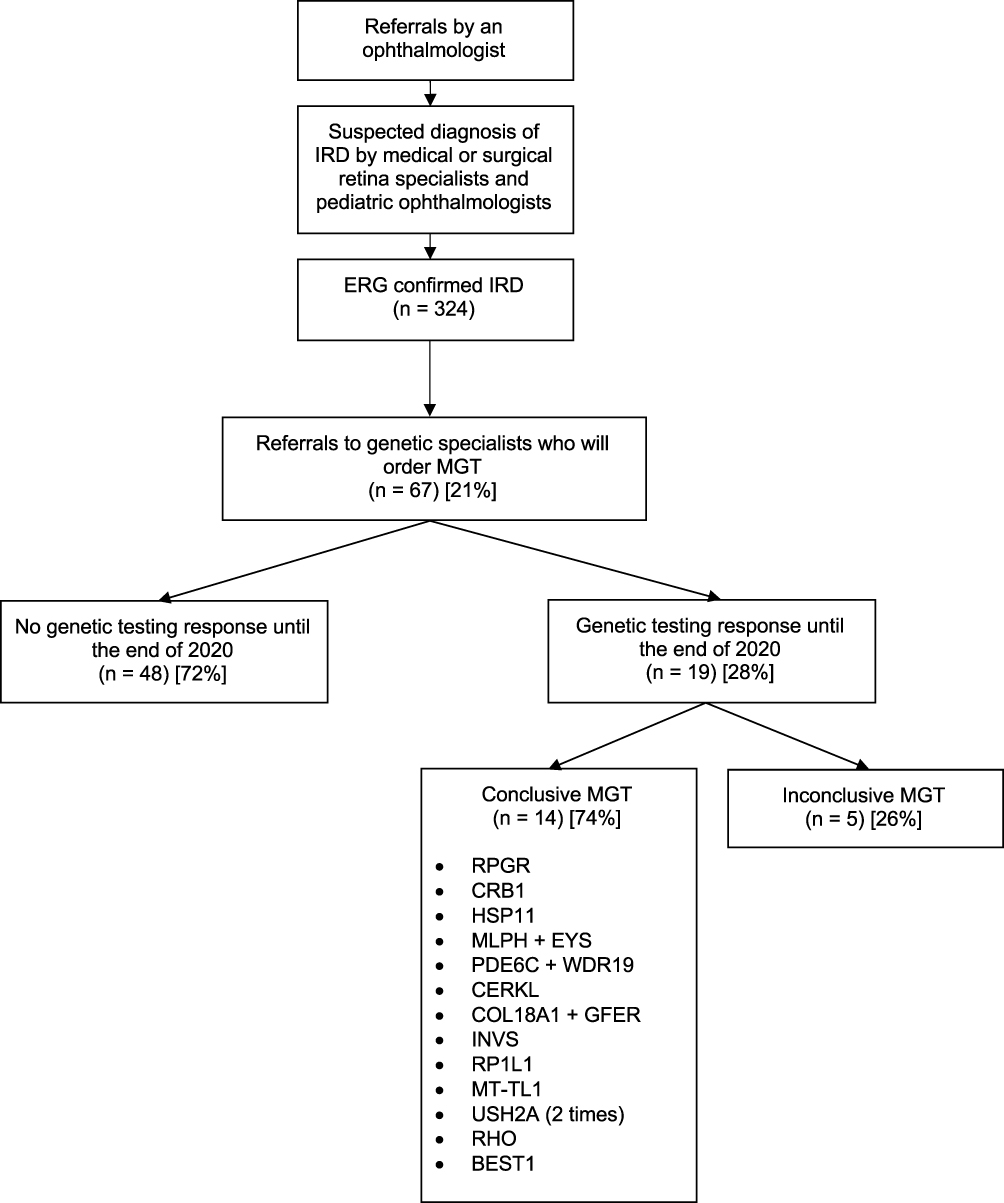 |
Figure 2 Flowchart representing the process and number of patients undergoing genetic referrals for suspicion of IRD. Abbreviations: IRD, inherited retinal dystrophy; ERG, electroretinogram; MGT, molecular genetic testing. |
Discussion
These findings suggest the need for more genetic specialists in Quebec and suggest that the efficiency of communications and referrals between the ophthalmology and genetics departments should be addressed. A fairly small percentage of patients (n = 67, 21%) were referred for MGT. Currently, the main reasons for the small proportion of referrals to genetics are that MGT has not yet been integrated into the standard of care when performing the workup of a patient with IRD, partly due to the slow process and limited number of patients who can be seen in genetics. However, young patients who are pregnant, who plan to form a family, or who already have children are more likely to be referred given the implications for genetic counselling. With the growth of MGT in multiple fields of medicine in recent years, we should advocate for better access to MGT for our patients. Early genetic characterization is needed to properly manage these patients and to likely avoid preventable visual loss in the future.4,5 When treatments will be more widely available, it is expected that they will be most effective at earlier stages; therefore, timely MGT should be accessible at an institutional level in all university hospitals. According to Fighting Blindness Canada, the waiting time for an appointment in genetics with referral is a maximum of 2 years in Quebec and the results are generally communicated within 6 weeks after the appointment.6
MGT and medical staff with expertise in genetics should be increased to meet the higher demand, and special collaborations should be implemented between departments to tailor care for patients with these rare diseases. The integration of genetic counselors in the workflow was piloted in other fields with good success, improving access to testing.7 At our institution, surgical and medical retina specialists and pediatric ophthalmologists follow patients with suspected IRD and refer them to a general genetic specialist covering all medical specialties. However, there is no ophthalmology genetic specialist or ophthalmologist with an IRD fellowship at our center.
Limitations
This is a retrospective cohort study which examined all patients with suspected IRD confirmed with ERG and the subset of patients who were referred to genetics. Not all IRD will have abnormal ERGs (eg, some macular dystrophies) which can slightly underestimate the total number of IRD reported. Moreover, this makes any conclusions regarding incidence of pathogenic variants out of the scope of this brief report. Likewise, some patients may have received calls from the hospital to be seen in genetics but failed to go to their hospital visit for several reasons; patient moved or out of town for example. This may therefore underestimate the proportion of patients who could have had a MGT result by the end of the study period. However, this allows us to have a global idea of the gap in care that need to be addressed to streamline genetics referrals and MGT results.
Conclusions
We believe that integrating genetic counsellors, ophthalmology-trained geneticists, or IRD-specialized ophthalmologists into the workflow may be helpful to accelerate MGT results and interpretation for patients with IRD. A genetic eye clinic should be established bringing together ophthalmologists, clinical geneticists, and genetic counsellors. Alternatively, giving direct access to MGT testing for retinal specialists or pediatric ophthalmologists who follow IRD patients could be beneficial to patients. The recent advances in gene therapy show that appropriate workup and genetic characterization of these patients are critical for their proper treatment.
Data Sharing Statement
All data relevant to the study are included in the article. The datasets used and/or analysed during the present study are available from the corresponding author on reasonable request.
Ethics Approval and Informed Consent
This study was approved by the Institutional Review Board of the Centre Hospitalier Universitaire de Québec – Université Laval (2022-6022) and adheres to the tenets of the Declaration of Helsinki.
Consent to Participate
This study was approved by the Institutional Review Board of the Centre Hospitalier Universitaire de Québec – Université Laval (2022-6022) who waived the necessity for signed informed consent from patients given the retrospective nature of the study. However, the data were anonymized to maintain patient confidentiality.
Acknowledgments
The authors would like to thank the Centre Universitaire d’Ophtalmologie clinical research team for their support of this research study (Marcelle Giasson and Johanne Doucet).
Funding
The authors did not receive support from any organization for the submitted work. Funded without commercial support by the authors.
Disclosure
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
1. RetNet [homepage on the Internet]. The retinal information network. Texas; 2021. Available from: https://sph.uth.edu/retnet/.
2. Chiu W, Lin TY, Chang YC, et al. An update on gene therapy for inherited retinal dystrophy: experience in Leber congenital amaurosis clinical trials. Int J Mol Sci. 2021;22(9):4534. doi:10.3390/ijms22094534
3. Stone EM, Andorf JL, Whitmore SS, et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. 2017;124(9):1314–1331. doi:10.1016/j.ophtha.2017.04.008
4. Souzeau E, Glading J, Ridge B, et al. Predictive genetic testing in minors for Myocilin juvenile onset open angle glaucoma. Clin Genet. 2015;88(6):584–588. doi:10.1111/cge.12558
5. Mensah A, Witting N, Duno M, et al. Delayed diagnosis of oculopharyngeal muscular dystrophy in Denmark: from initial ptosis to genetic testing. Acta Ophthalmol. 2014;92(3):e247–e249. doi:10.1111/aos.12243
6. Fighting Blindness Canada [homepage on the Internet]. Genetic testing for inherited retinal diseases. Canada; 2021. Available from: https://www.fightingblindness.ca/resources/genetic-testing-for-inherited-retinal-diseases/.
7. Amlie-Wolf L, Baker L, Hiddemen O, et al. Novel genetic testing model: a collaboration between genetic counselors and nephrology. Am J Med Genet A. 2021;185(4):1142–1150. doi:10.1002/ajmg.a.62088
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.