")
Back to Journals » International Medical Case Reports Journal » Volume 9
An unfortunate case of acquired hemochromatosis: a case report review of the clinical presentation, diagnosis, management, and prognosis
Authors Tariq A, Westra K, Santo A
Received 15 July 2016
Accepted for publication 23 September 2016
Published 8 December 2016 Volume 2016:9 Pages 385—387
DOI https://doi.org/10.2147/IMCRJ.S117322
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Anam Tariq,1 Kevin Westra,2 Arben Santo3
1Department of Internal Medicine, Pinnacle Health Internal Medicine, 2Department of Gastroenterology, Harrisburg Gastroenterology, Harrisburg, PA, 3Department of Pathology, Virginia College of Osteopathic Medicine-Virginia Tech, Blacksburg, VA, USA
Background: While blood transfusions are commonly used for prophylaxis and treatment for acute chest syndromes and strokes in sickle cell patients, accumulation of excess iron resulting in secondary hemochromatosis remains a rare disease. Chelation is the mainstay for preventing and treating iron overload to deter potential end-organ damages; it is rare when therapy fails.
Case report: A 52-year-old African American woman with chronic anemia secondary to sickle cell anemia and history of multiple blood transfusions presented with elevated serum ferritin (8000 ng/mL) and bilirubin (16.8 mg/dL). She had no previous personal or family history of liver disease. A magnetic resonance cholangiopancreatography (MRCP) and a liver biopsy confirmed the secondary hemochromatosis with marked fibrosis and 4+ iron deposits, but since she was therapeutically on deferasirox, her treatment regimen involved only closer monitoring. Her hemochromatosis led to readmission within a year for rapid progression of cardiac and hepatic failure.
Conclusion: Since chronically transfused sickle cell patients are at a significantly higher risk of mortality due to the secondary hemochromatosis and end-stage organ damage, knowledge of prophylactic iron chelation is important. Minimizing unnecessary transfusions should be strongly emphasized to reduce the sequelae as iron burden remains a threat. The effectiveness of iron-chelating therapy is best monitored via periodic magnetic resonance imaging, liver transaminases, bilirubin, creatinine, ferritin, and cardiac function tests. Despite the prophylactic treatment and quarterly blood work, in this case the initial presentation did not correlate with the severity of end-stage liver failure. The damage was not discovered until proven by liver biopsy and MRCP, too late to deter the sequelae and the mortality exactly 1 year after diagnosis of the secondary hemochromatosis.
Keywords: secondary hemochromatosis, sickle cell anemia, deferasirox
Introduction
Secondary hemochromatosis remains a rare, underdiagnosed disorder when presented atypically with “bronze diabetes” and cirrhosis in sickle cell patients. Blood transfusions serve as both prophylaxis and treatment for strokes and acute chest syndromes.1,2 However, excess transfusional iron accumulates in tissues and forms reactive oxygen species resulting in end-organ damages. Chelation is often initiated prophylactically in patients who have received greater than 10 units of packed red blood cells.3 In our case, despite prophylactic chelation and phlebotomy, our asymptomatic patient developed significant hepatocellular iron deposition with fibrosis leading to mortality within a year.
Case report
A 52-year-old African American woman with chronic anemia due to sickle cell disease, and history of multiple transfusions, presented for routine outpatient blood work and found to have elevated serum ferritin (8000 ng/mL) and bilirubin (16.8 mg/dL). The patient was asymptomatic. Her past medical history was significant only for cholecystectomy, with no previous personal or family history of liver disease. Written informed consent was obtained from the patient’s family for this case presentation. Her hematologist was concerned and consulted a gastroenterologist regarding the elevated bilirubin. A liver biopsy was also performed to rule out any autoimmune etiology. The hepatitis panel results were negative. An magnetic resonance cholangiopancreatography (MRCP) (Figure 1) was done to rule out an obstruction. To everyone’s surprise, it showed no obstruction, but showed evidence of hemochromatosis.
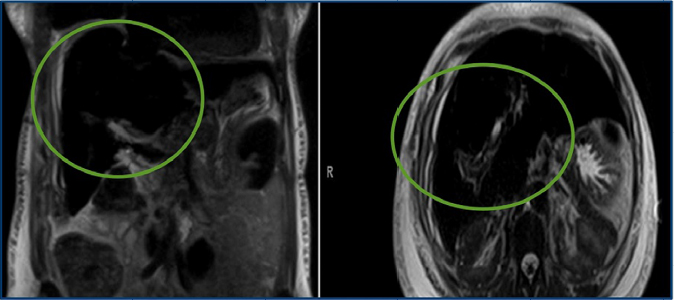 | Figure 1 Magnetic resonance cholangiopancreatography evaluation for possible obstructive jaundice due to the presence of elevated bilirubin on laboratory results. Note: The radiologist reported patient’s liver diffuse with decreased signal consistent with diagnosis of secondary hemochromatosis. The green circles highlight the blackened out liver in coronal (left) and axial (right) planes. |
The biopsy later confirmed the presumptive diagnosis of the secondary hemochromatosis with marked fibrosis and 4+ iron deposits (Figure 2). Since the patient was therapeutically receiving phlebotomies every 4–5 months in addition to her iron chelator deferasirox for the last 10 years, her treatment regimen post discharge involved further monitoring every 3 months with imaging and laboratory tests, including ferritin, transferrin, and hepatic panel.
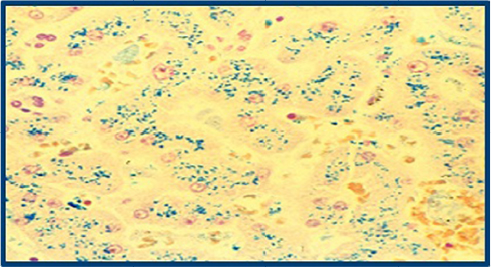 | Figure 2 Prussian blue stain of patient’s liver biopsy to rule out any presence of autoimmune disease or viral etiology. Note: Extensive iron staining (blue) of hepatocytes indicates the excess deposition confirming the diagnosis of hemochromatosis. |
Her new diagnosis of hemochromatosis led to readmission within a year. Laboratory investigations represented significant cirrhosis secondary to hemochromatosis along with evidence of renal, pulmonary, and cardiac failures, severe anemia, and supratherapeutic international normalized ratio resulting in her death within 7 days of admission.
Discussion
Long-term transfusion therapy is now a routine, life saving treatment for patients with intractable anemia resulting from sickle cell disease, thalassemia, bone marrow failure, or aggressive treatment of cancer. Repeated transfusion leads to rapid iron loading: each unit of blood contains 200–250 mg of iron.4
The American Association for the Study of Liver Diseases recommends evaluating for hemochromatosis in patients with abnormal iron studies, even in the absence of symptoms, and in all patients with evidence of liver disease.5 They suggest diagnosing hemochromatosis if transferrin saturation is >45% in combination with elevated ferritin levels. However a previous clinical trial suggested that serum ferritin (range: 1500–3000 ng/mL) is not always accurate to interpret the hepatic iron store concentrations because it does not directly correlate with the degree of cirrhosis.5 The effectiveness of iron-chelating therapy is best monitored via periodic magnetic resonance imaging (MRI), liver transaminases, bilirubin, creatinine, ferritin, and cardiac function tests. While this patient presented with an atypical, asymptomatic presentation, her routine labs conveyed only increased ferritin and bilirubin so hepatic failure was not considered. After the liver biopsy, a concern was raised for hemochromatosis even though she was prophylactically on iron chelator.
This case raises important questions to consider: does initiating therapy early really help to increase the life expectancy of a patient with the secondary hemochromatosis? Despite chelation therapy, iron burden in chronically transfused patients remains a threat. Since her chronic conditions were well-known, would a liver biopsy earlier in the course have indicated the degree of her serious hepatic and cardiac failures? Should routine quarterly MRI and echocardiogram be used in asymptomatic patients to prevent some sequelae of end-organ damages?
Conclusion
Since chronically transfused sickle cell patients are at significantly higher risk of mortality due to the secondary hemochromatosis and end-organ damages, knowledge of prophylactic iron chelation is of paramount importance.2,3 Minimizing unnecessary transfusions should be strongly emphasized early. The effectiveness of iron-chelating therapy may be best monitored via periodic MRI, liver transaminases, bilirubin, creatinine, ferritin, and cardiac function tests. In this atypical case, the initial presentation did not correlate with the severity of end-stage liver failure until proven by liver biopsy and MRI, which still could not deter the sequelae and the mortality exactly 1 year after the diagnosis of secondary hemochromatosis.
Disclosure
The authors report no conflicts of interest in this work.
References
Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS; American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54(1):328–343. | ||
Raghupathy R, Manwani D, Little JA. Iron overload in sickle cell disease. Adv Hematol. 2010;2010:272940. | ||
Brittenham GM. Iron-chelating therapy for transfusional iron overload. N Engl J Med. 2011;364(2):146–156. | ||
Barton JC, Acton RT, Leiendecker-Foster C, et al. Characteristics of participants with self-reported hemochromatosis or iron overload at HEIRS study initial screening. Am J Hematol. 2008;83(2):126–132. | ||
Harmatz P, Butensky E, Quirolo K, et al. Severity of iron overload in patients with sickle cell disease receiving chronic red blood cell transfusion therapy. Blood. 2000;96(1):76–79. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.