Back to Journals » OncoTargets and Therapy » Volume 15
An Innovative Site-Specific Anti-HER2 Antibody-Drug Conjugate with High Homogeneity and Improved Therapeutic Index
Authors Hui X, Yuan C, Cao W, Ge W, Zhang D, Dan M, Zhao Q, Liu B, Yao B
Received 19 January 2022
Accepted for publication 28 March 2022
Published 8 April 2022 Volume 2022:15 Pages 331—343
DOI https://doi.org/10.2147/OTT.S357326
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Xiwu Hui,1,* Can Yuan,1,* Weirong Cao,1 Wenli Ge,1 Di Zhang,1 Mo Dan,2 Qian Zhao,2 Boning Liu,1,* Bing Yao1,*
1Institute of Quality Analysis, CSPC Megalith Biopharmaceutical Co., Ltd., Shijiazhuang, Hebei, People’s Republic of China; 2Pharmacology Center, CSPC Pharmaceutical Group Co., Ltd., Shijiazhuang, Hebei, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Boning Liu; Bing Yao, Institute of Quality Analysis, CSPC Megalith Biopharmaceutical Co., Ltd., No. 226 Huanghe Street, Shijiazhuang, Hebei, People’s Republic of China, Tel +8613284452520 ; +8613930148328, Fax +86031169085667, Email [email protected]; [email protected]
Purpose: Antibody-drug conjugates (ADCs) have emerged as a potent cancer therapeutic option in recent years. DP303c is a HER2-targeting ADC with a cleavable linker-MMAE payload. The current study aimed to evaluate the therapeutic potentials of DP303c in vitro as well as in vivo.
Materials and Methods: Size exclusion chromatography (SEC), reverse-phase high-performance liquid chromatography (RP-HPLC), and liquid chromatography-tandem mass spectrometry (LC-MS/MS) were used to analyze the physicochemical characterization of DP303c. An enzyme-linked immunosorbent assay (ELISA), a cell-based assay, and bio-layer interferometry (BLI) were used to evaluate DP303c’s affinity with HER2 and Fc receptors. A confocal laser scanning microscopy was used to observe the internalization of DP303c. Antibody-dependent cell-mediated cytotoxicity (ADCC) and cytotoxicity assays were used to investigate the activity of DP303c in vitro. The antitumor activity of DP303c was assessed in vivo in the HER2-positive cell-derived xenograft model.
Results: DP303c was a site-specific anti-HER2 antibody-drug conjugate with a monomethyl auristatin E (MMAE) with an average drug-to-antibody ratio (DAR) of 2.0. DP303c showed a high affinity with HER2 and could be effectively internalized. In vitro and in vivo, DP303c showed stronger antitumor activity as compared to trastuzumab-DM1 (T-DM1) in a series of HER2-positive cancer cells and cell-derived xenograft (CDX) models, especially in the lower HER2-expressing cells. DP303c also exhibited high serum stability and a good PK profile.
Conclusion: DP303c was a steady and homogenous DAR 2 ADC that was predicted to deliver MMAE inhibitor to tumor cells. DP303c demonstrated remarkable anticancer efficacy against T-DM1 in xenograft models. DP303c was a strong candidate for the treatment of patients with HER2-positive cancer.
Keywords: antibody-drug conjugates, ADCs, site-specific conjugation, engineered microbial transglutaminase, monomethyl auristatin E
Introduction
Antibody-drug conjugates (ADCs) have been studied for decades as a promising cancer treatment. As compared to conventional chemotherapy, ADCs have a larger therapeutic window because of their ability to target antigen-expressing tumor cells.1 As ADC drug development technology continues to evolve, several ADC drugs are being introduced to the market. FDA approved a total of 11 ADC drugs until now, including brentuximab Adcetris, T-DM1, POLIVY, Besponsa, Mylotarg, Lumoxiti, Enhertu, Padcev, Trodelvy, Blenrep, and Akalux.2–8 However, the majority of them are used in linker-payload systems like T-DM1 or Adcetris, which have resulted in dose-limiting toxicities in several human trials.9 Given these circumstances, an advancement in linker-payload systems would be desirable to produce next-generation ADCs with a higher safety profile.
ADC is composed of a small molecule cytotoxic drug coupled to a tumor-specific monoclonal antibody (mAb) through a cleavable or non-cleavable linker. The mechanism of ADCs relies on the binding of antibodies to specific antigens expressed on the surface of tumor cells. The specificity is provided by the mAb portion, which targets cell surface receptors expressed on tumor cells. After the binding of ADC to the cell surface receptor, the antigen/ADC complex is then endocytosed into the cell, where the toxin is released to kill the cancer cells.10
The third generation ADC technology is currently under development. In first-generation ADCs, drug linker conjugation to surface-exposed lysine or cysteine residues was random and uncontrolled, leading to heterogeneous ADCs mixtures having an elevated average value of the drug-antibody ratio (DAR).11,12 The efficacy and safety of the second generation of ADC drugs have been demonstrated in clinical trials. However, some still have limitations, including off-target toxicity, the presence of unbound antibodies, and a large drug-antibody ratio (DAR) (greater than 4) that causes ADC aggregation or rapid clearance, and an overall low therapeutic window.1,13–15 The development of third-generation ADC drugs focuses on overcoming the limitations of the first two generations to optimize monoclonal antibodies, linkers, and hazardous small chemical compounds. ADCs with DARs of 2 or 4 are formed by coupling a small molecule drug with the site features of a monoclonal antibody, reducing drug toxicity, unbound antibodies, and improving drug stability and pharmacokinetic (PK) efficiency.16–18
We developed a unique conjugation technology, which enables site-specific modification of certain glutamine (Gln, Q) amino acids in proteins by an engineered microbial transglutaminase (mTgase). ADC drugs have been covalently linked to Gln of mAb through mTgase to generate ADC drugs. Unlike existing third-generation ADC technologies, we used native mAb without the need for protein engineering, resulting in minimal impact on the physical and chemical properties of the antibodies. The highlighted procedure yields a high coupling efficiency. Furthermore, the processing technique is simple to use and extend. The ADC product is highly purified, active, and homogeneous, which is beneficial to the quality characterization and control of drugs, and effectively reduces the production cost.
We developed DP303c, a novel HER2-targeting ADC made up of a humanized anti-HER2 antibody (DP001), an enzyme-based cleavable peptide-linker, and two tubulin polymerization inhibitors (MMAE), using our original conjugation and linker-payload technique. DP303c is a steady and homogenous DAR 2 ADC that was predicted to deliver a significant amount of the highly active MMAE inhibitor to tumor cells regularly. The current study explored the efficacy, pharmacokinetics, and safety of DP303c. DP303c demonstrated remarkable anticancer efficacy against T-DM1 in xenograft models (having a high and low expression of HER2, accordingly), as well as a favorable PK and safety profile, implying that it has the potential to serve as a candidate drug in patients suffering from HER2 positive cancer.
Materials and Methods
Generation of DP303c
The materials used for DP303c production included anti-HER2 mAb (DP001), linker-MMAE (LND1002) and mTgase. DP001 was manufactured using a stable Chinese Hamster Ovary (CHO) cell line by ourselves. The amino acid sequence of DP001 was the same with that of Herceptin. This stable cell line was constructed by transfecting CHO cells with an expression plasmid containing DP001 coding gene. LND1002 was manufactured by a CRO, Levena. The mTgase was expressed in E. coli BL21. DP001, LND1002, and mTgase were incubated in sterile plastic bags at 30°C for 120 hours. Once the reaction was complete, a sanitized protein A column was used to remove excess LND1002 and mTGase, and followed by Capto Adhere polishing step to remove impurities.
Various Types of Cell Lines and Culture
The American Type Culture Collection (ATCC, Manassas, VA, USA) supplied the HER2-positive human breast cancer cell lines (SK-BR-3, BT474, HCC1954, JIMT-1), the HER2-positive human gastric cancer cell line (NCI-N87) and human ovarian cell line (SK-OV-3), as well as the HER2-negative breast cancer cell line (MDA-MB-468). All these cell lines were cultured according to the manufacturer’s protocol.
Size Exclusion Chromatography (SEC) Analysis
The SEC-HPLC method was used to determine the molecular size and heterogeneity of monomers, polymers, and residues. The SEC analyses were carried out under the following conditions: XBridge®BEH200Å (7.8 × 300 mm, particle size 3.5μm; Waters); Mobile phase: 100 mM PB, 100 mM NaCl, pH: 6.7 ± 0.1; Flow rate: 0.8 mL/min. UV absorbance was recorded at 280 nm.
Reversed-Phase HPLC (RP- HPLC) Analysis
The DAR of ADCs were measured by RP-HPLC. The RP-HPLC analyses were carried out under the following conditions: XBridge®Protein BEH C4 column (4.6 × 250 mm, particle size 3.5μm; Waters); Mobile phase A: 0.1% (v/v) TFA in water; Mobile phase B: 0.1% (v/v) TFA in acetonitrile; Flow rate: 1.0 mL/min; UV absorbance at 280 nm; Column temperature: 50°C; Mobile phase B flowed 10%-45% gradient elution in 0–33 min.
Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Analysis
The number of conjugated drugs was determined on the bases of molecular weight using LC-MS/MS. The following conditions were used to perform the LC-MS analyses: Equipment: Waters vion Q-TOF high-resolution mass spectrometer; Column: BioResolve RP mAb Polyphenyl chromatographic column; Sample treatment: N-glycosidase (PNGaseF) was added to a 100μg sample, which was then reacted for 10 minutes before being diluted to 0.1 mg/mL for sample loading; Mass spectrometry conditions: Positive ion scanning mode; Quality analysis range was 50–4000 Da; Capillary voltage was 3.0 KV; Taper hole voltage was 40 V; MSE collision energy was 20–45Ev; The ion source temperature was 120°C; Atomization temperature was 500°C. The atomization flow rate was 800 L/Hr.
ELISA Based-Binding Assay
The ELISA was used to determine HER2-binding capability of DP303c. HER2-ECD was diluted in PBS to a final concentration of 500 ng/mL and plated into 96-well plates (100 µL/well) for 24hrs at 4 °C, followed by removing the coating solution. Next, the remaining protein binding sites were blocked with 3% (V/V) bovine serum albumin in PBS for 2hrs, followed by adding gradient concentrations of DP303c. The plate was rinsed with PBST (four times) after reacting for 2 hrs. 100 µL of Goat anti-human IgG Fc antibody, conjugated with horseradish peroxidase (HRP), was added into each well, followed by incubating for 60 min at room temperature before being washed again. Finally, TMB was added for color development. A plate reader (Tecan/M200) was used to record the absorbance of each well at 450 nm/650 nm.
Fab and Fc Receptor Binding Affinity
An Octet RED96 instrument was used to conduct multi-concentration binding kinetic experiments at 30°C. CD32a, CD64, HER2, CD16a, and C1q binding experiments were performed with 1 × PBS (with pH 7.4), while FcRn binding tests were performed with 1 × PBS buffer (with pH 6.0). Anti-Penta-HIS Biosensors (PALL/ForteBio, 18–0038) were used to immobilize CD64, CD32a, CD16a, FcRn, and HER2. For association phase monitoring, DP001 and DP303c were diluted to 78.125–5000 nmol/L (CD32a and CD16a), between 7.8125 and 500 nmol/L (FcRn), and between 1.5625 and 100 nmol/L (CD64 and HER2)) and then allowed to bind loaded biosensors for 1 min. It took approximately 2 minutes for the dissociation phase. For C1q, DP001 and DP303c were immobilized on Protein L Biosensors (PALL/ForteBio, 18–0023). C1q was diluted to concentrations ranging between 12.5 and 800 nmol/L. In the Octet data analysis software, data was retrieved via a parallel buffer blank subtraction. The Langmuir model, which specifies a 1:1 binding stoichiometry, was used to fit the computed binding curves across the globe.
Flow Cytometric Evaluations
SK-BR-3, NCI-N87, HCC1954, BT474, SK-OV-3, JIMT-1, and MDA-MB-468 cells were trypsinized and treated with DP303c on ice in the dark (for 45 minutes). After incubation, the cells were rinsed twice with PBS (containing 2% FBS). Each well received 100 mL of fluorescent 488-conjugated Goat anti-human IgG Fc antibody, which was incubated on ice for 45 minutes. After incubation, the cells were washed twice and resuspended in 100 µL of PBS with 2% FBS and examined using a flow cytometer (Tecan/M200).
Endocytosis Assay Method
To obtain a single-cell suspension, the SK-BR-3 cells were harvested and resuspended in DMEM with 10% FBS. The cell density was adjusted to 1×105 cells/mL and seeded in a 96-well cell culture plate at 100 µL/well (1×104 cells/well). DP303c labeled with DyLight 488 was added into the 96-well plate with a final concentration of 2 µg/mL. After that, the cells were incubated for 24 hours at 37 °C with 5% CO2. Lysosome was labeled with a red fluorescence probe (Beyotime/C1046). A confocal laser scanning microscopy (Olympus/FV3000) was used to observe and photograph the cells in each well.
In vitro Cytotoxicity Assay
SK-BR-3, NCI-N87, HCC1954, BT474, SK-OV-3, and JIMT-1 cell lines were used to evaluate the ADCs’ cell cytotoxicity in vitro. The ADCs were also examined in MDA-MB-468 breast cancer line (with no expression of HER2). Cells were collected and resuspended at a density of 1×105 cells/well in a 96 well plate. The cells were incubated overnight at 37 °C followed by co-incubation with various concentrations of ADCs or antibodies for 72 hours. Resazurin solution (0.03%) was then added to examine the effect of antibodies and ADCs on cell growth. A microplate reader (Tecan/M200) was used to record the absorbance at 550 nm/610 nm. The obtained data was represented as mean ± standard deviation (SD) of triplicate data points.
ADCC Assay
The SK-BR-3 target cells (1.0 × 104 cells/well) were seeded (in triplicates) in 96-well plates, followed by incubation at 37 °C for 24 hrs. Next, the supernatant of the target cells’ culture medium was discarded. The Jurkat/hFcγRIIIa-NFAT effector cells were added to the culture plate of target cells with 1.5×105 cells/well in triplicate. Different concentration of ADCs or antibodies were co-incubated with the target and effector cells for 20–22 hrs. A plate reader (Tecan/M200) was used to read the chemiluminescence of each well after adding the luciferase assay substrate solution (Promega/G7940).
Inhibitory Effect of DP303c on the Growth of Xenograft Models
Pathogen-free female nude mice were injected subcutaneously with each cell suspension. According to tumor volumes, the mice were randomly divided in two groups: treatment and control groups. When the tumor had grown to an adequate size, medication was started on day 0. Each drug was given to the animals intravenously. The dosing frequency was once a week (qw), qw × 2 or qw × 3. The following equation was used to compute tumor growth inhibition (TGI, %): 100 × Average tumor volume (treated group)/Average tumor volume (control group). The experimental animals used in this study were purchased from Beijing Weitong Lihua Experimental Animal Technology Co., LTD. They were about 4–6 weeks old.
In vitro Stability of DP303c in Plasma
The MMAE release rate of DP303c was measured at 0.1 mg/mL in rat, monkey and human plasma for 2 weeks at 37 °C.
Pharmacokinetics of DP303c in Cynomolgus Monkey
The concentrations of DP303c and total antibody DP001 in monkey plasma were measured using a validated ligand-binding ELISA assay; 0.781 ng/mL was the lower limit of quantification of ELISA. The concentrations of MMAE in monkey plasma were measured using a validated liquid chromatography-tandem mass spectrometry (LC/MS-MS) method; 0.05 ng/mL was the lower limit of quantification. Male cynomolgus monkeys were given DP303c intravenously at 4.0 mg/kg. DP303c, total antibody, and MMAE plasma concentrations were determined up to 948 hrs. after the treatment.
Statistics
All statistical results are shown as the mean ± standard deviation (SD). Unpaired t-test was used to analyze differences in affinity, ADCC, cytotoxicity assay and pharmacokinetics between two groups. The antitumor activity of DP303c in vivo was evaluated by one-way ANOVA among multiple groups. GraphPad Prism 8.00 was used for data analysis. A p value of less than 0.05 was considered as statistically significant.
Results
HER2-Targeting Antibody-Drug Conjugate with DAR 2.0
DP303c is a DAR 2.0 antibody-drug conjugate that is designed to target HER2-positive cancer in humans. The product was an IgG1 monoclonal antibody against HER2 (DP001) with two MMAE molecules attached site-specifically through transamidation to residue Q295 in the antibody heavy chain’s constant region (Figure 1A). The anti-HER2 monoclonal antibody, DP001, was manufactured using a stable Chinese Hamster Ovary (CHO) cell line. DP001 had the same amino acid sequence with Trastuzumab (Herceptin). An enzyme-cleavable conjugate of DP001 and LND1002 was designed to release MMAE by lysosomal degradation. The DP303c was prepared as described previously (WO 2015/191883). The resulting ADCs were characterized in terms of their homogeneity and conjugation specificity. The DP303c chromatograms obtained from SEC-HPLC revealed that the aggregation of DP303c was 0.3% HMW species and 99.7% monomers, as indicated in Figure 1B. Only monomeric ADCs were found to have distinguishable single peaks. Furthermore, it was found that the antibody-to-drug conjugates were completely and quantitatively converted from antibody to ADC, with three major peaks representing 1, 2, and 3 drug molecules for each antibody (Figure 1C), with an approximated average DAR of 2. Additionally, a 3rd peak indicated the presence of a DAR 3 species, which revealed that another conjugation site exists in this antibody. LC-MS/MS analysis of DP303c showed the deconvoluted mass spectrum and was consistent with RP-HPLC analysis (Figure 1D).
 |
Figure 1 Structure and characterization of DP303c. Molecular structures of DP303c. (A) DP303c was composed of a HER2 targeting monoclonal antibody IgG1 (DP001) with two cleavable MMAE molecules attached to Glutamine residue 295 in the constant region of the antibody heavy chain. The linker-drug molecule LND1002 was an MMAE derivative with an amine-PEG linker. (B) There was no formation of aggregates according to the SEC analysis. (C) Three distinct peaks resulted from RP-HPLC analysis, referring to mAbs with one, two, and three-drug molecules, respectively. (D) The LC-MS/MS analysis of the DAR of DP303c. |
Antibody Binding is Unaffected by LND1002 Conjugation (at Glutamine 295)
The DP303c’s affinity for HER2 was determined by ELISA, cell-based assay, and BLI analysis. ELISA (Figure 2A) and cell-based binding assay (Figure 2B) revealed that both DP001 and DP303c had a similar affinity (p value >0.05) that was higher than T-DM1 (p value<0.05). DP001 and DP303c bind to human HER2 with similar affinity (p value >0.05), with KD values in a 1.2-fold range (Figure 2C). Since LND1002 was conjugated at glutamine 295, this did not affect DP303c’s affinity for HER2 antigen. Further characterization studies (Figure 2C) did not reveal any difference between the C1q and Fc gamma receptors (ie, CD16a, CD32a, and CD64) or FcRn binding affinities of DP001 and DP303c (p value >0.05).
 |
Figure 2 Binding activity, endocytosis, and ADCC activity of DP303c. (A) ELISA-based binding assay for the affinity with HER2 ECD (EC50 value of DP001, DP303c, and T-DM1 was 4.1 ng/mL, 4.5 ng/mL, and 10.5 ng/mL, accordingly). (B) Cell-based binding assay for the affinity with SK-BR-3 cells (EC50 value of DP001, DP303c, and T-DM1 was 124.6 ng/mL, 132.5 ng/mL, and 345.2 ng/mL, respectively). (C) The binding affinity of DP001 and DP303c with human HER2, FcRn, CD64, CD32a, CD16a, and C1q, was evaluated by Octet. (D) ADCC effects of DP001 and DP303c on HER2-positive human breast cancer SK-BR-3 cells that were incubated with the Jurkat/hFcγRIIIa-NFAT effector cell. (E) SK-BR-3 cells were treated for 24 hours at 37 °C with green fluorescence-labeled DP303c (2μg/mL). DP303c was localized in the cell membrane and then endocytosed into the cell and found in lysosomes after 24 hours. The data is presented as the mean ± SD from three independent experiments. |
The DP001 underlying mechanism is believed to be associated with ADCC activity via interacting with CD16a (an Fc gamma receptor) on immune effector cells,19–21 we also investigated whether DP303c retained the same mechanisms of action with DP001. The luciferase produced as a result of NFAT pathway activation was used to quantify ADCC activity in a bioluminescent reporter assay. The results showed that there was no significant difference in ADCC activity between DP303c and DP001 (p value >0.05), with EC50 of 47.3 ± 5.2 ng/mL and 35.6 ± 4.2 ng/mL, respectively, as shown in Figure 2D. The evaluation of ADCC indicated that DP303c retained the functions of DP001 after MMAE conjugation.
Immunofluorescence microscopy was performed to evaluate the DP303c internalization and lysosomal distribution in SK-BR-3 cells. Firstly, P303c was observed on the cellular membrane. However, when DP303c was incubated for 24hrs at 37 °C, the intrinsic ADC signals were found to be co-localized with lysosomes (Figure 2E).
In Comparison to T-DM1, DP303c Exhibits Potent Activity in vitro
The anticancer activity of DP303c, T-DM1, and DP001 (an anti-HER2 antibody) were evaluated using human cancer cell lines including breast, gastric, and ovarian cancer cell lines with an elevated level of HER2 expression. Flow cytometry was employed to assess the expression of HER2 on the surface of these cell lines, as indicated in Figure 3A. The DP303c anticancer activity was compared with T-DM1 and DP001 activities against the underlined cancer cell lines ie, breast, gastric, and ovarian cancer cell lines expressing variant levels of HER2. The MFIs ranged from 157,231 to 1,106,494 as compared to the isotype control ranged from 125 to 176, as represented in Table 1.
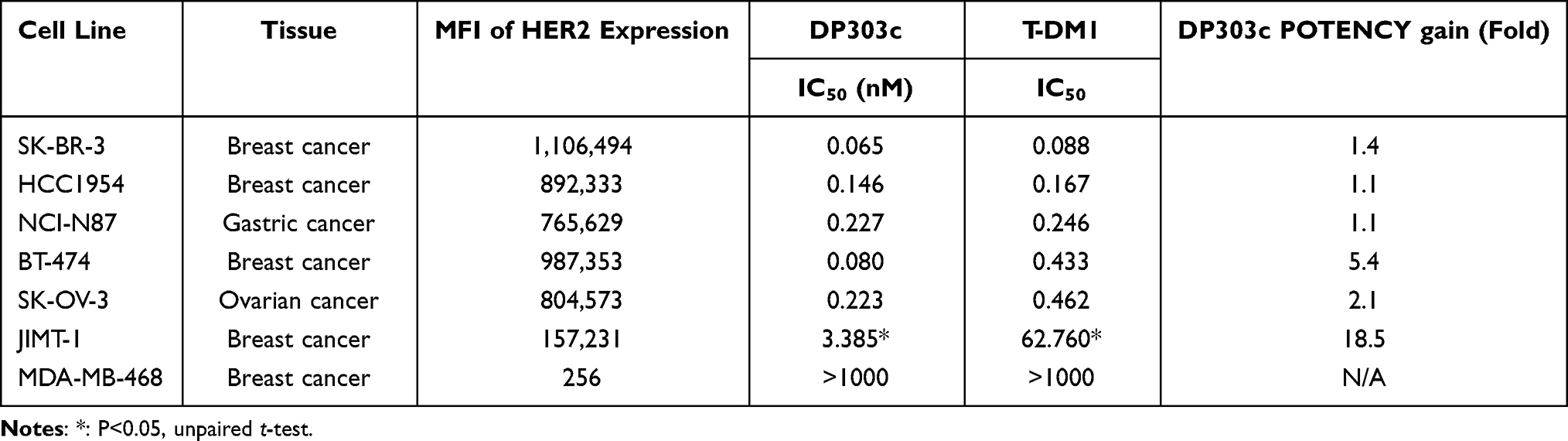 |
Table 1 Comparison of in vitro Activity Between DP303c and T-DM1 in Variable HER2 Cell Lines |
 |
Figure 3 In vitro cell growth inhibitory activity and expression level of HER2 in various cancer cell lines. (A) Binding of DP001 (Red) and DP303c (Green) to several cancer cell lines. Anti-human IgG1 was used as a negative control (Grey). The X-axis represents the fluorescence signal value of the antibody after binding with HER2 on the cell surface and secondary antibody (Goat anti-Human Antibody, Alexa Fluor 488) (B) After 72 hrs. of co-incubation, the cytotoxicity was evaluated in vitro using Resazurin (0.03%). The data is shown as the mean ± SD from three independent experiments. |
DP303c potently suppressed cellular proliferation in HER2-expressing cancer cells (having ≥ 157,231 MFI). Furthermore, the potency of DP303c against cancer increased with increasing expression level of HER2. DP303c was inactive against a HER2-negative cell line MDA-MB-468 as the IC50 concentration exceeded 10,000nM (Table 1). Based on the obtained results, the DP303c inhibitory potency against cellular proliferation was considerably elevated by drug conjugation to DP001. In addition, the growth inhibitory activity of DP303c against HER2 expressing cells were observed in a target-specific manner (Figure 3B). When compared to T-DM1, DP303c displayed comparable or greater effect against cells with overexpression of HER2 while potent growth inhibitory activity was also found in the cells with a reduced level of HER2 expression. In the cell line JIMT-1, DP303c showed more potent activity than T-DM1 (p value<0.05), with an IC50 value of 3.385 and 62.760nM, respectively (Table 1). This cell line was reported as HER2 IHC2 + or 1,22,23 suggesting that in cancers with a reduced level of HER2, DP303c activity may be more distinct from T-DM1 activity.
In vitro, DP303c Has High Plasma Stability and Enhanced Pharmacokinetics
ADCs with two drugs per antibody were effectively constructed using the site-specific conjugation technique. The stability of ADC can be reflected by the detection of ADC and total antibody at different time points. Firstly, the in vitro plasma stability of DP303c was evaluated. DP303c’s release of MMAE ranged from 0.003% to 0.599% on day 14 in plasma from rats, monkeys, and humans (Figure 4A), which was significantly lowered than the release rates of other VC-MMAE ADCs.24–26 The obtained results suggest that the stability of DP303c in plasma remains unaffected.
 |
Figure 4 In vitro stability in plasma of DP303c and PK characteristics of DP303c in the cynomolgus monkey. (A) In vitro stability of DP303c in plasma. (B) Concentration (μg/mL)-time profiles of DP303c, the total antibody DP001 and unconjugated MMAE (MMAE) post the first dose of DP303c at 4.0mg/kg. |
For the evaluation of DP303c stability in the bloodstream and to assess their pharmacokinetic (PK) characteristics, DP303c (4 mg per kg of total body weight) was injected into cynomolgus monkeys (n = 3 monkeys per group). The blood samples were taken from each monkey, followed by testing for total antibodies, ADC, and MMAE. The concentration-time curves for total antibody and intact Low concentrations of MMAE can only be detected at certain points. ADC intersected, indicating that DP303c was very stable in the bloodstream and remained intact as a DAR2 ADC all through the 42 days (Figure 4B). The DP001 half-life (138.4 hrs.) and DP303c concentration-time curves (136.8 hrs.) were found to be significantly similar (p value >0.05). Furthermore, a comparable clearance (ie, 0.57 and 0.65 mL/hr/kg, respectively) was recorded with exposure (AUC of 7333 and 6417hrs*g/mL, respectively) (p value >0.05) (Table 2). A low concentration of MMAE were identified at certain points, as shown in Figure 4B. MMAE exposure in cynomolgus monkey plasma was at least 3000 and 6000 times lower than DP303c at all time points, respectively.
 |
Table 2 PK Parameters (Mean ± SD) of DP303C, Total Antibody DP001, and Unconjugated MMAE After Administration of the First Dose of DP303C at 4.0 mg/Kg (90 min Intravenously) |
Anticancer Activity of DP303c and T-DM1 in Cell Line-Derived Xenograft Models
The cell lines with elevated levels of HER2 expression (ie, HCC1954, SK-OV-3, and NCI-N87) and those with reduced levels of HER2 expression (ie, JIMT-1) were used to investigate the anticancer activity of T-DM1 and DP303c in xenograft models. In the gastric carcinoma NCI-N87 xenograft model, DP303c, T-DM1, and DP001 were given to each mouse (n = 9 nude mice per group). As shown in Figure 5A, DP303c induced obvious tumor growth inhibition at a single dose of 2.5, 5, and 10 mg/kg, respectively. DP303c showed potent anticancer efficacy at a dose of 2.5 mg/kg, showing 119.7% of tumor growth inhibition (TGI) that was similar to that of 10 mg/kg of T-DM1. In addition, 10 mg/kg administration of DP001 showed moderate inhibition of the tumor growth, indicating 51.1% of TGI.
 |
Figure 5 Antitumor activity of DP303c in vivo. (A) Effect of in vivo antitumor activity on NCI-N87 xenograft model. DP303c was administered intravenously as a single dose of 2.5, 5 or 10 mg/kg on day 0. DP001 (10 mg/kg) was injected intravenously on days 0. T-DM1 (10 mg/kg) was given through intravenous administration on day 0. (B) In antitumor activity effect on HCC1954 xenograft model. The effects of DP303c (3, 10 or 15 mg/kg), DP001 (15 mg/kg), and T-DM1 at 15 mg/kg were evaluated. After randomized grouping, the mice in each group were administered drugs on days 0 and 7. (C) In vivo tumor inhibition effect on SK-OV-3 xenograft model. Different doses (1, 3 or 10 mg/kg) of DP303c were given via intravenous injection at (mention days). DP001 (10 mg/kg) and T-DM1 (10 mg/kg) were injected intravenously. After randomized grouping, the mice in each group were administered drugs on days 0 and 7. (D) In vivo antitumor efficacy on JIMT-1 xenograft model. The effects of DP303c (0.3, 1, 3 or 10 mg/kg), DP001 (10 mg/kg), and T-DM1 (10mg/kg) were also evaluated as a single intravenous injection on day 0. Each point indicates the tumor volume, which was represented as the average tumor volume ± SD (n = 9). |
In the breast cancer HCC1954 xenograft model, the mice in each group were given drugs on day 0 and 7 after randomized grouping. Doses of 3 mg/kg DP303c demonstrated obvious tumor shrinkage with a TGI of 117.3% (Figure 5B), which was similar to the effect of T-DM1 at doses of 15 mg/kg with a TGI of 119.4%. There were no inhibitory effects on tumor growth when treated with DP001 at doses of 15 mg/kg.
In the ovarian cancer SK-VO-3 xenograft models (Figure 5C), DP303c significantly inhibited tumor growth in a dose-dependent manner at a dose of 1, 3, and 10 mg/kg on days 0, 7 and 14, with a 52.0%, 97.8%, and 113.0% tumor inhibition rate, respectively. T-DM1 clearly showed obvious potent antitumor efficacy at the dose of 10 mg/kg, indicating 111.2% of TGI that was similar to the dose of 10 mg/kg (qw × 3) of DP303c with the same dosing frequency. DP001 of 10 mg/kg (qw × 3) showed no obvious tumor growth.
In the breast cancer JIMT-1 xenograft model, DP303c at doses of 0.3, 1, 3, 10 (qw × 3) and 10 mg/kg (single dose) showed significant anti-tumor growth, indicating 56.1%, 125.1%, 129.3%, 129.1% and 128.4% of TGI, respectively (Figure 5D). T-DM 10 mg/kg (qw × 3) had similar antitumor efficacy as DP303c 0.3 mg/kg (qw × 3), with a 52.5% and 56.1% tumor inhibition rate, respectively. DP001 at 10 mg/kg (qw × 3) showed no obvious tumor growth. The underlined data also confirmed DP303c’s HER2 specificity in a HER2 low-expressing model.
Exploratory Toxicology of DP303c
In repeated-dose investigations, the nonclinical safety profile of DP303c was characterized in nonhuman primates (up to 20 mg/kg). In all treatment groups, there were no aberrant findings associated with DP303c in ophthalmoscopic, body temperature, electrocardiogram (ECG), urinalysis, bone marrow smear, lymphocyte phenotypic assay, or local irritation. The plasma exposure of LND1002 and MMAE was low, with MMAE AUC of no more than 0.03% (normalized to molecular weight) as that of serum DP303c. During clinical trials, free MMAE-related toxicity such as thrombocytopenia and anemia was not found in monkeys or patients. In cynomolgus monkeys, the major safety signals were minimum to moderate lymphocyte depletion in the spleen and lymph node, as well as minimal to moderate inflammation in the lungs, which were all regained in the recovery phase. The no observed adverse effect level (NOAEL) of DP303c after five repeated doses once every three weeks in cynomolgus monkeys is 6.0 mg/kg. The highest non-severely toxic dose (HNSTD) is 20.0 mg/kg.
Discussion
Given HER2ʹs importance in tumor formation and prognosis, as well as its expression in several types of malignant tumors, HER2 has been considered a significant target in the treatment of carcinoma. The Her2 receptor is over-expressed in many tumors (25% of breast cancer, 20% of ovarian cancer, 30% of intestinal-type gastric cancer, and 20% of lung cancers) and such over-expression is correlated with aggressive tumors and bad prognosis.27–29
HER2 is a clinically validated therapeutic target against breast carcinoma progression. Currently, several monoclonal antibodies, such as mAbs and ADCs, against HER2 are available which block and show downstream signaling cascades of HER2.9,18,23,29 T-DM1 and DS-8201a are the only two ADCs available for HER2-targeting-based medications. For the first time, an ADC has been approved for the treatment of HER2-positive breast carcinoma patients that integrates trastuzumab’s anti-tumor potential with DM1.8,18,30 The efficacy and therapeutics window of T-DM1 is limited by its low thioester bond stability between mAb and linker, and the heterogeneity of conjugation. Off-target toxicity has also resulted in concerns about safety, such as thrombocytopenia, neutropenia, and peripheral neuropathy, which are all prevalent side effects associated with premature drug release.31–34 Although T-DM1 has been approved for advanced HER2-positive breast cancer in which Trastuzumab and taxoid therapy have failed, primary (congenital) and secondary (acquired) resistance is also emerging with T-DM1 as a major clinical challenge.35 DS-8201a is a potential HER2-targeting ADC that has shown promising efficacy in cancer patients and robust anticancer activity in multiple tumor xenograft models. Although DS-8201a overcomes heterogeneity to provide an effective therapeutic outcome in patients suffering from gastric carcinoma, the linked membrane-permeable payload Dxds inflict bystander killing on cancerous cells with negative expression of the target antigens.23,36–38 Permeable loads have potential to provide bystander action in heterogeneous tumors, meanwhile they can cause off-target damage in healthy tissue as well.
Using enzyme-based site-specific conjugation platform technology and a cleavable linker design, DP303c was developed for chemical stability and homogeneity to solve challenges encountered by T-DM1 and other ADCs. The enzymatic site-specific conjugation platform can more effectively control DAR and avoid changes in antigen binding affinity, resulting in homogenous products with excellent pharmacokinetic and pharmacodynamic characteristics. In killing MDR-tumors, the hydrophilicity of the DP303c PEG linker outperformed that of SMCC linked ADCs.39 In the current study, it was shown that enzyme-based site-conjugated ADC, ie, DP303c, has high homogeneity (DAR), high antitumor activity (in vitro and in vivo studies), high PK stability, and high safety (toxicology). In addition, DP303c has shown significant antitumor activity in HER2-specific and payload drug-derived antitumor models.
In the non-reduced RP-HPLC analysis, DP303c was initially considered as a DAR2 ADC having a mean DAR value of 2.0. It showed the high homogeneity of DP303c. In addition, DP303c exhibited significant HER2-specific efficacy both in vitro and in vivo studies. In vitro studies with HER2-positive gastric and breast cancer cells revealed that DP303c is associated with maintaining the HER2 binding. When DP303c was compared to T-DM1, it was shown that DP303c had more potent in vitro activity than T-DM1. In addition, in xenograft models, DP303c has also displayed high and long-lasting tumor suppression effects at a comparatively low concentration. This was especially relevant in tumors with lower HER2 expression levels compared to T-DM1. There could be relatively different mechanism for the payload shown by internalization of DP303c, which accumulates more efficiently in cells than the lys-MCC-DM1 payload released by T-DM1, which could explain the differential activity of DP303c.40 DP001 without conjugation was found to be insufficient to exhibit the same antitumor activity in vivo when compared to DP303c and T-DM1. The underlined finding implies that conjugating DP001 with MMAE significantly boosted the anticancer activity of MMAE.
DP303c was shown to be well tolerated in rats and cynomolgus monkeys. Monkeys were given a repeated dose of DP303c and showed no signs of thrombocytopenia or peripheral neuropathy, as seen in the T-DM1 experiments (monkeys). The increased stability of DP303c in plasma is likely to account for these favorable characteristics. The linker drug LND1002 is a compound with an NH2-PEG3-val-cit linker that connects it to MMAE. PEG3 is utilized to boost solubility in solution, which improves stability. DP303c has improved serum stability due to its stable linker (low release rates of MMAE). Aside from T-DM1, SGN-35 (Brentuximab vedotin) has been approved, and currently there are more than 30 different ongoing ADC clinical trials.41 The majority of them are used in linker-payload systems with microtubule polymerization inhibitors, such as T-DM1 or SGN-35 (DM1 and MMAE, respectively). In clinical trials of this tubulin inhibitor conjugate ADCs, numerous dose-limiting toxicities, including thrombocytopenia, neutropenia, and neuropathy, were observed, some of them were thought to be caused by free drugs in plasma.42 Our newly established drug linker method allowed the anti-HER2 ADC stable in plasma, have superior pharmacokinetics, and display significant anticancer activity in vitro and in vivo.
Pharmacokinetic studies on the Cynomolgus Monkey showed that DP303c remained stable as a DAR2 ADC with a terminal half-life of 5.7 days. DP303c has a similar half-life and exposure time as unconjugated anti-HER2 mAb, while the randomly conjugated hydrophobic loaded ADC has a lower exposure time and shorter half-life than its unconjugated antibody.12,43 The monkey pharmacokinetic result was used to establish human pharmacokinetic profiling after a repeated dose of DP303c every 3 weeks for three cycles. In a clinical trial, 0.5 mg/kg of DP303c every 3 weeks is predicted to show some efficacy (data not shown). The repeated dosage in monkeys revealed that DP303c caused no serious side effects at doses up to 20 mg/kg (HNSTD), suggesting a broad therapeutic window.
To conclude, a novel third-generation site-specific DP303c was found to be a highly effective agent for treating HER2-positive malignancies, laying the groundwork for further improvement of this ADC for the treatment of HER2-positive breast cancer. This product is currently being studied in clinical trials (NCT04146610, NCT04828616, and NCT04826107). The obtained results revealed that it is effective and safe (data not shown). Furthermore, it was able to construct a broad range of ADCs that included a variety of different mAb, linker, and drugs.
Ethics Statement
All the animals were acclimated under standard laboratory conditions (ventilated room, 25±1 °C, 60±5 % humidity, 12 h light/dark cycle) and had free access to standard water and food (SYXK-2016-006). All procedures were conducted in accordance with the “Guiding Principles in the Care and Use of Animals” (China) and were approved by the Laboratory Animal Ethics Committee of CSPC Megalith Biopharmaceutical Co., Ltd. (P20160515-2).
Funding
There is not any sources of financial assistance were used to conduct the study described in the manuscript and/or used to assist with the preparation of the manuscript.
Disclosure
The authors are all employees of CSPC Megalith Biopharmaceutical Co., Ltd. The author report no conflicts of interest in this work.
References
1. Dan N, Setua S, Kashyap VK, et al. Antibody-Drug Conjugates for Cancer Therapy: chemistry to Clinical Implications. Pharmaceuticals. 2018;11(2):66.
2. de Claro RA, McGinn K, Kwitkowski V, et al. Drug Administration approval summary: brentuximab vedotin for the treatment of relapsed Hodgkin lymphoma or relapsed systemic anaplastic large-cell lymphoma. Clin Cancer Res. 2012;18(21):5845–5849.
3. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard of care in relapsed or refractory acute lymphoblastic leukemia: final report and long‐term survival follow‐up from the randomized, Phase 3 INO‐VATE study. Cancer. 2019;125(14):2474–2487.
4. Modi S, Saura C, Yamashita T, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N Engl J Med. 2020;382(7):610–621.
5. Rosenberg JE, O’Donnell PH, Balar AV, et al. Pivotal Trial of Enfortumab Vedotin in Urothelial Carcinoma After Platinum and Anti-Programmed Death 1/Programmed Death Ligand 1 Therapy. J Clin Oncol. 2019;37(29):2592–2600.
6. Sehn LH, Herrera AF, Flowers CR, et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J Clin Oncol. 2020;38(2):155–165.
7. Norsworthy KJ, Ko CW, Lee JE, et al. Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncologist. 2018;23(9):1103–1108.
8. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(19):1783–1791.
9. Bang Y-J, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376(9742):687–697.
10. King HD, Dubowchik GM, Mastalerz H, et al. Monoclonal antibody conjugates of doxorubicin prepared with branched peptide linkers: inhibition of aggregation by methoxytriethyleneglycol chains. J Med Chem. 2002;45(19):4336–4343.
11. Nejadmoghaddam MR, Minai-Tehrani A, Ghahremanzadeh R, Mahmoudi M, Dinarvand R, Zarnani AH. Antibody-Drug Conjugates: possibilities and Challenges. Avicenna J Med Biotechnol. 2019;11(1):3–23.
12. Lyon RP, Bovee TD, Doronina SO, et al. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol. 2015;33(7):733–735.
13. Fisher JE. Considerations for the Nonclinical Safety Evaluation of Antibody-Drug Conjugates. Antibodies. 2021;10:2.
14. Shen BQ, Xu K, Liu L, et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol. 2012;30(2):184–189.
15. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315–337.
16. van der Lee MM, Groothuis PG, Ubink R, et al. The Preclinical Profile of the Duocarmycin-Based HER2-Targeting ADC SYD985 Predicts for Clinical Benefit in Low HER2-Expressing Breast Cancers. Mol Cancer Ther. 2015;14(3):692–703.
17. Li JY, Perry SR, Muniz-Medina V, et al. HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell. 2016;29(1):117–129.
18. Lewis Phillips GD, Li G, Dugger DL, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280–9290.
19. Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. 2010;10(5):328–343.
20. Guy DG, Uy GL. Bispecific Antibodies for the Treatment of Acute Myeloid Leukemia. Curr Hematol Malig Rep. 2018;13(6):417–425.
21. Kubota T, Niwa R, Satoh M, Akinaga S, Shitara K, Hanai N. Engineered therapeutic antibodies with improved effector functions. Cancer Sci. 2009;100(9):1566–1572.
22. Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. Arch Pathol Lab Med. 2014;138(2):241–256.
23. Ogitani Y, Aida T, Hagihara K, et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin Cancer Res. 2016;22(20):5097–5108.
24. Li C, Zhang C, Li Z, et al. Clinical pharmacology of vc-MMAE antibody-drug conjugates in cancer patients: learning from eight first-in-human Phase 1 studies. MAbs. 2020;12(1):1699768.
25. Deslandes A. Comparative clinical pharmacokinetics of antibody-drug conjugates in first-in-human Phase 1 studies. MAbs. 2014;6(4):859–870.
26. Mahmood I. Clinical Pharmacology of Antibody-Drug Conjugates. Antibodies. 2021;10(2):76.
27. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137.
28. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–182.
29. Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244(4905):707–712.
30. LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res. 2011;17(20):6437–6447.
31. Mahalingaiah PK, Ciurlionis R, Durbin KR, et al. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol Ther. 2019;200:110–125.
32. Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs. 2016;8(4):659–671.
33. Zhao H, Gulesserian S, Malinao MC, et al. Mechanism for ADC-Induced Neutropenia: role of Neutrophils in Their Own Demise. Mol Cancer Ther. 2017;16(9):1866–1876.
34. Lee HE, Park KU, Yoo SB, et al. Clinical significance of intratumoral HER2 heterogeneity in gastric cancer. Eur J Cancer. 2013;49(6):1448–1457.
35. Hunter FW, Barker HR, Lipert B, et al. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br J Cancer. 2020;122(5):603–612.
36. Li F, Emmerton KK, Jonas M, et al. Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical Models. Cancer Res. 2016;76(9):2710–2719.
37. Ogitani Y, Hagihara K, Oitate M, Naito H, Agatsuma T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016;107(7):1039–1046.
38. Doi T, Shitara K, Naito Y, et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody–drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: a phase 1 dose-escalation study. Lancet Oncol. 2017;18(11):1512–1522.
39. Kovtun YV, Audette CA, Mayo MF, et al. Antibody-maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res. 2010;70(6):2528–2537.
40. Erickson HK, Lewis Phillips GD, Leipold DD, et al. The effect of different linkers on target cell catabolism and pharmacokinetics/pharmacodynamics of trastuzumab maytansinoid conjugates. Mol Cancer Ther. 2012;11(5):1133–1142.
41. Hamilton GS. Antibody-drug conjugates for cancer therapy: the technological and regulatory challenges of developing drug-biologic hybrids. Biologicals. 2015;43(5):318–332.
42. Hinrichs MJ, Dixit R. Antibody Drug Conjugates: nonclinical Safety Considerations. AAPS J. 2015;17(5):1055–1064.
43. Boswell CA, Mundo EE, Zhang C, et al. Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1 antibody-drug conjugates in rats. Bioconjug Chem. 2011;22(10):1994–2004.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.