Back to Journals » Journal of Pain Research » Volume 12
Analgesic effects of amiodarone in mouse models of pain
Authors Kotoda M ![]() , Ino H, Kumakura Y, Iijima T, Ishiyama T
, Ino H, Kumakura Y, Iijima T, Ishiyama T ![]() , Matsukawa T
, Matsukawa T
Received 29 November 2018
Accepted for publication 6 May 2019
Published 6 June 2019 Volume 2019:12 Pages 1825—1832
DOI https://doi.org/10.2147/JPR.S196480
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Erica Wegrzyn
Masakazu Kotoda,1 Hirofumi Ino,1 Yasutomo Kumakura,1 Tetsuya Iijima,1 Tadahiko Ishiyama,2 Takashi Matsukawa1
1Department of Anesthesiology, Faculty of Medicine, University of Yamanashi, Chuo, Yamanashi 409-3898, Japan; 2Surgical Center, University of Yamanashi Hospital, University of Yamanashi, Chuo, Yamanashi 409-3898, Japan
Purpose: Although amiodarone is classified as a Vaughan-Williams class Ⅲ antiarrhythmic drug, it has inhibitory effects on voltage-gated sodium and calcium channels and on β-adrenergic receptors. Given these pharmacological profiles, amiodarone may have analgesic properties. Most patients who are prescribed amiodarone possess multiple cardiovascular risk factors. Despite the fact that pain plays a crucial role as a clinical indicator of cardiovascular events, the effects of amiodarone on pain have not been investigated. The aim of the current study was to investigate the analgesic effects of amiodarone by using mouse models of pain in an effort to elucidate underlying mechanisms.
Methods: Adult male C57B6 mice received single bolus intraperitoneal injections of amiodarone at doses of 25, 50, 100, and 200 mg/kg, while the mice in the control group received only normal saline. The analgesic effects of amiodarone were evaluated using the acetic acid-induced writhing test, formalin test, and tail withdrawal test. In addition, the potassium channel opener NS1643, voltage-gated sodium channel opener veratrine, calcium channel opener BAYK8644, and selective β-adrenergic agonist isoproterenol were used to uncover the underlying mechanism.
Results: During the acetic acid-induced writhing test, formalin test, and tail withdrawal test, amiodarone induced analgesic responses in a dose-dependent manner. The analgesic effects of amiodarone were abolished by veratrine but not by NS1643, BAYK8644, or isoproterenol.
Conclusion: Amiodarone induced analgesic responses in a dose-dependent manner, likely by blocking voltage-gated sodium channels. These results indicate that clinical doses of amiodarone can affect nociception and may mask or attenuate pain induced by acute cardiovascular events.
Keywords: amiodarone, antinociception, pain, sodium channels
Introduction
Voltage-gated ion channels play crucial roles in pain, and various types of ion channel antagonists have been developed for pain management. Sodium channel blockers have been widely used as local anesthetics. These types of drugs inhibit the generation and propagation of neuronal membrane action potentials by blocking voltage-gated sodium channels.1 In addition to their usefulness as local anesthetics, sodium channel blockers have also been demonstrated to have systemic analgesic effects.2–4 Voltage-gated calcium channels mediate intracellular calcium levels and play a pivotal role in synaptic transmission.5,6 When the action potential reaches the nerve terminal, presynaptic voltage-gated calcium channels open. The influx of calcium ions triggers the release of neurotransmitters such as glutaminate and substance P into the synaptic cleft. The neurotransmitters cross the synapse and bind to neuroreceptors, resulting in a signal in the post-synaptic neuron.5,6 Gabapentin and pregabalin mediate the function of voltage-gated calcium channels, and block neurotransmission. They are commercially available drugs, and have been widely used to treat chronic pain.
Amiodarone is one of the most effective drugs for the treatment of life-threatening arrhythmia. Although it is classified as a Vaughan-Williams class Ⅲ antiarrhythmic drug, amiodarone acts as a multiple channel blocker and its characteristics cover all four classes.7–9 In addition to its inhibitory effects on potassium channels, amiodarone also blocks voltage-gated sodium and calcium channels7–9 and β-adrenergic receptors.10 Previous studies have shown that a blockage of β-adrenergic receptors can also induce analgesic effect.11,12 Given these considerations and the multimodal pharmacological profile of amiodarone, there is a possibility that amiodarone possesses antinociceptive properties.
Patients who present with life-threatening arrhythmia and receive amiodarone frequently have various cardiovascular risk factors. Although pain plays an important role as a clinical indicator of acute cardiovascular events, the analgesic property of amiodarone and its mechanism have been poorly understood.
Therefore, the aims of this study were to investigate antinociceptive effects of amiodarone and the associated underlying mechanisms using experimental murine models of pain.
Methods
All experiments were conducted in accordance with the International Association for the Study of Pain and National Institutes of Health guidelines. The experimental protocol was reviewed and approved by the University of Yamanashi Animal Care Committee.
Animals
Male C57B6 mice (age, 8–10 weeks; weight, 20–25 g) were purchased from Japan SLC (Tokyo, Japan). The mice were housed at 23±2 °C under a 12 h light–dark cycle with free access to standard food and water. All experiments were performed between 09:00 and 17:00, under normal room light and at 23±2 °C.
Drugs
Amiodarone (Sanofi K.K., Tokyo, Japan), veratrine (Santa Cruz Biotechnology, Dallas, TX), NS1643 (Abcam, Cambridge, England), and isoproterenol (Kowa Pharmaceutical, Tokyo, Japan) were dissolved or suspended in normal saline. BAYK8644 (Abcam, Cambridge, England) was dissolved in dimethyl sulfoxide and diluted with normal saline (the final concentration of dimethyl sulfoxide was 0.5%). All drugs were freshly prepared before use and administered intraperitoneally. The dose of amiodarone (50 mg/kg) was decided based on the estimated body surface area of mice used in the present study (0.008 m2)13 and the average body surface area of an adult human (1.86 m2),14 and is considered equivalent to the clinical dosage for an adult human (300 mg). Other doses of amiodarone (25, 100, and 200 mg/kg), and antagonists were chosen based on previously published data.15–19
Antinociceptive activity tests
In the acetic acid-induced writhing test,20 mice were injected intraperitoneally with 0.01 ml/g of 1% acetic acid (Hayashi Pure Chemical, Osaka, Japan) in normal saline and were placed in an observation chamber. The number of nociceptive movements (writhing movements) was counted during a 15-min period starting 5 min after the injection of acetic acid. Typical nociceptive movements observed in this test are arching of the back, body stretching, and extension of the forelimbs.21
In the formalin test,22 mice were gently restrained in a cloth pocket, and 0.01 ml of 5% formalin (Muto Pure Chemicals, Tokyo, Japan) in normal saline was injected subcutaneously into their right dorsal hind paws of mice by using a 30-gauge needle. The mice were then put back into an observation chamber, and the observation period was started. The durations of licking and biting responses directed at the right hind paw were measured using a stopwatch from 0 to 5 min (phase I) and from 15 to 30 min (phase II) immediately after the formalin injection. Phase I represents a period of direct pain response caused by the chemical effect of formalin on sensory C fibers, while phase II response is caused by local inflammation and the release of algesic mediators.23
Nociceptive response to thermal stimuli was assessed using the tail withdrawal test.20 In this test, the mice were gently restrained in a cloth pocket, and the distal half of the tail was immersed into a thermostatically-controlled water bath. The mice were tested successively at three different temperatures in an increasing order (45, 50, and 55 °C). Tail withdrawal latencies were measured using a stopwatch with an interval time of 10 min between each trial. A cut-off time of 25 s for withdrawal was set to avoid the tissue damage.
Hemodynamic measurements
To evaluate the effects of amiodarone on hemodynamic functions, heart rate and non-invasive blood pressure were measured using a tail-cuff monitor (Softron, Tokyo, Japan). Values were recorded before the administration of amiodarone or saline, and 1 h thereafter.
Spontaneous activity test
Possible sedative effects of amiodarone were evaluated using the open-field test.24,25 Mice were placed into a 50×50×50-cm observation chamber, the floor of which was demarcated with a 5×5 grid containing a total of twenty-five 10×10 cm-sized squares. The number of squares crossed with all paws during a 5 min period was counted.
Dose responses
Mice were randomly assigned to either the control group or to one of the amiodarone groups (25, 50, 100, or 200 mg/kg). First, mice were habituated to an observation chamber for 30 min. They then received single bolus intraperitoneal injections of amiodarone at doses of 25, 50, 100, or 200 mg/kg (amiodarone groups), or of normal saline (control group, n=10 each). One hour thereafter, the antinociceptive activity of amiodarone was evaluated via the acetic acid-induced writhing test, formalin test, and tail withdrawal test. The effects of amiodarone on heart rate, mean arterial pressure, and spontaneous activity were also investigated before the antinociceptive activity tests. Ten mice per group were used in these experiments.
Mechanism investigations
The potassium channel agonist NS1643, voltage-gated sodium channel agonist veratrine, calcium channel agonist BAYK8644, and the selective β-adrenergic agonist isoproterenol were used to investigate potential mechanisms of the antinociceptive properties of amiodarone. Mice were habituated to an observation chamber for 30 min, then received a single bolus intraperitoneal injection of either normal saline (control), 100 mg/kg amiodarone alone, agonist alone, or 100 mg/kg amiodarone + agonist (n=8 each). The effects of each agonist on amiodarone-induced analgesic responses were assessed via the acetic acid-induced writhing test in separate groups using the following different doses: NS1643, 1.5, 6, and 24 mg/kg; veratrine, 0.125, 0.5, and 2 mg/kg; BAYK8644, 0.5, 2, and 8 mg/kg; and isoproterenol, 0.04, 0.16, and 0.56 mg/kg. In the agonist alone group, each drug was used at the highest dose (NS1643, 24 mg/kg; veratrine, 2 mg/kg; BAYK8644, 8 mg/kg; isoproterenol, 0.56 mg/kg).
As a next step, the effects of veratrine on amiodarone-induced analgesic responses were tested via the formalin test and tail withdrawal test. Mice received a single bolus intraperitoneal injection of either normal saline (control), 100 mg/kg amiodarone alone, or amiodarone 100 mg/kg + veratrine 2 mg/kg (n=4 each).
Statistical analysis
All values are presented as means ± standard deviations. Statistical analysis was performed using Prism 6 software (GraphPad Software, San Diego, CA). Subgroup comparisons were analyzed via one-way (the writhing test and formalin test) or two-way (tail withdrawal test) analysis of variance, followed by Dunnett test. p<0.05 was considered statistically significant. Each experimental sample size was calculated to detect a difference in pain responses of 10% while providing 80% power with an α level of 0.05 (G*Power 3.1.9.3).
Results
In the acetic acid-induced writhing test, intraperitoneal administration of amiodarone significantly reduced pain responses in a dose-dependent manner (control, 46.0±12.1; amiodarone 25 mg/kg, 47.3±9.8, p=0.998; amiodarone 50 mg/kg, 31.1±13.7, p<0.05; amiodarone 100 mg/kg, 20.6±11.4, p<0.001; amiodarone 200 mg/kg, 1.4±2.1, p<0.0001; n=10 each) (Figure 1A). Amiodarone at doses of 100 and 200 mg/kg also reduced pain responses in the formalin test (phase I: control, 81.9±19.8; amiodarone 100 mg/kg, 55.5±12.1, p<0.01; amiodarone 200 mg/kg, 4.3±4.6, p<0.0001; phase II: control, 127.6±18.9; amiodarone 100 mg/kg, 72.0±44.8, p<0.01; amiodarone 200 mg/kg, 8.0±5.1, p<0.0001; n=10 each) (Figure 1B and C). In the tail withdrawal test, amiodarone at doses of 100 and 200 mg/kg also increased the withdrawal latency (50 °C: control, 2.2±1.0; amiodarone 200 mg/kg, 6.5±3.0, p<0.01; 55 °C: control, 0.9±0.3; amiodarone 100 mg/kg, 3.9±0.5, p<0.05; amiodarone 200 mg/kg, 4.8±1.2, p<0.01; n=8 each) (Figure 1D),
 | Figure 1 Analgesic responses induced by amiodarone. (A) Acetic acid-induced writhing test. At doses of 50 mg/kg and higher, amiodarone significantly reduced nociceptive responses in a dose-dependent manner. (B) Formalin test, phase I. At doses of 100 and 200 mg/kg, amiodarone significantly reduced nociceptive response. (C) Formalin test, phase II. At doses of 100 and 200 mg/kg, amiodarone significantly reduced nociceptive response. (D) Tail withdrawal test. At doses of 100 and 200 mg/kg, amiodarone significantly increased the withdrawal latency. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Abbreviation: AMD, amiodarone. |
As shown in Figure 2, while amiodarone at doses of 100 mg/kg or lower did not affect hemodynamic status or spontaneous activity, amiodarone at the highest dose (200 mg/kg) significantly reduced heart rate, mean arterial pressure, and spontaneous activity (heart rate: control, 473±64; amiodarone 200 mg/kg, 399±37 beats/min, p<0.05; mean arterial pressure: control, 76±12; amiodarone 200 mg/kg, 59±10 mmHg, p<0.05; spontaneous activity: control, 249±41; amiodarone 200 mg/kg, 134±97, p<0.01; n=10 each).
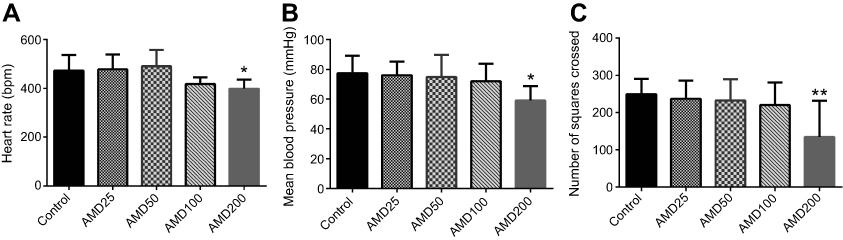 | Figure 2 Effects of amiodarone on hemodynamic status and spontaneous activity. (A) Heart rate 1 h after administration of either normal saline (control group) or amiodarone (amiodarone groups). (B) Mean blood pressure 1 h after the administration of either normal saline (control group) or amiodarone (amiodarone groups). (C) Number of squares crossed during the open-field test. *p<0.05, **p<0.01. Abbreviation: AMD, amiodarone. |
As a next step, the voltage-gated sodium channel opener veratrine, the calcium channel opener BAYK8644, the potassium channel opener NS1643, and the selective β-adrenergic agonist isoproterenol were used to investigate mechanisms potentially involved in the above-described results. As shown in Figure 3, the analgesic effects of amiodarone were abolished by veratrine (amiodarone 100 mg/kg, 19.5±6.9; amiodarone 100 mg/kg + veratrine 0.5 mg/kg, 35.6±7.4, p<0.05; amiodarone 100 mg/kg + veratrine 2 mg/kg, 50.1±14.1, p<0.0001; n=8 each) but not by BAYK8644, NS-1643, or isoproterenol. When administered alone, veratrine, BAYK8644, NS1643, and isoproterenol did not affect nociceptive responses in the writhing test. Veratrine 2 mg/kg also reversed the antinociceptive effects of amiodarone in the formalin test and tail withdrawal test (formalin test phase I: control, 94.0±9.6; amiodarone 100 mg/kg, 58.5±8.3, p<0.01; amiodarone 100 mg/kg + veratrine 2 mg/kg, 90.8±15.8, p=0.892; formalin test phase II: control, 127.0±27.1; amiodarone 100 mg/kg, 77.3±23.8, p<0.05; amiodarone 100 mg/kg + veratrine 2 mg/kg, 120.5±12.0, p=0.882; tail withdrawal test (55 °C): control, 1.0±0.2; amiodarone 100 mg/kg, 3.2±1.3, p<0.05; amiodarone 100 mg/kg + veratrine 2 mg/kg, 1.8±0.9, p=0.435; n=4 each) (Figure 4).
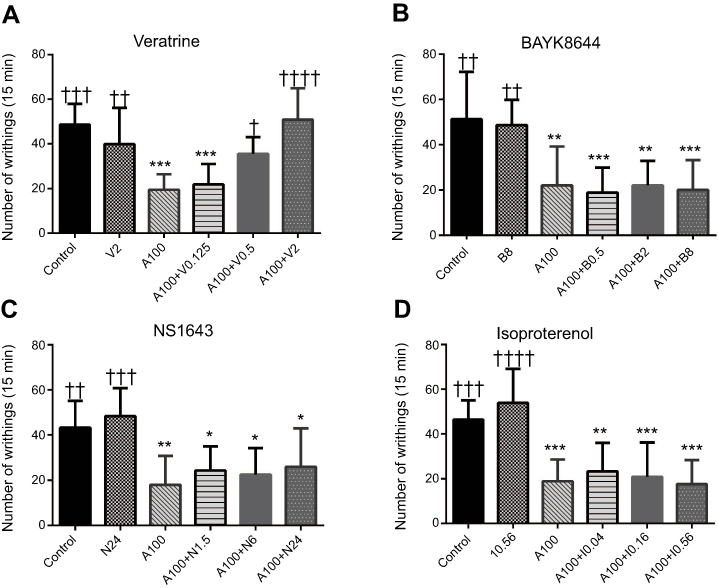 | Figure 3 Effects of each selective agonist on analgesic responses induced by amiodarone during the acetic acid-induced writhing test. (A) Veratrine, a voltage-gated sodium channel opener. (B) BAYK8644, an L-type calcium channel opener. (C) NS1643, voltage-gated potassium channel (Kv 11.1) opener. (D) Isoproterenol, a β-adrenergic receptor agonist. The analgesic effects of amiodarone (100 mg/kg) were abolished by veratrine but not by BAYK8644, NS1643, or isoproterenol. When administered alone, veratrine, BAYK8644, NS1643, and isoproterenol did not affect nociceptive responses in the writhing test. *p<0.05, **p<0.01, ***p<0.001 compared with the control group. †p<0.05, ††p<0.01, †††p<0.001, ††††p<0.0001 compared with the amiodarone alone group. Abbreviations: N, NS1643; A, amiodarone; V, veratrine; B, BAYK8644; I, isoproterenol. |
 | Figure 4 Effects of veratrine on analgesic responses induced by amiodarone during the formalin test and tail withdrawal test. (A) Formalin test, phase I. (B) Formalin test, phase II. (C) Tail withdrawal test at 55 °C. Veratrine 2 mg/kg reversed the antinociceptive effects of amiodarone in the formalin test and tail withdrawal test. *p<0.05 . Abbreviations: A, amiodarone; V, veratrine. |
Discussion
In the present study, amiodarone demonstrated dose-dependent analgesic effects in experimental murine pain models. The highest dose of amiodarone (200 mg/kg) induced strong analgesic responses accompanied by reduced spontaneous activity. This dose of amiodarone also affected hemodynamic state and reduced the heart rate and arterial blood pressure. In contrast, 50 and 100 mg/kg amiodarone doses induced milder analgesic responses without affecting the spontaneous activity or hemodynamic state. Based on body surface area, 50 mg/kg amiodarone dose is considered equivalent to the adult human dosage used during cardiopulmonary resuscitation.13,14 Thus, the present results suggest that amiodarone at the clinical dose can exert mild analgesic effects without affecting heart rate or blood pressure. The results also suggest that amiodarone may have strong antinociceptive activity when used at doses that exceed the clinical dose. In addition, at very high doses such as 200 mg/kg, amiodarone may have sedative effects that may result in the antinociceptive effects observed in this study.
The pharmacological profile of amiodarone is complex.7–9 In addition to affecting potassium channels, amiodarone exerts inhibitory effects on voltage-gated sodium channels (Nav), L-type calcium channels (Cav 1), and β-adrenergic receptors. Therefore, in the current study we investigated the effects of selective agonists of these ion channels and receptors in an effort to clarify mechanisms underlying the antinociceptive activity of amiodarone. The results indicated that amiodarone exerted analgesic effects by blocking voltage-gated sodium channels, because only veratrine, a selective opener of voltage-gated sodium channels, abolished amiodarone’s analgesic effects.
Systemic analgesic effects of voltage-gated sodium channel blockers, including lidocaine, mexiletine, and flecainide, have been demonstrated in both experimental and clinical studies.2,26–29 It is known that the inhibitory effects of amiodarone on voltage-gated sodium channels are mainly on the fast inward sodium current and are both use- and dose-dependent,7 thus resembling the pharmacological characteristics of lidocaine. Therefore, it is not surprising that amiodarone can exert analgesic effects by blocking voltage-gated sodium channels, as does lidocaine. Datta et al have reported that amiodarone attenuates hyperalgesia in an experimental neuropathic pain model and suggested that this effect may come from sodium channel blockage.30 Our results support their hypothesis and demonstrated that amiodarone exerts its analgesic effects via the blockage of voltage-gated sodium channels and that the inhibitory effects of amiodarone on calcium and potassium channels and on β-adrenergic receptors are unlikely to be involved in its analgesic effects. Our study also demonstrates that systemically administered amiodarone can attenuate not only neuropathic pain but also nociceptive pain.
With regard to calcium channels, amiodarone inactivates the L-type voltage-gated calcium channels (Cav 1) and blocks inward calcium current.7,8,31 Accumulating evidence suggests that voltage-gated calcium channels have pivotal roles in pain signaling, and that the blockage of voltage-gated calcium channels leads to antinociception.32–34 Notably, however, these effects are mainly associated with the blockage of N-type channels (Cav 2.2) and T-type channels (Cav 3.2).5,6,32,34 While some studies have reported the involvement of L-type channels in pain and antinociception at the spinal level,35,36 systemic L-type channel blockage is not likely to have any analgesic effect.37
With regard to potassium channels, acute application of amiodarone mainly inactivates the voltage-gated potassium channel 11.1 (Kv 11.1), and blocks the delayed rectifier potassium current.7,8,38 Previous studies have revealed the involvement of several types of voltage-gated potassium channels, such as Kv 1, Kv 2, Kv 3, Kv 4, and Kv 7.1 in chronic and neuropathic pain.39,40 Enhancement of these types of potassium channels may suppress cellular hyperexcitability and reduce pain.39,40 In contrast, the involvement of Kv 11.1 and delayed rectifier potassium current on pain has not been reported in the literature. In the present study, we therefore used a selective Kv 11.1 channel opener to investigate the involvement of this channel in the analgesic activity of amiodarone. However, the results did not indicate any involvement of Kv 11.1 in the analgesic effects of amiodarone, as neither NS 1643 alone nor coadministration of amiodarone and NS1643 affected the nociceptive responses.
In addition to electrophysiological activity, amiodarone also has inhibitory effects on β-adrenergic receptors.7,8 Some evidence suggests that β-adrenergic receptor antagonists have antinociceptive effects.11,12 However, amiodarone exerts inhibitory effects on β-adrenergic receptors via downregulation of the receptors and not via ligand interaction.10 This process takes some time, and the inhibitory effect on β-adrenergic receptors is more predominant in the chronic phase than in the acute phase.7,10
The most clinically important result in the current study is that clinical doses of amiodarone affected nociception. Patients who receive amiodarone often have multiple cardiovascular risk factors, and pain plays an important role as a clinical indicator of acute cardiovascular events. Based on the present results, there is a possibility that the analgesic properties of amiodarone can mask pain induced by acute cardiovascular events.
The current study has some limitations. We used relatively young male mice that had no degenerative vascular or cellular changes. Future studies should test aged mice, because cardiovascular events and life-threatening arrhythmia are more common in older subjects. Additionally, although neither 50 nor 100 mg/kg amiodarone significantly reduced spontaneous activity, we cannot exclude the possible involvement of amiodarone’s hypnotic effect in amiodarone-induced analgesic responses observed in the present study.
Conclusions
Amiodarone induced dose-dependent analgesic responses during the acetic acid-induced writhing test, the formalin test, and the tail withdrawal test. The analgesic effects of amiodarone observed in the current study may be attributable to the inhibition of voltage-gated sodium channels. The results of the study indicate that amiodarone exerts analgesic effects via voltage-gated sodium channel blockage.
Acknowledgments
This work was supported by the Japan Society for Promotion of Science (JSPS KAKENHI grant number 17K11044).
Author contributions
Masakazu Kotoda conceived and designed the study, performed experiments and statistical analysis, and drafted the manuscript. Hirofumi Ino performed experiments, analyzed the data, and helped prepare the manuscript. Yasutomo Kumakura collected and analyzed the data and helped prepare the manuscript. Tetsuya Iijima analyzed the data and revised the manuscript for important intellectual content. Tadahiko Ishiyama analyzed the data and revised the manuscript for important intellectual content. Takashi Matsukawa analyzed the data and revised the manuscript for important intellectual content. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Scholz A. Mechanisms of (local) anaesthetics on voltage-gated sodium and other ion channels. Br J Anaesth. 2002;89:52–61.
2. Eipe N, Gupta S, Penning J. Intravenous lidocaine for acute pain: an evidence-based clinical update. BJA Educ. 2016;16:292–298. doi:10.1093/bjaed/mkw008
3. Groudine SB, Fisher HA, Kaufman RP
4. Farag E, Ghobrial M, Sessler DI, et al. Effect of perioperative intravenous lidocaine administration on pain, opioid consumption, and quality of life after complex spine surgery. Anesthesiology. 2013;119:932–940. doi:10.1097/ALN.0b013e318297d4a5
5. Park J, Luo ZD. Calcium channel functions in pain processing. Channels (Austin). 2010;4:510–517.
6. Bourinet E, Altier C, Hildebrand ME, Trang T, Salter MW, Zamponi GW. Calcium-permeable ion channels in pain signaling. Physiol Rev. 2014;94:81–140. doi:10.1152/physrev.00023.2013
7. Kodama I, Kamiya K, Toyama J. Cellular electropharmacology of amiodarone. Cardiovasc Res. 1997;35:13–29.
8. Kodama I, Kamiya K, Toyama J. Amiodarone: ionic and cellular mechanisms of action of the most promising class III agent. Am J Cardiol. 1999;84:20R–28R.
9. Riera AR, Uchida AH, Ferreira C, et al. Relationship among amiodarone, new class III antiarrhythmics, miscellaneous agents and acquired long QT syndrome. Cardiol J. 2008;15:209–219.
10. Drvota V, Haggblad J, Blange I, Magnusson Y, Sylven S. The effect of amiodarone on the beta-adrenergic receptor is due to a downregulation of receptor protein and not to a receptor-ligand interaction. Biochem Biophys Res Comm. 1999;255:515–520. doi:10.1006/bbrc.1998.0138
11. Chia YY, Chan MH, Ko NH, Liu K. Role of beta-blockade in anaesthesia and postoperative pain management after hysterectomy. Br J Anaesth. 2004;93:799–805. doi:10.1093/bja/aeh268
12. Zhao H, Sugawara T, Miura S, Iijima T, Kashimoto S. Intrathecal landiolol inhibits nociception and spinal c-Fos expression in the mouse formalin test. Can J Anaesth. 2007;54:201–207. doi:10.1007/bf03022641
13. Cheung MC, Spalding PB, Gutierrez JC, et al. Body surface area prediction in normal, hypermuscular, and obese mice. J Surg Res. 2009;153:326–331. doi:10.1016/j.jss.2008.05.002
14. Baker SD, Verweij J, Rowinsky EK, et al. Role of body surface area in dosing of investigational anticancer agents in adults, 1991–2001. J Natl Cancer Inst. 2002;94:1883–1888. doi:10.1093/jnci/94.24.1883
15. Ozbakis-Dengiz G, Bakirci A. Anticonvulsant and hypnotic effects of amiodarone. J Zhejiang Univ Sci B. 2009;10:317–322. doi:10.1631/jzus.B0820316
16. Kotoda M, Ishiyama T, Mitsui K, Hishiyama S, Matsukawa T. Neuroprotective effects of amiodarone in a mouse model of ischemic stroke. BMC Anesthesiol. 2017;17:168. doi:10.1186/s12871-017-0459-3
17. Bourin M, Chenu F, Hascoet M. Topiramate and phenytoin anti-immobility effect in the mice forced swimming test is reversed by veratrine: implication for bipolar depression treatment. Behav Brain Res. 2009;205:421–425. doi:10.1016/j.bbr.2009.07.030
18. O’Neill SK, Bolger GT. Enantiomer selectivity and the development of tolerance to the behavioral effects of the calcium channel activator BAY K 8644. Brain Res Bull. 1988;21:865–872.
19. Fukushiro-Lopes DF, Hegel AD, Rao V, et al. Preclinical study of a Kv11.1 potassium channel activator as antineoplastic approach for breast cancer. Oncotarget. 2017;9:3321–3337. doi:10.18632/oncotarget.22925
20. Leo S, D’Hooge R, Meert T. Exploring the role of nociceptor-specific sodium channels in pain transmission using Nav1.8 and Nav1.9 knockout mice. Behav Brain Res. 2010;208:149–157. doi:10.1016/j.bbr.2009.11.023
21. Park Y, Jung SM, Yoo SA, et al. Antinociceptive and anti-inflammatory effects of essential oil extracted from chamaecyparis obtusa in mice. Int Immunopharmacol. 2015;29:320–325. doi:10.1016/j.intimp.2015.10.034
22. Moore PK, Oluyomi AO, Babbedge RC, Wallace P, Hart SL. L-NG-nitro arginine methyl ester exhibits antinociceptive activity in the mouse. Br J Pharmacol. 1991;102:198–202.
23. Hunskaar S, Hole K. The formalin test in mice: dissociation between inflammatory and non-inflammatory pain. Pain. 1987;30:103–114.
24. Rodrigues AL, Da Silva GL, Mateussi AS, et al. Involvement of monoaminergic system in the antidepressant-like effect of the hydroalcoholic extract of siphocampylus verticillatus. Life Sci. 2002;70:1347–1358.
25. Yin ZY, Li L, Chu SS, Sun Q, Ma ZL, Gu XP. Antinociceptive effects of dehydrocorydaline in mouse models of inflammatory pain involve the opioid receptor and inflammatory cytokines. Sci Rep. 2016;6:27129. doi:10.1038/srep27129
26. Kurabe M, Furue H, Kohno T. Intravenous administration of lidocaine directly acts on spinal dorsal horn and produces analgesic effect: an in vivo patch-clamp analysis. Sci Rep. 2016;6:26253. doi:10.1038/srep26253
27. Lauretti GR. Mechanisms of analgesia of intravenous lidocaine. Rev Bras Anestesiol. 2008;58:280–286.
28. von Gunten CF, Eappen S, Cleary JF, et al. Flecainide for the treatment of chronic neuropathic pain: a phase II trial. Palliat Med. 2007;21:667–672. doi:10.1177/0269216307083031
29. Dejgard A, Petersen P, Kastrup J. Mexiletine for treatment of chronic painful diabetic neuropathy. Lancet. 1988;1:9–11.
30. Datta S, Waghray T, Torres M, Glusman S. Amiodarone decreases heat, cold, and mechanical hyperalgesia in a rat model of neuropathic pain. Anesth Analg. 2004;98:178–184.
31. Nishida A, Takizawa T, Matsumoto A, Miki T, Seino S, Nakaya H. Inhibition of ATP-sensitive K+ channels and L-type Ca2+ channels by amiodarone elicits contradictory effect on insulin secretion in MIN6 cells. J Pharmacol Sci. 2011;116:73–80.
32. Yekkirala AS, Roberson DP, Bean BP, Woolf CJ. Breaking barriers to novel analgesic drug development. Nat Rev Drug Discov. 2017;16:545–564. doi:10.1038/nrd.2017.87
33. Bauer CS, Nieto-Rostro M, Rahman W, et al. The increased trafficking of the calcium channel subunit alpha2delta-1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. J Neurosci. 2009;29:4076–4088. doi:10.1523/jneurosci.0356-09.2009
34. Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev. 2015;67:821–870. doi:10.1124/pr.114.009654
35. Chang E, Chen X, Kim M, Gong N, Bhatia S, Luo ZD. Differential effects of voltage-gated calcium channel blockers on calcium channel alpha-2-delta-1 subunit protein-mediated nociception. Eur J Pain. 2015;19:639–648. doi:10.1002/ejp.585
36. Fossat P, Dobremez E, Bouali-Benazzouz R, et al. Knockdown of L calcium channel subtypes: differential effects in neuropathic pain. J Neurosci. 2010;30:1073–1085. doi:10.1523/jneurosci.3145-09.2010
37. Kumar R, Mehra R, Ray SB. L-type calcium channel blockers, morphine and pain: newer insights. Indian J Anaesth. 2010;54:127–131. doi:10.4103/0019-5049.63652
38. Kamiya K, Nishiyama A, Yasui K, Hojo M, Sanguinetti MC, Kodama I. Short- and long-term effects of amiodarone on the two components of cardiac delayed rectifier K(+) current. Circulation. 2001;103:1317–1324.
39. Tsantoulas C, McMahon SB. Opening paths to novel analgesics: the role of potassium channels in chronic pain. Trends Neurosci. 2014;37:146–158. doi:10.1016/j.tins.2013.12.002
40. Du X, Gamper N. Potassium channels in peripheral pain pathways: expression, function and therapeutic potential. Curr Neuropharmacol. 2013;11:621–640. doi:10.2174/1570159x113119990042
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.