")
Back to Journals » Journal of Experimental Pharmacology » Volume 13
An Overview of Investigational and Experimental Drug Treatment Strategies for Marfan Syndrome
Authors Deleeuw V, De Clercq A , De Backer J , Sips P
Received 1 May 2021
Accepted for publication 19 July 2021
Published 11 August 2021 Volume 2021:13 Pages 755—779
DOI https://doi.org/10.2147/JEP.S265271
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Paola Rogliani
Violette Deleeuw, 1 Adelbert De Clercq, 1 Julie De Backer, 1, 2 Patrick Sips 1
1Center for Medical Genetics, Department of Biomolecular Medicine, Ghent University, Ghent, 9000, Belgium; 2Department of Internal Medicine and Pediatrics, Ghent University Hospital, Ghent, 9000, Belgium
Correspondence: Patrick Sips
Center for Medical Genetics, Department of Biomolecular Medicine, Ghent University, Corneel Heymanslaan 10, Ghent, 9000, Belgium
Tel +32 9 332 13 45
Email [email protected]
Abstract: Marfan syndrome (MFS) is a heritable connective tissue disorder caused by pathogenic variants in the gene coding for the extracellular matrix protein fibrillin-1. While the disease affects multiple organ systems, the most life-threatening manifestations are aortic aneurysms leading to dissection and rupture. Other cardiovascular complications, including mitral valve prolapse, primary cardiomyopathy, and arrhythmia, also occur more frequently in patients with MFS. The standard medical care relies on cardiovascular imaging at regular intervals, along with pharmacological treatment with β-adrenergic receptor blockers aimed at reducing the aortic growth rate. When aortic dilatation reaches a threshold associated with increased risk of dissection, prophylactic surgical aortic replacement is performed. Although current clinical management has significantly improved the life expectancy of patients with MFS, no cure is available and fatal complications still occur, underscoring the need for new treatment options. In recent years, preclinical studies have identified a number of potentially promising therapeutic targets. Nevertheless, the translation of these results into clinical practice has remained challenging. In this review, we present an overview of the currently available knowledge regarding the underlying pathophysiological processes associated with MFS cardiovascular pathology. We then summarize the treatment options that have been developed based on this knowledge and are currently in different stages of preclinical or clinical development, provide a critical review of the limitations of current studies and highlight potential opportunities for future research.
Keywords: thoracic aortic aneurysm and dissection, rare genetic disease, pharmacological treatment, pathophysiology, preclinical research
Plain Language Summary
Marfan syndrome (MFS) is a rare heritable disorder caused by defects in the fibrillin-1 protein. This protein plays an important role in the structure and maintenance of connective tissue, which provides support to many organ systems in the body. Healthy connective tissue is important for bones, eyes, skin, lungs, nervous system, heart and blood vessels, which can all be affected by MFS. One of the most important consequences of MFS is a gradual enlargement of the aorta, which can lead to potentially fatal rupture of this large blood vessel. Currently, there is no cure for patients with MFS. In order to reduce the risk of life-threatening complications, patients with MFS rely on regular medical check-ups, treatment with blood pressure lowering medication, and surgery to repair the aorta when indicated. Because the risk of rupture of the aorta is still present in all patients with MFS, researchers are continuously trying to find new ways to treat the disease. To achieve this goal, many research efforts are focused on trying to understand how the disease exactly develops and progresses. In this review paper, we summarize the known disease mechanisms and provide an overview of the current treatment strategies for patients with MFS. In addition, we highlight and discuss new research based on animal models of MFS that have shown promising results and might lead to the development of new treatment options.
Introduction
Marfan Syndrome
Marfan syndrome (MFS, OMIM #154700) is a rare connective tissue disorder with a reported prevalence ranging from 1.5 to 17.2 per 100,000 individuals.1 It is an autosomal dominantly inherited disease caused by pathogenic variants in the fibrillin-1 gene (FBN1).2 Fibrillin-1 is an important extracellular matrix (ECM) protein, which is widely distributed throughout the body and plays an essential role in microfibril assembly.3–5 Patients with MFS show pleiotropic manifestations affecting the ocular, skeletal and cardiovascular system. For the purpose of this review, we will focus on the cardiovascular system. The most common cardiovascular manifestation is aortic root dilatation, which is estimated to be present in over 80% of adults with MFS, and is associated with an increased risk of aortic dissection or rupture.6,7 Other cardiovascular manifestations requiring surveillance include mitral valve prolapse (MVP), cardiomyopathy and arrhythmias.2,7–15 Cardiovascular manifestations are major causes of mortality and morbidity in MFS. In order to minimize the impact of these complications, correct and timely diagnosis of MFS is a key feature in patient management. The diagnosis of MFS is based on a distinct set of clinical criteria and may be complemented with molecular confirmation of pathogenic variants in FBN1 as described in the revised Ghent nosology.8 After confirmation of the diagnosis, close cardiovascular follow-up is mandatory in all patients with MFS.
The primary goal of medical and surgical treatment of cardiovascular features in patients with MFS is to reduce the risk of often fatal aortic dissection. Currently, standard medical treatment of aortic disease in patients with MFS is limited to the administration of β-adrenergic receptor blockers (β-blockers) and/or angiotensin receptor blockers (ARBs), and prophylactic aortic root surgery when indicated. Although improved (early) diagnosis and optimization of treatment have substantially increased life expectancy over the past 40 years, fatal aortic dissections still occur in patients with MFS.16–18 Hence, there is a need for novel insights into the underlying pathophysiological mechanisms of MFS, which in turn could contribute to improved diagnostics and better clinical management of patients with MFS. Today, multiple studies conducting research into the pathophysiological processes of Marfan disease are moving beyond the use of antihypertensive agents in the search for new treatment strategies.
Aims of This Review
In this review, we will summarize the currently available and experimental drug treatments in MFS. Considering that cardiovascular manifestations mainly determine the life expectancy in patients with MFS, we will focus on the treatment of cardiovascular disease. First, in the light of discussing potential targets for therapy, we will briefly highlight the pathophysiological mechanisms of MFS. Second, we will review the current knowledge on medical treatment of patients with MFS. Finally, we will discuss different experimental treatment strategies being evaluated in preclinical research, predominantly using murine animal models.
Mouse Models for Marfan Syndrome
Over the past two decades, mouse models have been used extensively to study the effects of fibrillin-1 deficiency and have led to many new insights into the molecular mechanisms that drive MFS pathology. Multiple mouse models of MFS have been generated, characterized by various genetic mutations of the gene coding for fibrillin-1, leading to a range of severity in phenotypic presentation (Table 1). The most widely used models to study MFS-related pathologies are the Fbn1C1039G/+ and Fbn1mgR/mgR mouse models. The heterozygous Fbn1C1039G/+ mouse model carries a missense mutation affecting a conserved cysteine reside (Cys1039Gly), and presents a mild but classic MFS phenotype.19 Although these mice have a normal life span, they display cardiovascular manifestations including aortic dilatation, fragmentation of the elastic fibers, and MVP. Homozygous Fbn1mgR/mgR mice, on the other hand, display an early onset and severe MFS phenotype with an average lifespan of less than 4 months due to premature mortality caused by aortic rupture.20 The Fbn1mgR/mgR mouse model is a hypomorphic model, with an approximately 5-fold reduced expression of wild-type fibrillin-1 due to the introduction of a neomycin resistance gene in the genomic Fbn1 locus. Apart from severe aortic dilatation and elastic fiber fragmentation, initial reports showed that these mice also show left ventricular dilatation and systolic dysfunction.21 On the other hand, recent results from our group did not show any dilated cardiomyopathy in these mice, but instead identified spontaneous right ventricular pseudoaneurysms as a new previously unreported cardiac manifestation in the Fbn1mgR/mgR model.22 The discrepancy between both studies can likely be explained by a difference in genetic background, which has previously been shown to strongly affect the clinical phenotype in a related MFS mouse model.23 Table 1 provides an overview of the genetic backgrounds in which the MFS mouse models were originally generated when their cardiovascular phenotypes were first described. If for a specific follow-up experiment discussed in this review the genetic background was different from that listed in Table 1, it will be indicated in the text. Interestingly, MFS mice with a C57BL/6 genetic background seem less susceptible to TAA, AAA and ventricular dysfunction compared to 129 mice.22–26 These observations underscore the importance of taking into account the effects of potential genetic modifiers, which might complicate the interpretation of results obtained in specific inbred mouse strains.
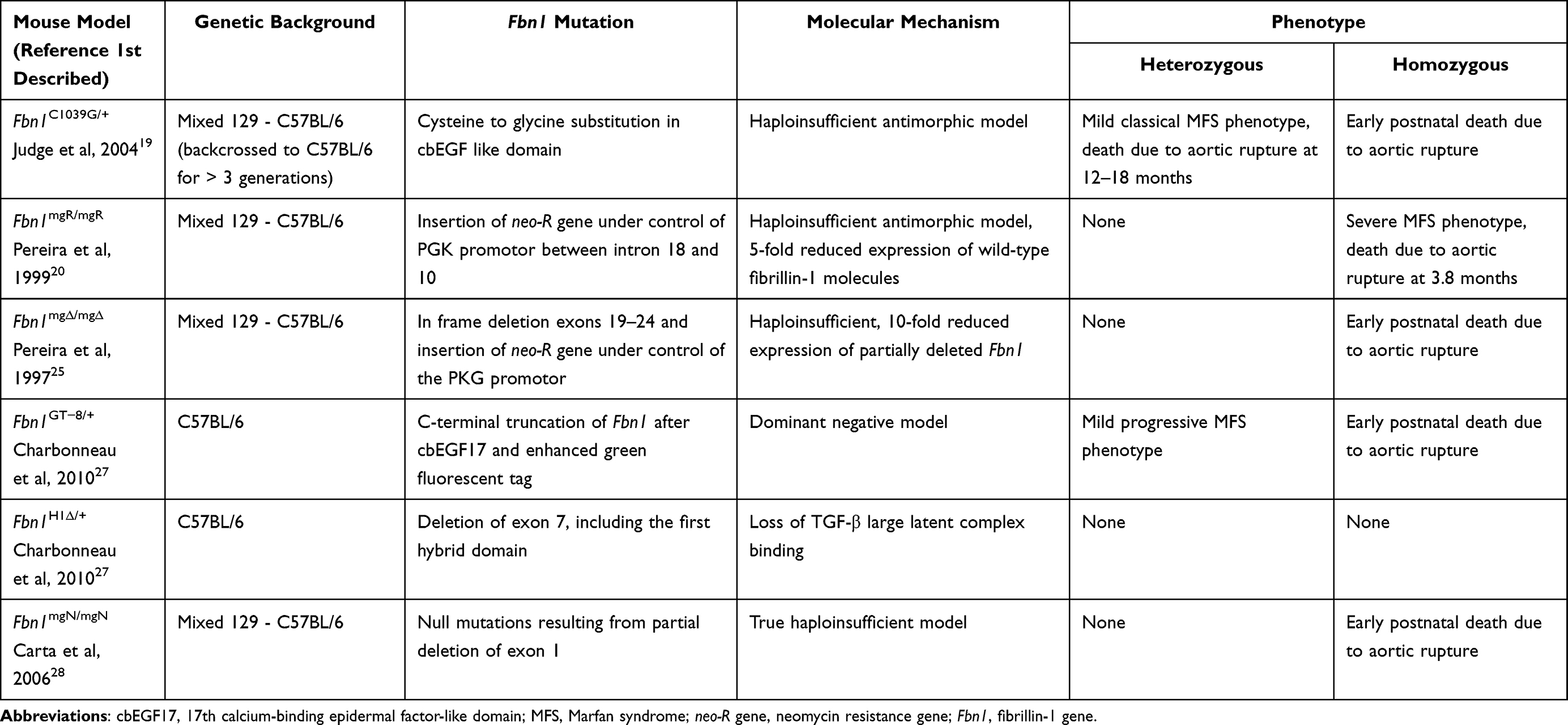 |
Table 1 Murine Models for Marfan Syndrome |
The Fbn1mg∆/mg∆ mouse model is another antimorphic model, caused by an in-frame deletion of exons 19–24 and insertion of the neomycin resistance gene.25 Fbn1mg∆/mg∆ mice have a 10-fold reduced expression of partially deleted fibrillin-1 and show early postnatal mortality due to aortic rupture. The heterozygous Fbn1GT−8/+ mouse model is a dominant-negative model characterized by a C-terminal truncation, followed by a fusion of the green fluorescent protein reporter to track the localization of the mutant fibrillin-1 protein.27 Similar to the Fbn1C1039G/+ mouse model, Fbn1GT−8/+ mice have a normal lifespan but display a progressive MFS cardiovascular phenotype. Homozygous Fbn1GT−8/GT−8 mice, on the other hand, all die within the first weeks after birth. This correlates with the early postnatal death observed in other homozygous Fbn1-deficient mouse models, including the Fbn1mgN/mgN model, which carries a partial deletion of exon 1 resulting in a null mutation.28 Fbn1H1∆/H1∆ mice carrying a homozygous deletion of the first hybrid domain, which contains the binding site for LTBP, however have a normal life span and do not seem to display a cardiovascular phenotype.27
Pathophysiology of Marfan Syndrome
Several pathophysiological models have been proposed to explain the cardiovascular phenotypes of MFS, and have been under investigation as potential targets for treatment. A summary of the cardiovascular manifestations encountered in MFS and the pathophysiological mechanisms involved is presented in Figures 1 and 2.
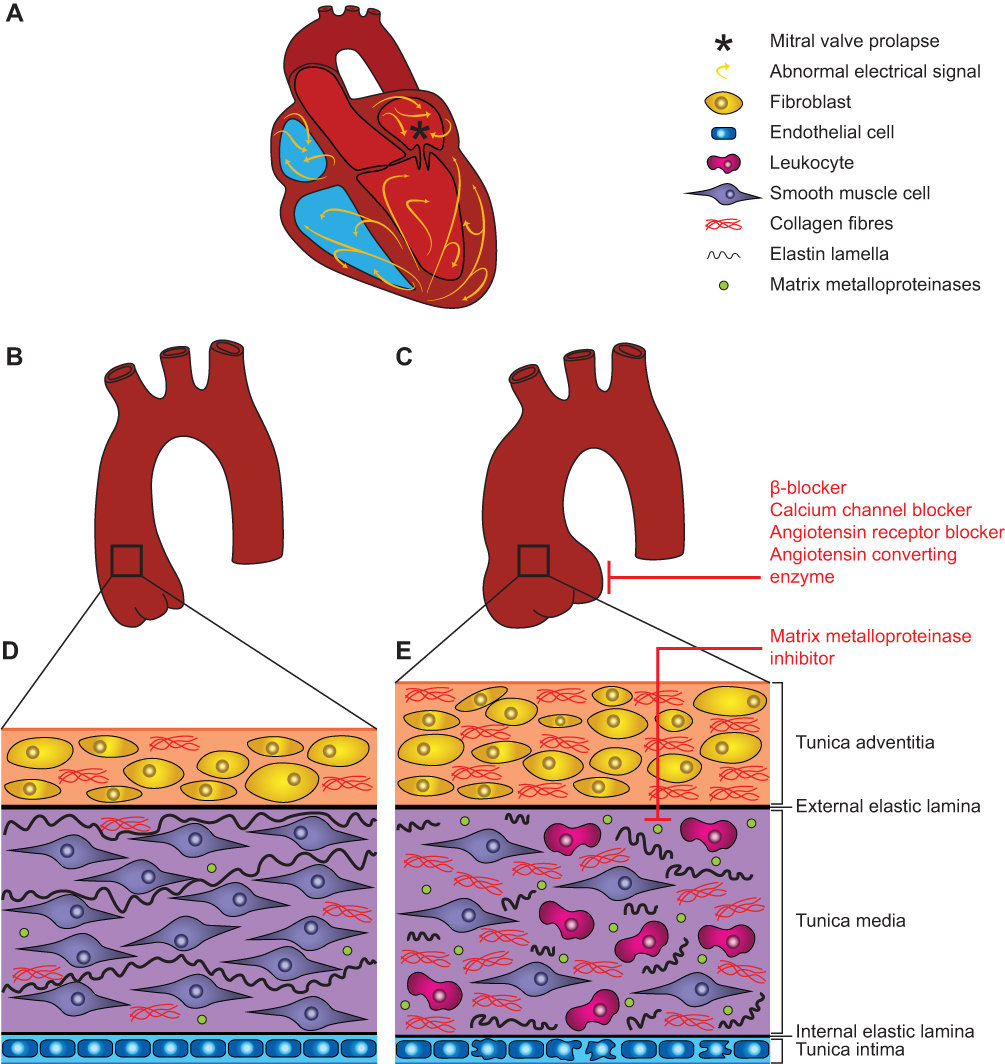 |
Figure 1 Pathophysiology of aortic and cardiac manifestations in Marfan syndrome. (A) Cardiovascular manifestations in the MFS heart include mitral valve prolapse (indicated by *), ventricular dysfunction and arrhythmia. Except for the aorta, the large blood vessels have been omitted from this scheme for clarity. (B) Healthy aorta. (C) Aortic aneurysm development in a MFS aorta, increasing the risk for dissection and rupture. (D) Aortic wall architecture of healthy tissue. (E) Aortic wall architecture of MFS diseased tissue, characterized by adventitia thickening, increased collagen deposition, smooth muscle cell apoptosis, elastic fiber fragmentation, increased MMP expression, leukocyte infiltration and endothelial dysfunction. Therapeutic approaches for the treatment of cardiovascular manifestations in MFS are indicated in red and discussed in the manuscript. |
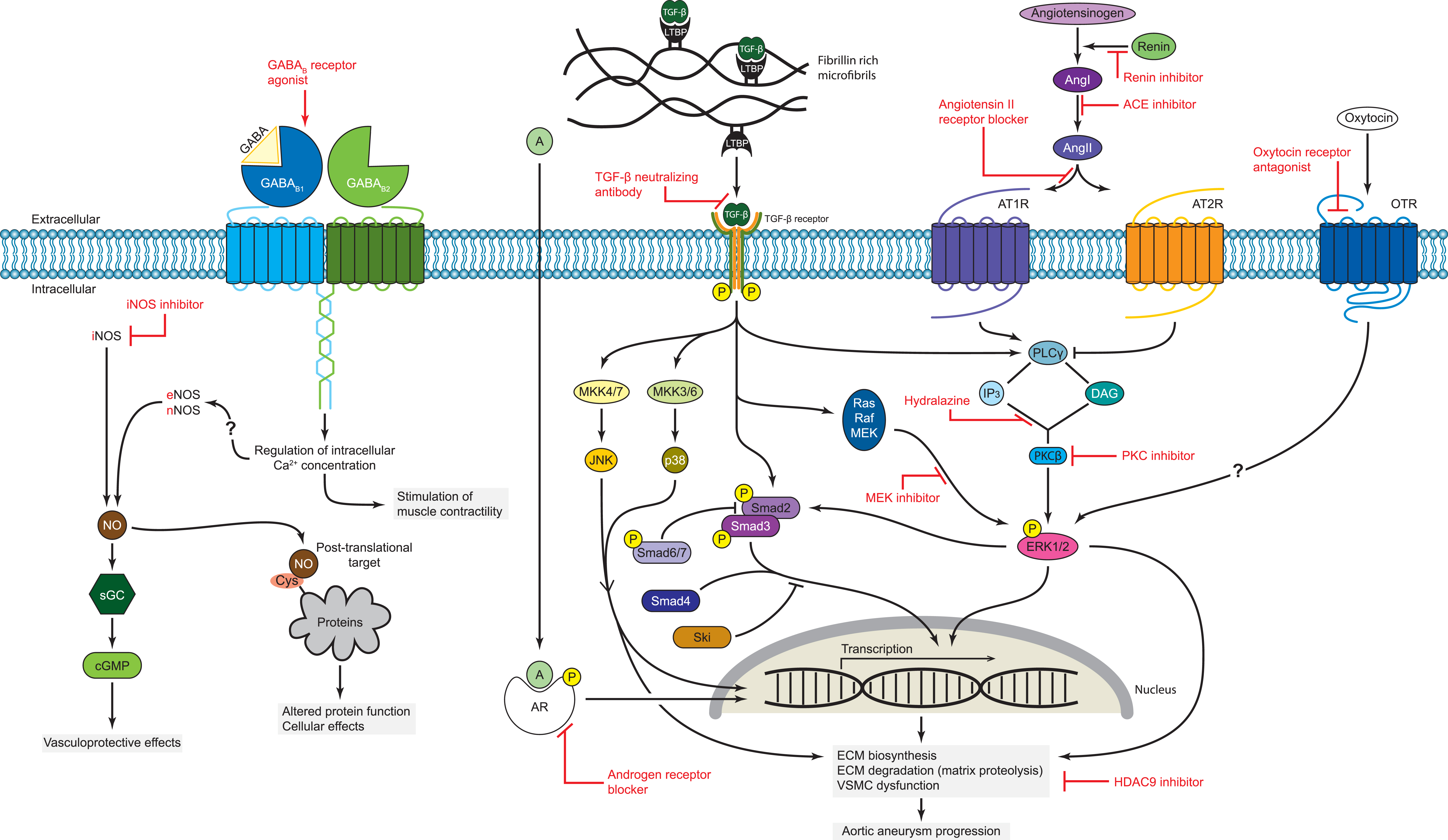 |
Figure 2 Proposed molecular mechanisms involved in MFS pathology and potential treatment targets. Mechanisms for which experimental evidence exists include: (1) increased bioavailability of TGF-β and binding to its receptor, activating both the canonical Smad-dependent as well as the noncanonical ERK1/2 signaling pathways; (2) activation of AT1R-dependent pathways; (3) crosstalk between RAS- and TGF-β-mediated signaling pathways; (4) JNK/p38-MAPK signaling activity; (5) oxytocin-mediated ERK1/2 signaling; (6) GABAB receptor signaling; (7) androgen-induced ERK1/2 and Smad2 signaling; (8) increased iNOS expression levels; (9) increased HDAC9 expression levels. Specific pharmacological approaches for the treatment of cardiovascular manifestations in MFS are indicated in red and discussed in the manuscript. |
Microfibril Structure
When pathogenic variants in FBN1 were discovered as the cause of MFS, the generally accepted hypothesis was that structural defects in fibrillin-rich microfibrils lead to the observed manifestations of the disease. FBN1 mutations alter the structural properties of microfibrils in connective tissues, which will affect elastogenesis, leading to a weakening of connective tissue support. This structural weakness has been proposed as a direct cause of the aortic aneurysms in MFS.20,25,29,30 When the increasing wall stress exceeds the capacity of the weakened aortic wall, aortic dissection or rupture can occur. A typical histopathological finding of the aortic wall observed in patients with MFS is medial degeneration, which is an indication of the underlying aortic pathology.31,32 Cystic medial degeneration of the aortic wall in MFS is characterized by accumulation of basophilic ground substance with cyst-like lesions and extracellular matrix (ECM) degradation. The latter includes fibrosis, proteoglycan accumulation, vascular smooth muscle cell (VSMC) apoptosis, and elastic fiber disorganization and fragmentation.31–33 This concept of weakened tissue being the predominant factor in MFS pathogenesis does not explain many other MFS-related manifestations and might be an oversimplification, especially in light of the additional molecular mechanisms, which have been discovered later and are discussed below.
Further research, meanwhile, has shown that fibrillin-1 also plays an important role in mechanotransduction from the endothelium and ECM to the VSMCs.34 It has previously been suggested that abnormalities in mechanotransduction and mechanosensing contribute to the pathophysiology in MFS.21,34–37 This hypothesis is based on the concept that weakness of the aortic wall is caused by abnormal mechanical homeostasis. Defects in the mechanosensing abilities of intramural cells affect their ability to appropriately regulate ECM production and turnover in response to changes in their environment.37,38 Abnormalities in (i) matrix stiffness, (ii) transmembrane receptors of ECM components, or (iii) cytoskeletal structures (including the actomyosin apparatus), or in the signaling ability to express and assemble these 3 elements, are thought to affect the mechanical stimuli, sensing of these stimuli and/or (resulting) mechanosignaling, which in turn can lead to thoracic aortic aneurysm formation. These concepts have been reviewed extensively by Humphrey et al.34 In addition, ECM-induced abnormal mechanosignaling by cardiomyocytes can in turn give rise to dilated cardiomyopathy and ventricular dysfunction, regardless of valvular pathology.12,21,39,40 Disruption of cardiac microfibril structures due to fibrillin-1 deficiency has been suggested to lead to a loss of myocardial compaction, which is believed to affect myocardial signal conduction, leading to the development of arrhythmias.22,41–43
Transforming Growth Factor-β
An important discovery leading to a better understanding of the mechanisms involved in MFS pathophysiology is that fibrillin-1-rich microfibrils can bind the latent form of transforming growth factor-β (TGF-β).44–46 As such, dysfunctional fibrillin-1 can lead to a reduced capacity of the latent TGF-β binding protein (LTBP) to sequester TGF-β in the ECM, resulting in an increased TGF-β release in the ECM. Upon release and activation, TGF-β binds the TGF-β receptor type II (TβR2), a transmembrane serine/threonine kinase located at the cell surface, which is encoded by the TGFBR2 gene. In turn, TβR2 recruits and activates TGF-β receptor type I (TβR1), encoded by the TGFBR1 gene. Subsequently, activated TβR1 initiates the canonical and multiple non-canonical TGF-β signaling pathways.47–49
The canonical TGF-β signaling pathway, also known as the small mothers against decapentaplegic (Smad)-dependent TGF-β pathway, is initiated when TβR1 phosphorylates the receptor-regulated Smads (R-Smads), ie Smad2 and Smad3. In turn, the R-Smads form a trimeric complex with Smad4, which translocates to the nucleus, where it binds TGF-β responsive DNA sequences and regulates target gene expression.50,51 Inhibitory Smads (I-Smads), ie Smad6 and Smad7, provide a negative feedback loop to regulate TGF-β signaling.52
Besides the Smad-dependent TGF-β pathway, TGF-β activation can also induce other non-canonical downstream signaling cascades, ie mitogen-activated protein kinase (MAPK) cascades. These include the c-Jun N-terminal kinase (JNK), extracellular regulated kinase-1 and 2 (ERK1/2), and p38 MAPK pathway.49 Both canonical and non-canonical TGF-β signaling pathways are known to highly interact with each other and mutually regulate each other’s activity or expression levels.53 Monitoring the status of both canonical and non-canonical TGF-β signaling cascades is usually performed by analysis of Smad2/3 or ERK1/2 phosphorylation levels, respectively.
Elevated TGF-β-induced signaling can cause further ECM degradation through increased transcription of matrix metalloproteinases (MMPs) and inflammatory genes, degradation of elastic fibers, excessive collagen secretion and deposition, and increased proliferation and migration of VSMCs.54–60 Based on these new insights, it has been proposed that the aortic aneurysm development in MFS is caused by altered regulation of TGF-β signaling as a result of impaired sequestration by mutant fibrillin-1.6,61–63
Seminal findings from animal studies confirmed the hypothesis that MFS is associated with excessive activation of TGF-β signaling, leading to the consideration of TGF-β antagonism as a possible treatment strategy for MFS.54,64–67 Indeed, the treatment of the Fbn1C1039G/+ MFS mouse model with anti-TGF-β antibodies resulted in the amelioration of pulmonary emphysema and the rescue of the mitral valve phenotype.54,66 Moreover, Habashi et al demonstrated that aortic aneurysm development was prevented by treatment with TGF-β neutralizing antibodies.67 Mice that were treated postnatally, after aortic root aneurysm development, displayed normalized aortic root growth rate and elastic fiber fragmentation in the aortic wall. However, further research has indicated that the timing of TGF-β neutralization is of fundamental importance. Another study using the Fbn1mgR/mgR MFS mouse model showed that TGF-β neutralizing antibodies only had a beneficial effect when treatment was started after onset of the aneurysm development, whereas aortic aneurysm formation was accelerated when treatment was started prior to the onset of aneurysm development.68 Moreover, basal TGF-β signaling in VSMCs has been found to be beneficial to preserve postnatal aortic wall homeostasis and to limit aortic aneurysm progression in Fbn1C1039G/+ mice.65,69 These results again highlight the complex role of TGF-β signaling in the aortic disease progression of MFS and suggest a dual role for TGF-β demonstrating a protective role in the early developmental stages before aortic aneurysm development and a more detrimental role in later stages as a contributor to aorta pathology. On the other hand, TGF- β has been identified as a potential mediator of myxomatous mitral valve disease in MFS. Mitral valves of Fbn1C1039G/+ mice displayed significantly increased TGF-β activity and signaling, thereby suggesting a correlation to the mitral valve pathology in MFS. In fact, in vivo TGF-beta antagonism was able to rescue the mitral valve phenotype, ie reducing the mitral valve length and thickness.66
Adding to the complexity of TGF-β signaling in MFS is the fact that different isoforms of the ligand exist. Most studies do not discriminate between the three isoforms of TGF-β (ie TGF-β1, TGF-β2, and TGF-β3), even though they may exert differential effects on aortic disease. As such, Tgfβ2 haploinsufficiency in Fbn1C1039G/+ MFS mice resulted in a significantly worse aortic phenotype compared to either Fbn1C1039G/+ mice or Tgfβ2± mice at 2 and 4 months of age, which coincided with an accumulation of significantly increased Tgfβ1 expression in the proximal aorta.70 Taken together, these data highlight the need to take TGF-β isoform-specific effects on aortic disease progression into account and suggest that TGF-β2 might have protective rather than pathogenic effects in MFS.
Interestingly, a recent study by Hara et al reported on the role of myeloid cell-specific TGF-β signaling in the disease progression of MFS.71 Myeloid-specific deletion of Tgfbr2 in Fbn1C1039G/+ mice attenuated aortic aneurysm development, decreased aortic medial thickening and reduced elastic fiber fragmentation in the aortic wall. Moreover, ablation of Tgfbr2 in myeloid cells reduced macrophage infiltration into the aortic wall of MFS mice, and attenuated phosphorylation of Smad2/3 and ERK1/2. Continuous TGF-β stimulation of in vitro cultured macrophages was shown to enhance their capacity to migrate, but not to proliferate. Taken together, these data indicate that TGF-β-driven Smad2/3 and ERK1/2 activation in myeloid cells plays an important role in promoting aortic aneurysm development in MFS, and is not essential in normal aortic development. Therefore, these findings suggest that cell type-specific inhibition of TGF-β signaling in myeloid cells might be a potential new treatment option that can be considered for MFS.
The Renin-Angiotensin System
Substantial evidence from both preclinical and clinical studies revealed the involvement of the renin–angiotensin system (RAS) in aortic aneurysm pathogenesis. The RAS is known to be a key regulator of the cardiovascular system. It plays a crucial role in regulating blood pressure, extracellular fluid homeostasis and electrolyte balance. Impaired function of the RAS can lead to the development of a number of cardiovascular diseases, usually associated with renal disease.72,73 Angiotensin II (Ang II) is the main effector molecule of the RAS, which mediates vasoconstriction, renal water and salt retention, fibrosis and inflammation through stimulation of the Ang II type 1 receptor (AT1R). In contrast, the Ang II type 2 receptor (AT2R) is thought to function as an endogenous antagonist of the AT1R, which mediates vasodilation, natriuresis, and suppression of fibrosis and inflammation.72 Although evidence points to a detrimental role of AT1R-dependent signaling, the exact role of AT2R in aortic pathology is less well established.
Stimulation with high levels of exogenous Ang II is commonly used as a trigger for abdominal aortic aneurysm development in mouse models, leading to a cascade of events with macrophage accumulation in the vascular wall, resulting in medial elastin degeneration, structural weakening of the aortic wall, dissections and vascular hematomas.74,75 Interestingly, Ang II infusion can also trigger thoracic aortic pathology.76 While the pathogenic mechanisms likely involve local immune cell recruitment, in vitro data also suggest a role of increased oxidative stress due to activation of the NADH/NADPH oxidase system.77 Observations in MFS suggest that Ang II also leads to increased TGF-β-dependent activation of MMPs, resulting in medial degradation.64,78,79 Increasing evidence suggests that the RAS interacts with TGF-β signaling pathways on multiple levels in MFS.55,78,80,81 Several studies have suggested that increased RAS activity can lead to stimulation of TGF-β gene expression as well as activation of latent TGF-β protein.82,83 A summary of the role of TGF-β–RAS mechanisms in aortic pathology and their clinical implications has recently been reviewed by van Dorst et al.65
Matrix Metalloproteinase
Several lines of evidence have shown that aortic aneurysm development in MFS is correlated with increased expression of MMPs in the vascular wall.84,85 These enzymes are involved in the degradation of matrix and non-matrix proteins in the extracellular space, and are involved in the regulation of both inflammatory and normal physiological processes.86 MMPs are zinc endopeptidases that are capable of degrading the ECM surrounding cells, thereby affecting various cellular processes, such as cell proliferation, signaling, differentiation, cell–cell interactions, migration, and death.87–89 ECM degradation in the aorta is associated with extensive elastic fiber degeneration, endothelial dysfunction, reduced smooth muscle contractility and modified mechanical properties of the aorta, therefore strongly contributing to the development of aortic aneurysms.90–93 More specifically, aortic degradation in MFS has been associated with increased aortic VSMC expression of MMP-2 and MMP-9.94,95 Interestingly, while MMP upregulation is induced by non-canonical signaling downstream of the TGF-β receptor,89,96 MMP activity further induces TGF-β activation,97,98 creating a positive feedback loop. MMP-mediated aortic damage and aneurysm development are believed to be caused by an imbalance between MMPs and their endogenous inhibitors (TIMPs).85,99 Reduced TIMP-3 protein expression levels have been observed in the aortae of patients with MFS compared to controls, hereby suggesting a distinct MMP/TIMP profile in MFS.85
Nitric Oxide Signaling
Recent evidence has emerged to implicate nitric oxide (NO) signaling in MFS aortic pathophysiology. NO is an endogenously produced gasotransmitter, which has a large range of physiological effects, particularly in the cardiovascular system.100 The discovery that NO produced by endothelial cells can lead to smooth muscle relaxation by activation of cyclic guanosine monophosphate (cGMP) production by soluble guanylate cyclase (sGC) was awarded with the 1998 Nobel Prize in Physiology or Medicine.101 NO is produced by three isoforms of the NO synthase (NOS) enzyme: two of which are present constitutively and are named according to their original source of purification, neuronal NOS (nNOS) and endothelial NOS (eNOS), and one which is an inducible NOS (iNOS) enzyme.102 Despite their original nomenclature, both nNOS and eNOS have been shown to be present and often co-expressed in multiple other cell types, including cardiomyocytes and skeletal myocytes.
The current concept is that NO produced by the constitutive nNOS and eNOS isoforms has protective effects, while large amounts of NO produced by iNOS in inflammatory conditions can cause cell and tissue damage due to increased nitrosative stress. Increased iNOS levels have been found in aortic sections of MFS patients and animal models,103–105 and are often associated with increased oxidative stress markers in this tissue. Studies on animal models indicate that iNOS contributes to the aortic phenotype of MFS, potentially by decreasing vascular smooth muscle cell contractility.103,104 This is consistent with the observation that a gain-of-function mutation leading to persistent activation of cGMP-dependent protein kinase 1, a downstream target of NO regulating myosin function, leads to familial thoracic aortic aneurysms.106
Countering the proposed detrimental effects of iNOS-derived NO, a provocative study by Sellers et al showed that losartan does not exert its beneficial effects in an MFS mouse model via activation of the AT1R, but rather through activation of NO production by eNOS.107 Genetic or pharmacological eNOS activation significantly reduced aortic root and ascending aorta dilatation in the MFS mouse model, suggesting that NO produced by this enzyme has protective properties. Considering that most vasculoprotective effects of NO can be attributed to activation of sGC-dependent signaling,108 the authors next tested whether increasing cGMP levels by sildenafil-induced phosphodiesterase inhibition could improve the MFS phenotype. This strategy failed to inhibit aortic root dilatation and even led to worsened elastic fiber degradation, even though the lung emphysema phenotype was prevented.109 While it is difficult to reconcile both studies, it is possible that the sildenafil experiment was not performed under ideal conditions since the authors observed a paradoxical rise in blood pressure.
Another aspect of NO-dependent signaling is the sGC-independent, direct post-translational modification of specific protein targets. Previous studies have highlighted the importance of protein S-nitrosylation, a process tightly regulated by enzymatic denitrosylating mechanisms, in a number of cardiovascular effects of NO.110,111 It has already been shown that, depending on the specific target modified, protein S-nitrosylation has important effects on aortic physiology, affecting vascular smooth muscle contractility112,113 or even leading to increased aneurysms and dissections due to endothelial barrier dysfunction.114
Taken together, the presently available evidence suggests that NO is a double-edged sword in MFS aortic pathology, having both potentially detrimental and protective effects depending on the situation. This concept has been observed previously in different aspects of NO physiology,115,116 and complicates the interpretation of the exact role of this gasotransmitter in disease etiology.
Inflammation and Oxidative Stress
Data from mouse models as well as from the clinic have shown that inflammation and oxidative stress play an important role in the pathophysiology of MFS.117 A study by He et al on biopsies from patients with MFS undergoing elective aortic root repair demonstrated infiltration of immune cells into the aortic media and adventitia.118 A significant influx of T cells and macrophages was found in the aortic segments. A similar observation was made in samples from patients who underwent surgery for aortic root aneurysm but who did not have MFS.119 He et al also demonstrated that inflammatory cell infiltration, as well as activation of apoptotic pathways, was greater in the aortic media of patients who had an aortic dissection than those who only had an aneurysm.120 Later studies confirmed an increased influx of CD4+ T-cells in the media and CD8+ T-cells in the adventitia of patients with MFS, as well as increased levels of inflammatory markers.58 Based on these data, immune cell recruitment and inflammation were identified as a possible contributor to smooth muscle cell apoptosis and medial degradation in the aortic wall during the development of aortic aneurysms and dissection. Although the role of inflammation and the immune response has been extensively studied in abdominal aortic aneurysms, little is known about their contribution to the pathogenesis of thoracic aneurysms in MFS. It would be interesting to elucidate the exact role of initiation and propagation of the inflammatory response in the development of aortic aneurysm and the risk of dissection. Interestingly, immune cell infiltration has also been associated with myxomatous valve degeneration (MVD) in MFS.121 A recent study by Kim et al demonstrated that diseased mitral valves of Fbn1C1039G/+ mice showed both increased infiltrating and resident macrophages, as well as elevated chemokine activity and inflammatory ECM modification. Mitral valve specimens from human patients with MFS similarly showed increased presence of monocytes and macrophages. Deletion of CCR2 in Fbn1C1039G/+ mice, leading to loss of monocyte recruitment, ameliorated MVD progression resulting in preserved matrix integrity and minimal leaflet thickening. Taken together, these data suggest monocyte infiltration as a potential therapeutic target for the prevention of MVD progression in patients with MFS.
Reactive oxygen species (ROS), likely produced in pro-inflammatory conditions, also contribute to aortic pathology in MFS. It is known that ROS lead to weakening of the aortic wall by increasing expression and activity of MMPs and inducing VSMC apoptosis.122,123 Increased oxidative stress was observed in the wall of aortic root tissues in both patients with MFS and mouse models.104,124,125 An imbalance between pro-oxidant and antioxidant enzymes is proposed to play a prominent role in increasing ROS levels. The levels of pro-oxidant enzymes, including NAD(P)H oxidase (NOX), xanthine oxidase, and iNOS were upregulated in aortic tissues of Fbn1C1039G/+ mice, while protein expression of the antioxidant superoxide dismutase (SOD) was decreased.104 Interestingly, NOX4, which is strongly induced by TGF-β,126 was shown to contribute to aortic aneurysm development in a mouse model of MFS. Genetic deletion of NOX4 in this mouse model resulted in a significant reduction in aortic elastic fiber fragmentation, aortic root growth and endothelial dysfunction.125 Taken together, these data suggest that antioxidant treatment might offer a potential strategy to improve aortic disease in MFS.
Current Medical Management
Medical treatment in MFS aspires to limit aortic growth rate, thus preventing or delaying the need for surgical interventions and fatal complication. Currently, the first-line medical treatment of MFS consists of β-blockers and/or ARBs. Multiple studies were aimed at comparing the effect of both drugs – either head-to-head or in protocols comparing the combination versus β-blockers alone. Overall, the effect of both drugs is comparable, and some studies suggest a combined regimen is beneficial.64,79,127,128 Dosing of the drugs is done on an individual basis and will be different for each compound, taking heart rate (in case of β-blockers) and blood pressure as well as symptoms into account. With beta-blockers, dosage should be titrated to maximum effect, typically to a resting heart rate of <60 bpm if blood pressure allows.129 Blood pressure thresholds are typically kept slightly below conventional values (130/85 mmHg). Available medical treatment does not provide a cure for MFS and predominantly serves to delay progression of cardiovascular manifestations. As such, the development of more effective medical treatment options remains a major focus of research.
β-adrenergic Receptor Blockers
The rationale for using β-blockers to reduce aortic root growth and prevent aortic dissection in patients with MFS is based on the knowledge that these drugs have the capacity to reduce hemodynamic stress in the proximal aorta. Currently, the β-blocker atenolol is most commonly used to treat pediatric patients with MFS, while atenolol, metoprolol, and bisoprolol are used in adult patients. This treatment strategy traces back to the 1970s, when it was shown that reducing the maximum acceleration rate of aortic pressure increase (dP/dt) was more efficient than lowering blood pressure alone in limiting the development of aortic dissection.130–132 A randomized control trial in 1994 first demonstrated the beneficial effects of using β-blockers in patients with MFS.133 Although this study concluded that propranolol treatment aided in slowing down the rate of aortic growth, it did not conclusively demonstrate an impact on dissections or mortality. Since then, multiple studies have described the beneficial effect of β-blockers in MFS.16,79,134–141 Nevertheless, the exact mechanisms by which this treatment contributes to a beneficial effect in MFS are still unknown and the efficacy of β-blocker therapy is not consistent in all studies. Furthermore, a recent systematic review highlighted the lack of solid evidence to support the efficacy of β-blocker treatment in reducing mortality or morbidity, including aortic dissection, heart failure, and prophylactic surgery.127 In addition, a number of side effects are known to occur with the use of β-blockers, including bronchospasms, bradyarrhythmia, fatigue, depression, sexual dysfunction, and mood disturbance.133,142 Therefore, even though β-blockers can be considered as the primary therapy for the prevention of aortic complications in patients with MFS, a significant margin for improvement remains.143–145
Calcium Channel Blockers
Multiple studies have investigated alternatives for β-blockade treatment to treat patients with MFS. Considering the hemodynamic effects of β-blocker treatment, the effects of calcium channel blockers (CCBs), another class of antihypertensive agents, were evaluated for the treatment of cardiovascular manifestations in patients with MFS. Initially, a small clinical study suggested that CCBs had an equally beneficial therapeutic effect compared to β-blockers.146 However, recent reports on MFS mouse models contradicted these findings and demonstrated alarming effects of CCB treatment. Mice treated with CCBs displayed increased rates of aortic aneurysm development, rupture and premature deaths.147 These findings were also observed in retrospective analysis of clinical trials, which assessed the effect of CCBs in patients with MFS compared to β-blocker therapy. Notably, patients with MFS who received CCB treatment were more likely to have acute aortic dissection and aortic surgery.147 Taken together, there is a lack of solid evidence of CCB efficacy in treating Marfan disease and considering the potential detrimental effects CCBs should be avoided in patients with MFS.
Angiotensin Receptor Blockers
Increasing evidence supports the role of pharmacological interference with the RAS as a treatment for aortic aneurysms. One approach to reduce the Ang II–ATR1 interaction of the RAS is the use of ARBs. Habashi et al reported that Losartan, an ATR1 blocker, normalized the aortic root growth rate and restored elastic lamellae fragmentation in the Fbn1C1039G/+ MFS mouse model.67 Remarkably, ATR1 blockade with losartan displayed a full correction of the phenotypic abnormalities in the aortic wall. Shortly afterwards, a small clinical study observed a significant reduction in the rate of progressive aortic root dilatation in pediatric patients with MFS with ARB treatment.81 Subsequently, other studies on different MFS mouse models also highlighted the beneficial effect of losartan in reducing aortic aneurysm formation.68,148–151 Surprisingly, the efficacy of losartan treatment was not consistent amongst murine studies and studies using phenotypically more severe MFS mouse models often displayed a more modest or even no amelioration of the aortic phenotype after administration.68,149,151
The initial success led to several clinical trials in patients with MFS verifying the efficacy of ARBs as monotherapy or in combination with β-blockers, compared to β-blocker treatment alone.128,141,152–157 While losartan is used in most trials, some studies also evaluate the efficacy of the other ARB irbesartan.158,159 Unfortunately, losartan treatment did not present the same level of protection as observed in mice studies, nor could they provide evidence for superiority over β-blocker monotherapy. Various explanations for this difference between mice and men have been suggested, one of which may be dose related.160 The losartan dose given to mice was exponentially higher than the dose given in the human trials—extrapolating this dose to humans would have required potentially lethal doses of >1 g/day.
Interestingly, a recent review by van Dorst et al highlighted the importance of careful interpretation of the results of these clinical trials, considering several limitations of the comparison studies, such as small groups, dosage and start of treatment variances, low statistical power, and lack of long-term follow-ups.65 Nevertheless, recent evidence suggests a potential synergistic effect of combining β-blocker treatment with an ARB in syndromic thoracic aneurysms. After verifying the long-term clinical outcomes of the losartan treatment in patients originally enrolled in the COMPARE trial, a potential clinical benefit of a combined treatment strategy of both ARBs and β-blockers was demonstrated.128,152 These data support the assumption of a favorable combined treatment strategy to limit aortic aneurysm growth and prevent long-term adverse outcomes in patients with MFS. Upcoming data from larger trials should provide more conclusive evidence supporting this hypothesis.161,162 In addition, a prospective collaborative meta-analysis is currently ongoing, based on individual patient data from all previous randomized trials in MFS, including ARB treatments versus placebo (or open-label control) and ARB treatments versus β-blockers.160 This study could provide a more reliable estimate of the effects of the particular treatment and limit bias, as it allows for analysis of the treatment outcomes of ARB treatment in particular patient subgroups. Moreover, it is estimated that this study would provide more statistical power to address the effects of treatment on a composite end point of several clinical outcomes.
One interesting perspective to look at subgroups is based on the underlying genotype. It has been evidenced that the beneficial outcome of losartan treatment varies between genetically classified subgroups of patients with MFS.157,163,164 A pre-defined sub-study of the COMPARE trial152 investigated the effectiveness of losartan treatment on aortic root dilatation in patients with MFS having a FBN1 mutation leading to either haploinsufficiency or to a dominant negative fibrillin-1 effect.157 Losartan treatment in patient with MFS carrying a haploinsufficient FBN1 mutation demonstrated a significant reduction of aortic root growth rate, whereas only a small insignificant reduction was observed in dominant negative patients. Another sub-study of the COMPARE trial,152 verified the effect of losartan on ventricular volume and function in patients with MFS carrying a haploinsufficient or dominant negative FBN1 mutation, without significant valvular regurgitation.163 Similarly to the previous sub-study, biventricular end-diastolic volume and stroke volume were only significantly increased in haploinsufficient patients after losartan treatment, independent of change in blood pressure. Taken together, these data highlight the relevance of genetic categorization of patients with MFS in clinical trials and provide insights for a potentially more beneficial personal-based treatment strategy.
ARBs have been used in cardiovascular medicine for the treatment of other conditions, including heart failure. Recently, a new class of drugs was developed, taking advantage of combined neprilysin inhibition and angiotensin receptor blockade, which has proven very successful in the treatment of heart failure with reduced ejection fraction.165 The efficacy of this new approach relies on the synergistic beneficial effects of accumulation of protective endogenous natriuretic peptides together with inhibition of Ang II signaling. Preliminary data suggest that a low dose of the angiotensin receptor neprilysin inhibitor (ARNI) sacubitril/valsartan can inhibit ascending aorta dilation in the Fbn1C1039G/+ mouse model to a greater extent than the ARB valsartan alone.166 Since the doses of both the ARNI and the ARB were titrated to a level not affecting blood pressure, additional neprilysin inhibition is likely to afford additional protection due to its anti-remodeling effects. A first case report of the use of sacubitril/valsartan in an MFS patient showed that this treatment can improve MFS-related cardiomyopathy.167 Taken together, ARNI treatment could represent a new therapeutic avenue that can be explored for MFS.
Angiotensin Converting Enzyme Inhibitors
Another approach to reduce RAS signaling and potentially abrogate aortic aneurysm development, is to interfere with angiotensin converting enzyme (ACE) activity. ACE inhibitors (ACEIs) block the conversion of angiotensin I to Ang II, subsequently reducing downstream signaling of both AT1R and AT2R. The protective effects of ACEIs in MFS are believed to be attributed to blood pressure control and reduced aortic wall stiffness.64,79,168–170 A first trial of ACEI treatment in patients with MFS provided promising results including improved aortic distensibility and reduced aortic stiffness.139 The results of a follow-up trial showing improved aortic root diameter associated with reduced circulating TGF-β levels were later retracted due to inadequate validation of primary data sources and data misrepresentation.171 Other clinical studies comparing the effect of ACEIs and β-blockers in MFS nevertheless concluded that ACEIs exert a beneficial effect on central aortic pressure, heart rate and aortic growth rate albeit to a lesser extent.140,172
Renin Inhibitor
A small prospective randomized clinical trial was designed to evaluate the potentially added benefit of combining renin inhibition with standard β-blocker treatment, using aortic parameters as a primary readout.173 Patients with MFS receiving the renin inhibitor aliskiren together with atenolol did not show any improvement in aortic diameter or central aortic stiffness compared to patients receiving atenolol alone. In spite of the small number of patients investigated, the data from this study indicate that renin is likely not a suitable target for the treatment of MFS.
Experimental Treatments Tested in Preclinical Studies
Matrix Metalloproteinase Inhibitors
The observation of increased MMP expression in vascular tissue samples taken from aneurysms of the ascending aorta of patients with MFS suggested that inhibition of MMPs could have a potential therapeutic effect on MFS treatment.84,85 Since then, multiple preclinical studies have assessed the effects of doxycycline, a general and nonspecific inhibitor of MMPs, on aortic pathology in MFS mouse models.95,148,151,174–176 Besides directly inhibiting MMP enzyme activity, doxycycline treatment is also highly efficient in reducing aortic MMP-2 and MMP-9 expression levels, thereby delaying aortic aneurysm rupture in Fbn1mgR/mgR and Fbn1C1039G/+ mice by reducing aortic elastic fiber fragmentation.174,175 A recent long-term treatment study demonstrated that doxycycline not only ameliorated aortic pathology in Fbn1C1039G/+ mice but also improved skin elasticity and structural integrity.176 In addition, long-term doxycycline treatment also demonstrated superior efficacy over the β-blocker atenolol in preventing aortic aneurysm development in Fbn1C1039G/+ mice, along with improved endothelial function, elastic fiber properties and overall aortic wall structure.95 Tests of combined administration of losartan and doxycycline provided very encouraging results in both the Fbn1mgR/mgR and Fbn1C1039G/+ mouse models, showing a greater suppression of aneurysm development than either drug treatment alone.148,151 Combination therapy improved elastic fiber structural properties, normalized vascular integrity and cell function, decreased MMP-2 and MMP-9 expression and reduced TGF-β activation. Of note, Fbn1mgR/mgR mice carrying a deletion of the gene coding for MMP-2 demonstrated a prolonged survival, which was associated with decreased activation of TGF-β and downstream signaling pathways including phosphorylation of ERK1/2 and Smad2.151 Interestingly, two small randomized trials testing the effects of 2-week treatment with doxycycline prior to elective abdominal aortic aneurysmal repair demonstrated promising results. Not only was the proteolytical balance improved but vascular inflammation was also reduced, associated with reduced neutrophil contents in the aortic wall.177,178 Taken together, these data suggest that MMPs might play an important role in aortic pathology, potentially warranting future clinical studies testing the efficacy of MMP inhibition in patients with MFS.
Inducible Nitric Oxide Synthase Inhibitors
Based on the elevated levels of aortic iNOS expression in the Fbn1C1039G/+ mouse model as well as in patients with MFS, the inhibition of this enzyme was evaluated as a target to ameliorate the cardiovascular phenotype.103 Genetic or pharmacologic inhibition of iNOS significantly ameliorated the aortic dilatation and elastic fiber fragmentation phenotypes in Fbn1C1039G/+ mice. Importantly, iNOS-specific inhibition by 1400W did not raise blood pressure, which did occur when blocking all NOS isoforms using Nω-nitro-L-arginine-methyl ester (L-NAME). Considering these data, specific iNOS inhibition could be considered as a novel treatment option for the aortic pathology associated with MFS. This treatment might provide beneficial effects by preventing tissue damage and vascular dysfunction due to nitrosative stress caused by local pro-inflammatory conditions in the aortic wall of patients with MFS.117 An earlier study also confirmed the efficacy of 1400W to improve the contractile function of thoracic aorta segments isolated from Fbn1C1039G/+ mice,104 lending further support to the rationale for testing iNOS inhibition for the treatment of MFS. Nevertheless, previous attempts to translate similar findings of protection afforded by (i)NOS inhibition in mouse models of sepsis, septic shock, and cardiogenic shock to the clinic have not been met with great success,179,180 indicating that targeting NO production might not be an ideal strategy, and perhaps targeting upstream or downstream processes might be a more promising approach.
Inhibitors of the Mitogen-Activated Protein Kinase Cascade
The identification of the central role of TGF-β in MFS pathogenesis has led to scrutiny of the downstream signaling pathways as potential targets for the treatment of MFS. While the canonical signaling pathways downstream of TGF-β act via Smad-dependent cascades, noncanonical signaling leads to the activation of MAPK cascades.181 Experimental evidence showed that ERK1/2, as well as JNK activation, was associated with aortic aneurysm formation in the Fbn1C1039G/+ mouse model,169,182 while p38 kinase activity was increased in the, aorta of the Fbn1mgN/mgN model.183 Cardiac ERK1/2 activation has also been shown to be associated with cardiomyopathy in Fbn1C1039G/+ and Fbn1mgR/mgR mouse models.12,21 Interestingly, Fbn1mgR/mgR mice backcrossed on a pure C57BL/6 genetic background, which did not show a similar cardiac phenotype as in the previous study, also did not show ERK1/2 activation.22
Considering the experimental evidence of the detrimental role of ERK1/2 signaling in mouse models of MFS, the therapeutic potential of inhibition of the upstream MAPK kinase (MEK) was further explored. The highly selective allosteric MEK1/2 inhibitor refametinib (also known as RDEA119 or BAY 86–9766) was shown to significantly inhibit aortic root growth in the Fbn1C1039G/+ mouse model, associated with selective inhibition of ERK1/2 activation without affecting JNK or p38 activity.182 Recent improvements in the formulation of refametinib, with confirmed inhibition of ERK1/2 activity in mouse aortic and myocardial tissue, will facilitate future experiments with this inhibitor.184
Via an alternative noncanonical pathway, the TGF-β receptor complex signals through TGF-β-activated kinase 1 (TAK1) to activate the MAPK kinase (MKK) 4/7 and MKK3/6, which are upstream of JNK and p38 kinase, respectively.185 Treatment of Fbn1C1039G/+mice with the SP600125 JNK inhibitor led to a significant reduction of aortic root growth without affecting ERK1/2 signaling activity.182 On the other hand, early postnatal exposure of Fbn1mgN/mgN mice to the p38 MAPK inhibitor FR167653 was shown to lessen the level of activation of Smad2/3 signaling in aortic tissue,183 suggesting that early cross-talk between these pathways might be involved in the development of the aortic phenotype.
Recently, the therapeutic potential of the MAP kinase signaling pathway in MFS was highlighted at a more upstream level.26 Increased expression of MAP3K4, which is a MEK kinase activating both MKK4 and MKK6, was associated with MFS in cultured dermal fibroblasts. In addition, Map3k4 haploinsufficiency in Fbn1C1039G/+ MFS mice significantly ameliorated aortic root growth and was associated with reduced activation of phospho-ERK1/2 and phospho-p38. Moreover, MKK6, an activator of p38 kinase, has also been identified as a strong modulator of aortic aneurysm development in Fbn1C1039G/+ MFS mice.
Taken together, these data reveal great potential in identifying novel specific therapeutic targets upstream of the MAP kinase signaling pathway in MFS treatment. Nonetheless, the translation of these findings from mouse models to a clinical setting remains to be proven, especially considering the fact that the contribution of the noncanonical signaling pathways in human MFS is not yet well established.186
Androgen Receptor Blockers
Sex is a well-known risk factor for cardiovascular disease. Clinical studies from our group and others have shown that sex also strongly influences the outcome of MFS patients, with men having a higher risk for aortic dilatation and aortic events (dissection and need for prophylactic surgery).187–190 Likewise, in the Fbn1GT−8/+ MFS mouse model, our group was the first to demonstrate that male mice have more severe aortic disease, with a greater extent of major elastic fiber fragmentation in the aortic wall and aortic aneurysms, compared to females.187 In vitro experiments showed that human aortic smooth muscle cells increase fibrillin-1 production upon stimulation with the female hormone 17β-estradiol, providing a possible mechanism for the observed sex differences. An independent study confirmed that similar sex discrepancies can also be identified in the aortic phenotype of the Fbn1C1039G/+ MFS mouse model.191 Taken together, these data provide a dual hypothesis: (i) males are more prone to aortic disease, and/or (ii) females exert a protective effect against aortic disease in MFS.
To date, the pathophysiological mechanisms that govern sex differences in MFS are not fully elucidated. A recent study by Tashima et al reported the potential detrimental role of androgens in the aortic phenotype of MFS.192,193 Enhanced phospho-ERK1/2 and phospho-Smad2 signaling was reported in the aortic root and ascending aorta of Fbn1C1039G/+ MFS male mice, which was associated with increased MMP-2 activity. Interestingly, TGF-β-induced phospho-ERK1/2 and phospho-Smad2 signaling was enhanced by dihydrotestosterone and reversed by androgen receptor blocker flutamide in vitro in mouse ascending aorta-derived smooth muscle cells. Administration of flutamide in male Fbn1C1039G/+ mice recapitulated the inhibition of ERK1/2 and Smad2 signaling, associated with reduced aortic aneurysm growth and extracellular matrix remodeling. This study was the first to provide solid evidence for the detrimental effects of androgens on the aortic pathology of MFS. Although the specific mechanisms by which androgens contribute to a worse aortic phenotype in MFS have not yet been clarified, these data suggest a potential link between androgen signaling and ERK activation in aorta pathology. Considering these preclinical results, it might be worthwhile to further explore androgen-induced mechanisms as potential targets to reduce aortic aneurysm progression in male patients with MFS, which might be preferable to direct androgen receptor blockade considering the unwanted side effects.
Oxytocin Receptor Antagonists
Besides the more severe phenotype observed in male mice, pregnancy was also shown to adversely affect aortic disease in female Fbn1GT−8/+ MFS mice.187 Similarly, over the years, multiple clinical studies in patients with MFS have identified pregnancy as a risk factor for aortic dissection.194–199 Pregnant MFS women have an 8-fold greater risk for a type A or B dissection compared to MFS women who were never pregnant, with a significantly increased risk particularly during the immediate postpartum period.200
A recent study by Habashi et al, using the Fbn1mgR/mgR MFS mouse model on a pure C57BL/6 background, reported pregnancy-related increased ascending aortic growth rates and postpartum mortality due to dissections, associated with increased ERK1/2 phosphorylation. Interestingly, these phenotypes were reversed by preventing lactation or administering an oxytocin receptor antagonist during the third trimester and first month postpartum, indicating a detrimental effect of this peptide hormone.201 Inhibition of downstream ERK1/2 activation using the MEK inhibitor trametinib also improved the pregnancy-related phenotypes, indicating that this signaling pathway is an effector of oxytocin activity. Nevertheless, administration of oxytocin agonists was not sufficient to accelerate aortic dissection in never-pregnant or pregnant female Fbn1mgR/mgR mice, indicating that other pregnancy-associated factors besides oxytocin, likely including oxytocin receptor upregulation, are involved in pregnancy-related aortic dissection.
While hemodynamic stress has generally been considered as an important factor in the predisposition for pregnancy-related aortic dissection, the vast majority of dissections do not occur during labor but rather during the postpartum period.202 Currently, the only medical treatment option to slow the aortic root growth rate and potentially reduce the risk of cardiovascular events during pregnancy in MFS consists of β-blocker treatment, as ARBs and ACE inhibitors increase the risk of birth defects.203–205 Interestingly, while administration of the blood pressure-lowering drugs propranolol and hydralazine had previously been shown to suppress aortic root growth in Fbn1C1039G/+ mice,67,147 only hydralazine was able to significantly reduce the ascending aortic growth rate in lactating Fbn1mgR/mgR mice (on a C57BL/6 background).201 This discrepancy is likely the result of the additional inhibition of protein kinase C by hydralazine (see below). Taken together, these results suggest that hemodynamic stress is not an important factor in the risk of postpartum aortic dissection in MFS mice, but rather that specific pregnancy-associated signaling events, including increased oxytocin release leading to ERK activation, play an important role. This insight might lead to the development of novel intervention strategies to reduce pregnancy-related vascular events in women with MFS.
Protein Kinase C Inhibitors
Considering the mounting evidence for a detrimental role of noncanonical TGF-β signaling in aortic pathology in MFS, protein kinase C (PKC), which was previously known to mediate TGF-β expression in human fibroblasts and aortic VSMCs,206,207 was considered as a potential target for the treatment of MFS. Activation of the PKCβ isoform was increased in the aortic root and ascending aorta of Fbn1C1039G/+ mice and was reduced by administration of TGF-β neutralizing antibody or losartan, indicating that this kinase is downstream of TGF-β in MFS mice. Interestingly, the specific inhibition of PKCβ by enzastaurin improved the aortic root growth rate and the aortic wall architecture in the MFS mouse model, associated with decreased ERK1/2 activation.147
As highlighted previously, hydralazine is known to not only have antihypertensive properties by causing smooth muscle relaxation but also to inhibit PKC-mediated ERK1/2 signaling activity in vivo.208,209 Therefore, the beneficial effects of hydralazine on aortic root growth and aortic wall phenotype in Fbn1C1039G/+ mice are thought to be mediated at least partially via inhibition of this signaling cascade.147 This is further supported by the observation that hydralazine but not propranolol could improve the aortic phenotype in lactating Fbn1mgR/mgR mice, even though both drugs caused a similar decrease in blood pressure.201 Taken together, these findings provide evidence for the identification of PKCβ as an important upstream activator of ERK1/2 in mouse models of MFS, suggesting that this kinase might be an alternative therapeutic target for the treatment of aortic pathology in MFS.
γ-aminobutyric Acid Receptor Agonists
γ-aminobutyric acid (GABA) is an important inhibitory neurotransmitter of the central nervous system, which regulates multiple physiological processes.210 It is widely distributed in the brain and peripheral tissues and consists of two classes of receptors: GABAA and GABAB. The former are ligand-gated ion channels, while the latter are G protein-coupled receptors.211 Recent studies have associated GABA receptor agonists with a potential role in attenuating aortic aneurysm progression.212–214 In vitro data suggested that activation of the GABAB receptor in human aortic endothelial cells by the agonist baclofen leads to a transient increase in intracellular calcium concentration and translocation of eNOS.212 Based on these observations the hypothesis was generated that GABAB receptors can modulate aortic NO production and vascular permeability.
A recent preclinical in vivo study identified the GABAB receptor agonist baclofen as a possible therapeutic drug to treat muscle contractility defects, which contribute to aortic aneurysm development in MFS.213 Using transcriptomic data from both Fbn1mgR/mgR MFS mouse (on a pure C57BL/6 background) and human MFS aortic samples, reduced muscle contractility was identified as a common affected subcellular pathway, with baclofen as a predicted treatment to correct the dysregulated transcription profile. Treatment of Fbn1mgR/mgR mice with baclofen indeed demonstrated a reduced aneurysm growth rate, resulting in a significant extension of the median survival rate. Histological analysis of the aortic wall showed normalization of elastic fiber fragmentation, improved medial cellularity and mitigated vessel wall fibrosis compared to vehicle-treated mutant mice. These effects were associated with normalized aortic phosphorylation levels of Smad2 and ERK1/2 in MFS mice.
On the other hand, GABAA signaling has also recently been reported to have therapeutic potential in abdominal aortic aneurysms (AAA).214 GABAA receptor signaling is associated with the regulation of immune and inflammatory responses,215 in particular by stimulating the switch from the pro-inflammatory M1 to the pro-resolving M2 macrophage phenotype.216 A similar immunomodulatory effect of the GABAA receptor agonist topiramate was observed in the aortic wall of Ang II/ApoE−/- mice, while also improving abdominal aorta phenotype.214 Taken together, these studies have identified GABA signaling as a new potential modulator of the pathology in MFS and suggest novel potential therapeutic candidates for drug treatment.
Histone Deacetylase 9 Inhibitors
A recent study showed that patients with MFS and other types of sporadic or familial TAA had increased aortic expression of the epigenetic repressor histone deacetylase 9 (HDAC9), associated with destabilization of the actin cytoskeleton and decreased contractile protein expression in VSMC. In vitro experiments in VSMC indicated that HDAC9 forms a complex with the long non-coding RNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) and the chromatin-remodeling protein Brahma-related gene 1 (BRG1).217 MALAT1 is essential to transport BRG1 and HDAC9 into the nucleus. In turn, the HDAC9-MALAT1-BRG1 complex is recruited to the promoters of VSMC-specific contractile genes where transcriptional repression leads to a deleterious VSMC phenotype.217,218 Repression of contractile genes was associated with trimethylation of histone 3 on lysine 27 (H3K27me3), which is mediated by polycomb repressive complex 2 (PRC2) via its enzymatic subunit EZH2, recruited by the HDAC9-MALAT1-BRG1 complex. Dysregulation of other HDAC isoforms has also been shown in other forms of thoracic as well as abdominal aorta disease,219,220 suggesting that epigenetic mechanisms might play an important role in the regulation of vascular wall homeostasis.
Interestingly, increased co-association of the HDAC9-MALAT1-BRG1 complex was confirmed in aortic VSMCs from MFS Fbn1C1039G/+ mice,217 and deletion of either HDAC9 or MALAT1 resulted in an improved thoracic aortic phenotype in these mice, suggesting that this complex might be a valuable therapeutic target. The beneficial effects of GSK343, a small-molecule inhibitor of the methyltransferase EZH2, on the thoracic aorta phenotype of Fbn1C1039G/+ mice supports this hypothesis.221 Interestingly, while treatment with GSK34 improved aortic architecture and restored contractile protein expression in aortic VSMC, TGF-β signaling activity was not altered as Smad2 and ERK1/2 protein phosphorylation remained unchanged. Altogether, these data offer new possibilities for targeting VSMC dysfunction as potential treatment strategy for MFS.
Current Challenges and Future Directions
Although the protective effects of losartan administration in mouse models of MFS were very promising based on the efficacy of this treatment to prevent aortic aneurysm and dissection,67 the same level of protection was not observed and inconsistent results were obtained when translation of these mouse studies to a clinical setting was attempted.128,141,152–157,159 Several potential reasons can be proposed to explain these discrepancies, which should also be taken into account when attempting to translate the results of other experimental drugs into clinical practice.
First of all, when interpreting the pre-clinical results of losartan treatment, the timing of treatment initiation should be carefully considered. In experimental mouse models of MFS, the efficacy of losartan treatment varies profoundly when administration starts perinatally compared to a later postnatal age. No protective effects were demonstrated when losartan treatment was initiated on postnatal day 21 in the Fbn1mgR/mgR mouse model, while treatment initiation at birth via maternal lactation resulted in a full rescue of the aortic phenotype.151,222 Losartan therapy initiation as late as postnatal day 16, however, still showed efficacy in improving MFS phenotypes.68,149 The effects of losartan have also been tested in a pediatric and young adult patient cohort aged from 6 months to 25 years. While earlier treatment initiation was associated with a greater decrease in aortic root growth, this effect was also present to a similar extent in atenolol-treated patients, and full protection was still not obtained by losartan.154
Importantly, the level of exposure of the ARB used in clinical trials with MFS patients, which is affected by the dosage strategy as well as the half-life of the drug administered, has a significant impact on treatment outcome.156,159 In the AIMS trial, Irbesartan, which has a longer half-life than losartan, demonstrated better outcomes compared to the Marfan Sartan Trial. In addition, it is important to note that the daily dosage of losartan normalized to body weight was much higher in mouse studies than in human trials (by a factor of almost 100). It is therefore possible that sufficient drug exposure levels were not obtained in patients with MFS to be able to observe stronger effects on aortic pathology.
Finally, an important unanswered question is whether the available repertoire of genetically modified mouse models carrying specific mutations in Fbn1 sufficiently represents the range of pathogenic FBN1 variants observed in patients with MFS. This might have an important impact on the translation of findings in mouse models to treatments for patients with MFS, as evidenced by the observation that only patients with haploinsufficient FBN1 variants were responsive to losartan treatment, while patients carrying dominant negative missense mutations were not.157 In addition, several lines of evidence suggest that the genetic background may have a very strong influence on the type and severity of the manifestations of MFS.21–23,223 Considering the inbred nature of the mouse models used in research, caution needs to be exercised when extrapolating preclinical mouse data to genetically diverse human patient populations.
Despite several setbacks, the search for novel treatment targets to cure or to ameliorate cardiovascular manifestations in MFS continues. Recently, new innovative strategies have been implemented to identify novel treatment targets. Hansen et al used an unbiased approach to identify potential biological treatment targets using computation analysis of transcriptomic data derived from both pre-clinical experiments and from clinical trials.213 The goal was to investigate common subcellular pathways involved in MFS pathology and use this information to predict the potential efficacy of repurposed FDA-approved drugs for treating the disease. This approach has already successfully led to the identification of the GABAB receptor as a potential therapeutic target, and might uncover more novel mechanisms in the future.
Another avenue that can be explored for the identification of novel potential treatment targets is to make use of alternative animal models or more advanced in vitro models. Zebrafish models of cardiovascular disease have already proven to be useful tools for successful unbiased in vivo screening of large amounts of pharmacological compounds.224–226 Using a different approach, several studies have shown the feasibility to generate human induced pluripotent stem cell (iPSC)-derived in vitro models of MFS,227–229 representing a big step forward towards the realization of precise medicine for MFS.230 Using novel zebrafish and/or iPSC models of MFS could help to identify new, currently unrecognized drug targets that can be exploited for medical treatment.
Conclusions
Currently, no medical treatment is available to completely cure MFS or to definitively prevent fatal complications. This is likely a consequence of the still incomplete understanding of the complex mechanisms involved in MFS pathophysiology. Nevertheless, research aimed at discovering new treatment strategies is moving forward, and has already led to the discovery of several novel pathophysiological processes. Although experience has taught us that caution needs to be exercised when attempting to translate preclinical data into clinical practice, the new research avenues currently being explored hold promise for the development of future successful treatments of MFS.
Abbreviations
A, androgen; AAA, abdominal aortic aneurysms; ACE, angiotensin converting enzyme; ACEi, angiotensin converting enzyme inhibitor; Ang II, angiotensin II; AR, androgen receptor; ARB, angiotensin receptor blocker; ARNI, angiotensin receptor neprilysin inhibitor; AT1R, angiotensin II type 1 receptor; AT2R, angiotensin II type 2 receptor; BRG1, Brahma-related gene 1; Ca2+, Calcium2+; CCB, calcium channel blocker; cGMP, cyclic guanosine monophosphate; Cys, cysteine; ECM, extracellular matrix; eNOS, endothelial nitric oxide synthase; ERK, extracellular regulated kinase; FBN1, Fibrillin-1; GABA, γ-aminobutyric acid; HDAC9, Histone deacetylase 9; H3K27me3, trimethylation of histone 3 on lysine 27; iNOS, inducible nitric oxide synthase; JNK, c-Jun N-terminal kinase; L-NAME, Nω-nitro-L-arginine-methyl ester; LTBP, latent transforming growth factor-β binding protein; MALAT1, metastasis-associated lung adenocarcinoma transcript 1; MAPK, mitogen activated protein kinase; MEK or MKK, mitogen activated protein kinase kinase; MFS, Marfan syndrome; MMP, matrix metalloproteinase; MVD, Myxomatous valve degeneration; MVP, mitral valve prolapse; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; NOS, nitric oxide synthase; NOX, NAD(P)H oxidase; OTR, oxytocin receptor; PKCβ, protein kinase C β; PRC2, polycomb repressive complex 2; RAS, renin–angiotensin system; ROS, reactive oxygen species; sGC, soluble guanylate cyclase; Smad, small mothers against decapentaplegic; r-Smad, Receptor-regulated smad; SOD, superoxide dismutase; TAK1, TGF-β activated kinase 1; TβR1, TGF-β receptor type 1; TβR2, TGF-β receptor type 2; TGF-β, transforming growth factor-β; TIMP, tissue inhibitor of metalloproteinases; VSMC, vascular smooth muscle cell; β-blockers, β adrenergic receptor blockers.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work.
Funding
This work was supported by the Baillet-Latour Grant for Medical Research and a Concerted Research Action of the Special Research Fund from Ghent University (JDB).
Disclosure
The authors reported no conflicts of interest for this work and declare that the research was conducted in the absence of any commercial of financial relationships that could be construed as a potential conflict of interest.
References
1. von Kodolitsch Y, De Backer J, Schuler H, et al. Perspectives on the revised Ghent criteria for the diagnosis of Marfan syndrome. Appl Clin Genet. 2015;8:137–155. doi:10.2147/TACG.S60472
2. Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337–339. doi:10.1038/352337a0
3. Sakai LY, Keene DR, Engvall E. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol. 1986;103(6 Pt 1):2499–2509. doi:10.1083/jcb.103.6.2499
4. Maslen CL, Corson GM, Maddox BK, Glanville RW, Sakai LY. Partial sequence of a candidate gene for the Marfan syndrome. Nature. 1991;352(6333):334–337. doi:10.1038/352334a0
5. Sakai LY, Keene DR, Renard M, De Backer J. FBN1: the disease-causing gene for Marfan syndrome and other genetic disorders. Gene. 2016;591(1):279–291. doi:10.1016/j.gene.2016.07.033
6. Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005;366(9501):1965–1976. doi:10.1016/S0140-6736(05)67789-6
7. De Backer J, Renard M, Campens L, et al. Marfan syndrome and related heritable thoracic aortic aneurysms and dissections. Curr Pharm Des. 2015;21(28):4061–4075. doi:10.2174/1381612821666150826093152
8. Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476–485. doi:10.1136/jmg.2009.072785
9. Taub CC, Stoler JM, Perez-Sanz T, et al. Mitral valve prolapse in Marfan syndrome: an old topic revisited. Echocardiography. 2009;26(4):357–364. doi:10.1111/j.1540-8175.2008.00825.x
10. De Backer JF, Devos D, Segers P, et al. Primary impairment of left ventricular function in Marfan syndrome. Int J Cardiol. 2006;112(3):353–358. doi:10.1016/j.ijcard.2005.10.010
11. Rybczynski M, Koschyk DH, Aydin MA, et al. Tissue Doppler imaging identifies myocardial dysfunction in adults with Marfan syndrome. Clin Cardiol. 2007;30(1):19–24. doi:10.1002/clc.3
12. Campens L, Renard M, Trachet B, et al. Intrinsic cardiomyopathy in Marfan syndrome: results from in-vivo and ex-vivo studies of the Fbn1C1039G/+ model and longitudinal findings in humans. Pediatr Res. 2015;78(3):256–263. doi:10.1038/pr.2015.110
13. Demolder A, von Kodolitsch Y, Muino-Mosquera L, De Backer J. Myocardial function, heart failure and arrhythmia in Marfan syndrome: a systematic literature review. Diagnostics (Basel). 2020;10(10):751. doi:10.3390/diagnostics10100751
14. Hetzer R, Siegel G, Delmo Walter EM. Cardiomyopathy in Marfan syndrome. Eur J Cardiothorac Surg. 2016;49(2):
15. Hoffmann BA, Rybczynski M, Rostock T, et al. Prospective risk stratification of sudden cardiac death in Marfan’s syndrome. Int J Cardiol. 2013;167(6):2539–2545. doi:10.1016/j.ijcard.2012.06.036
16. Silverman DI, Burton KJ, Gray J, et al. Life expectancy in the Marfan syndrome. Am J Cardiol. 1995;75(2):157–160. doi:10.1016/S0002-9149(00)80066-1
17. Pyeritz RE. Marfan syndrome: 30 years of research equals 30 years of additional life expectancy. Heart. 2009;95(3):173–175. doi:10.1136/hrt.2008.160515
18. Vanem TT, Geiran OR, Krohg-Sorensen K, Roe C, Paus B, Rand-Hendriksen S. Survival, causes of death, and cardiovascular events in patients with Marfan syndrome. Mol Genet Genomic Med. 2018;6(6):1114–1123. doi:10.1002/mgg3.489
19. Judge DP, Biery NJ, Keene DR, et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114(2):172–181. doi:10.1172/JCI200420641
20. Pereira L, Lee SY, Gayraud B, et al. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci U S A. 1999;96(7):3819–3823. doi:10.1073/pnas.96.7.3819
21. Cook JR, Carta L, Benard L, et al. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J Clin Invest. 2014;124(3):1329–1339.
22. Steijns F, Renard M, Vanhomwegen M, et al. Spontaneous right ventricular pseudoaneurysms and increased arrhythmogenicity in a mouse model of Marfan syndrome. Int J Mol Sci. 2020;21(19):7024. doi:10.3390/ijms21197024
23. Lima BL, Santos EJ, Fernandes GR, et al. A new mouse model for Marfan syndrome presents phenotypic variability associated with the genetic background and overall levels of Fbn1 expression. PLoS One. 2010;5(11):e14136. doi:10.1371/journal.pone.0014136
24. Dale MA, Suh MK, Zhao S, et al. Background differences in baseline and stimulated MMP levels influence abdominal aortic aneurysm susceptibility. Atherosclerosis. 2015;243(2):621–629. doi:10.1016/j.atherosclerosis.2015.10.006
25. Pereira L, Andrikopoulos K, Tian J, et al. Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet. 1997;17(2):218–222. doi:10.1038/ng1097-218
26. Dietz H, Doyle JJ, Doyle AJ, Dietz H, Doyle JJ; Johns Hopkins University, assignee. MAP kinase pathway targets for the treatment of Marfan syndrome. US patent Pending; Publication No. US2020/0247881A1; March 2, 2018.
27. Charbonneau NL, Carlson EJ, Tufa S, et al. In vivo studies of mutant fibrillin-1 microfibrils. J Biol Chem. 2010;285(32):24943–24955. doi:10.1074/jbc.M110.130021
28. Carta L, Pereira L, Arteaga-Solis E, et al. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J Biol Chem. 2006;281(12):8016–8023. doi:10.1074/jbc.M511599200
29. Fleischer KJ, Nousari HC, Anhalt GJ, Stone CD, Laschinger JC. Immunohistochemical abnormalities of fibrillin in cardiovascular tissues in Marfan’s syndrome. Ann Thorac Surg. 1997;63(4):1012–1017. doi:10.1016/S0003-4975(97)00061-1
30. Wanga S, Hibender S, Ridwan Y, et al. Aortic microcalcification is associated with elastin fragmentation in Marfan syndrome. J Pathol. 2017;243(3):294–306. doi:10.1002/path.4949
31. Yuan SM, Jing H. Cystic medial necrosis: pathological findings and clinical implications. Rev Bras Cir Cardiovasc. 2011;26(1):107–115. doi:10.1590/S0102-76382011000100019
32. Gillis E, Van Laer L, Loeys BL. Genetics of thoracic aortic aneurysm: at the crossroad of transforming growth factor-beta signaling and vascular smooth muscle cell contractility. Circ Res. 2013;113(3):327–340. doi:10.1161/CIRCRESAHA.113.300675
33. Perrucci GL, Rurali E, Gowran A, et al. Vascular smooth muscle cells in Marfan syndrome aneurysm: the broken bricks in the aortic wall. Cell Mol Life Sci. 2017;74(2):267–277. doi:10.1007/s00018-016-2324-9
34. Humphrey JD, Schwartz MA, Tellides G, Milewicz DM. Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections. Circ Res. 2015;116(8):1448–1461. doi:10.1161/CIRCRESAHA.114.304936
35. Horiguchi M, Ota M, Rifkin DB. Matrix control of transforming growth factor-beta function. J Biochem. 2012;152(4):321–329.
36. Pannu H, Tran-Fadulu V, Milewicz DM. Genetic basis of thoracic aortic aneurysms and aortic dissections. Am J Med Genet C Semin Med Genet. 2005;139C(1):10–16. doi:10.1002/ajmg.c.30069
37. Humphrey JD, Milewicz DM, Tellides G, Schwartz MA. Cell biology. Dysfunctional mechanosensing in aneurysms. Science. 2014;344(6183):477–479. doi:10.1126/science.1253026
38. Milewicz DM, Guo DC, Tran-Fadulu V, et al. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283–302. doi:10.1146/annurev.genom.8.080706.092303
39. Ramirez F, Dietz HC. Fibrillin-rich microfibrils: structural determinants of morphogenetic and homeostatic events. J Cell Physiol. 2007;213(2):326–330. doi:10.1002/jcp.21189
40. Muino-Mosquera L, De Wilde H, Devos D, et al. Myocardial disease and ventricular arrhythmia in Marfan syndrome: a prospective study. Orphanet J Rare Dis. 2020;15(1):300. doi:10.1186/s13023-020-01581-8
41. Awerbach JD, Khatib S, Moodie DS, Snyder CS. Atrial ectopic tachycardia in a patient with Marfan syndrome. Ochsner J Summer. 2011;11(2):125–127.
42. Steijns F, van Hengel J, Sips P, De Backer J, Renard M. A heart for fibrillin: spatial arrangement in adult wild-type murine myocardial tissue. Histochem Cell Biol. 2018;150(3):271–280. doi:10.1007/s00418-018-1686-5
43. Veeraraghavan R, Gourdie RG, Poelzing S. Mechanisms of cardiac conduction: a history of revisions. Am J Physiol Heart Circ Physiol. 2014;306(5):H619–H627. doi:10.1152/ajpheart.00760.2013
44. Chaudhry SS, Cain SA, Morgan A, Dallas SL, Shuttleworth CA, Kielty CM. Fibrillin-1 regulates the bioavailability of TGFbeta1. J Cell Biol. 2007;176(3):355–367. doi:10.1083/jcb.200608167
45. Dallas SL, Keene DR, Bruder SP, et al. Role of the latent transforming growth factor beta binding protein 1 in fibrillin-containing microfibrils in bone cells in vitro and in vivo. J Bone Miner Res. 2000;15(1):68–81. doi:10.1359/jbmr.2000.15.1.68
46. Isogai Z, Ono RN, Ushiro S, et al. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem. 2003;278(4):2750–2757. doi:10.1074/jbc.M209256200
47. Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370(6488):341–347. doi:10.1038/370341a0
48. Derynck R, Feng XH. TGF-beta receptor signaling. Biochim Biophys Acta. 1997;1333(2):F105–F150.
49. Zhang YE. Non-smad signaling pathways of the TGF-beta family. Cold Spring Harb Perspect Biol. 2017;9(2):a022129. doi:10.1101/cshperspect.a022129
50. Heldin CH, Miyazono K, Ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390(6659):465–471. doi:10.1038/37284
51. Xu L, Chen YG, Massague J. The nuclear import function of Smad2 is masked by SARA and unmasked by TGFbeta-dependent phosphorylation. Nat Cell Biol. 2000;2(8):559–562. doi:10.1038/35019649
52. Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19(8):1745–1754. doi:10.1093/emboj/19.8.1745
53. Luo K. Signaling cross talk between TGF-beta/smad and other signaling pathways. Cold Spring Harb Perspect Biol. 2017;9(1):a022137. doi:10.1101/cshperspect.a022137
54. Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33(3):407–411. doi:10.1038/ng1116
55. Yu C, Jeremy RW. Angiotensin, transforming growth factor beta and aortic dilatation in Marfan syndrome: of mice and humans. Int J Cardiol Heart Vasc. 2018;18:71–80.
56. El-Hamamsy I, Yacoub MH. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat Rev Cardiol. 2009;6(12):771–786. doi:10.1038/nrcardio.2009.191
57. Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18(7):816–827. doi:10.1096/fj.03-1273rev
58. Radonic T, de Witte P, Groenink M, et al. Inflammation aggravates disease severity in Marfan syndrome patients. PLoS One. 2012;7(3):e32963. doi:10.1371/journal.pone.0032963
59. MacFarlane EG, Haupt J, Dietz HC, Shore EM. TGF-beta family signaling in connective tissue and skeletal diseases. Cold Spring Harb Perspect Biol. 2017;9(11):a022269. doi:10.1101/cshperspect.a022269
60. Jones A, Deb R, Torsney E, et al. Rosiglitazone reduces the development and rupture of experimental aortic aneurysms. Circulation. 2009;119(24):3125–3132. doi:10.1161/CIRCULATIONAHA.109.852467
61. Robinson PN, Arteaga-Solis E, Baldock C, et al. The molecular genetics of Marfan syndrome and related disorders. J Med Genet. 2006;43(10):769–787. doi:10.1136/jmg.2005.039669
62. Matt P, Schoenhoff F, Habashi J, et al. Circulating transforming growth factor-beta in Marfan syndrome. Circulation. 2009;120(6):526–532. doi:10.1161/CIRCULATIONAHA.108.841981
63. Nataatmadja M, West J, West M. Overexpression of transforming growth factor-beta is associated with increased hyaluronan content and impairment of repair in Marfan syndrome aortic aneurysm. Circulation. 2006;114(1 Suppl):I371–377. doi:10.1161/CIRCULATIONAHA.105.000927
64. Canadas V, Vilacosta I, Bruna I, Fuster V. Marfan syndrome. Part 2: treatment and management of patients. Nat Rev Cardiol. 2010;7(5):266–276. doi:10.1038/nrcardio.2010.31
65. van Dorst DCH, de Wagenaar NP, van der Pluijm I, Roos-Hesselink JW, Essers J, Danser AHJ. Transforming growth factor-beta and the renin-angiotensin system in syndromic thoracic aortic aneurysms: implications for treatment. Cardiovasc Drugs Ther. 2020. doi:10.1007/s10557-020-07116-4
66. Ng CM, Cheng A, Myers LA, et al. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114(11):1586–1592.
67. Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312(5770):117–121. doi:10.1126/science.1124287
68. Cook JR, Clayton NP, Carta L, et al. Dimorphic effects of transforming growth factor-beta signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35(4):911–917. doi:10.1161/ATVBAHA.114.305150
69. Li W, Li Q, Jiao Y, et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014;124(2):755–767. doi:10.1172/JCI69942
70. Lindsay ME, Schepers D, Bolar NA, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44(8):922–927. doi:10.1038/ng.2349
71. Hara H, Maemura S, Fujiwara T, et al. Inhibition of transforming growth factor-beta signaling in myeloid cells ameliorates aortic aneurysmal formation in Marfan syndrome. PLoS One. 2020;15(11):e0239908. doi:10.1371/journal.pone.0239908
72. Ferrario CM, Strawn WB. Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am J Cardiol. 2006;98(1):121–128. doi:10.1016/j.amjcard.2006.01.059
73. Steckelings UM, Bader M. Renin-angiotensin system in aortic aneurysm. Hypertension. 2018;72(3):579–581. doi:10.1161/HYPERTENSIONAHA.118.11238
74. Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2004;24(3):429–434. doi:10.1161/01.ATV.0000118013.72016.ea
75. Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105(11):1605–1612. doi:10.1172/JCI7818
76. Daugherty A, Rateri DL, Charo IF, Owens AP, Howatt DA, Cassis LA. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE-/- mice. Clin Sci (Lond). 2010;118(11):681–689. doi:10.1042/CS20090372
77. Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74(6):1141–1148. doi:10.1161/01.RES.74.6.1141
78. Moltzer E, Essers J, van Esch JH, Roos-Hesselink JW, Danser AH. The role of the renin-angiotensin system in thoracic aortic aneurysms: clinical implications. Pharmacol Ther. 2011;131(1):50–60. doi:10.1016/j.pharmthera.2011.04.002
79. Bin Mahmood SU, Velasquez CA, Zafar MA, et al. Medical management of aortic disease in Marfan syndrome. Ann Cardiothorac Surg. 2017;6(6):654–661. doi:10.21037/acs.2017.11.09
80. Forrester SJ, Booz GW, Sigmund CD, et al. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev. 2018;98(3):1627–1738.
81. Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC
82. Zhou Y, Poczatek MH, Berecek KH, Murphy-Ullrich JE. Thrombospondin 1 mediates angiotensin II induction of TGF-beta activation by cardiac and renal cells under both high and low glucose conditions. Biochem Biophys Res Commun. 2006;339(2):633–641. doi:10.1016/j.bbrc.2005.11.060
83. Gibbons GH, Pratt RE, Dzau VJ. Vascular smooth muscle cell hypertrophy vs. hyperplasia. Autocrine transforming growth factor-beta 1 expression determines growth response to angiotensin II. J Clin Invest. 1992;90(2):456–461. doi:10.1172/JCI115881
84. Segura AM, Luna RE, Horiba K, et al. Immunohistochemistry of matrix metalloproteinases and their inhibitors in thoracic aortic aneurysms and aortic valves of patients with Marfan’s syndrome. Circulation. 1998;98(19Suppl):
85. Ikonomidis JS, Jones JA, Barbour JR, et al. Expression of matrix metalloproteinases and endogenous inhibitors within ascending aortic aneurysms of patients with Marfan syndrome. Circulation. 2006;114(1 Suppl):I365–370. doi:10.1161/CIRCULATIONAHA.105.000810
86. Manicone AM, McGuire JK. Matrix metalloproteinases as modulators of inflammation. Semin Cell Dev Biol. 2008;19(1):34–41. doi:10.1016/j.semcdb.2007.07.003
87. Palombo D, Maione M, Cifiello BI, Udini M, Maggio D, Lupo M. Matrix metalloproteinases. Their role in degenerative chronic diseases of abdominal aorta. J Cardiovasc Surg (Torino). 1999;40(2):257–260.
88. Van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol. 2007;82(6):1375–1381. doi:10.1189/jlb.0607338
89. Krstic J, Santibanez JF. Transforming growth factor-beta and matrix metalloproteinases: functional interactions in tumor stroma-infiltrating myeloid cells. ScientificWorldJournal. 2014;2014:521754. doi:10.1155/2014/521754
90. Wang C, Chang Q, Sun X, et al. Angiotensin II induces an increase in matrix metalloproteinase 2 expression in aortic smooth muscle cells of ascending thoracic aortic aneurysms through JNK, ERK1/2, and p38 MAPK activation. J Cardiovasc Pharmacol. 2015;66(3):285–293. doi:10.1097/FJC.0000000000000276
91. Yang CQ, Li W, Li SQ, et al. MCP-1 stimulates MMP-9 expression via ERK 1/2 and p38 MAPK signaling pathways in human aortic smooth muscle cells. Cell Physiol Biochem. 2014;34(2):266–276. doi:10.1159/000362997
92. Rabkin SW. The role matrix metalloproteinases in the production of aortic aneurysm. Prog Mol Biol Transl Sci. 2017;147:239–265.
93. Nagasawa A, Yoshimura K, Suzuki R, et al. Important role of the angiotensin II pathway in producing matrix metalloproteinase-9 in human thoracic aortic aneurysms. J Surg Res. 2013;183(1):472–477. doi:10.1016/j.jss.2012.12.012
94. Chung AW, Au Yeung K, Sandor GG, Judge DP, Dietz HC, van Breemen C. Loss of elastic fiber integrity and reduction of vascular smooth muscle contraction resulting from the upregulated activities of matrix metalloproteinase-2 and −9 in the thoracic aortic aneurysm in Marfan syndrome. Circ Res. 2007;101(5):512–522. doi:10.1161/CIRCRESAHA.107.157776
95. Chung AW, Yang HH, Radomski MW, van Breemen C. Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in Marfan syndrome through the inhibition of matrix metalloproteinase-2 and −9. Circ Res. 2008;102(8):e73–85. doi:10.1161/CIRCRESAHA.108.174367
96. Kim ES, Kim MS, Moon A. TGF-beta-induced upregulation of MMP-2 and MMP-9 depends on p38 MAPK, but not ERK signaling in MCF10A human breast epithelial cells. Int J Oncol. 2004;25(5):1375–1382.
97. Kobayashi T, Kim H, Liu X, et al. Matrix metalloproteinase-9 activates TGF-beta and stimulates fibroblast contraction of collagen gels. Am J Physiol Lung Cell Mol Physiol. 2014;306(11):L1006–L1015. doi:10.1152/ajplung.00015.2014
98. Wang M, Zhao D, Spinetti G, et al. Matrix metalloproteinase 2 activation of transforming growth factor-beta1 (TGF-beta1) and TGF-beta1-type II receptor signaling within the aged arterial wall. Arterioscler Thromb Vasc Biol. 2006;26(7):1503–1509. doi:10.1161/01.ATV.0000225777.58488.f2
99. Tamarina NA, McMillan WD, Shively VP, Pearce WH. Expression of matrix metalloproteinases and their inhibitors in aneurysms and normal aorta. Surgery. 1997;122(2):
100. Farah C, Michel LYM, Balligand JL. Nitric oxide signalling in cardiovascular health and disease. Nat Rev Cardiol. 2018;15(5):292–316.
101. Nicholls M. Nitric oxide discovery Nobel prize winners. Eur Heart J. 2019;40(22):1747–1749. doi:10.1093/eurheartj/ehz361
102. Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33(7):
103. Oller J, Mendez-Barbero N, Ruiz EJ, et al. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat Med. 2017;23(2):200–212. doi:10.1038/nm.4266
104. Yang HH, van Breemen C, Chung AW. Vasomotor dysfunction in the thoracic aorta of Marfan syndrome is associated with accumulation of oxidative stress. Vascul Pharmacol. 2010;52(1–2):37–45. doi:10.1016/j.vph.2009.10.005
105. Soto ME, Soria-Castro E, Lans VG, et al. Analysis of oxidative stress enzymes and structural and functional proteins on human aortic tissue from different aortopathies. Oxid Med Cell Longev. 2014;2014:760694. doi:10.1155/2014/760694
106. Guo DC, Regalado E, Casteel DE, et al. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet. 2013;93(2):398–404. doi:10.1016/j.ajhg.2013.06.019
107. Sellers SL, Milad N, Chan R, et al. Inhibition of Marfan syndrome aortic root dilation by losartan: role of angiotensin II receptor type 1-independent activation of endothelial function. Am J Pathol. 2018;188(3):574–585. doi:10.1016/j.ajpath.2017.11.006
108. Buys E, Sips P. New insights into the role of soluble guanylate cyclase in blood pressure regulation. Curr Opin Nephrol Hypertens. 2014;23(2):135–142. doi:10.1097/01.mnh.0000441048.91041.3a
109. White Z, Milad N, Tehrani AY, et al. Sildenafil prevents Marfan-associated emphysema and early pulmonary artery dilation in mice. Am J Pathol. 2019;189(8):1536–1546. doi:10.1016/j.ajpath.2019.05.003
110. Sips PY, Irie T, Zou L, et al. Reduction of cardiomyocyte S-nitrosylation by S-nitrosoglutathione reductase protects against sepsis-induced myocardial depression. Am J Physiol Heart Circ Physiol. 2013;304(8):H1134–H1146. doi:10.1152/ajpheart.00887.2012
111. Irie T, Sips PY, Kai S, et al. S-Nitrosylation of calcium-handling proteins in cardiac adrenergic signaling and hypertrophy. Circ Res. 2015;117(9):793–803. doi:10.1161/CIRCRESAHA.115.307157
112. Choi H, Tostes RC, Webb RC. S-nitrosylation Inhibits protein kinase C-mediated contraction in mouse aorta. J Cardiovasc Pharmacol. 2011;57(1):65–71. doi:10.1097/FJC.0b013e3181fef9cb
113. Choi H, Allahdadi KJ, Tostes RC, Webb RC. Augmented S-nitrosylation contributes to impaired relaxation in angiotensin II hypertensive mouse aorta: role of thioredoxin reductase. J Hypertens. 2011;29(12):2359–2368. doi:10.1097/HJH.0b013e32834d2554
114. Pan L, Lin Z, Tang X, et al. S-Nitrosylation of plastin-3 exacerbates thoracic aortic dissection formation via endothelial barrier dysfunction. Arterioscler Thromb Vasc Biol. 2020;40(1):175–188. doi:10.1161/ATVBAHA.119.313440
115. Mocellin S, Bronte V, Nitti D. Nitric oxide, a double edged sword in cancer biology: searching for therapeutic opportunities. Med Res Rev. 2007;27(3):317–352. doi:10.1002/med.20092
116. Calabrese V, Cornelius C, Rizzarelli E, Owen JB, Dinkova-Kostova AT, Butterfield DA. Nitric oxide in cell survival: a janus molecule. Antioxid Redox Signal. 2009;11(11):2717–2739. doi:10.1089/ars.2009.2721
117. Malecki C, Hambly BD, Jeremy RW, Robertson EN. The role of inflammation and myeloperoxidase-related oxidative stress in the pathogenesis of genetically triggered thoracic aortic aneurysms. Int J Mol Sci. 2020;21(20):7678. doi:10.3390/ijms21207678
118. He R, Guo DC, Sun W, et al. Characterization of the inflammatory cells in ascending thoracic aortic aneurysms in patients with Marfan syndrome, familial thoracic aortic aneurysms, and sporadic aneurysms. J Thorac Cardiovasc Surg. 2008;136(4):922–929, 929 e921. doi:10.1016/j.jtcvs.2007.12.063
119. Tang PC, Yakimov AO, Teesdale MA, et al. Transmural inflammation by interferon-gamma-producing T cells correlates with outward vascular remodeling and intimal expansion of ascending thoracic aortic aneurysms. FASEB J. 2005;19(11):1528–1530. doi:10.1096/fj.05-3671fje
120. He R, Guo DC, Estrera AL, et al. Characterization of the inflammatory and apoptotic cells in the aortas of patients with ascending thoracic aortic aneurysms and dissections. J Thorac Cardiovasc Surg. 2006;131(3):671–678. doi:10.1016/j.jtcvs.2005.09.018
121. Kim AJ, Xu N, Umeyama K, et al. Deficiency of circulating monocytes ameliorates the progression of myxomatous valve degeneration in Marfan syndrome. Circulation. 2020;141(2):132–146. doi:10.1161/CIRCULATIONAHA.119.042391
122. Li PF, Dietz R, von Harsdorf R. Reactive oxygen species induce apoptosis of vascular smooth muscle cell. FEBS Lett. 1997;404(2–3):249–252. doi:10.1016/S0014-5793(97)00093-8
123. McCormick ML, Gavrila D, Weintraub NL. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2007;27(3):461–469. doi:10.1161/01.ATV.0000257552.94483.14
124. Emrich F, Penov K, Arakawa M, et al. Anatomically specific reactive oxygen species production participates in Marfan syndrome aneurysm formation. J Cell Mol Med. 2019;23(10):7000–7009. doi:10.1111/jcmm.14587
125. Jimenez-Altayo F, Meirelles T, Crosas-Molist E, et al. Redox stress in Marfan syndrome: dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic Biol Med. 2018;118:44–58. doi:10.1016/j.freeradbiomed.2018.02.023
126. Martin-Garrido A, Brown DI, Lyle AN, et al. NADPH oxidase 4 mediates TGF-beta-induced smooth muscle alpha-actin via p38MAPK and serum response factor. Free Radic Biol Med. 2011;50(2):354–362. doi:10.1016/j.freeradbiomed.2010.11.007
127. Koo HK, Lawrence KA, Musini VM. Beta-blockers for preventing aortic dissection in Marfan syndrome. Cochrane Database Syst Rev. 2017;11:CD011103.
128. van Andel MM, Indrakusuma R, Jalalzadeh H, et al. Long-term clinical outcomes of losartan in patients with Marfan syndrome: follow-up of the multicentre randomized controlled COMPARE trial. Eur Heart J. 2020;41(43):4181–4187. doi:10.1093/eurheartj/ehaa377
129. Keane MG, Pyeritz RE. Medical management of Marfan syndrome. Circulation. 2008;117(21):2802–2813. doi:10.1161/CIRCULATIONAHA.107.693523
130. Prokop EK, Palmer RF, Wheat MW
131. Halpern BL, Char F, Murdoch JL, Horton WB, McKusick VA. A prospectus on the prevention of aortic rupture in the Marfan syndrome with data on survivorship without treatment. Johns Hopkins Med J. 1971;129(3):123–129.
132. McKusick VA. Historical introduction. The Marfan syndrome: from clinical delineation to mutational characterization, a semiautobiographic account. In: Robinson PN, Godfrey M, editors. Marfan Syndrome: A Primer for Clinicians and Scientists. Texas, USA: Landes Bioscience/Eurekah.com and Kluwer Academic/Plenum Publishers; 2004:1–12.
133. Shores J, Berger KR, Murphy EA, Pyeritz RE. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan’s syndrome. N Engl J Med. 1994;330(19):1335–1341. doi:10.1056/NEJM199405123301902
134. Salim MA, Alpert BS, Ward JC, Pyeritz RE. Effect of beta-adrenergic blockade on aortic root rate of dilation in the Marfan syndrome. Am J Cardiol. 1994;74(6):629–633. doi:10.1016/0002-9149(94)90762-5
135. Rios AS, Silber EN, Bavishi N, et al. Effect of long-term beta-blockade on aortic root compliance in patients with Marfan syndrome. Am Heart J. 1999;137(6):1057–1061. doi:10.1016/S0002-8703(99)70362-5
136. Ladouceur M, Fermanian C, Lupoglazoff JM, et al. Effect of beta-blockade on ascending aortic dilatation in children with the Marfan syndrome. Am J Cardiol. 2007;99(3):406–409. doi:10.1016/j.amjcard.2006.08.048
137. Tahernia AC. Cardiovascular anomalies in Marfan’s syndrome: the role of echocardiography and beta-blockers. South Med J. 1993;86(3):305–310. doi:10.1097/00007611-199303000-00012
138. Groenink M, de Roos A, Mulder BJ, Spaan JA, van der Wall EE. Changes in aortic distensibility and pulse wave velocity assessed with magnetic resonance imaging following beta-blocker therapy in the Marfan syndrome. Am J Cardiol. 1998;82(2):203–208. doi:10.1016/S0002-9149(98)00315-4
139. Yetman AT, Bornemeier RA, McCrindle BW. Usefulness of enalapril versus propranolol or atenolol for prevention of aortic dilation in patients with the Marfan syndrome. Am J Cardiol. 2005;95(9):1125–1127. doi:10.1016/j.amjcard.2005.01.032
140. Williams A, Kenny D, Wilson D, et al. Effects of atenolol, perindopril and verapamil on haemodynamic and vascular function in Marfan syndrome - a randomised, double-blind, crossover trial. Eur J Clin Invest. 2012;42(8):891–899. doi:10.1111/j.1365-2362.2012.02668.x
141. Mueller GC, Stierle L, Stark V, et al. Retrospective analysis of the effect of angiotensin II receptor blocker versus beta-blocker on aortic root growth in paediatric patients with Marfan syndrome. Heart. 2014;100(3):214–218. doi:10.1136/heartjnl-2013-304946
142. Gersony DR, McClaughlin MA, Jin Z, Gersony WM. The effect of beta-blocker therapy on clinical outcome in patients with Marfan’s syndrome: a meta-analysis. Int J Cardiol. 2007;114(3):303–308. doi:10.1016/j.ijcard.2005.11.116
143. Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: executive summary: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. Circulation. 2014;130(24):2215–2245. doi:10.1161/CIR.0000000000000105
144. Anekwe L. New guideline on beta blockers challenges ESC advice. Lancet. 2014;383(9918):682. doi:10.1016/S0140-6736(14)60243-9
145. Nowbar AN, Cole GD, Shun-Shin MJ, Finegold JA, Francis DP. International RCT-based guidelines for use of preoperative stress testing and perioperative beta-blockers and statins in non-cardiac surgery. Int J Cardiol. 2014;172(1):138–143. doi:10.1016/j.ijcard.2013.12.309
146. Rossi-Foulkes R, Roman MJ, Rosen SE, et al. Phenotypic features and impact of beta blocker or calcium antagonist therapy on aortic lumen size in the Marfan syndrome. Am J Cardiol. 1999;83(9):1364–1368. doi:10.1016/S0002-9149(99)00101-0
147. Doyle JJ, Doyle AJ, Wilson NK, et al. A deleterious gene-by-environment interaction imposed by calcium channel blockers in Marfan syndrome. Elife. 2015;4:e08648. doi:10.7554/eLife.08648
148. Yang HH, Kim JM, Chum E, van Breemen C, Chung AW. Effectiveness of combination of losartan potassium and doxycycline versus single-drug treatments in the secondary prevention of thoracic aortic aneurysm in Marfan syndrome. J Thorac Cardiovasc Surg. 2010;140(2):305–312 e302. doi:10.1016/j.jtcvs.2009.10.039
149. Lee JJ, Galatioto J, Rao S, Ramirez F, Costa KD. Losartan attenuates degradation of aorta and lung tissue micromechanics in a mouse model of severe marfan syndrome. Ann Biomed Eng. 2016;44(10):2994–3006. doi:10.1007/s10439-016-1616-4
150. Yang HH, Kim JM, Chum E, van Breemen C, Chung AW. Long-term effects of losartan on structure and function of the thoracic aorta in a mouse model of Marfan syndrome. Br J Pharmacol. 2009;158(6):1503–1512. doi:10.1111/j.1476-5381.2009.00443.x
151. Xiong W, Meisinger T, Knispel R, Worth JM, Baxter BT. MMP-2 regulates Erk1/2 phosphorylation and aortic dilatation in Marfan syndrome. Circ Res. 2012;110(12):e92–e101. doi:10.1161/CIRCRESAHA.112.268268
152. Groenink M, den Hartog AW, Franken R, et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J. 2013;34(45):3491–3500. doi:10.1093/eurheartj/eht334
153. Pees C, Laccone F, Hagl M, Debrauwer V, Moser E, Michel-Behnke I. Usefulness of losartan on the size of the ascending aorta in an unselected cohort of children, adolescents, and young adults with Marfan syndrome. Am J Cardiol. 2013;112(9):1477–1483. doi:10.1016/j.amjcard.2013.06.019
154. Lacro RV, Dietz HC, Sleeper LA, et al. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med. 2014;371(22):2061–2071. doi:10.1056/NEJMoa1404731
155. Forteza A, Evangelista A, Sanchez V, et al. Efficacy of losartan vs. atenolol for the prevention of aortic dilation in Marfan syndrome: a randomized clinical trial. Eur Heart J. 2016;37(12):978–985. doi:10.1093/eurheartj/ehv575
156. Milleron O, Arnoult F, Ropers J, et al. Marfan Sartan: a randomized, double-blind, placebo-controlled trial. Eur Heart J. 2015;36(32):2160–2166. doi:10.1093/eurheartj/ehv151
157. Franken R, den Hartog AW, Radonic T, et al. Beneficial outcome of losartan therapy depends on type of FBN1 mutation in marfan syndrome. Circ Cardiovasc Genet. 2015;8(2):383–388. doi:10.1161/CIRCGENETICS.114.000950
158. Mullen MJ, Flather MD, Jin XY, et al. A prospective, randomized, placebo-controlled, double-blind, multicenter study of the effects of irbesartan on aortic dilatation in Marfan syndrome (AIMS trial): study protocol. Trials. 2013;14:408. doi:10.1186/1745-6215-14-408
159. Mullen M, Jin XY, Child A, et al. Irbesartan in Marfan syndrome (AIMS): a double-blind, placebo-controlled randomised trial. Lancet. 2019;394(10216):2263–2270. doi:10.1016/S0140-6736(19)32518-8
160. Pitcher A, Emberson J, Lacro RV, et al. Design and rationale of a prospective, collaborative meta-analysis of all randomized controlled trials of angiotensin receptor antagonists in Marfan syndrome, based on individual patient data: a report from the marfan treatment trialists’ collaboration. Am Heart J. 2015;169(5):605–612. doi:10.1016/j.ahj.2015.01.011
161. Gambarin FI, Favalli V, Serio A, et al. Rationale and design of a trial evaluating the effects of losartan vs. nebivolol vs. the association of both on the progression of aortic root dilation in Marfan syndrome with FBN1 gene mutations. J Cardiovasc Med (Hagerstown). 2009;10(4):354–362. doi:10.2459/JCM.0b013e3283232a45
162. Di Toro A, Klersy C, Giuliani L, et al. Losartan vs nebivolol vs the association of both on the progression of aortic root dilation in genotyped marfan syndrome: 48 months open label randomized controlled Phase III trial. Paper presented at: ESC Congress together with World Congress of Cardiology; 31 August – 4 September. Paris: France; 2019.
163. den Hartog AW, Franken R, van den Berg MP, et al. The effect of losartan therapy on ventricular function in Marfan patients with haploinsufficient or dominant negative FBN1 mutations. Neth Heart J. 2016;24(11):675–681. doi:10.1007/s12471-016-0905-8
164. Jondeau G, Milleron O, Boileau C. Marfan sartan saga, episode X. Eur Heart J. 2020;41(43):4188–4190. doi:10.1093/eurheartj/ehaa418
165. Jhund PS, McMurray JJ. The neprilysin pathway in heart failure: a review and guide on the use of sacubitril/valsartan. Heart. 2016;102(17):1342–1347. doi:10.1136/heartjnl-2014-306775
166. Bernatchez P, Sauge E, White Z Attenuation of aortic remodeling with low doses of neprilysin/angiotensin ii receptor blockers sacubitril/valsartan in a fibrillin-1-deficient model of marfan syndrome. Faseb J; 2020;34:1.
167. Spoto S, Valeriani E, Locorriere L, et al. Use of sacubitril/valsartan in Marfan syndrome-related cardiomyopathy: the first case report. Medicine (Baltimore). 2019;98(47):e17978. doi:10.1097/MD.0000000000017978
168. Nagashima H, Sakomura Y, Aoka Y, et al. Angiotensin II type 2 receptor mediates vascular smooth muscle cell apoptosis in cystic medial degeneration associated with Marfan’s syndrome. Circulation. 2001;104(12 Suppl 1):I282–287. doi:10.1161/hc37t1.094856
169. Habashi JP, Doyle JJ, Holm TM, et al. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;332(6027):361–365. doi:10.1126/science.1192152
170. Seva Pessoa B, van der Lubbe N, Verdonk K, Roks AJ, Hoorn EJ, Danser AH. Key developments in renin-angiotensin-aldosterone system inhibition. Nat Rev Nephrol. 2013;9(1):26–36. doi:10.1038/nrneph.2012.249
171. Kingwell BA, Aggarwal A, D’Orsa KM, et al. Effect of perindopril on large artery stiffness and aortic root diameter in patients with marfan syndrome: a randomized controlled trial. JAMA. 2007;298(13):1539–1547. doi:10.1001/jama.298.13.1539
172. Phomakay V, Huett WG, Gossett JM, Tang X, Bornemeier RA, Collins RT
173. Hwang JW, Kim EK, Jang SY, et al. Comparison of the effect of aliskiren versus negative controls on aortic stiffness in patients with marfan syndrome under treatment with atenolol. Rev Esp Cardiol (Engl Ed). 2018;71(9):743–749. doi:10.1016/j.recesp.2017.10.040
174. Xiong W, Knispel RA, Dietz HC, Ramirez F, Baxter BT. Doxycycline delays aneurysm rupture in a mouse model of Marfan syndrome. J Vasc Surg. 2008;47(1):
175. Cui JZ, Lee L, Sheng X, et al. In vivo characterization of doxycycline-mediated protection of aortic function and structure in a mouse model of Marfan syndrome-associated aortic aneurysm. Sci Rep. 2019;9(1):2071. doi:10.1038/s41598-018-38235-6
176. Tehrani AY, Cui JZ, Bucky Jones T, et al. Characterization of doxycycline-mediated inhibition of Marfan syndrome-associated aortic dilation by multiphoton microscopy. Sci Rep. 2020;10(1):7154. doi:10.1038/s41598-020-64071-8
177. Lindeman JH, Abdul-Hussien H, van Bockel JH, Wolterbeek R, Kleemann R. Clinical trial of doxycycline for matrix metalloproteinase-9 inhibition in patients with an abdominal aneurysm: doxycycline selectively depletes aortic wall neutrophils and cytotoxic T cells. Circulation. 2009;119(16):2209–2216. doi:10.1161/CIRCULATIONAHA.108.806505
178. Abdul-Hussien H, Hanemaaijer R, Verheijen JH, van Bockel JH, Geelkerken RH, Lindeman JH. Doxycycline therapy for abdominal aneurysm: improved proteolytic balance through reduced neutrophil content. J Vasc Surg. 2009;49(3):741–749. doi:10.1016/j.jvs.2008.09.055
179. Bailey A, Pope TW, Moore SA, Campbell CL. The tragedy of TRIUMPH for nitric oxide synthesis inhibition in cardiogenic shock: where do we go from here? Am J Cardiovasc Drugs. 2007;7(5):337–345. doi:10.2165/00129784-200707050-00003
180. Bredan AS, Cauwels A. Is there no treatment for severe sepsis? Libyan J Med. 2008;3(1):34–38. doi:10.3402/ljm.v3i1.4748
181. Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011;473(7347):308–316. doi:10.1038/nature10145
182. Holm TM, Habashi JP, Doyle JJ, et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332(6027):358–361. doi:10.1126/science.1192149
183. Carta L, Smaldone S, Zilberberg L, et al. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J Biol Chem. 2009;284(9):5630–5636. doi:10.1074/jbc.M806962200
184. Steijns F, Bracke N, Renard M, et al. MEK1/2 inhibition in murine heart and aorta after oral administration of refametinib supplemented drinking water. Front Pharmacol. 2020;11:1336. doi:10.3389/fphar.2020.01336
185. Shen YH, LeMaire SA, Webb NR, Cassis LA, Daugherty A, Lu HS. Aortic aneurysms and dissections series: part II: dynamic signaling responses in aortic aneurysms and dissections. Arterioscler Thromb Vasc Biol. 2020;40(4):e78–e86.
186. Pedroza AJ, Koyano T, Trojan J, et al. Divergent effects of canonical and non-canonical TGF-beta signalling on mixed contractile-synthetic smooth muscle cell phenotype in human Marfan syndrome aortic root aneurysms. J Cell Mol Med. 2020;24(3):2369–2383. doi:10.1111/jcmm.14921
187. Renard M, Muino-Mosquera L, Manalo EC, et al. Sex, pregnancy and aortic disease in Marfan syndrome. PLoS One. 2017;12(7):e0181166. doi:10.1371/journal.pone.0181166
188. Detaint D, Faivre L, Collod-Beroud G, et al. Cardiovascular manifestations in men and women carrying a FBN1 mutation. Eur Heart J. 2010;31(18):2223–2229. doi:10.1093/eurheartj/ehq258
189. Holmes KW, Maslen CL, Kindem M, et al. GenTAC registry report: gender differences among individuals with genetically triggered thoracic aortic aneurysm and dissection. Am J Med Genet A. 2013;161A(4):779–786. doi:10.1002/ajmg.a.35836
190. Meijboom LJ, Timmermans J, Zwinderman AH, Engelfriet PM, Mulder BJ. Aortic root growth in men and women with the Marfan’s syndrome. Am J Cardiol. 2005;96(10):1441–1444. doi:10.1016/j.amjcard.2005.06.094
191. Jimenez-Altayo F, Siegert AM, Bonorino F, et al. Differences in the thoracic aorta by region and sex in a murine model of marfan syndrome. Front Physiol. 2017;8:933. doi:10.3389/fphys.2017.00933
192. Tashima Y, He H, Cui JZ, et al. Androgens accentuate TGF-beta dependent erk/smad activation during thoracic aortic aneurysm formation in marfan syndrome male mice. J Am Heart Assoc. 2020;9(20):e015773. doi:10.1161/JAHA.119.015773
193. Anderson NK, Juzwiak EE, Dietz HC. A seX(X/Y) article on marfan syndrome. J Am Heart Assoc. 2020;9(20):e018814. doi:10.1161/JAHA.120.018814
194. Lind J, Wallenburg HC. The Marfan syndrome and pregnancy: a retrospective study in a Dutch population. Eur J Obstet Gynecol Reprod Biol. 2001;98(1):28–35. doi:10.1016/S0301-2115(01)00314-1
195. Lipscomb KJ, Smith JC, Clarke B, Donnai P, Harris R. Outcome of pregnancy in women with Marfan’s syndrome. Br J Obstet Gynaecol. 1997;104(2):201–206. doi:10.1111/j.1471-0528.1997.tb11045.x
196. Pyeritz RE. Maternal and fetal complications of pregnancy in the Marfan syndrome. Am J Med. 1981;71(5):784–790. doi:10.1016/0002-9343(81)90365-X
197. Pacini L, Digne F, Boumendil A, et al. Maternal complication of pregnancy in Marfan syndrome. Int J Cardiol. 2009;136(2):156–161. doi:10.1016/j.ijcard.2008.04.035
198. Meijboom LJ, Drenthen W, Pieper PG, et al. Obstetric complications in Marfan syndrome. Int J Cardiol. 2006;110(1):53–59. doi:10.1016/j.ijcard.2005.07.017
199. Rahman J, Rahman FZ, Rahman W, al-Suleiman SA, Rahman MS. Obstetric and gynecologic complications in women with Marfan syndrome. J Reprod Med. 2003;48(9):723–728.
200. Regitz-Zagrosek V, Roos-Hesselink JW, Bauersachs J, et al. 2018 ESC Guidelines for the management of cardiovascular diseases during pregnancy. Eur Heart J. 2018;39(34):3165–3241.
201. Habashi JP, MacFarlane EG, Bagirzadeh R, et al. Oxytocin antagonism prevents pregnancy-associated aortic dissection in a mouse model of Marfan syndrome. Sci Transl Med. 2019;11(490):eaat4822. doi:10.1126/scitranslmed.aat4822
202. Roman MJ, Pugh NL, Hendershot TP, et al. Aortic complications associated with pregnancy in marfan syndrome: the NHLBI national registry of genetically triggered thoracic aortic aneurysms and cardiovascular conditions (GenTAC). J Am Heart Assoc. 2016;5(8):e004052. doi:10.1161/JAHA.116.004052
203. Goland S, Elkayam U. Cardiovascular problems in pregnant women with marfan syndrome. Circulation. 2009;119(4):619–623. doi:10.1161/CIRCULATIONAHA.104.493569
204. Elkayam U, Ostrzega E, Shotan A, Mehra A. Cardiovascular problems in pregnant women with the Marfan syndrome. Ann Intern Med. 1995;123(2):117–122. doi:10.7326/0003-4819-123-2-199507150-00007
205. Goland S, Elkayam U. Pregnancy and Marfan syndrome. Ann Cardiothorac Surg. 2017;6(6):642–653. doi:10.21037/acs.2017.10.07
206. Mulsow JJ, Watson RW, Fitzpatrick JM, O’Connell PR. Transforming growth factor-beta promotes pro-fibrotic behavior by serosal fibroblasts via PKC and ERK1/2 mitogen activated protein kinase cell signaling. Ann Surg. 2005;242(6):
207. Ryer EJ, Hom RP, Sakakibara K, et al. PKCdelta is necessary for Smad3 expression and transforming growth factor beta-induced fibronectin synthesis in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2006;26(4):780–786. doi:10.1161/01.ATV.0000209517.00220.cd
208. Deng C, Lu Q, Zhang Z, et al. Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum. 2003;48(3):746–756. doi:10.1002/art.10833
209. Gorelik G, Fang JY, Wu A, Sawalha AH, Richardson B. Impaired T cell protein kinase C delta activation decreases ERK pathway signaling in idiopathic and hydralazine-induced lupus. J Immunol. 2007;179(8):5553–5563. doi:10.4049/jimmunol.179.8.5553
210. Cryan JF, Kaupmann K. Don’t worry ‘B’ happy!: a role for GABA(B) receptors in anxiety and depression. Trends Pharmacol Sci. 2005;26(1):36–43.
211. Kuriyama K, Hirouchi M, Nakayasu H. Structure and function of cerebral GABAA and GABAB receptors. Neurosci Res. 1993;17(2):91–99. doi:10.1016/0168-0102(93)90087-7
212. Wang XP, Cheng ZY, Schmid KL. GABAB receptors expressed in human aortic endothelial cells mediate intracellular calcium concentration regulation and endothelial nitric oxide synthase translocation. Biomed Res Int. 2014;2014:871735.
213. Hansen J, Galatioto J, Caescu CI, et al. Systems pharmacology-based integration of human and mouse data for drug repurposing to treat thoracic aneurysms. JCI Insight. 2019;4(11):e127652. doi:10.1172/jci.insight.127652
214. Chen X, Li Y, Xiao J, et al. Modulating neuro-immune-induced macrophage polarization with topiramate attenuates experimental abdominal aortic aneurysm. Front Pharmacol. 2020;11:565461. doi:10.3389/fphar.2020.565461
215. Crowley T, Cryan JF, Downer EJ, O’Leary OF. Inhibiting neuroinflammation: the role and therapeutic potential of GABA in neuro-immune interactions. Brain Behav Immun. 2016;54:260–277. doi:10.1016/j.bbi.2016.02.001
216. Reyes-Garcia MG, Hernandez-Hernandez F, Hernandez-Tellez B, Garcia-Tamayo F. GABA (A) receptor subunits RNA expression in mice peritoneal macrophages modulate their IL-6/IL-12 production. J Neuroimmunol. 2007;188(1–2):64–68. doi:10.1016/j.jneuroim.2007.05.013
217. Lino Cardenas CL, Kessinger CW, Cheng Y, et al. An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat Commun. 2018;9(1):1009. doi:10.1038/s41467-018-03394-7
218. Portelli SS, Robertson EN, Malecki C, Liddy KA, Hambly BD, Jeremy RW. Epigenetic influences on genetically triggered thoracic aortic aneurysm. Biophys Rev. 2018;10(5):1241–1256. doi:10.1007/s12551-018-0460-1
219. Guo X, Fang ZM, Wei X, et al. HDAC6 is associated with the formation of aortic dissection in human. Mol Med. 2019;25(1):10. doi:10.1186/s10020-019-0080-7
220. Galan M, Varona S, Orriols M, et al. Induction of histone deacetylases (HDACs) in human abdominal aortic aneurysm: therapeutic potential of HDAC inhibitors. Dis Model Mech. 2016;9(5):541–552.
221. Lino Cardenas CL, Kessinger CW, MacDonald C, et al. Inhibition of the methyltranferase EZH2 improves aortic performance in experimental thoracic aortic aneurysm. JCI Insight. 2018;3(5):e97493. doi:10.1172/jci.insight.97493
222. Dale M, Fitzgerald MP, Liu Z, et al. Premature aortic smooth muscle cell differentiation contributes to matrix dysregulation in Marfan Syndrome. PLoS One. 2017;12(10):e0186603. doi:10.1371/journal.pone.0186603
223. Fernandes GR, Massironi SM, Pereira LV. Identification of loci modulating the cardiovascular and skeletal phenotypes of marfan syndrome in mice. Sci Rep. 2016;6:22426. doi:10.1038/srep22426
224. Asnani A, Peterson RT. The zebrafish as a tool to identify novel therapies for human cardiovascular disease. Dis Model Mech. 2014;7(7):763–767. doi:10.1242/dmm.016170
225. Asimaki A, Kapoor S, Plovie E, et al. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci Transl Med. 2014;6(240):240ra274. doi:10.1126/scitranslmed.3008008
226. MacRae CA, Peterson RT. Zebrafish as tools for drug discovery. Nat Rev Drug Discov. 2015;14(10):721–731. doi:10.1038/nrd4627
227. Granata A, Serrano F, Bernard WG, et al. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet. 2017;49(1):97–109. doi:10.1038/ng.3723
228. Aalders J, Leger L, Van der Meeren L, et al. Effects of fibrillin mutations on the behavior of heart muscle cells in Marfan syndrome. Sci Rep. 2020;10(1):16756. doi:10.1038/s41598-020-73802-w
229. Iosef C, Pedroza AJ, Cui JZ, et al. Quantitative proteomics reveal lineage-specific protein profiles in iPSC-derived Marfan syndrome smooth muscle cells. Sci Rep. 2020;10(1):20392. doi:10.1038/s41598-020-77274-w
230. Rurali E, Perrucci GL, Pilato CA, et al. Precise therapy for thoracic aortic aneurysm in marfan syndrome: a puzzle nearing its solution. Prog Cardiovasc Dis. 2018;61(3–4):328–335. doi:10.1016/j.pcad.2018.07.020
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.