Back to Journals » Journal of Blood Medicine » Volume 9
An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B
Received 22 May 2018
Accepted for publication 10 July 2018
Published 22 August 2018 Volume 2018:9 Pages 135—140
DOI https://doi.org/10.2147/JBM.S159297
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Martin H Bluth
Nicoletta Machin, Margaret V Ragni
Department of Medicine, University of Pittsburgh, Hemophilia Center of Western Pennsylvania, Pittsburgh, PA, USA
Abstract: Fitusiran is an RNA interference therapeutic that targets antithrombin (AT) in the liver and interferes with AT translation by binding and degrading messenger RNA-AT, thereby silencing AT gene expression and preventing AT synthesis. In both preclinical and clinical studies, AT knockdown results in dose-dependent AT lowering when fitusiran is given weekly or monthly subcutaneously. In clinical trials, fitusiran dose escalation has resulted in improved thrombin generation and clinical hemostasis as measured by reduction in annualized bleed rate. Unlike currently licensed drugs, this improvement was not only in patients with hemophilia A but in also those with hemophilia B, with or without inhibitors. In dental and surgical procedures, fitusiran also provided perioperative hemostasis in association with AT lowering. Fitusiran is well tolerated, with minor local injection site reactions, but in one subject with severe hemophilia A, the concomitant use of daily high-dose factor VIII, inconsistent with trial guidance to avoid high, repeat doses of clotting factor, was associated with fatal thrombosis, suggesting the need for caution when using hemostatic agents in conjunction with fitusiran. Preclinical in vitro and in silico studies indicate improvement in thrombin generation in rare bleeding disorder plasmas, including in plasmas from patients with severe factors V, VII, and X deficiency, suggesting potential therapeutic benefit.
Keywords: congenital bleeding disorder, clotting factor, fitusiran, mRNA, novel bypass
Background
The burden of disease for individuals with hemophilia is high, particularly in those who develop inhibitors. Fitusiran, an RNA interference (RNAi) therapy, presents a novel mechanism to treat these patients, and in those with inhibitors, provides a more effective bypass agent than currently available. The purpose of this review is to provide an update of completed and ongoing clinical trials in hemophilia A and B patients, with and without inhibitors, receiving fitusiran prophylaxis for bleed prevention. In particular, the efficacy and safety data in hemophilia with or without inhibitors will be reviewed, as well as preclinical data in rare bleeding disorders.
Hemophilia is an X-linked bleeding disorder resulting from a deficiency of factor VIII, for hemophilia A, or a deficiency of factor IX, for hemophilia B. Clotting factor deficiency results in clinical bleeding, spontaneous or traumatic, primarily in joints and muscles.1 Recurrent bleeding into joints results in significant morbidity, with chronic arthritis, disability, and pain.1 The current standard of care to prevent bleeds is through prophylactic intravenous infusion of concentrated factor products multiple times per week.2 In the 30% who develop inhibitors from infused factor VIII, the inhibitor neutralizes factor VIII, so that bypass therapy with activated prothrombin complex concentrates (APCCs) or rFVIIa, is required. Response to bypass is poor, however, resulting in significantly greater cost, hospitalization, and mortality than in those without inhibitors.3,4 Treatment with clotting factor is costly and patients’ quality-of-life is reduced, with limitations on career choice, sports, and life activities. Prophylaxis, although effective in preventing bleeds and reducing joint disease, is associated with a high rate of noncompliance, so that although begun in childhood, over half of affected adults with hemophilia no longer practice prophylaxis due to its invasiveness and inconvenience, and revert to less-effective, on-demand therapy.3 Even with perfect compliance on prophylaxis therapy, spontaneous or traumatic bleeding may occur which may be life-threatening, such as central nervous system bleeding or retroperitoneal bleeding. Thus, the burden of disease and invasiveness of treatment are high, and better approaches to treatment are needed. The novel bypass agents under development, in particular those utilizing RNAi technology, provide potential for simpler, less invasive and more effective treatment than currently available.
RNAi utilizes a normal cellular process, first identified in plants, which results in highly specific gene silencing.5 Gene silencing occurs through a cellular degradation mechanism for messenger RNA (mRNA) transcripts, which are targeted for elimination by double-stranded RNA (dsRNA; Figure 1).6 Most commonly, fully complementary dsRNAs, minimally 19–23 bp, are used to interfere with a target mRNA, with optimal binding achieved through the particular nucleotide composition or modification of nucleotides.7 This RNAi specificity makes it attractive for therapeutic development in rare and orphan diseases, including hemophilia.7 Initially, its clinical use was stymied by difficulties with targeting the correct cell population, RNAi (siRNA) instability, limited cell uptake, and pharmacokinetics issues.6 Recent breakthroughs, however, in lipid-based nanoparticle formulations have solved many of the remaining problems in cellular targeting and delivery.7 The availability of novel therapeutics, in particular siRNA targeting antithrombin (AT) for those with hemophilia, has the potential to reduce bleeding and the burden of treatment, and improve quality-of-life.
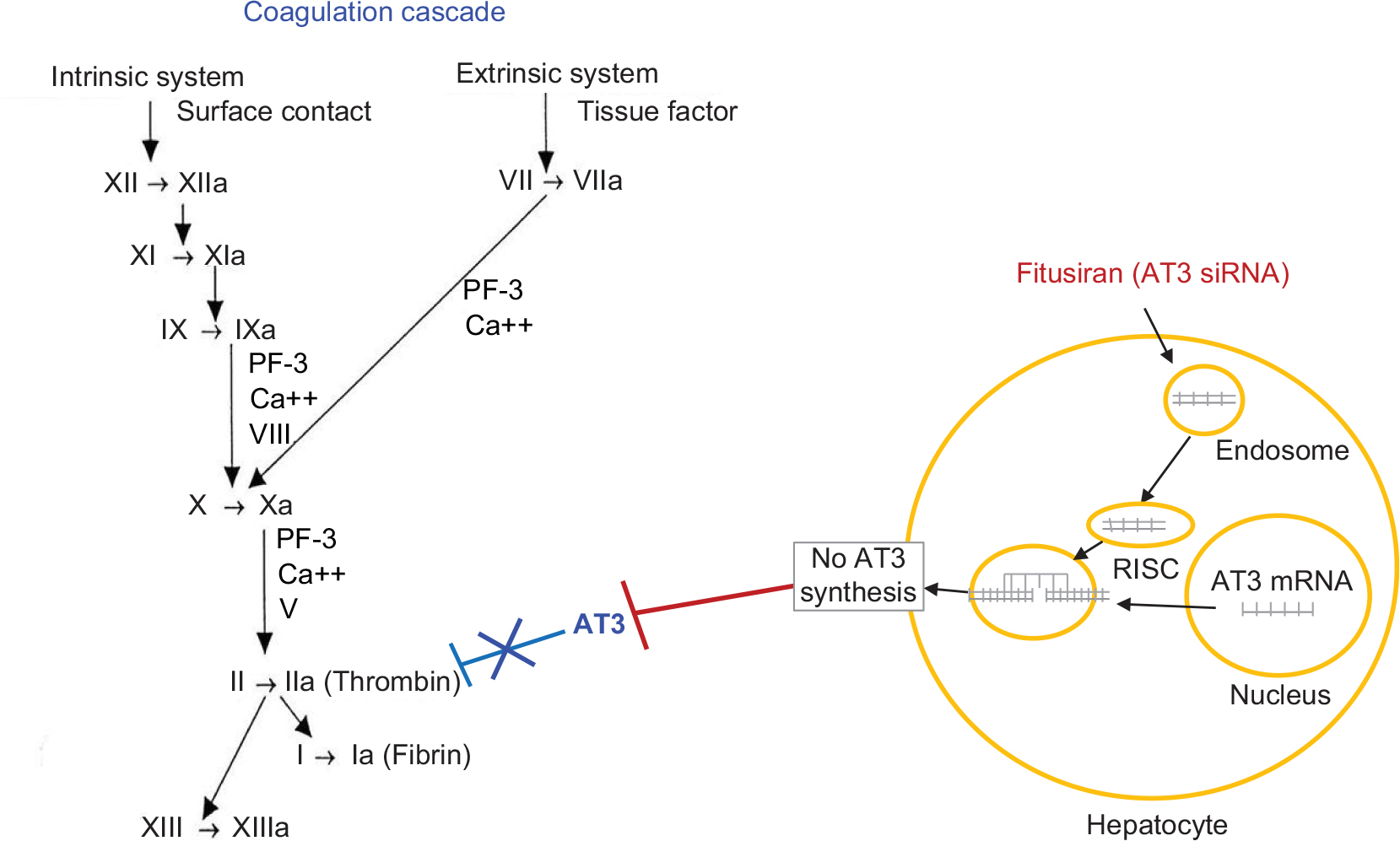 | Figure 1 Site of action of fitusiran on coagulation cascade. Note: Fitusiran is an RNAi therapeutic that targets AT in the liver and interferes with AT translation by binding and degrading mRNA-AT, silencing AT gene expression, and preventing AT synthesis. Abbreviations: AT, antithrombin; mRNA, messenger RNA; RISC, RNA-induced silencing complex; RNAi, RNA interference. |
The evolution of siRNA-AT
An important step for the development of siRNA therapy as a therapeutic agent for individuals with hemophilia was the identification of an appropriate therapeutic target. Pro- and antithrombotic factors present within the coagulation system tightly control the generation of thrombin, the key terminal enzyme which converts fibrinogen to fibrin. Hemophilia patients who have a concomitant prothrombotic mutation such as factor V Leiden, protein C deficiency, protein S deficiency, or AT deficiency have less severe bleeding phenotypes.8,9 Of these, AT deficiency was deemed an attractive target for siRNA therapy, as studies in mice with hemophilia A and AT deficiency had decreased bleeding, and studies with AT inhibitors normalized coagulation parameters in hemophilic blood.10,11
An RNAi therapeutic (ALN-AT3) was developed that targeted AT mRNA in the liver and interfered with mRNA AT translation by binding to and degrading mRNA AT to prevent AT synthesis, and thereby promote hemostasis (Figure 1). In preclinical studies, AT knockdown demonstrated a potent response in mice and nonhuman primates.12 In these studies, AT knockdown led to a dose-dependent lowering of AT levels, which corresponded to a dose-dependent increase in thrombin generation and reduction in bleeding. Based on animal models and in vivo tail clip studies, it was shown that achieving a >50% reduction in AT levels was hemostatic and could predictably reduce the bleeding phenotype in patients with hemophilia, with or without inhibitors.12 ALN-AT3 targets hepatocytes by conjugation of N-acetyl-galactosamine, a ligand for the asialoglycoprotein receptor located on hepatocytes,13 and also the site of synthesis and release of AT into the circulation.11 This approach was considered to be an ideal means to accomplish prophylaxis, or bleed prevention, in individuals with hemophilia by providing a simpler subcutaneous mode of administration with a prolonged duration of action.14
Phase I and I/II clinical trials
Fitusiran has been evaluated in hemophilia patients with and without inhibitors in Phase I and II trials and is currently under evaluation in Phase III trials (Table 1). A Phase I, open-label dose-escalation trial of ALN-AT3 (fitusiran) was conducted in four healthy volunteers and 30 patients with hemophilia A or B.15 In healthy volunteers, a single subcutaneous dose of fitusiran resulted in maximum AT lowering (19%) by day 21, with AT recovery into the normal range by day 56. Alternate dosing schedules were evaluated in hemophilia patients with weekly and monthly subcutaneous dosing. A fixed dose of fitusiran 80 mg monthly was demonstrated to achieve a mean maximum AT lowering of 87%. The association between AT lowering and increased thrombin generation was also confirmed and shown to be similar between subjects with hemophilia A and B.
 | Table 1 Fitusiran for hemophilia A and B Notes: aAsymptomatic LFT elevations occurred in patients with untreated chronic hepatitis C. bFatal event occurred with concomitant fitusiran and bypass agent use. cPhase III trials are ongoing, and planned enrollment figures are provided. Abbreviations: ABR, annualized bleed rate; AT, antithrombin; HA, hemophilia A; HA-I, hemophilia A with inhibitor; HB, hemophilia B; HB-I, hemophilia B with inhibitor; LFT, liver function test. |
In an extension Phase I and Phase I/II studies conducted in 25 hemophilia A and B patients without inhibitors, a monthly fixed dose of fitusiran was evaluated.15 Initial dosing with 50 mg in six patients resulted in AT lowering of 81% and a preliminary, post-hoc analysis suggested this resulted in a 49%–100% reduction in bleeding frequency. Trials also assessed the 80 mg dose in six hemophilia A and B patients with inhibitors.16 This dose is under evaluation in the ongoing Phase III trials. To date, fitusiran appears to be a promising investigational therapy for hemophilia patients complicated by inhibitors.
Efficacy
While the Phase I trial was neither designed nor powered to examine efficacy, a post-hoc exploratory analysis was conducted to examine the impact of monthly administration of fitusiran on bleeding rates. This analysis demonstrated a decrease in bleeding rates for patients receiving fitusiran, which correlated with the reduction in AT levels.15 More robust efficacy data were observed in a Phase II open-label extension trial which enrolled 33 patients, including 14 with inhibitors.17,18 Before treatment with fitusiran, patients without inhibitors receiving prophylaxis had an annualized bleeding rate (ABR) of 2, compared with an ABR of 1.7 after receiving fitusiran prophylaxis for a median of 13 months. Those without inhibitors receiving on-demand therapy who had a greater ABR of 12, were similarly able to reduce ABR to 1.7 during fitusiran prophylaxis during the same period. Inhibitor patients showed an even more striking change in ABR, with a pre-fitusiran ABR of 38 and a 6-month post-fitusiran ABR of zero.17,18 Overall, with an 80% AT lowering, a median ABR of 1 was observed for all patients, 48% of whom were bleed-free and 67% of whom experienced no spontaneous bleeds. Observed bleeding events in noninhibitor patients were mostly traumatic and located in joints, all of which were successfully treated with FVIII therapy.17,18 Current efficacy data for fitusiran appear encouraging, particularly for inhibitor patients, and it will be instructive to follow the ongoing Phase III trials for additional data on clinical endpoints including ABR, joint ABR, spontaneous ABR, as well as quality-of-life.
Safety, adverse events
In Phase I dose-escalation trial no serious adverse events or discontinuations occurred in healthy volunteers. Five mild adverse events were reported in two volunteers, four of which were not deemed by investigators as drug related. There was one event of spontaneously resolving headache, which was considered possibly related to fitusiran.15 In 25 participating hemophilia patients receiving fitusiran, 19 reported an adverse event of which nine were deemed possibly related to the drug. The most common mild adverse events were injection site pain in six participants and injection site erythema in four participants. Two patients experienced an increase in alanine aminotransferase level which resolved following cessation of the drug. Three participants experienced a severe adverse event including an episode of viral pneumonia, tooth fracture, and noncardiac chest pain. The patient who experienced chest pain also experienced an elevation in C-reactive protein, alanine aminotransferase, aspartate aminotransferase, and d-dimer. Extensive medical evaluation, however, was unable to identify an underlying event. The participant improved with drug cessation, analgesics, and antacids.15
In ongoing Phase I/II trials, fitusiran was generally well tolerated in hemophilia A and B patients, including those with inhibitors.16–18 Interim data from the Phase II open-label extension, which enrolled 33 patients, again demonstrated elevations in liver enzymes in 11 participating patients. In this analysis, it was noted that all were asymptomatic, and in eight, the liver enzymes resolved without dose interruption.19 Whether untreated chronic hepatitis C with detectable HCV viral load (PCR) played a role is not known. To date, no antibody development to fitusiran has been seen in study participants.16–18
After release of the Phase II interim data, one subject on the trial suffered a fatal cerebral sinus thrombosis.20 This severe adverse event occurred after repeated infusions of high-dose factor VIII for >24 hours, which had been advised against in the trial. Following this event, the Food and Drug Administration placed a hold on the Phase II trials, which was subsequently lifted after risk mitigation measures were implemented to reduce risk of bleeding, thrombosis, and liver disease and to standardize management of these potential risks. These included safety precautions for concomitant use of clotting factor with fitusiran.
Mitigation procedures in ongoing trials
Procedures have been put in place in ongoing and new clinical trials, including ALN-AT3SC-003 for hemophilia A or B inhibitor patients and ALN-AT3SC-004 for hemophilia A or B patients without inhibitors, to help mitigate risk and standardize management of bleeding, thrombosis, and liver disease.
For bleeding, an individualized bleed management plan with lower doses of factor or bypass agents must be used, specifically, FVIII, 10–20 IU/kg, FIX 20–30 IU/kg, APCC 30–50 IU/kg, or rFVIIa≤45 µg/kg, and antifibrinolytics cannot be combined with the latter. If a second dose is required, subjects must be in contact with their center, and, if multiple repeated doses are required, they must be evaluated clinically. In addition, a patient education module and brochure, diary, home storage and administration guidelines, and emergency cards are provided. Thrombin generation and other coagulation parameters will be assessed pre- and posttreatment with factor or bisphenol A.
For thrombosis, there is a low threshold to evaluate any signs and symptoms of thrombosis, including appropriate imaging studies, laboratory studies including prothrombin activation fragment PF 1+2, and consideration for AT reversal and anticoagulation.
For liver safety, liver function test (LFT) monitoring for fibrosis will be assessed by Fibrotest or Fibroscan, and in those with past hepatitis C infection, treatment with direct-acting antiviral agents will be required prior to participating in fitusiran trials. Dosing was similar in those with liver disease.
Concomitant treatment with bypass agents
Experience with the use of bypass agents to treat breakthrough bleeds in hemophilia patients receiving fitusiran is limited and based on expert opinion. However, recent reports of in vitro spiking studies of hemophilia plasmas offer some guidance.21 In these studies, platelet-poor plasma, obtained from hemophilia patients with and without inhibitors, before and after treatment with fitusiran, was spiked with bypass agents, including APCC (eg, factor VIII inhibitor bypass activity) and rFVIIa over a wide dose range. Heightened increases in peak thrombin were observed after the addition of each of these bypass agents, even with low-dose APCCs. The latter was observed to be equivalent to peak thrombin increases observed after full-dose APCC in pre-fitusiran-treated plasma samples from the same patient. These data, therefore, suggest that reduced-dose bypass agents may be a safer approach than full-dose bypass agents for breakthrough bleeding during fitusiran treatment.21
Surgery
Surgical experience with fitusiran is limited. In early clinical trials several participants required surgical procedures while receiving fitusiran and provided some information in this area.22 Four patients with severe hemophilia A, including two with inhibitors, have undergone surgical procedures while on fitusiran. These procedures included endoscopic cholecystectomy, thoracotomy/partial lung segmentectomy, molar tooth extraction, and premolar tooth extraction. Before these procedures, the AT level of each patient was <20% of baseline. The choice of factor therapy, including FVIII, APCC, or rFVIIa, was left to the discretion of the investigators. In four of the five procedures, hemostatic treatment of variable duration was provided based on the procedure, including 15 hours for the premolar tooth extraction and up to 13 days for the thoracotomy. There was minimal blood loss with all the procedures. While limited, the clinical experience in patients receiving fitusiran therapy and undergoing surgical procedures suggests it is possible to manage patients receiving fitusiran with hemostatic agents, and that AT levels when available may be helpful in guiding perioperative management. For example, elective surgery might be scheduled for 2 weeks after fitusiran dosing, when maximum AT lowering occurs, so that little if any additional hemostatic agent would be needed. For complex or emergency surgeries, reversing or stopping fitusiran with AT replacement might be considered to enable concomitant use of additional hemostatic agents, eg, factor concentrate, temporarily, with a plan to resume fitusiran following postoperative achievement of surgical hemostasis.
Other rare bleeding disorders
An ongoing area of research is the potential hemostatic effect of fitusiran on other rare bleeding disorders. Similar to hemophilia, rare bleeding disorders are characterized by insufficient thrombin generation due to deficiencies of other rare clotting factors. In vitro studies of plasmas from patients with severe deficiency of factor V, VII, or X have shown that thrombin generation improves with the addition of fitusiran.23,24 This suggests fitusiran may provide potential therapeutic benefit in individuals with rare bleeding disorders, but confirmation of this hypothesis will require future clinical trials.
Current challenges
The optimal use of fitusiran in the setting of bleeding events, trauma, or emergency procedures is still not robust. Moreover, while in vitro and in silico studies suggest the need for lower dosing of bypass agents when used during fitusiran therapy, what dose and what hemostatic agent(s) to use for breakthrough bleeds in hemophilia A or B patients with inhibitors remains unclear and will require further study in ongoing clinical trials.
Conclusion
Fitusiran is unique among potential hemostatic agents for congenital bleeding disorders, as it provides hemostasis and annualized bleed rate lowering in hemophilia A and B patients, with and without inhibitors. Further, as its mechanism of action is not specific for hemophilia, and based on preclinical in vitro studies, it may have potential utility in the hemostatic treatment of rare bleeding disorders, eg, severe factor V, VII, and X deficiency. Safety will be critical, not only in hemophilia patients requiring concomitant hemostatic agents for breakthrough bleeds, but also in those with rare bleeding disorders of varying severity. As more experience and data accrue in the ongoing Phase III (ATLAS) trials, the optimal patient management of those with bleeding disorders receiving this novel bypass agent will be informed.
Acknowledgments
This study was supported by NHLBI 1-T32 110849-01-A1; HRSA Federal Hemophilia Treatment Centers, Grant H30MC24050-04-00; and PA-DOH State Support of Hemophilia Center of Western PA, SAP #41000058531. MVR received research funding from Alnylam, Bayer, Biomarin, Bioverativ, Novo Nordisk, Sangamo, Shire, and Spark and has served on Advisory Boards for Alnylam, Bayer, Biomarin, Bioverativ, and Spark. The first preliminary results on fitusiran were presented online as a PowerPoint at http://www.alnylam.com/wpcontent/uploads/2017/07/ISTH-2017_Pasi_Fitusiran-OLE_CAPELLA-Deck_vFINAL_2017.07.10-1.pdf & http://www.alnylam.com/web/assets/ASH-2016_Fitusiran_NONINHIBITORS_04Dec2016_CAPELLA2.pdf.
Author contributions
Both authors contributed to conception and design, drafting and revising the manuscript, and both authors are accountable for all aspects of the work.
Disclosure
Both authors report no conflicts of interest in this work.
References
Mannucci PM, Tuddenham EG. The hemophilias--from royal genes to gene therapy. N Engl J Med. 2001;344(23):1773–1779. | ||
Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1–e47. | ||
Oladapo AO, Epstein JD, Williams E, Ito D, Gringeri A, Valentino LA. Health-related quality of life assessment in haemophilia patients on prophylaxis therapy: a systematic review of results from prospective clinical trials. Haemophilia. 2015;21(5):e344–e358. | ||
Gouw SC, van den Berg HM, Fischer K, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. Blood. 2013;121(20):4046–4055. | ||
Shuey DJ, Mccallus DE, Giordano T. RNAi: gene-silencing in therapeutic intervention. Drug Discov Today. 2002;7(20):1040–1046. | ||
Gao K, Huang L. Achieving efficient RNAi therapy: progress and challenges. Acta Pharm Sin B. 2013;3(4):213–225. | ||
Haussecker D. Current issues of RNAi therapeutics delivery and development. J Control Release. 2014;195:49–54. | ||
Shetty S, Vora S, Kulkarni B, et al. Contribution of natural anticoagulant and fibrinolytic factors in modulating the clinical severity of haemophilia patients. Br J Haematol. 2007;138(4):541–544. | ||
Franchini M, Montagnana M, Targher G, et al. Interpatient phenotypic inconsistency in severe congenital hemophilia: a systematic review of the role of inherited thrombophilia. Semin Thromb Hemost. 2009;35(3):307–312. | ||
Bolliger D, Szlam F, Suzuki N, Matsushita T, Tanaka KA. Heterozygous antithrombin deficiency improves in vivo haemostasis in factor VIII-deficient mice. Thromb Haemost. 2010;103(6):1233–1238. | ||
Patnaik MM, Moll S. Inherited antithrombin deficiency: a review. Haemophilia. 2008;14(6):1229–1239. | ||
Sehgal A, Barros S, Ivanciu L, et al. An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat Med. 2015;21(5):492–497. | ||
Nair JK, Willoughby JL, Chan A, et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. 2014;136(49):16958–16961. | ||
Ragni MV. Targeting antithrombin to treat hemophilia. N Engl J Med. 2015;373(4):389–391. | ||
Ragni MV, Georgiev P, Mant T. Fitusiran, an investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia: updated results from a Phase 1 and Phase 1,2 extension study in patients without inhibitors. Blood. 2016;128:2572. | ||
Pasi KJ, Georgiev P, Mant T, et al. Fitusiran, an investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia: updated results from a phase 1 and phase 1,2 extension study in patients with inhibitors. Blood. 2016;128:1397. | ||
Pasi KJ, Rangarajan S, Georgiev P, et al. Targeting of antithrombin in hemophilia A or B with RNAi therapy. N Engl J Med. 2017;377(9):819–828. | ||
Pasi KJ, Georgiev P, Fitusiran MT. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia: interim results from a phase 2 extension study in patients with hemophilia A or B with and without inhibitors. Res Pract Thromb Haemost. 2017;1(Suppl 1):25. | ||
Rocha P, Carvalho M, Lopes M, Araújo F. Costs and utilization of treatment in patients with hemophilia. BMC Health Serv Res. 2015; 15:484. | ||
Alnylam. Alnylam Reports Patient Death in Fitusiran Clinical Study. National Hemophilia Foundation, 2017. Available from: https://www.hemophilia.org/Newsroom/Industry-News/Alnylam-Reports- Patient-Death-in-Fitusiran-Clinical-Study. Accessed August 8, 2018. | ||
Qian K, Liu J, Huang S, Akin A. Thrombin generation response with the addition of bypassing agents in plasma of patients treated with fitusiran. Hemophilia. 2018;24(S5):MP-219. | ||
Negrier C, Ragni MV, Georgiev P, et al. Perioperative management in patients with hemophilia receiving fitusiran, an investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia. Blood. 2017;130(Suppl 1):2366. | ||
Sridharan G, Liu J, Qian K, Goel V, Huang S, Akinc A. In silico modeling of the impact of antithrombin lowering on thrombin generation in rare bleeding disorders. Blood. 2017;130:3659. | ||
Liu J, Qian K, Huang S, Colberg T. Effect of antithrombin lowering on thrombin generation in rare bleeding disorder patient plasma. Hemophilia. 2018;24(S5):TP-121. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.