")
Back to Journals » Clinical Ophthalmology » Volume 8
An observational study of bimatoprost 0.01% in patients on prior intraocular pressure-lowering therapy: the Canadian Lumigan® RC Early Analysis Review (CLEAR) trial
Authors Crichton A, Nixon D, Simonyi S, Bhogal M, Sigouin C, Discepola M, Hutnik C, Baptiste D, Yan D
Received 5 April 2013
Accepted for publication 30 October 2013
Published 23 May 2014 Volume 2014:8 Pages 1031—1038
DOI https://doi.org/10.2147/OPTH.S46298
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Andrew C Crichton,1 Donald R Nixon,2 Susan Simonyi,3 Meetu Bhogal,3 Christopher S Sigouin,4 Marino J Discepola,5 Cindy ML Hutnik,6 Darryl C Baptiste,3 David B Yan7
On behalf of the CLEAR Study Group
1Division of Ophthalmology, University of Calgary, Calgary, AB, Canada; 2Private practice, Barrie, ON, Canada; 3Medical Affairs, Allergan Inc., Markham, ON, Canada; 4CLINWest Research, Burlington, ON, Canada; 5Department of Ophthalmology, McGill University, Montreal, QC, Canada; 6Department of Ophthalmology and Pathology, Ivey Eye Institute, London, ON, Canada; 7Department of Ophthalmology, University of Toronto, Toronto, ON, Canada
Purpose: To evaluate the ocular hyperemia and intraocular pressure (IOP)-lowering efficacy of bimatoprost 0.01% in subjects with elevated IOP due to primary open-angle glaucoma (POAG) or ocular hypertension (OHT) in a real-world clinical setting.
Subjects and methods: This open-label, 12-week, observational study was conducted at 67 centers in Canada. Subjects with elevated IOP due to POAG or OHT instilled bimatoprost 0.01% as monotherapy once daily. Ocular hyperemia was graded by the investigator at baseline, week 6, and week 12 using a standardized photographic 5-point grading scale. Change in IOP from baseline was also evaluated at these time points. This analysis includes the subgroup of 268 subjects who had been previously treated with latanoprost 0.005%, bimatoprost 0.03%, travoprost 0.004%, and travoprost 0.004% with SofZia™ or nonselective beta-adrenergic receptor blockers prior to the study.
Results: After 12 weeks of treatment with 0.01% bimatoprost, ocular hyperemia was graded as none-to-mild hyperemia (grades 0, +0.5, or +1) for 94.1% of subjects and as moderate-to-severe hyperemia (grades +2 or +3) for 5.9%. No statistically significant shifts in ocular hyperemia ratings were observed at week 12 for any of the prior IOP-lowering therapies except bimatoprost 0.03%, in which 20.8% of subjects experienced an improvement. The mean percentage change from baseline IOP at week 12 following the switch to bimatoprost 0.01% monotherapy ranged from –2.3%±17.3% to –26.3%±12.4%. Furthermore, the decreased mean percentage change from baseline IOP was statistically significant across all prior IOP-lowering medications, except for bimatoprost 0.03% at the 6- and 12-week visits and travoprost 0.004% at the 6-week visit.
Conclusion: This observational study demonstrates that bimatoprost 0.01% was well tolerated among POAG and OHT subjects who switched from prior IOP-lowering medication. Furthermore, a switch in ocular hypertensive treatment to bimatoprost 0.01% was associated with an additional 10%–15% reduction in IOP.
Keywords: glaucoma, intraocular pressure, hyperemia, bimatoprost
Introduction
Prostaglandin analogs (PGAs) and prostamides (PMs) are considered first-line therapies for the treatment of glaucoma and ocular hypertension (OHT), because they effectively lower intraocular pressure (IOP) and have a good safety profile.1 The most common adverse event (AE) associated with both PGAs and PMs is conjunctival hyperemia. This side effect may be bothersome to patients to the extent that it reduces adherence to treatment.2
Bimatoprost 0.03% (Allergan, Irvine, CA, USA) is a PM approved for the safe and effective lowering of IOP in patients with open-angle glaucoma (OAG) or OHT.3–5 Several studies have found that bimatoprost 0.03% leads to greater reductions in IOP than latanoprost 0.005% or travoprost 0.004%, and may be useful for patients who do not respond to latanoprost.4,6–8 However, in comparison to PGA options, bimatoprost 0.03% is associated with a higher rate of conjunctival hyperemia, which in some cases may lead to discontinuation.4 Bimatoprost 0.01% was developed with the goal of creating a formulation of bimatoprost that would maintain the IOP-lowering efficacy achieved with bimatoprost 0.03% while offering an improved overall safety profile, particularly, improved ocular surface tolerability.3
In a Phase III, randomized, double-masked, multicenter clinical trial, bimatoprost 0.01% proved to have equivalent IOP-lowering efficacy to bimatoprost 0.03% through 12 months of treatment, while also showing improved tolerability, including less frequent and severe conjunctival hyperemia.3 Although these data provided evidence of the safety and efficacy of bimatoprost 0.01%, these findings were derived from a glaucoma and OHT patient population that met defined inclusion criteria that may not represent the diversity of patients typical of a given clinical practice. As a result, the “real-world” safety and efficacy of bimatoprost 0.01% as an ocular hypotensive therapy may be different from the results reported by Katz et al3 if bimatoprost 0.01% is administered to a more diverse patient population. The aim of the present study – the Canadian Lumigan RC Early Analysis Review (CLEAR) Trial – was to determine the external validity of bimatoprost 0.01% in a real-world clinical setting. We reported the safety and efficacy of bimatoprost 0.01% in treatment-naive primary OAG (POAG) and OHT subjects in a separate paper.9 Herein, we report the safety and efficacy of switching POAG and OHT subjects from a single IOP-lowering therapy to bimatoprost 0.01% monotherapy.
Subjects and methods
Study design
This 12-week, open-label, noncontrolled, prospective, observational, multicenter (67 Canadian centers) study evaluated the occurrence and severity of ocular hyperemia following a switch to bimatoprost 0.01% (Lumigan RC; Allergan) in subjects with POAG or OHT. This study was approved by the respective institutional review boards or independent ethics committees at each site, and followed the regulations of the Personal Information Protection and Electronic Documents Act and the Good Clinical Practice guidelines of the Declaration of Helsinki.
Study population
Eligible subjects were at least 18 years of age, diagnosed with elevated IOP due to either POAG or OHT, and determined by the treating physician to require treatment with bimatoprost 0.01% either as first-line monotherapy, as a switch of prior IOP-lowering medication, or as adjunctive IOP-lowering medication. This paper focuses on the subgroup of subjects who switched to bimatoprost 0.01% from a prior IOP-lowering medication. Subjects who were willing to return for all scheduled study visits signed an informed consent prior to or concurrent with the screening visit.
Subjects were excluded if they had hypersensitivity to benzalkonium chloride (BAK), any PGA, PM, or any other component of the study medication, or had any other abnormal ocular condition or symptom that would prevent them from entering the trial according to the treating physician’s judgment. Female subjects who were pregnant, nursing, or of childbearing potential and not using adequate contraception, as well as any subject with an inability to adhere to the treatment or study visit plan, or subjects who had participated in any other clinical trial involving an investigational product of a new chemical entity within 6 months prior to the screening or baseline visit were also excluded.
Treatment
Bimatoprost 0.01% was obtained by the patient through commercial means, and not as investigational study drug provided by Allergan. At the investigator’s discretion, the patient’s prior IOP-lowering medication was switched to bimatoprost 0.01% (baseline visit), either because the patient required additional IOP lowering or the patient could not tolerate the prior IOP-lowering therapy. All subjects were instructed to self-instill bimatoprost 0.01% into the affected eye(s) in the evening at approximately 8 pm. No washout period was applied on switching to bimatoprost 0.01% to reflect more closely real-world practice patterns. Other therapy considered necessary for the patient’s welfare was given at the discretion of the treating physician and was documented.
Outcome variables
Outcome variables were evaluated at baseline and at weeks 6 and 12 at 10 am (±2 hours). The primary outcome variable was the occurrence and severity of ocular hyperemia at week 12, using a photographic 5-point grading scale: 0= none – normal; +0.5= trace – trace flush reddish pink; +1= mild – mild flush, reddish color; +2= moderate – bright red color; +3= severe – deep, bright, diffuse redness.3
Secondary outcome variables included IOP change and IOP percentage change from baseline at weeks 6 and 12. Each participating site had an assigned examiner to measure IOP at each study visit. Although the type of tonometer was not specified in the protocol, all sites were instructed to use the same tonometer throughout the study for consistency. AEs were documented throughout the study, including AE seriousness and severity, action taken and relationship of the AE to treatment with bimatoprost 0.01%.
Statistical analyses
All subjects who provided informed consent and completed the screening/baseline visit (visit 1) were included in the intent-to-treat analysis. The intent-to-treat analysis used only data from the eye with the higher IOP at baseline (worse eye). If both eyes had the same IOP at baseline, the right eye was used.
Hyperemia grading was reported individually as 0, +0.5, +1, +2, and +3. Subsequently, the hyperemia scores were collapsed into two categories: 1) none to mild = subjects with hyperemia scores of 0, +0.5, or +1, and 2) moderate to severe = subjects with hyperemia scores of +2 or +3. These data were summarized as frequency counts and percentages at baseline and at weeks 6 and 12. The changes in collapsed hyperemia grading response from baseline at weeks 6 and 12 were summarized as improved, no change or worsened.
An improvement in hyperemia following a switch of IOP-lowering therapy to bimatoprost 0.01% was defined as a change in hyperemia grouping from moderate-to-severe hyperemia to none-to-mild hyperemia. No change in hyperemia was defined as staying within the same hyperemia grouping at baseline and on treatment with bimatoprost 0.01%. A worsening in hyperemia following a switch of IOP-lowering therapy to bimatoprost 0.01% was defined as a change in hyperemia grouping from none-to-mild hyperemia to moderate-to-severe hyperemia. Two-sided McNemar tests were used to evaluate treatment effects. IOP change and IOP percentage change from baseline were analyzed at weeks 6 and 12 using two-sided paired t-tests.
The present study was exploratory in nature, and as such no formal sample-size calculations were carried out. SAS for Windows version 9.2 (SAS Institute, Cary, NC, USA) was used for the analysis. All statistical tests were performed as two-sided tests with the significance level set at P≤0.05. P-values were not adjusted for multiple comparisons.
Results
Demographics
A total of 1,137 subjects were enrolled in the CLEAR Trial across 67 Canadian centers. Of the total population of subjects, 522 were naive to IOP-lowering treatment, and these were considered in a separate paper.9 The remaining 615 subjects had been treated with a prior IOP-lowering therapy and were switched to bimatoprost 0.01% as adjunctive IOP-lowering therapy (n=165) or as monotherapy (n=450). A complete list of the 450 subjects who switched prior IOP-lowering therapy to receive bimatoprost 0.01% monotherapy is presented in Table 1.
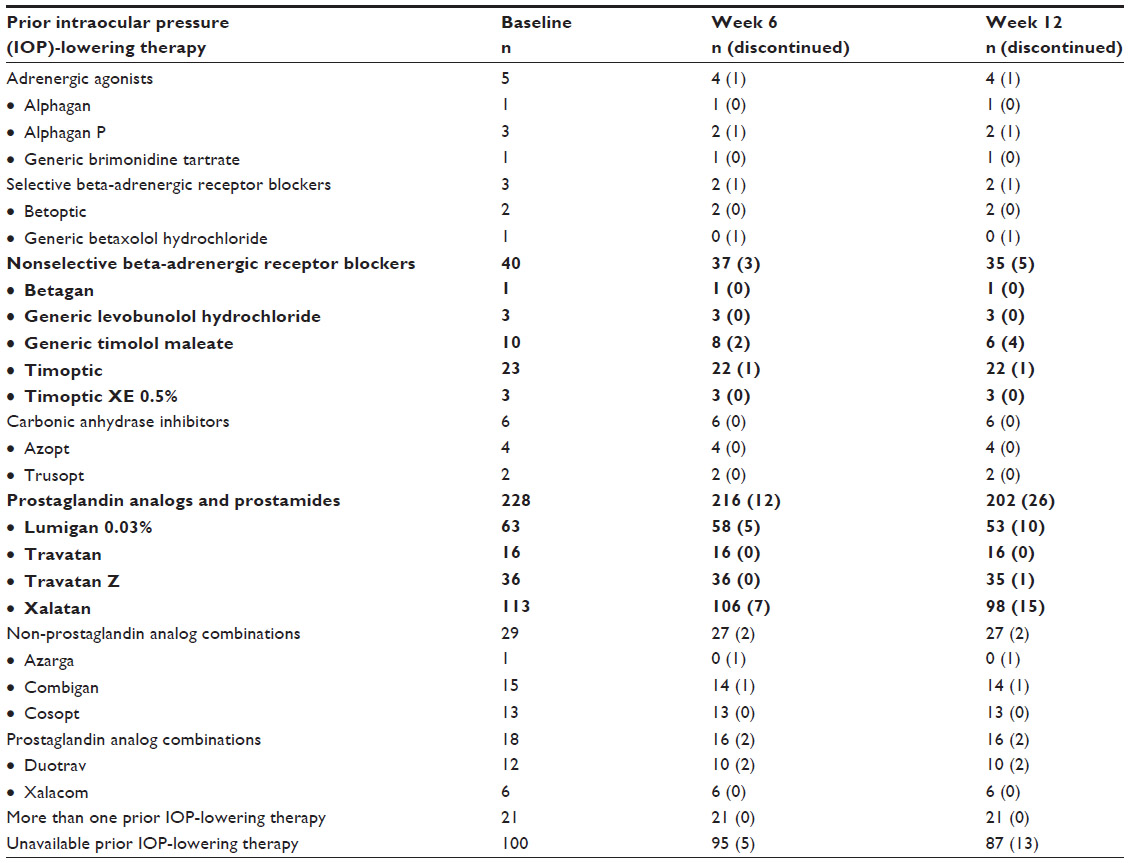 | Table 1 Switch-subject disposition |
The present report focuses on the subset of 268 subjects who were switched from a prior IOP-lowering monotherapy to bimatoprost 0.01% monotherapy and had a sample size greater than 15 (ie, latanoprost 0.005% [Xalatan®]; Pfizer, New York, NY, USA; n=113), bimatoprost 0.03% (Lumigan; Allergan; n=63), travoprost 0.004% (Travatan® original version preserved with BAK; Alcon Laboratories, Fort Worth, TX, USA; n=16), travoprost 0.004% with SofZia™ (Travatan Z; Alcon Laboratories; n=36), and nonselective beta-adrenergic blockers (n=40). Thirty-two of the 268 subjects discontinued for the following reasons: lost to follow-up, n=13 (4.9%); ocular AE, n=5 (1.9%); other AE, n=2 (0.7%); and reasons not related to AEs, n=12 (4.5%). Of the five monotherapy-switch subjects who discontinued the study due to ocular AEs, a total of nine ocular AEs related to bimatoprost 0.01% monotherapy were reported (Table 2).
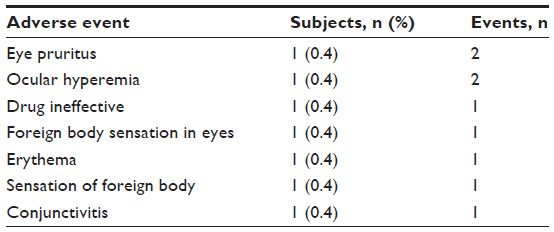 | Table 2 Early discontinuation due to treatment-related ocular adverse events |
Of the 268 subjects who received bimatoprost 0.01% monotherapy, 142 (53.0%) were female (mean age 67.5 years) and 126 (47.0%) were male (mean age 67.3 years). Common associated medical comorbidities among the enrolled 268 switch patients included hypertension (n=125, 46.6%), cardiovascular disease (n=48, 17.9%), diabetes (n=25, 9.3%), asthma (n=22, 8.2%), and pulmonary disease (n=15, 5.6%).
Ocular hyperemia
Among those who were switched from a prior IOP-lowering therapy to bimatoprost 0.01% monotherapy, the baseline numbers (and percentages) of subjects presenting with each hyperemia grade (ie, 0, +0.5, +1, +2, and +3) are presented in Table 3.
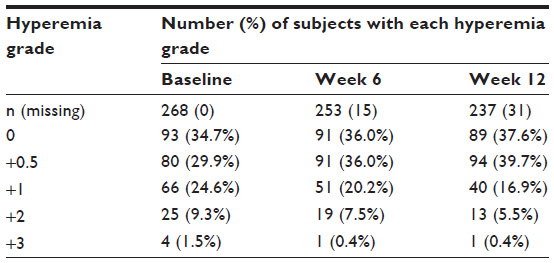 | Table 3 Overall rates of occurrence and severity of ocular hyperemia |
The numbers (and percentages) of subjects who were switched from either latanoprost 0.005%, travoprost 0.004%, travoprost 0.004% with SofZia or a nonselective beta-adrenergic receptor blocker to bimatoprost 0.01% and demonstrated none-to-mild or moderate-to-severe hyperemia at baseline, week 6, and week 12 are presented in Table 4.
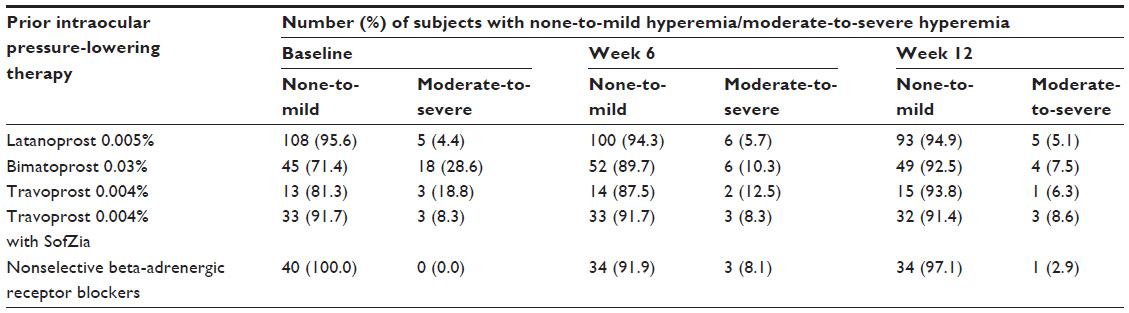 | Table 4 Rates of ocular hyperemia by prior intraocular pressure-lowering therapy |
The percentages of subjects who switched from one prior IOP-lowering therapy and showed improved, no change, or worsened ocular hyperemia grading at the week 6 and week 12 visits are displayed in Table 5. At week 6, 96.2% of subjects who were previously treated with latanoprost 0.005% showed no change, and 3.8% demonstrated worsened hyperemia grade. This change in ocular hyperemia grade was statistically significant (P=0.046). By week 12, the change in ocular hyperemia grade for subjects previously treated with latanoprost 0.005% was no longer statistically significant (P=0.180). Subjects previously treated with bimatoprost 0.03% who switched to bimatoprost 0.01% demonstrated improved (19.0% and 20.8%), no change (77.6% and 77.4%), or worsened (3.4% and 1.9%) ocular hyperemia grades at week 6 and week 12, respectively. These changes in ocular hyperemia grade observed at both study visits were statistically significant (P=0.013 and P=0.004 at week 6 and week 12, respectively). Analysis of ocular hyperemia following the switch to bimatoprost 0.01% from the other prior IOP-lowering therapies (travoprost 0.004% original version preserved with BAK, travoprost 0.004% preserved with SofZia, and nonselective beta-adrenergic receptor blockers) failed to reveal any statistically significant changes at the week 6 and week 12 study visits (Table 5).
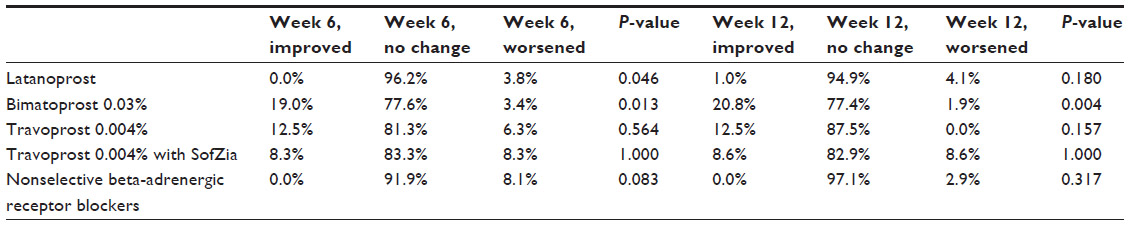 | Table 5 Change in ocular hyperemia following switch from prior intraocular pressure-lowering therapy |
Intraocular pressure
The mean IOP at baseline and weeks 6 and 12 and the mean percentage changes in IOP from baseline following switching to bimatoprost 0.01% at week 6 and week 12 are shown in Table 6. The additional decrease in mean percentage change in IOP achieved by switching to bimatoprost 0.01% monotherapy was statistically significant at week 6 and week 12 for all prior IOP-lowering therapies, except bimatoprost 0.03% at week 6 and week 12 and travoprost 0.004% (original version preserved with BAK) at week 6 (Table 6).
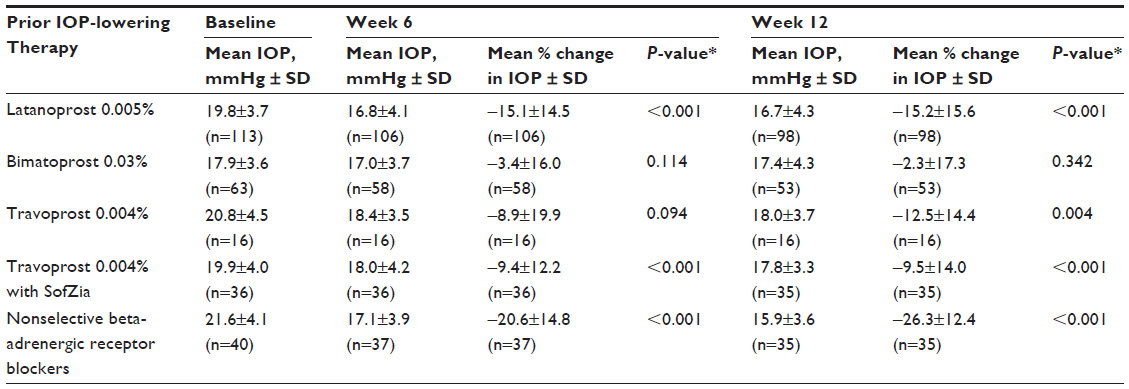 | Table 6 Intraocular pressure (IOP) values by prior IOP-lowering therapy |
Safety
Thirteen subjects reported 22 AEs, for an overall incidence of 4.9% (13 of 268). A total of 19 AEs reported by eleven subjects were considered related to bimatoprost 0.01%, for an incidence of 4.1% (eleven of 268). The most frequent reported treatment-related AEs associated with bimatoprost 0.01% were ocular hyperemia (1.1%), eye irritation (0.7%), and skin hyperpigmentation (0.7%). No serious AEs were observed in this study population.
Discussion
An ideal first-line IOP-lowering treatment for glaucoma and/or OHT should provide superior IOP-lowering efficacy with minimal side effects. In Canada, PGAs and PMs are recognized as first-line agents because of their superior efficacy and infrequent systemic side effects compared to other classes of topical ocular hypotensive therapies.1 Nonetheless, the most common AE experienced by PGA and PM users is hyperemia, a nonspecific clinical term implying vasodilation and increased blood supply to the conjunctival blood vessels that has been shown to be an important factor affecting patient adherence.2,10 While bimatoprost has been shown to have a unique pharmacology with respect to lowering IOP, bimatoprost and the PGAs are believed to share a common noninflammatory signaling mechanism leading to vasodilation.10–12
In the present study, the switch to bimatoprost 0.01% from prior IOP-lowering medication resulted in 94.1% and 5.9% of subjects presenting with none-to-mild and moderate-to-severe hyperemia, respectively, after 12 weeks. Of the prior IOP-lowering treatments, bimatoprost 0.03% was associated with the greatest percentage of subjects presenting with moderate-to-severe hyperemia at baseline (28.6%). Following the switch from bimatoprost 0.03% to bimatoprost 0.01%, the occurrence of moderate-to-severe hyperemia was reduced to 10.3% and 7.5% at week 6 and week 12, respectively. These results suggest that bimatoprost 0.01% was well tolerated following the switch from prior IOP-lowering medications, a finding that corroborates others.13,14 Additionally, our findings concerning the occurrence and severity of ocular hyperemia following the switch to bimatoprost 0.01% correlate well with the randomized, controlled trial results reported by Katz et al,3 which demonstrated a threefold-lower incidence of moderate-to-severe hyperemia in the bimatoprost 0.01% group compared to the bimatoprost 0.03% group.3
In terms of IOP-lowering efficacy, a number of studies have now been published comparing various PGAs and PMs, including latanoprost 0.005% with travoprost 0.004% and bimatoprost 0.03%. In 2003, a multicenter study involving 411 patients demonstrated that latanoprost 0.005%, travoprost 0.004%, and bimatoprost 0.03% were comparable in their ability to lower IOP, although differences were found in associated rates of conjunctival hyperemia.10 A recent observational study found that subjects who were switched to bimatoprost 0.01% from either latanoprost 0.005%, travoprost 0.004% preserved with 0.015% BAK (original version), tafluprost, or bimatoprost 0.03% achieved an additional IOP decrease of 2.8 mmHg, 3.1 mmHg, 2.8 mmHg, or 1.0 mmHg, respectively.15 It should be noted that for both the report by Pfennigsdorf et al15 and the present analysis, travoprost preserved with BAK was available in Europe and Canada, as well as in Argentina, Brazil, Chile, China, Colombia, India, Malaysia, Mexico, Taiwan, and Venezuela. As a result, the total number of patients who switched to bimatoprost 0.01% from prior travoprost preserved with BAK was low relative to other prior IOP-lowering therapies included in our analysis, due to the discontinuation of the original version of travoprost partway through study enrollment. A unique aspect of the present report is the inclusion of safety and efficacy data for the switch from prior travoprost preserved with SofZia to bimatoprost 0.01%, which demonstrated no statistically significant changes in hyperemia grade along with a −9.5%±14.0% change in IOP (P<0.001) at week 12.
In the present study, all of the patient subgroups on prior IOP-lowering therapies showed statistically significant improvements in IOP following the switch to bimatoprost 0.01% at week 12, except for the subgroup initially treated with bimatoprost 0.03%. The subgroup of patients previously treated with bimatoprost 0.03% was associated with the lowest IOP at baseline, which may explain why the IOP reductions following the switch to bimatoprost 0.01% were not statistically significant at the week 6 and 12 visits. Likely, the hyperemia side effect profile of bimatoprost 0.03% was the reason the investigators decided to switch the patient’s IOP-lowering regimen to bimatoprost 0.01%.
Some limitations of the present study should be acknowledged. Due to the open-label design of this study, the results presented may have been influenced by observer bias by the participating physician in the subjective grading of ocular hyperemia and the measurement of IOP, since neither the physician nor patients were masked to study treatment. The present study did not include a washout period between treatments to more closely resemble real-world practice. By not including a washout period, it is difficult to discern rates of ocular hyperemia caused by bimatoprost 0.01% and the prior IOP-lowering therapy.7 With 64 examiners involved in this study, we acknowledge the opportunity for interexaminer variability with hyperemia and IOP measurements. A Hawthorne effect for enhanced patient adherence to the study therapy may have contributed to the observed decrease in IOP when patients switched from prior therapy. Subject dropout can also affect a study’s results. Thirty-two of the 268 subjects discontinued for the following reasons: lost to follow-up, n=13 (4.9%); ocular AE, n=5 (1.9%); other AE, n=2 (0.7%); and reasons not related to AEs, n=12 (4.5%). Of the five monotherapy-switch subjects who discontinued the study due to ocular AEs, a total of nine ocular AEs were related to bimatoprost 0.01% monotherapy (Table 2). Exclusion of subjects with ocular AEs may have understated the primary outcome of this study, namely occurrence and severity of hyperemia attributed to a switch from prior IOP-lowering therapy to bimatoprost 0.01% monotherapy. Lastly, regression to the mean happens when a reduction in IOP occurs spontaneously rather than therapeutically. Since the present study did not include a second baseline IOP measurement prior to enrollment selection, bias causing a regression to the mean may have partially contributed to the significant IOP reductions observed.15
Overall, the results of this observational study provide real-world clinical evidence to support bimatoprost 0.01% as a suitable treatment option for POAG and OHT subjects. Additionally, further reduction in IOP may be achieved when switching from a prior IOP-lowering monotherapy to bimatoprost 0.01%.
Acknowledgments
The authors acknowledge the editorial assistance of Mary Ann Chapman, PhD and Jaspreet Grewal MSc, COMT, CCRP. This study was supported by Allergan Inc. The CLEAR Study Group: Iqbal Ike K Ahmed, Mississauga, ON; Rama Behki, Gatineau, QC; Robert Beldavs, Winnipeg, MB; Francois Bellefeuille, Trois-Rivieres, QC; Dan Belliveau, Halifax, NS; Raj Bindlish, Oakville, ON; FadiCalotti, Brantford, ON; Jeff Chambers, Kelowna, BC; Jason Cherry, Richmond, BC; Robert Chevrier, Gloucester, ON; Alan Coffey, Westmount, QC; Andy Crichton, Calgary, AB; Marino Discepola, Montreal, QC; Gordon Douglas, Calgary, AB; Stephane Dupont, Gramby, QC; Chaim Edelstein, Montreal, QC; Stephen Fichman, Montreal, QC; Bryce Ford, Calgary, AB; Michel Giunta, Sherbrooke, QC; Sheldon Goldhar, Scarborough, ON; Larry Green, Sidney, BC; Francine Guay, Lévis, QC; Eugene Hladky, Montreal, QC; Glen Hoar, Comox, BC; Muhammad Humayun, Dartmouth, NS; Cindy Hutnik, London, ON; Norman Hwang, Burnaby, BC; Fahim Ibrahim, Point Edward, ON; Carolyn Isbister, West Vancouver, BC; Helene Joyal, St-Jean-Sur-Richelieu, QC; Barry Kattleman, Montreal, QC; Baseer Khan, Vaughan, ON; Elie Khouri, Saint-Laurent, QC; Thomas Klein, Mississauga, ON; Alain Lachance, Saint-Georges, QC; Hesham Lakosha, Halifax, NS; Alain Lamoureux, Sorel, QC; Gisele Li, Montreal, QC; Derek Lui, Woodstock, ON; Mark Lukasik, Owen Sound, ON; Sam Markowitz, Toronto, ON; Jean Mayer, St-Charles Borrome, QC; Mohan Merchea, London, ON; Michael Miller, Mississauga, ON; Kam Mohaseb, North Vancouver, BC; Paul Murphy, Saskatoon, SK; Rashmi Nigam, Winnipeg, MB; Don Nixon, Barrie, ON; Patrick O’Keeffe, North Battleford, SK; Manuel Perrier, Lachine, QC; Carl Peters, Penticton, BC; Theodore Rabinovitch, Downsview, ON; Aaron Rifkind, Hamilton, ON; Suleiman Sefau, Charlottetown, PEI; Rob Semeniuk, Penticton, BC; Lesya Shuba, Halifax, NS; Barry Silver, Dorval, QC; Zakaria Tadrous, Abbotsford, BC; Eric Tam, Brampton, ON; Alexander Tan, Halifax, NS; Andrew Taylor, Niagara Falls, ON; Patricia Teal, Fort Erie, ON; David Yan, Mississauga, ON; Natasha Yepes, Kitchener, ON, Canada.
Disclosure
The authors report no conflicts of interest in this study.
References
Canadian Ophthalmological Society Glaucoma Clinical Practice Guideline Expert Committee. Canadian Ophthalmological Society evidence-based clinical practice guidelines for the management of glaucoma in the adult eye. Can J Ophthalmol. 2009;44 Suppl 1:S7–S93. | |
Zimmerman TJ, Hahn SR, Gelb L, Tan H, Kim EE. The impact of ocular adverse effects in patients treated with topical prostaglandin analogs: changes in prescription patterns and patient persistence. J Ocul Pharmacol Ther. 2009;25:145–152. | |
Katz LJ, Cohen JS, Batoosingh AL, Felix C, Shu V, Schiffman RM. Twelve-month, randomized, controlled trial of bimatoprost 0.01%, 0.0125%, and 0.03% in patients with glaucoma or ocular hypertension. Am J Ophthalmol. 2010;149:661–671. e1. | |
Aptel F, Cucherat M, Denis P. Efficacy and tolerability of prostaglandin analogs: a meta-analysis of randomized controlled clinical trials. J Glaucoma. 2008;17:667–673. | |
Noecker RS, Dirks MS, Choplin NT, Bernstein P, Batoosingh AL, Whitcup SM. A six-month randomized clinical trial comparing the intraocular pressure-lowering efficacy of bimatoprost and latanoprost in patients with ocular hypertension or glaucoma. Am J Ophthalmol. 2003;135:55–63. | |
Goldberg I, Li XY, Selaru P, Paggiarino D. A 5-year, randomized, open-label safety study of latanoprost and usual care in patients with open-angle glaucoma or ocular hypertension. Eur J Ophthalmol. 2008;18:408–416. | |
Craven ER, Liu CC, Batoosingh A, Schiffman RM, Whitcup SM. A randomized, controlled comparison of macroscopic conjunctival hyperemia in patients treated with bimatoprost 0.01% or vehicle who were previously controlled on latanoprost. Clin Ophthalmol. 2010;4: 1433–1440. | |
Law SK, Song BJ, Fang E, Caprioli J. Feasibility and efficacy of a mass switch from latanoprost to bimatoprost in glaucoma patients in a prepaid Health Maintenance Organization. Ophthalmology. 2005;112: 2123–2130. | |
Nixon DR, Simonyi S, Bhogal M, et al. An observational study of bimatoprost 0.01% in treatment-naïve patients with primary open angle glaucoma or ocular hypertension: the CLEAR Trial. Clin Ophthalmol. 2012;6:2097–2103. | |
Chen J, Dinh T, Woodward DF, et al. Bimatoprost: mechanism of ocular surface hyperemia associated with topical therapy. Cardiovasc Drug Rev. 2005;3:231–246. | |
Woodward DF, Krauss AH, Chen J, et al. The pharmacology of bimatoprost (Lumigan). Surv Ophthalmol. 2001;45 Suppl 4:S337–S345. | |
Astin M, Stjernschantz J, Selén G. Role of nitric oxide in PGF2α-induced ocular hyperemia. Exp Eye Res. 1994;59:401–408. | |
Kurtz S, Mann O. Incidence of hyperemia associated with bimatoprost treatment in naïve subjects and in subjects previously treated with latanoprost. Eur J Ophthalmol. 2009;19:400–403. | |
Abelson MB, Mroz M, Rosner SA, et al. Multicenter, open-label evaluation of hyperemia associated with use of bimatoprost in adults with open-angle glaucoma or ocular hypertension. Adv Ther. 2003;20: 1–13. | |
Pfennigsdorf S, Ramez O, von Kistowski G, et al. Multicenter, prospective, open-label, observational study of bimatoprost 0.01% in patients with primary open-angle glaucoma or ocular hypertension. Clin Ophthalmol. 2012;6:739–746. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.