Back to Journals » Drug Design, Development and Therapy » Volume 11
An in silico high-throughput screen identifies potential selective inhibitors for the non-receptor tyrosine kinase Pyk2
Authors Meirson T ![]() , Samson AO, Gil-Henn H
, Samson AO, Gil-Henn H
Received 10 March 2017
Accepted for publication 28 March 2017
Published 18 May 2017 Volume 2017:11 Pages 1535—1557
DOI https://doi.org/10.2147/DDDT.S136150
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Tomer Meirson, Abraham O Samson, Hava Gil-Henn
Faculty of Medicine in the Galilee, Bar-Ilan University, Safed, Israel
Abstract: The non-receptor tyrosine kinase proline-rich tyrosine kinase 2 (Pyk2) is a critical mediator of signaling from cell surface growth factor and adhesion receptors to cell migration, proliferation, and survival. Emerging evidence indicates that signaling by Pyk2 regulates hematopoietic cell response, bone density, neuronal degeneration, angiogenesis, and cancer. These physiological and pathological roles of Pyk2 warrant it as a valuable therapeutic target for invasive cancers, osteoporosis, Alzheimer’s disease, and inflammatory cellular response. Despite its potential as a therapeutic target, no potent and selective inhibitor of Pyk2 is available at present. As a first step toward discovering specific potential inhibitors of Pyk2, we used an in silico high-throughput screening approach. A virtual library of six million lead-like compounds was docked against four different high-resolution Pyk2 kinase domain crystal structures and further selected for predicted potency and ligand efficiency. Ligand selectivity for Pyk2 over focal adhesion kinase (FAK) was evaluated by comparative docking of ligands and measurement of binding free energy so as to obtain 40 potential candidates. Finally, the structural flexibility of a subset of the docking complexes was evaluated by molecular dynamics simulation, followed by intermolecular interaction analysis. These compounds may be considered as promising leads for further development of highly selective Pyk2 inhibitors.
Keywords: virtual screen, efficiency metrics, MM-GBSA, molecular dynamics
Introduction
The focal adhesion kinase (FAK) and its homologous FAK-related proline-rich tyrosine kinase 2 (Pyk2) define a distinct family of non-receptor tyrosine kinases that coordinate adhesion and cytoskeletal dynamics with survival and growth signaling. FAK and Pyk2 exhibit ~48% amino acid sequence identity, common phosphorylation sites, and a similar domain structure, which includes an N-terminal four-point one, ezrin, radixin, and moesin (FERM) domain, a kinase domain, three proline-rich regions, and a C-terminal focal adhesion-targeting (FAT) domain. Following integrin or growth factor stimulation, Pyk2 and FAK are autophosphorylated on a tyrosine residue (Y402 and Y397, respectively), which provides a critical binding site for Src kinase. Following its binding, Src phosphorylates additional tyrosines on Pyk2 or FAK, which are important for full activation of the kinases and for their binding to downstream signaling proteins.1 Although FAK is expressed in most cells, Pyk2 exhibits a more restricted expression pattern with the strongest expression in the central nervous system and in hematopoietic cells.2 FAK is a major intracellular signaling component of integrin-mediated cell adhesion and plays a role in signaling pathways mediated by growth factor receptors. PYK2, however, is activated by a variety of extracellular cues, including agonists of G protein-coupled receptors, increase in intracellular calcium concentration, inflammatory cytokines, and stress signals, as well as integrin-mediated cell adhesion.2–4 The differential expression and localization patterns of FAK versus Pyk2 might limit their functional redundancy and may suggest distinct and possibly antagonistic roles in cells.
Pyk2 appears to be important for the organization of cytoskeletal components in a polarized manner during directional motility in response to chemotactic gradients in macrophages5 and in B cells6 and for integrin-mediated degranulation response of neutrophils during infection.7 Deletion of Pyk2 in mice leads to increase in bone mass due to enhanced differentiation and activity of osteoprogenitor cells8 as well as impairment in osteoclast function.9 Pyk2 was also found to regulate skin wound healing by controlling epidermal keratinocyte migration via a pathway that requires the expression and function of matrix metalloproteinases.10 The gene encoding Pyk2 was recently found as one of 11 new susceptibility loci for late-onset Alzheimer’s disease11 and as an in vivo marker and modulator of tau toxicity.12 Upregulation of Pyk2 expression has been noted in several human tumors, including glioma, hepatocellular carcinoma, nonsmall cell lung carcinoma, prostate cancer,13 and early and advanced breast cancers.14 The different physiological and pathological roles of Pyk2 present it as a high-value therapeutic target for inflammatory cellular responses, osteoporosis, Alzheimer’s disease, and invasive cancers.
An increase in Pyk2 expression accompanied by a compensatory function for Pyk2 upon FAK loss has been described in FAK-deficient mouse embryonic fibroblasts (MEFs),4,15 following conditional deletion of FAK in macrophages;16 in the in vivo regulation of bone architecture and density;17 and in blood vessel formation during angiogenesis.18 The ability of Pyk2 to adopt a compensatory role in integrin-mediated signaling pathways suggests that strategies that will selectively target FAK might lack efficacy due to Pyk2 compensation, particularly in applications directed toward blocking inflammatory response, osteoporosis, and tumor angiogenesis.
Clinical translation of kinase inhibitors has mainly focused on competitive inhibition of the catalytic domain. While classical kinase inhibitors bind directly to the ATP binding site, this approach has been challenged by the significant sequence and structure conservation of the catalytic domains among different protein kinases. With the exception of cancer therapeutics, where inhibition of multiple kinase targets might gain additional therapeutic benefits, minimizing off-target activity is most often desired in drug design. Therefore, an alternative approach has been the use of bipartite inhibitors that target both the adenosine triphosphate (ATP)-binding site and a less conserved adjacent hydrophobic region formed by the altered conformation of the activation loop containing the DFG (Asp–Phe–Gly) motif. Inhibitors of this group stabilize the inactive conformation of the kinase (DFG-out conformation) and offer a potential of improved selectivity.
Despite significant efforts to develop a potent and selective inhibitor for Pyk2 over the last several years, the available inhibitors for Pyk2 lack the potency, selectivity, or have impaired pharmacokinetics,19,20 and no selective inhibitor has yet proceeded beyond pre-clinical trials, including Pfizer’s lead compound (S)-14a from the series of sulfoximine-substituted molecules. In spite of its increased selectivity for Pyk2 over FAK, N-methylsulfoximine (S)-14a lacks sufficient metabolic stability and is characterized by high in vivo clearance in rats.20
In this study, we used an in silico high-throughput screening approach in order to identify potential Pyk2 kinase inhibitors from a large library of small molecules. Results of these studies uncovered potential lead-like molecules that were subjected to comparative docking and free binding energy calculations, followed by molecular dynamics (MD) simulations in order to identify compounds that are predicted to have affinity and selectivity for Pyk2. A final set of potential candidates with predicted selectivity for Pyk2 was assembled. These candidates may be considered as lead compounds that could be further developed into potent and selective Pyk2 inhibitors.
Methods
Database selection and ligand preparation
We used the commercially available ZINC12 database (http://zinc.docking.org/)21 for virtual screening, which contains more than 35 million purchasable compounds in ready-to-dock, three-dimensional formats. The lead-like subset of compounds, containing six million compounds, was selected on the basis of their properties to allow further optimization. The lead-like criteria properties are molecular weight between 250 and 350 g/mol, predicted partition constant (xLogP) ≤3.5, and number of rotatable bonds (RBs) ≤7. The subset was processed by Raccoon22 utility where missing polar hydrogen atoms were added.
Target preparation and grid generation
Four high-resolution crystal structures of human Pyk2 kinase domain with distinct conformations of the active site and activation loop were selected for our high-throughput in silico screening: 1) apo-Pyk2 (Protein Data Bank [PDB] ID: 3FZO); 2) Pyk2 in complex with the inhibitor PF-431396 (PDB ID: 3FZR); 3) Pyk2 in complex with the inhibitor PF-4618433 (PDB ID: 3FZT);19 and 4) Pyk2 in complex with N-methylsulfonamide 2a (PDB ID: 3H3C).20 The resolutions of the target PDB structures 3FZO, 3FZR, 3FZT, and 3H3C were 2.2, 2.7, 1.95, and 2.0 Å, respectively. Prior to docking simulations, missing side chains and loops were filled using Prime23 and termini were capped. Hydrogen atoms were added, bond orders were assigned, and ions were removed using the Protein Preparation Wizard.24 Crystallographic water molecules were removed as they contained less than three hydrogen bonds after sampling their orientations at pH 7 using PROPKA25 and optimizing their hydrogen bonds. Following this step, the holo form of the structures was minimized by a restrained energy minimization using the OPLS2005 force field and default root-mean-square deviation (RMSD) constraint of 0.3 Å. Refined structures were imported to AutoDockTools26 where Gasteiger charges were assigned to all atoms and non-polar hydrogen atoms were merged. For each target, three-dimensional affinity grids of dimensions up to 30×18×26 Å were centered around the active site with 1.0 Å spacing.
In silico high-throughput screening
AutoDock Vina27 was used for the docking simulation. The settings of exhaustiveness for finding the global minimums were defined as 4, and only the best ranked poses were retrieved. The screening calculations ran on a double-threaded 480 CPU 2.67 GHz Xeon Linux cluster machine.
Efficiency metrics scoring
To promote hits with favorable affinity and pharmacokinetics, several indices were utilized. The mean accumulated binding (MAB) index was developed to assess the contribution of multiple conformational binding of a molecule without impairing other valid molecules that are predicted to bind fewer structures.
|
|
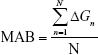
where ΔGn is the calculated free energy of a bound molecule to a specific conformation in kcal/mol and N denotes the total number of binding molecules. In this sense, binding is defined as any Vina docking score within the top 10,000 molecules of the corresponding structure. The binding efficiency index (BEI) quantifies the efficiency of the binding affinity based on a molecular weight scale.28
|
|
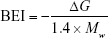
Surface-binding efficiency index (SEI) quantifies the efficiency of the binding affinity based on total polar surface area (TPSA).28
|
|
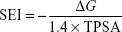
Lipophilic Ligand Efficiency (LLE) estimates the specificity of a molecule in binding to the target relative to the calculated partition constant (xLogP).29
|
|
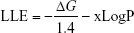
Pareto score optimizes multiple objectives, BEI versus SEI (BvS) simultaneously in a trade-off approach.28
|
|
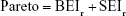
where BEIr and SEIr are the ranking of the respective index. Cheminformatic analysis was performed using MATLAB.
Visual inspection
The top-ranked molecules containing undesirable chemical groups and substructures,30 including toxic and highly metabolized molecules, were filtered via visual inspection. Molecules containing more than four aliphatic or aromatic rings were filtered as too many rings pose a liability in drug design.31,32 In addition, compounds containing more than two merged aromatic rings were excluded to avoid highly planar structures.33,34
Selectivity evaluation and screening
The 240 top filtered molecules were submitted for a second screen in AutoDock Vina and Glide35 to collect the compounds that are selective for Pyk2 compared to FAK. The crystal structures of Pyk2 (PDB ID: 3FZT and 3H3C) and FAK (PDB ID: 1MP8) were prepared as described earlier, including the grid generation for docking in Vina. As a preparation step for docking in Glide, the molecules were prepared using LigPrep Wizard36 and grids with similar box size were generated in the Receptor Grid Generation of Schrodinger.37
To assess the docking score more reliably, exhaustiveness in Vina was increased to 8 and the extra-precision (XP) mode was used in Glide. The resulting poses predicted in Glide XP for Pyk2 and FAK were rescored and compared using Prime MM-GBSA (molecular mechanics/generalized born surface area). Prime MM-GBSA is a physics-based method that calculates the force field energies of the bound and unbound states of the protein–ligand complex.38 The van der Waals surface-based surface generalized born 2.0 implicit solvation model was used with the OPLS2005 force field, and residue flexibility was defined throughout the structure. The MM-GBSA binding free energy is defined as follows:39,40
|
|
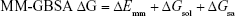
ΔEmm is the difference in energy between the complex structure and the sum of the energies of the unbound ligand and protein. ΔGsol is the difference in the solvation energy of the complex and the sum of the solvation energies for the unbound ligand and protein. ΔGsa is the difference in the surface area energy for the complex and the sum of the surface area energies for the unbound ligand and protein.40 Entropy terms related to the ligand and protein are not incorporated, due to the expensive computational demand and no apparent improvement in many cases.41,42
MD simulation
The final drug candidates were submitted to MD simulation performed using the Desmond package43 and OPLS2005 force field. The docking models of the most selective ligands as calculated by MM-GBSA were used as the initial coordinates for the MD calculation. The TIP3 explicit water model was used to define the solvent.44 The simulation solvent cell box with periodic boundary conditions was set to orthorhombic shape with a buffer distance of 10 Å to each dimension. The system was neutralized by placing counter ions of 6Na+ and 3Cl− for the structures of Pyk2 and FAK, respectively, and background salt was added to the solvent with a concentration of 0.15 M.45 Prior to the production of molecular dynamic simulations, the system was equilibrated using the default Desmond relaxation protocol. The final state of the system was set to 300 K and 1 atm,39 and the equations of motions were integrated with a 2 fs time step.46 The simulations were performed in the NPT (constant number of atoms, pressure, temperature) ensemble using Nosé–Hoover thermostat with 1 ps relaxation time and Martyana–Tobias–Klein barostat with a 2 ps relaxation time.47 The smooth particle mesh Ewald (PME) algorithm48 was used to deal with long-range electrostatic interactions with a cutoff radius of 9 Å.49 The production of 20 ns long MD simulations was performed on each complex system, and dynamic trajectories were analyzed in Desmonds’ Maestro simulation analysis tools and MATLAB. Quantitative analysis of the ligand–protein molecular interactions was calculated over the MD simulation. The distances as defined in Desmond for hydrogen bonds were <2.5 Å. General hydrophobic interaction distances were within 3.6 Å, and π–cation and π–π distances were within 4.5 Å. Ionic interaction distances were within 3.7 Å, and hydrogen bonding via water bridge molecule distance was <2.7 Å.
Results
In silico high-throughput screening
In order to identify potential inhibitors of Pyk2 kinase domain, an in silico high-throughput screening approach was used. To enhance the sampling of the target conformational space, four different Pyk2 kinase domain crystallographic structures with unique conformations were processed and prepared for in silico screening. The four crystal structures represent different conformations that were induced by ligands that have different selectivity profiles: 1) 3FZO is the apo, unbound state;19 2) 3FZR adopts an active conformation state (DFG-in) and contains an ATP-mimetic inhibitor, PF-431396, with a low selectivity to Pyk2;19 3) 3FZT adopts an inactive conformation (DFG-out) and contains an allosteric inhibitor, PF-4618433, selective to Pyk2;19 and 4) 3H3C adopts an active form (DFG-in) and contains the inhibitor N-methylsulfonamide 2a, which is selective to Pyk2.20 The two-dimensional interaction plots of the ligands are presented in Figure S1. A library of more than six million purchasable lead-like molecules from the ZINC database was screened against the binding pockets of each of the four structures (active site grids). The primary criterion used to select for initial set of molecules was binding free energy values (ΔG) calculated by AutoDock Vina. A histogram of the affinity scores for 10,000 molecules for each of the structures is plotted in Figure 1 and depicts the highest scores for 3FZT and 3H3C, followed by 3FZR and 3FZO in a semi-logarithmic scale.
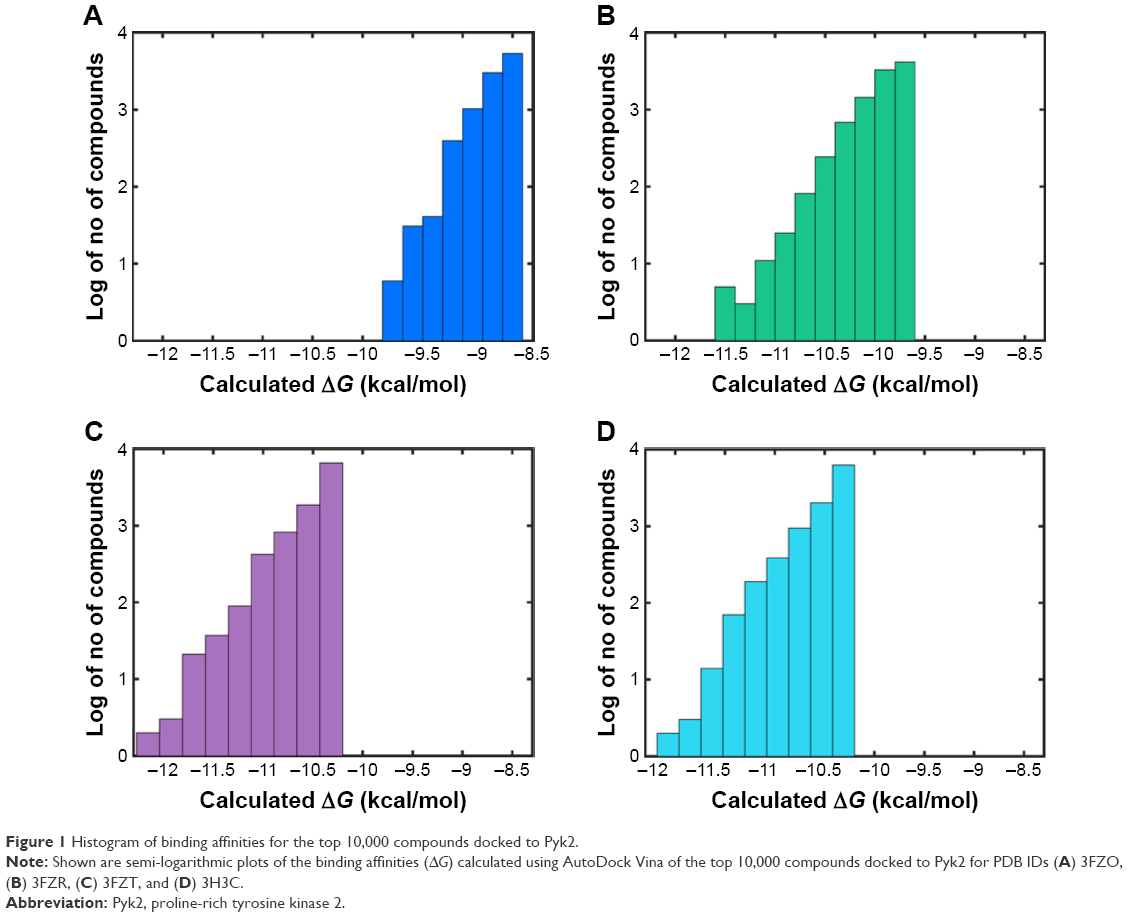 | Figure 1 Histogram of binding affinities for the top 10,000 compounds docked to Pyk2. |
To compare the affinities of the screened compounds to affinities of existing Pyk2 inhibitors, we docked the known inhibitors into their corresponding structures using AutoDock Vina. The calculated binding energies of the reference compounds ranged from −8.7 to −9.3 kcal/mol, which was similar to the range engaged by the top 10,000 molecules for the apo structure 3FZO and was higher than all the top compounds of the rest of the three structures. Therefore, the cutoff for our docking studies was set to include the top 10,000 molecules with the highest affinity for each of the four structures and considered for further evaluation, composing a pool of 40,000 docked modes.
To insure proper target preparation, we calculated the RMSD between the crystallographic poses and the docked poses of PDB IDs 3FZR, 3FZT, and 3H3C (Figure S2). The RMSD values of the reconstituted pose ranged from 0.41 to 2.57 Å, supporting the target preparation for docking protocols. To support the goodness of predictive docking screens, enrichment experiments were carried out. First, we collected a dataset comprising 124,000 decoys from the Directory of Useful Decoys (DUD) database50 and 18 actives known from the literature20 via Konstanz Information Miner (KNIME). In addition, we collected another dataset containing 800 decoys generated by the DUD-enhanced (DUD-E) server51 based on the 18 actives. These two datasets were then screened against all four Pyk2 target structures using AutoDock Vina. The docking enrichment plots for four protein targets are shown in Figure S3. The docking enrichment plots show that the percentage of true ligands found by docking, at any given percentage, of the docking-ranked database is almost always greater compared to being chosen by random selection.
Compound processing and ranking
We analyzed the retrieved compounds by combining the four sets of compounds to a single pool of compounds and scored via affinity and efficiency-based metrics: binding energy (ΔG), MAB, BEI, SEI, and LLE. The top 1,000 molecules for each index were ranked by the corresponding value and are presented as scatter plots in Figure 2. In an attempt to identify top hits, which are suitable for drug development, and to remove false positives, we used several sub-structural filters by manual review to remove unwanted groups.30 Owing to the potential detrimental effect of polycyclic compounds with a high ring count,31 molecules were additionally filtered out and restricted to a maximal limit of aromatic and aliphatic rings.
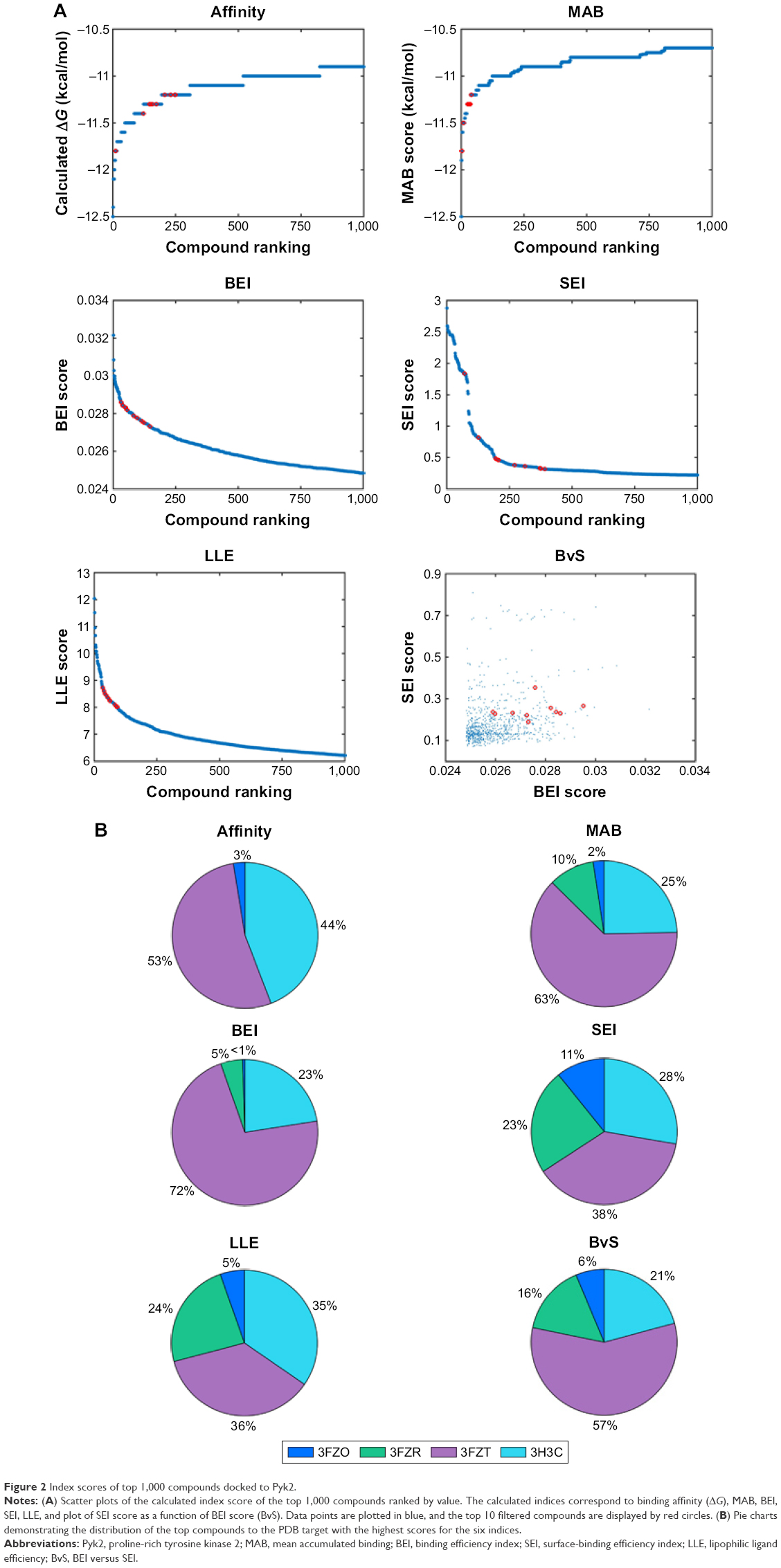 | Figure 2 Index scores of top 1,000 compounds docked to Pyk2. |
An additional approach for compound selection is the Pareto-based ranking scheme. The Pareto-based ranking scheme takes into account multiple objectives for the optimization/selection process52 and combines their separate contribution in the final ranking. The multiple objectives, BEI and SEI, were optimized by scoring each compound with the sum of molecules dominating it in any of the indices. The top 10 filtered molecules retrieved for every index and the mapping of BvS indices for the top 1,000 molecules are presented in Figure 2A. Pareto ranking is a strategy particularly well suited for competitive multi-objective problems. In many cases, the BEI and SEI objectives are correlated; however, this is not always true,53 and in this study, BEI and SEI were considered as partially competitive multi-object problems.
To visualize target distribution of compounds that displayed the best docking scores and efficiency values for every parameter, a chart summarizing the distribution of the top 1,000 compounds binding to a specific structure is depicted in Figure 2B, where binding is defined as any docking score within the top 10,000 molecules of the corresponding structure. The resulting distribution revealed that 3FZT that possessed a DFG-out conformation was a dominating structure for the majority of the compounds.
Selection of the highest-ranked compounds
From each index, 40 molecules were independently collected for further examination. The two-dimensional structures and molecular weights of the top 5 filtered candidates with the highest ranking per index are shown in Table 1, and their physicochemical properties along with the calculated indices are shown in Table 2. The total 240 compounds were evaluated for appearance in the literature using KNIME54 nodes, which facilitate querying and retrieving data from the ChEMBL55 bioactivity database via RESTful Web Services.56 These molecules were not detected as known inhibitors for Pyk2.
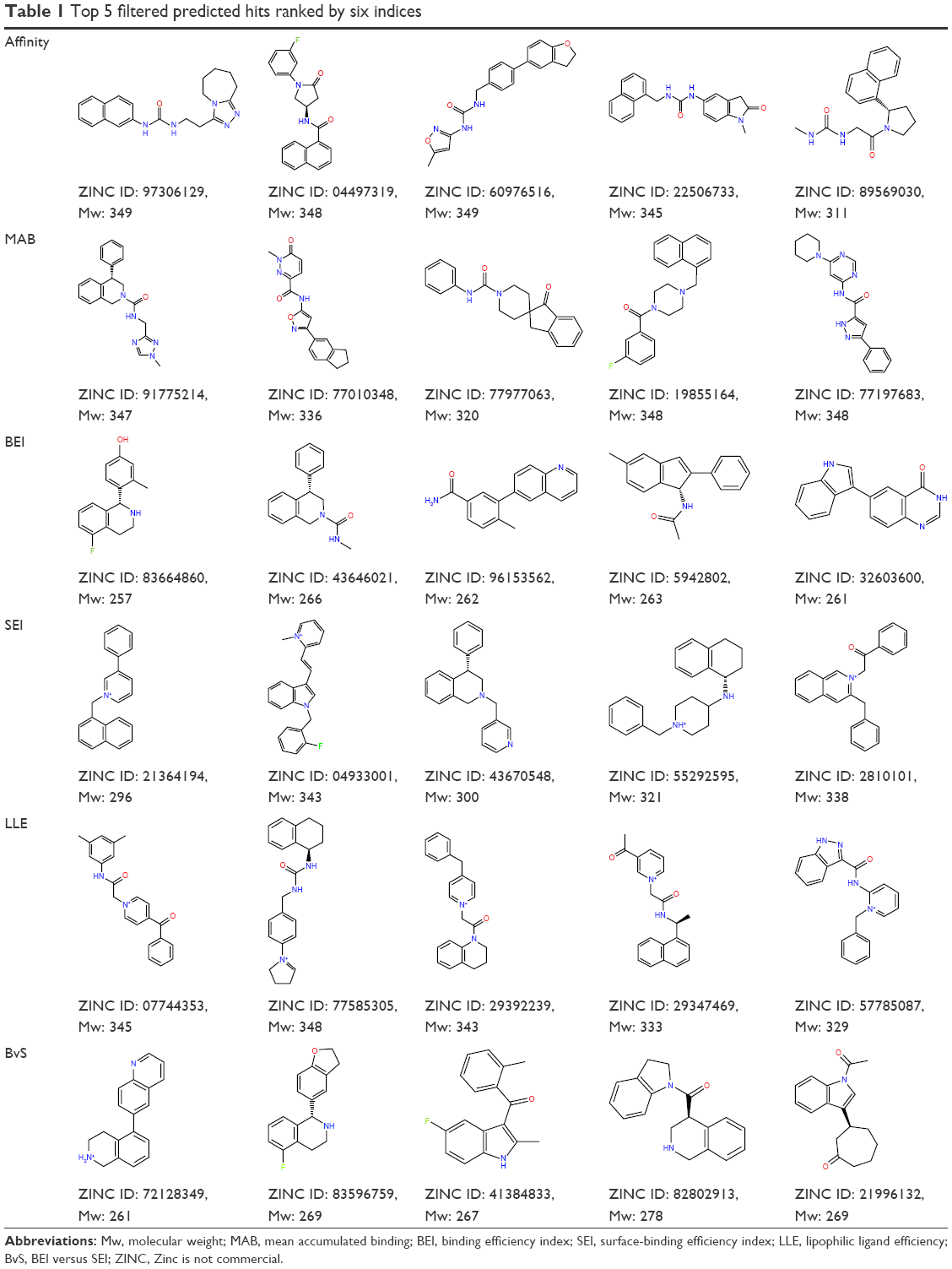 | Table 1 Top 5 filtered predicted hits ranked by six indices |
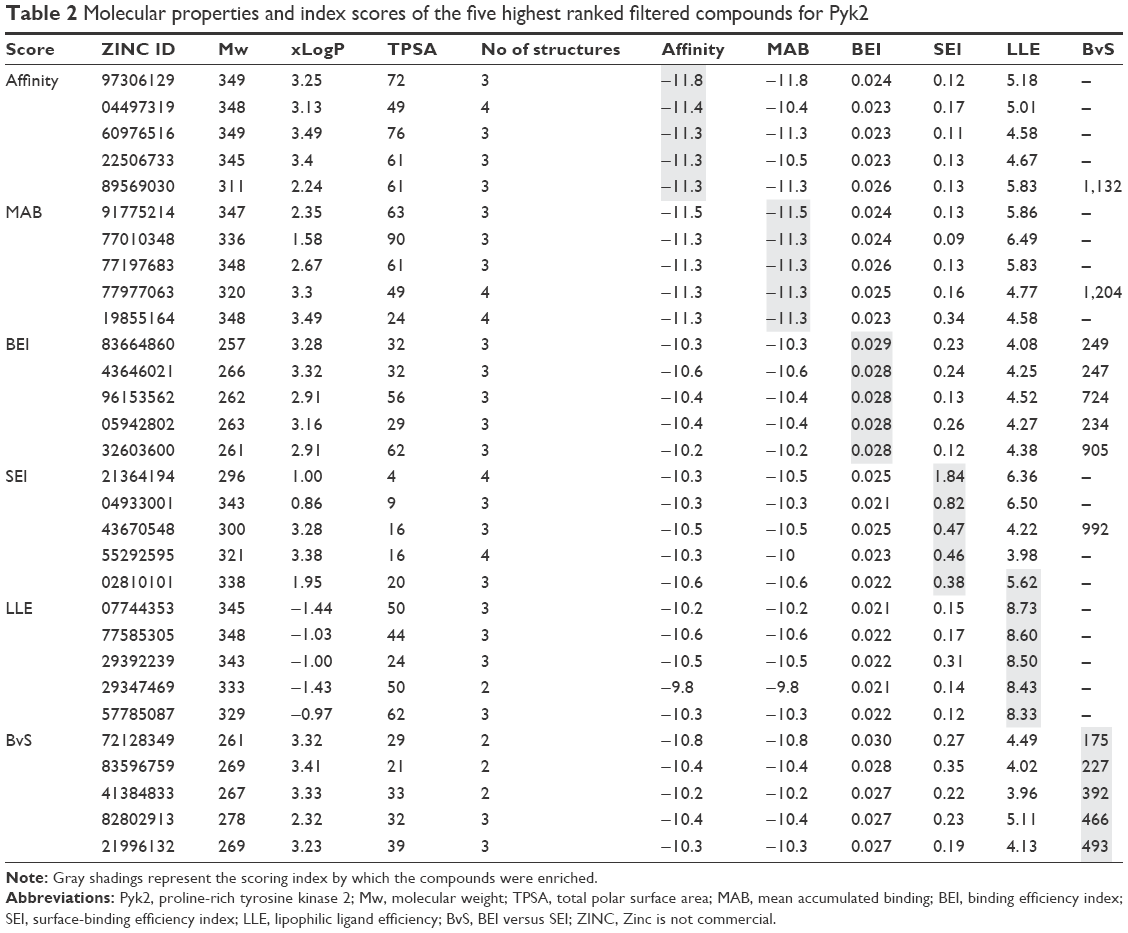 | Table 2 Molecular properties and index scores of the five highest ranked filtered compounds for Pyk2 |
Selectivity prediction by calculation of binding free energies
To evaluate the potential specificity for Pyk2 over FAK among the top retrieved predicted filtered hits, two different approaches were used. To predict the difference in binding association with the targets, we performed a second docking screen with a higher precision using increased exhaustiveness in Vina and Glide XP modes and the generated docked mode of Glide was utilized as an input to additional binding free energy scoring using Prime MM-GBSA. By implementing more precise docking methodologies and combining MM-GBSA, which outperforms docking in predicting the binding affinities to experimental data,57 we can predict more reliably the differences in ligand-binding efficiencies of Pyk2 versus FAK. The correlation coefficient between calculated and experimental binding of Prime MM-GBSA, based on a diverse set of 855 complexes, was reported to be R2=0.63.58 Since the predominant Pyk2 structure that bound the compounds contains a DFG-out conformation, a high-resolution crystal structure of FAK domain that possesses a DFG-out conformation was selected (PDB ID: 1MP8).59 The FAK structure was imported and prepared for docking and was then used as a reference for estimating the selectivity of the top 240 compounds that were selected as mentioned earlier. For selectivity prediction, both the DFG-in and DFG-out conformations were used (PDB ID: 3FZT and 3H3C), as the predicted Vina scores of cognate ligands for both conformations were similar. Only 40 compounds that resulted in favorable binding to Pyk2, as calculated by the difference in at least 2 out of 3 binding energies using Vina, Glide, and Prime MM-GBSA software, were further processed. Using these methods, we selected 20 potential inhibitors ranked by MM-GBSA for 3FZT (ZINC06232011, ZINC02529497, ZINC01646132, ZINC15952140, ZINC18700196, ZINC00173518, ZINC00217347, ZINC35514633, ZINC04975487, ZINC07503677, ZINC03358424, ZINC05078298, ZINC09550062, ZINC03421151, ZINC65845103, ZINC02644767, ZINC03341864, ZINC00269705, ZINC10556478, and ZINC06648526) and 20 potential inhibitors ranked by MM-GBSA for 3H3C (ZINC06232011, ZINC02529497, ZINC18700196, ZINC01646132, ZINC15952140, ZINC05244105, ZINC71894482, ZINC08104814, ZINC00094214, ZINC58514284, ZINC04842554, ZINC00470121, ZINC64790378, ZINC67630577, ZINC00004724, ZINC25251328, ZINC00782941, ZINC82137153, ZINC05626761, and ZINC00617844). Notably, the top 5 predicted selective candidates for 3H3C were among the top 8 selective compounds for 3FZT. Results showed that ZINC06232011 was predicted to be the most selective ligand based on MM-GBSA scoring with Pyk2 (PDB ID 3FZT) ΔG of −35.37 kcal/mol and FAK ΔG of −5.50 kcal/mol, which results in ΔΔG of −29.87 kcal/mol. The top eight candidates (PDB ID 3FZT) were ranked by the energy difference MM-GBSA ΔΔG, and their two-dimensional structures with calculated binding energies are presented in Table 3. Although MM-GBSA was the main method to rank the compounds, it should be pointed out that the procedure used a continuum model of the solvent and this approximation can strongly affect the calculated binding energies, which may result in unreliable results.57
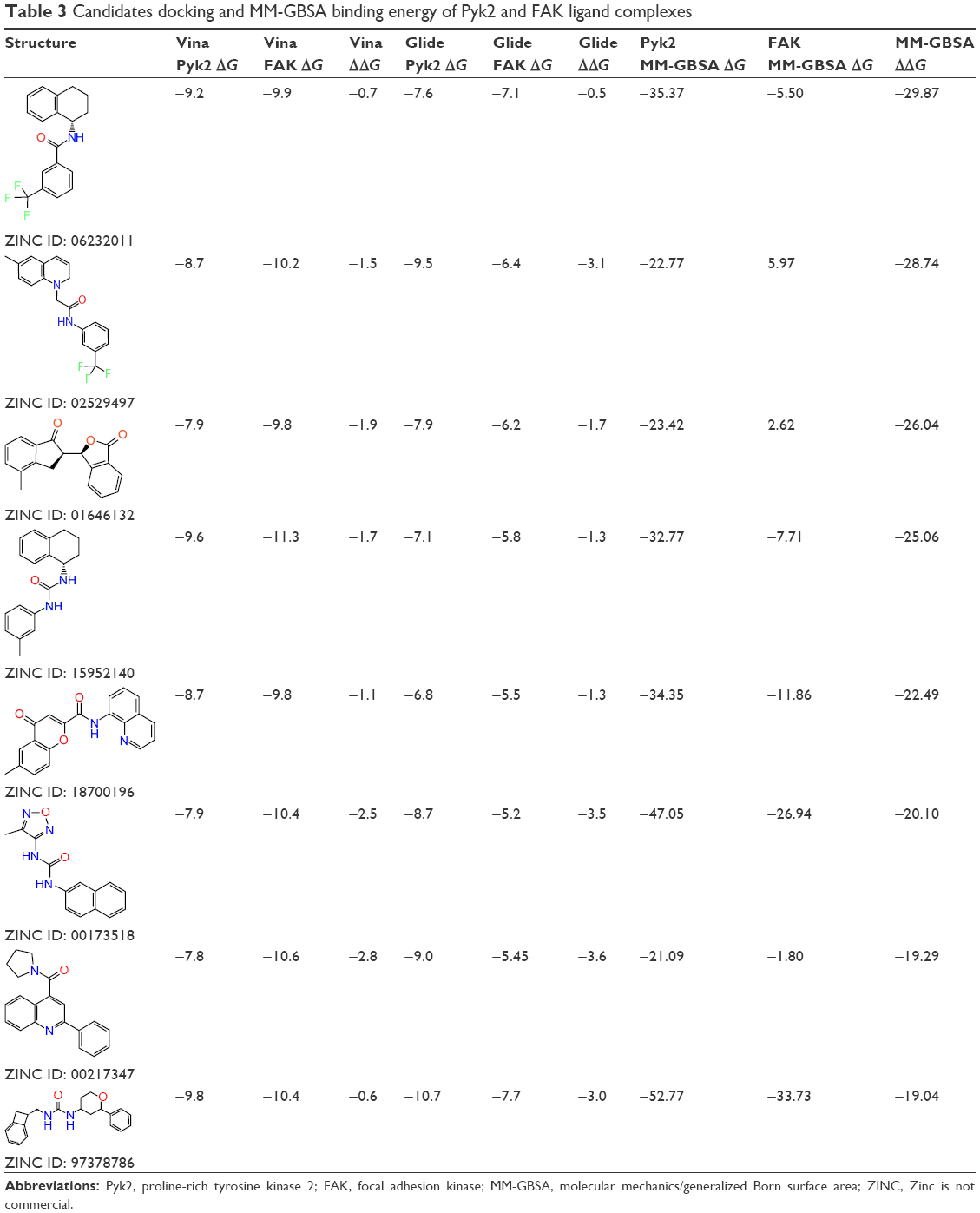 | Table 3 Candidates docking and MM-GBSA binding energy of Pyk2 and FAK ligand complexes |
To support the predictive goodness of this selectivity assay, we compared the predicted binding energy and interaction profile of the native control compounds of PDB IDs 3FZR (PF-431396), 3FZT (PF-4618433), and 3H3C (N-methylsulfonamide 2a) with those obtained using experimental data (Table S1). The predicted binding energies agreed with experimental activity assays, and the interaction profile was similar. 3FZR displayed the smallest predicted selectivity (largest ΔΔG), followed by 3H3C and 3FZT. By consensus, the predicted ΔΔG ranked the cognate ligands according to experimental data and thus substantiated our techniques for finding potential selective Pyk2 inhibitors.
Analysis of the identified molecules
The binding poses of the top candidate compounds bound to Pyk2 (3FZT) as predicted by MM-GBSA are given in Figure 3, and two-dimensional interaction plots are presented in Figure S4. Docking pose analysis revealed one hydrogen bond between Tyr505 and ZINC06232011, ZINC01646132, and ZINC00217347, in which the last two form π–π interactions with Phe568. Also observed were two hydrogen bonds of ZINC02529497 with Asp567 and with Glu474, respectively, as well as a cation–π interaction with Arg572. Compounds ZINC159521402, ZINC00173518 and ZINC97378786 were involved in a similar interaction forming two hydrogen bonds with Glu474 and one hydrogen bond to Asp657, while the last compound also formed π–π interaction with His547. Interestingly, ZINC18700196 was located furthest away from the ATP-binding site and formed a total of four hydrogen bonds with residues Lys457, Asp567, and Arg572, while still involved in π–π interaction with Phe436 and two cation–π interactions with Arg572. Molecular descriptors of physicochemical properties, ligand efficiency scores, and bound structures with the predicted highest binding affinity are presented in Table S2.
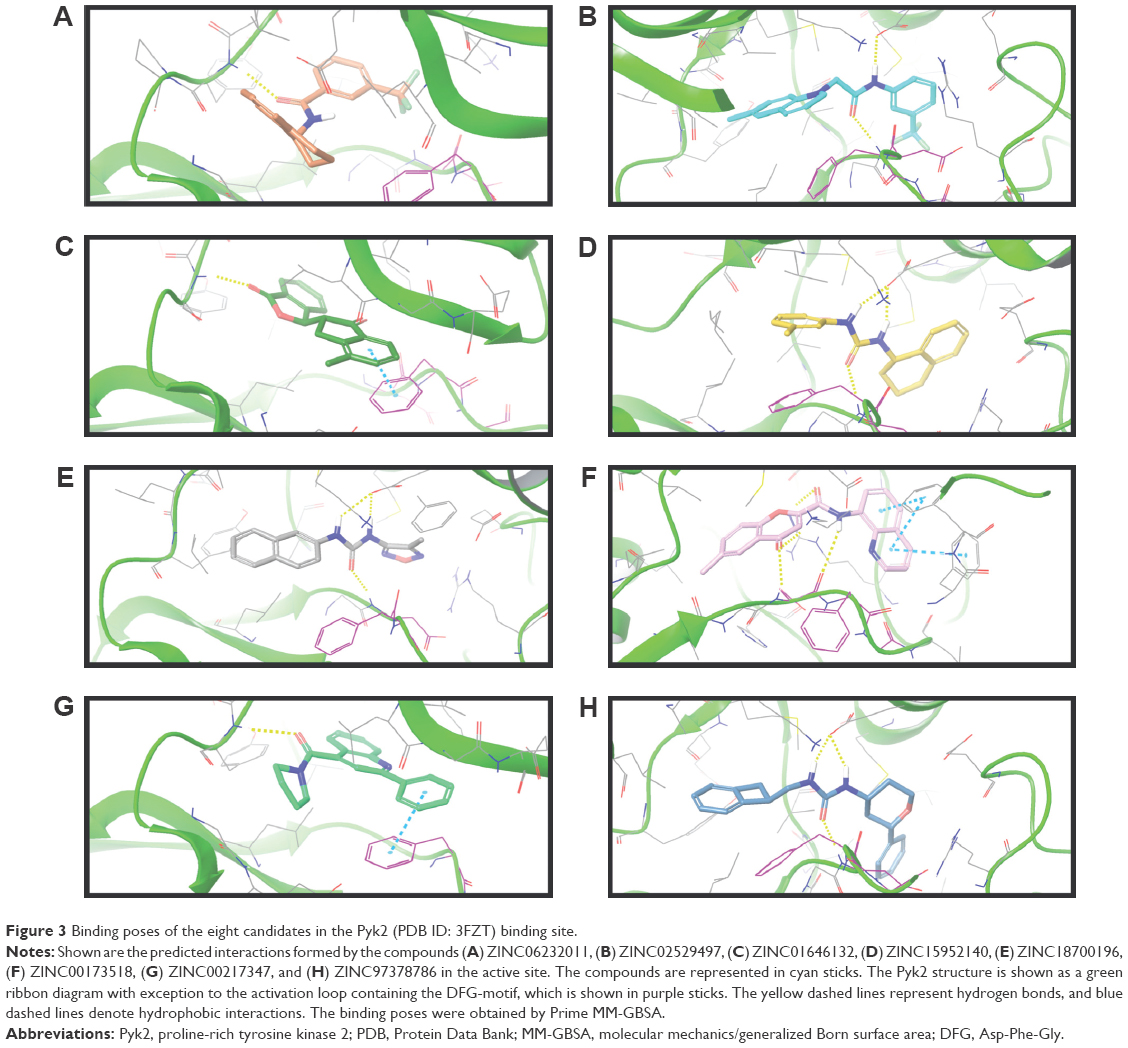 | Figure 3 Binding poses of the eight candidates in the Pyk2 (PDB ID: 3FZT) binding site. |
For selectivity prediction, both the DFG-in and DFG-out conformations were used. The predicted Vina scores of cognate ligands for the DFG-out and DFG-in were similar and differed by 1.0–1.5 kcal/mol (which is lower than Vina’s standard error of 2.85 kcal/mol).27 Thus, we decided to use both DFG-in (PDB ID 3FZT) and DFG-out (PDB ID 3H3C) conformations.
An alternative way to interpret the contribution of each scoring profile is to visualize the ranking of the compound instead of its scoring value. The information is displayed in Figure 4 by radar plots, where the value of each property corresponds to the ranking of the score; closer to the center indicates a property with a good result, while far from the center fails to compete with the rest of the compounds.
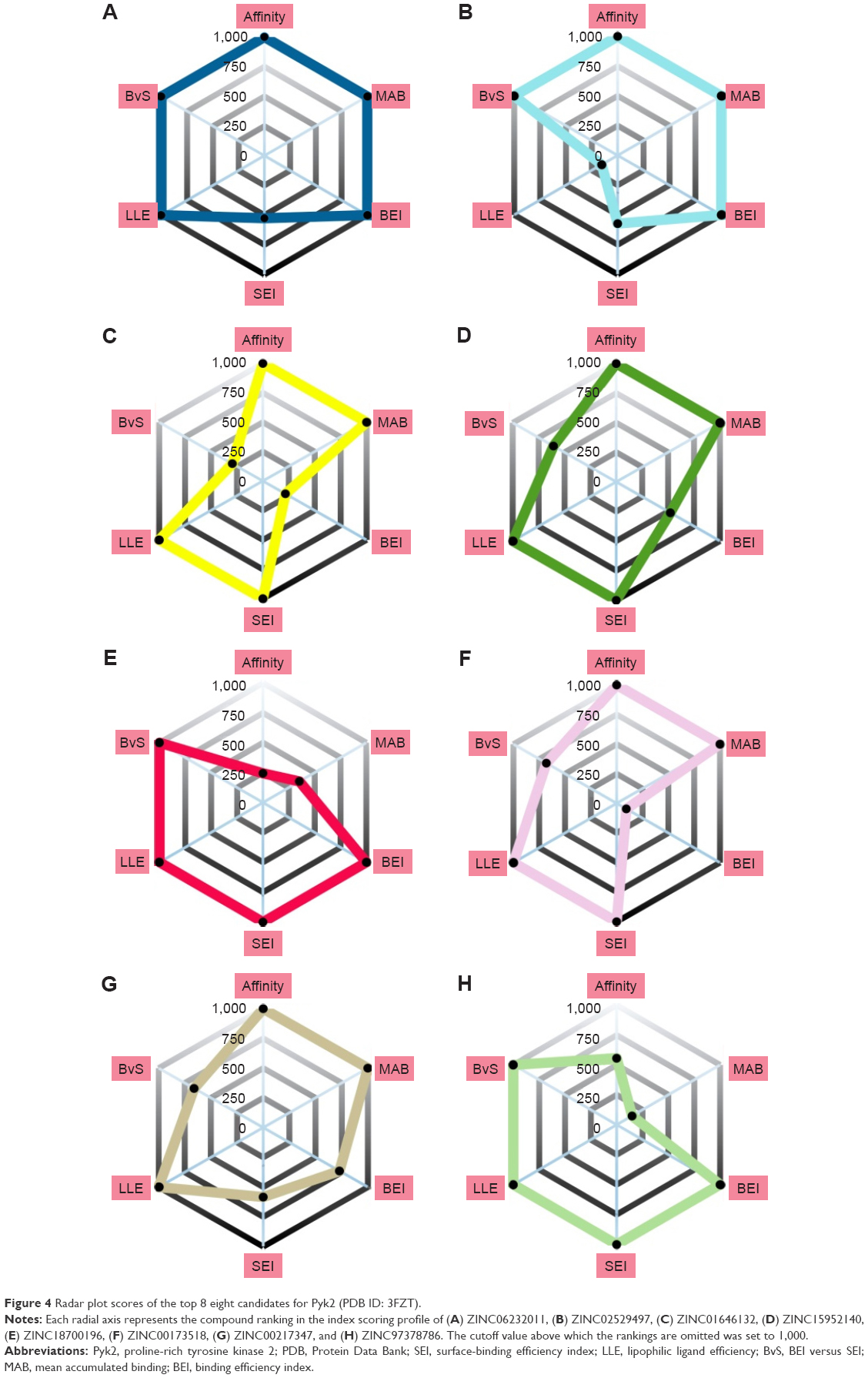 | Figure 4 Radar plot scores of the top 8 eight candidates for Pyk2 (PDB ID: 3FZT). |
MD simulation
To take into account structural flexibility, the behavior of a subset of the predicted complexes of Pyk2 and FAK was compared by MD simulation. The top 8 Pyk2 DFG-out candidates were incorporated in Desmond, and MD simulation was performed in explicit aqueous solution for 20 ns for each complex (Figure 5A). To explore the dynamic stability, RMSD of protein–ligand complexes of Pyk2 (3FZT) and FAK (1MP8) against their initial structure was generated and analyzed using MATLAB. The backbone RMSDs were stable throughout the simulations, with the exception of compound ZINC02529497, where there was a sudden increase in deviation at 9 ns within the FAK complex (Figure 5B).
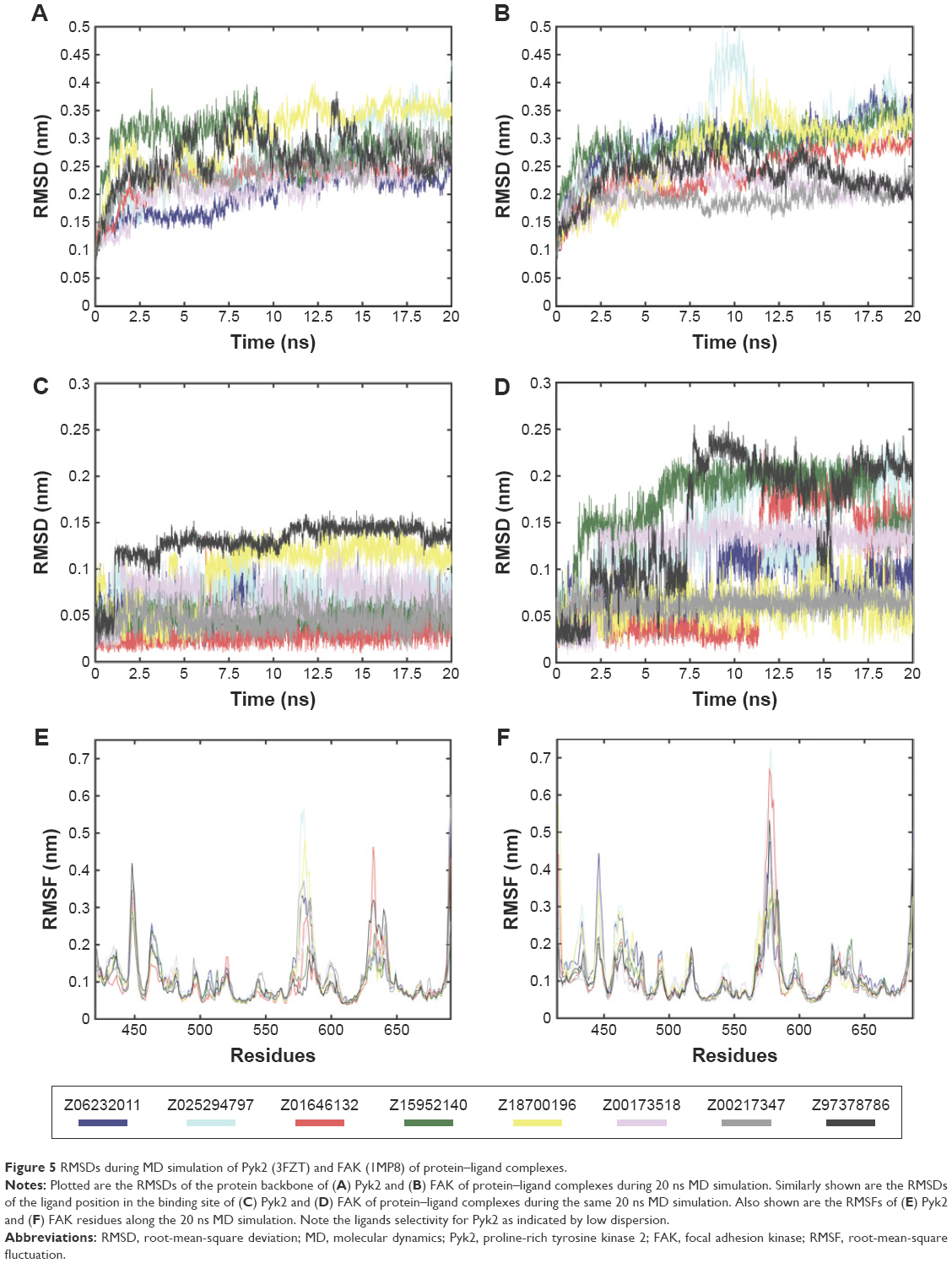 | Figure 5 RMSDs during MD simulation of Pyk2 (3FZT) and FAK (1MP8) of protein–ligand complexes. |
Ligand positional RMSDs were generated to evaluate and compare the binding stability of the lead molecules for each of the targets. Pyk2 complexes demonstrated conformational stability of the ligand in which the RMSD values remained within 0.15 nm (Figure 5C), whereas half of the FAK complexes showed higher RMSD values throughout most of the simulation time (Figure 5D). The computed RMSD values of the ligands ZINC02529497, ZINC01646132, ZINC159521402, and ZINC97378786 in complex with FAK were >0.15 nm and reached 0.25 nm, indicating lower stability.
Root-mean-square fluctuations (RMSFs) were generated to evaluate and compare the residual mobility of Pyk2 and FAK while bound to each of the lead compounds. The RMSFs were integrated along the MD simulation time for each protein–ligand complex and were plotted against the residue number (Figure 5E). In all cases, the computed RMSFs of the residues were lower than 0.6 nm and did not produce any abnormal fluctuations, with the exception of ZINC02529497 and ZINC01646132, which produced fluctuations >0.6 nm in FAK (Figure 5F).
Intermolecular interaction analysis
To determine the stability of protein–ligand binding during the trajectory period of the MD simulation, the intermolecular interactions of ligands in complex with Pyk2 and FAK were analyzed. The intermolecular interaction analysis of the complexes was generated in Desmond and processed in MATLAB, and the time-dependent data were integrated for each interaction type to compare the results as illustrated in Figure 6. The analysis revealed that ZINC18700196 was superior in binding to Pyk2 compared to FAK in all interaction types: hydrogen bond, hydrophobic, ionic, π–cation, π–π, and water bridge. ZINC159521402 exhibited a similar profile, excluding redundant ionic and π–π interactions, which are very low for Pyk2 and FAK. The compounds ZINC06232011, ZINC02529497, and ZINC97378786 bound preferably to Pyk2 in most of the interactions, particularly by hydrogen bonds and hydrophobic interactions, while π–π and water bridges had similar or lower values for Pyk2. The candidate molecules ZINC01646132 and ZINC00217347 maintained higher amount of hydrogen bonds to FAK, while ZINC00173518 showed more hydrophobic, ionic, and substantial increase in water bridge interactions to FAK. Overall, the MD simulations of the ligand complexes with Pyk2 display stability under dynamic conditions and the analysis supports the binding energy predictions.
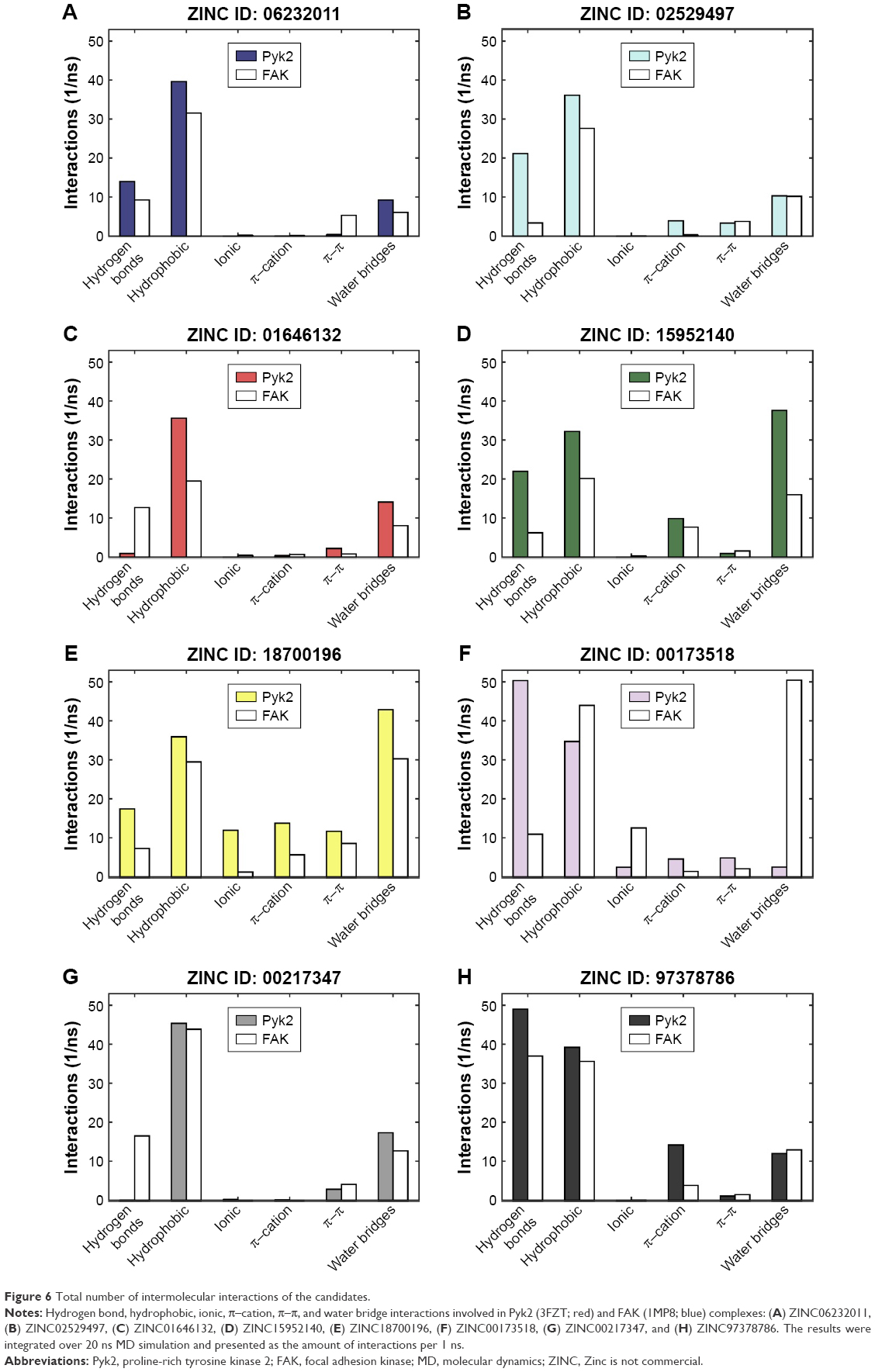 | Figure 6 Total number of intermolecular interactions of the candidates. |
Discussion
The identification of a proper lead compound for a given molecular target is a critical step in the process of drug discovery. Traditional high-throughput screening of large chemical libraries has been a primary source for identification of novel lead compounds. However, the high cost and low hit rate, which are associated with high-throughput screening, have stimulated the development of computational alternatives and the broad application of the cheaper and faster in silico screening approach.60 With an increasing number of targets being identified by high-throughput genomics and proteomics efforts, in silico screening may provide an excellent complementary approach to the traditional high-throughput screening and will potentially improve the speed and efficiency of drug discovery and development processes.
As part of our effort to identify novel candidates for Pyk2 (Figure 7), potency and several efficiency metrics that attempt to define aspects of compound quality have been used. The tendency to focus on enhancing potency rather than practicing a more holistic approach in drug discovery was suggested to be responsible for failing drug discovery projects with molecules burdened by excessive molecular weight and lipophilicity, referred to as molecular obesity.61 In order to avoid biasing toward molecules where the dominating attractor is potency, we allowed only part of the retrieved predicted hits to be beneficially prioritized for potency. Since the best performing single structure is not known in advance and using multiple fixed protein conformation is useful in predicting active compounds by docking calculations,62,63 we virtually screened four different Pyk2 structures and defined MAB to score the contribution of a well docking compound versus multiple conformations. With the increasing popularity of ligand efficiency indices (Figure S5) and increasing involvement in contemporary drug discovery,64 we aimed to balance potency and ADMET (absorption, distribution, metabolism, excretion and toxicity) with BEI, SEI, LLE, and the combination of BEI and SEI. These indices are among the most used and robust metrics,64–66 and we have implemented them computationally with calculating the binding energy as a surrogate for in vitro potency, as has been proposed by Abad-Zapatero.53 To our knowledge, this is the first study to test the efficiency metrics, including BEI, SEI, and LLE, in an in silico high-throughput screening approach that combines selectivity scoring methodologies and incorporates molecular docking, MM-GBSA, and MD.
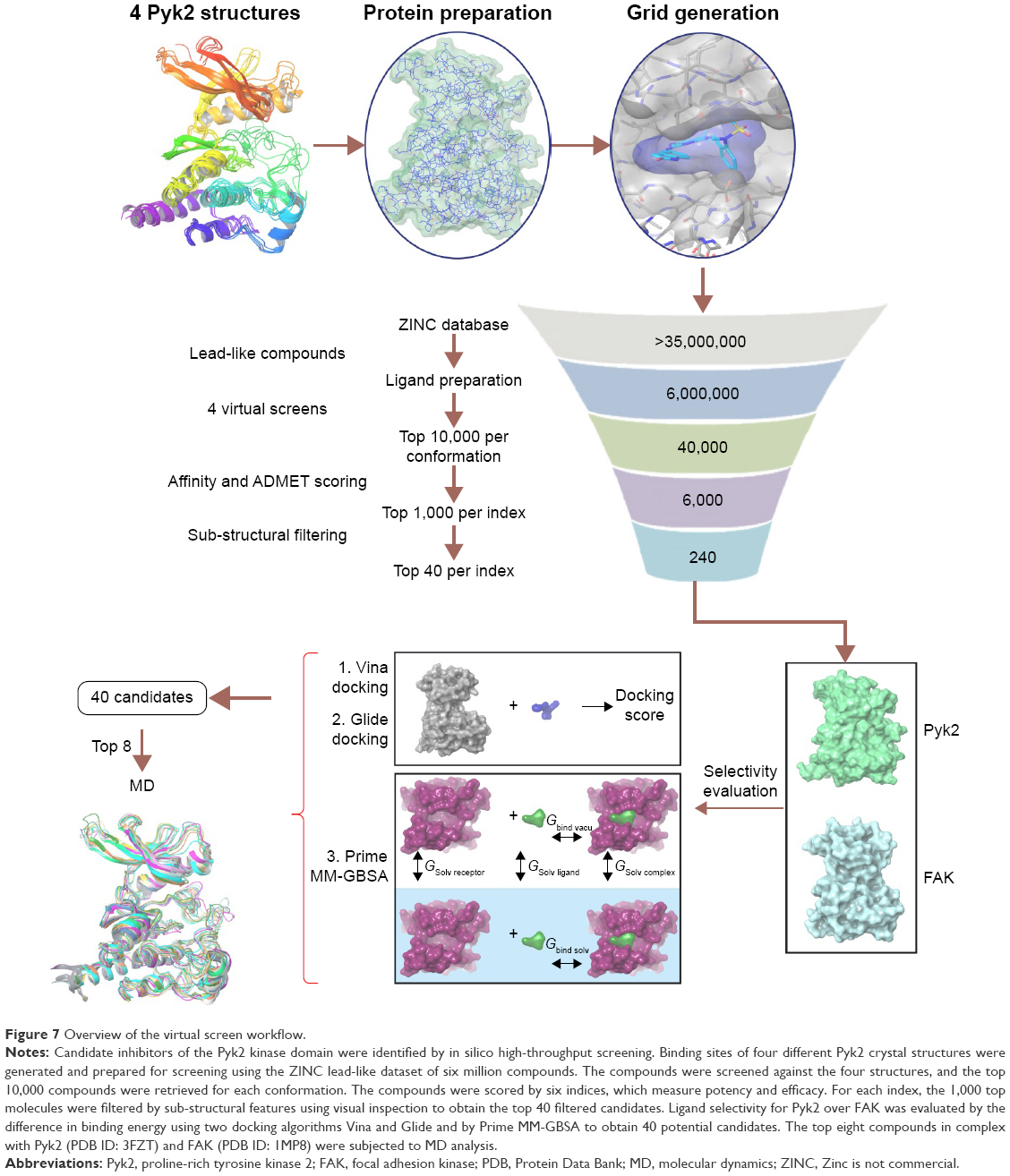 | Figure 7 Overview of the virtual screen workflow. |
Despite the advances in computational aided drug design and widespread application of docking methods, there are still limitations that affect the accuracy of the predictions. These limitations include unavoidable imprecision of crystal structures67 and the lack of information about the number and free energy of water molecules.57 In addition, the inhibition of kinase activity is not necessarily a direct indicator of binding affinity, and the idealized conditions of the docking simulations that predict binding free energies rarely reflect the conditions in experimental assays.68
The top predicted selective candidates exhibit fairly low SEI scores, and the Pareto-based BvS score was mostly dominated by BEI, which is also demonstrated in Figure 2A. Among these candidates, LLE was the least enriched metric. Although the initial compound enrichment in the virtual screen was scored by potency, the affinity and MAB rankings show that most of the top predicted selective candidates were not among the top predicted potent compounds.
The high structural similarity of Pyk2 and FAK kinase domains may pose a challenge in discovering novel selective and potent inhibitors. Selective ligands with reduced potency for Pyk2 such as PF-4618433 were observed, and other studies have identified selective compounds but at the expense of potency.69–71 We aimed to avoid focusing mainly on enhancing potency and to fulfill our preconditions by using efficiency metrics; the compounds were subjected to more stringent criteria affecting their affinity. In addition, the affinity of the identified ligands was compromised by small molecular weight, lipophilicity, and polarity.
Clinical experience with kinase inhibitors has demonstrated that inhibition of protein tyrosine kinases should not rely exclusively on modulation of catalytic activity due to specificity issues and the unexpected emergence of resistance. Thus, combining kinase inhibition with approaches that inhibit extra-catalytic modules that regulate effector functions of tyrosine kinases would be a welcome asset to the therapeutic arena. In addition to its tyrosine kinase activity, Pyk2 has scaffolding functions in the formation of multi-protein signaling complexes. Therefore, targeting extra-catalytic protein modules that regulate complex assembly may provide a complementary approach for efficient and specific inhibition of Pyk2. Future studies that will characterize Pyk2-mediated signaling complex formation will be necessary in order to achieve this significant goal.
Acknowledgments
This work was supported by the Israel Science Foundation (grant number 996/12), the Israel Cancer Research Fund (grant number 12-709-RCDA), the Israel Cancer Association (grant number 20141043), the Marie Curie Career Integration Grant (grant number 321556), the Helmsley Charitable Trust Fund (grant number 2012PG-ISL013; to H.G.-H.), and the Leir Foundation and Katz Foundation (to A.O.S.).
Disclosure
The authors report no conflicts of interest in this work.
References
Park SY, Avraham HK, Avraham S. RAFTK/Pyk2 activation is mediated by trans-acting autophosphorylation in a Src-independent manner. J Biol Chem. 2004;279(32):33315–33322. | ||
Lev S, Moreno H, Martinez R, et al. Protein tyrosine kinase PYK2 involved in Ca(2+) induced regulation of ion channel and MAP kinase functions. Nature. 1995;376(6543):737–745. | ||
Litvak V, Tian D, Shaul YD, Lev S. Targeting of PYK2 to focal adhesions as a cellular mechanism for convergence between integrins and G protein-coupled receptor signaling cascades. J Biol Chem. 2000;275(42):32736–32746. | ||
Sieg DJ, Ilic D, Jones KC, Damsky CH, Hunter T, Schlaepfer DD. Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK-cell migration. EMBO J. 1998;17(20):5933–5947. | ||
Okigaki M, Davis C, Falasca M, et al. Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. Proc Natl Acad Sci U S A. 2003;100(19):10740–10745. | ||
Guinamard R, Okigaki M, Schlessinger J, Ravetch JV. Absence of marginal zone B cells in Pyk-2-deficient mice defines their role in the humoral response. Nat Immunol. 2000;1(1):31–36. | ||
Kamen LA, Schlessinger J, Lowell CA. Pyk2 is required for neutrophil degranulation and host defense responses to bacterial infection. J Immunol. 2011;186(3):1656–1665. | ||
Buckbinder L, Crawford DT, Qi H, et al. Proline-rich tyrosine kinase 2 regulates osteoprogenitor cells and bone formation, and offers an anabolic treatment approach for osteoporosis. Proc Natl Acad Sci U S A. 2007;104(25):10619–10624. | ||
Gil-Henn H, Destaing O, Sims NA, et al. Defective microtubule-dependent podosome organization in osteoclasts leads to increased bone density in Pyk2(−/−) mice. J Cell Biol. 2007;178(6):1053–1064. | ||
Koppel AC, Kiss A, Hindes A, et al. Delayed skin wound repair in proline-rich protein tyrosine kinase 2 knockout mice. Am J Physiol Cell Physiol. 2014;306(10):C899–C909. | ||
Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–1458. | ||
Dourlen P, Fernandez-Gomez FJ, Dupont C, et al. Functional screening of Alzheimer risk loci identifies PTK2B as an in vivo modulator and early marker of Tau pathology. Mol Psychiatry. Epub 2016 Apr 26. | ||
Lipinski CA, Loftus JC. Targeting Pyk2 for therapeutic intervention. Expert Opin Ther Targets. 2010;14(1):95–108. | ||
Selitrennik M, Lev S. PYK2 integrates growth factor and cytokine receptors signaling and potentiates breast cancer invasion via a positive feedback loop. Oncotarget. 2015;6(26):22214–22226. | ||
Lim Y, Lim ST, Tomar A, et al. PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J Cell Biol. 2008;180(1):187–203. | ||
Owen KA, Thomas KS, Bouton AH. The differential expression of Yersinia pseudotuberculosis adhesins determines the requirement for FAK and/or Pyk2 during bacterial phagocytosis by macrophages. Cell Microbiol. 2007;9(3):596–609. | ||
Ray BJ, Thomas K, Huang CS, Gutknecht MF, Botchwey EA, Bouton AH. Regulation of osteoclast structure and function by FAK family kinases. J Leukoc Biol. 2012;92(5):1021–1028. | ||
Weis SM, Lim ST, Lutu-Fuga KM, et al. Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J Cell Biol. 2008;181(1):43–50. | ||
Han S, Mistry A, Chang JS, et al. Structural characterization of proline-rich tyrosine kinase 2 (PYK2) reveals a unique (DFG-out) conformation and enables inhibitor design. J Biol Chem. 2009;284(19):13193–13201. | ||
Walker DP, Zawistoski MP, McGlynn MA, et al. Sulfoximine-substituted trifluoromethylpyrimidine analogs as inhibitors of proline-rich tyrosine kinase 2 (PYK2) show reduced hERG activity. Bioorg Med Chem Lett. 2009;19(12):3253–3258. | ||
Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model. 2012;52(7):1757–1768. | ||
Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc. 2016;11(5):905–919. | ||
Jacobson MP, Pincus DL, Rapp CS, et al. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55(2):351–367. | ||
Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27(3):221–234. | ||
Sondergaard CR, Olsson MH, Rostkowski M, Jensen JH. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J Chem Theory Comput. 2011;7(7):2284–2295. | ||
Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDock Tools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Abad-Zapatero C, Metz JT. Ligand efficiency indices as guideposts for drug discovery. Drug Discov Today. 2005;10(7):464–469. | ||
Hopkins AL, Keseru GM, Leeson PD, Rees DC, Reynolds CH. The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov. 2014;13(2):105–121. | ||
Kuo LC. Fragment-based drug design – tools, practical approaches, and examples. Preface. Methods Enzymol. 2011;493:xxi–xxii. | ||
Ritchie TJ, Macdonald SJ. The impact of aromatic ring count on compound developability – are too many aromatic rings a liability in drug design? Drug Discov Today. 2009;14(21–22):1011–1020. | ||
Waring MJ, Arrowsmith J, Leach AR, et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov. 2015;14(7):475–486. | ||
Farrell N, Kelland LR, Roberts JD, Van Beusichem M. Activation of the trans geometry in platinum antitumor complexes: a survey of the cytotoxicity of trans complexes containing planar ligands in murine L1210 and human tumor panels and studies on their mechanism of action. Cancer Res. 1992;52(18):5065–5072. | ||
van Beusichem M, Farrell N. Activation of the trans geometry in platinum antitumor complexes. Synthesis, characterization, and biological activity of complexes with the planar ligands pyridine, N-methylimidazole, thiazole, and quinoline. Crystal and molecular structure of trans-dichlorobis(thiazole)platinum(II). Inorg Chem. 1992;31(4):634–639. | ||
Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49(21):6177–6196. | ||
Schrodinger. Schrodinger Release 2015-4: LipPrep, Version 3.6. New York, NY: LLC; 2015. | ||
Tripathi SK, Muttineni R, Singh SK. Extra precision docking, free energy calculation and molecular dynamics simulation studies of CDK2 inhibitors. J Theor Biol. 2013;334:87100. | ||
Sirin S, Kumar R, Martinez C, et al. A computational approach to enzyme design: predicting omega-aminotransferase catalytic activity using docking and MM-GBSA scoring. J Chem Inf Model. 2014;54(8):2334–2346. | ||
Banavath HN, Sharma OP, Kumar MS, Baskaran R. Identification of novel tyrosine kinase inhibitors for drug resistant T315I mutant BCR-ABL: a virtual screening and molecular dynamics simulations study. Sci Rep. 2014;4:6948. | ||
Lyne PD, Lamb ML, Saeh JC. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J Med Chem. 2006;49(16):4805–4808. | ||
Xu L, Sun H, Li Y, Wang J, Hou T. Assessing the performance of MM/PBSA and MM/GBSA methods. 3. The impact of force fields and ligand charge models. J Phys Chem B. 2013;117(28):8408–8421. | ||
Genheden S, Kuhn O, Mikulskis P, Hoffmann D, Ryde U. The normal-mode entropy in the MM/GBSA method: effect of system truncation, buffer region, and dielectric constant. J Chem Inf Model. 2012;52(8):2079–2088. | ||
Bowers KJ, Chow DE, Huafeng X, et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. Paper presented at: ACM/IEEE SC 2006 Conference; 2006; Tampa, FL. | ||
Jorgensen W, Chandrasekhar J, Madura J, Impey R, Klein M. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926. | ||
Hedger G, Sansom MS, Koldso H. The juxtamembrane regions of human receptor tyrosine kinases exhibit conserved interaction sites with anionic lipids. Sci Rep. 2015;5:9198. | ||
Lee HS, Qi Y, Im W. Effects of N-glycosylation on protein conformation and dynamics: protein data bank analysis and molecular dynamics simulation study. Sci Rep. 2015;5:8926. | ||
Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101(5):4177. | ||
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103(19):8577. | ||
Lawrenz M, Shukla D, Pande VS. Cloud computing approaches for prediction of ligand binding poses and pathways. Sci Rep. 2015;5:7918. | ||
Huang N, Shoichet BK, Irwin JJ. Benchmarking sets for molecular docking. J Med Chem. 2006;49(23):6789–6801. | ||
Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012;55(14):6582–6594. | ||
Nicolaou CA, Brown N, Pattichis CS. Molecular optimization using computational multiobjective methods. Curr Opin Drug Discov Devel. 2007;10(3):316–324. | ||
Abad-Zapatero C. Ligand efficiency indices for effective drug discovery. Expert Opin Drug Discov. 2007;2(4):469–488. | ||
Berthold MR, Cebron N, Dill F, et al. KNIME: The Konstanz Information Miner. Berlin, Heidelberg: Springer; 2008. | ||
Bento AP, Gaulton A, Hersey A, et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014;42(Database issue):D1083–D1090. | ||
Richardson L, Ruby S. Restful Web Services. First ed. Sebastopol: O’Reilly Media; 2007. | ||
Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10(5):449–461. | ||
Greenidge PA, Kramer C, Mozziconacci JC, Wolf RM. MM/GBSA binding energy prediction on the PDBbind data set: successes, failures, and directions for further improvement. J Chem Inf Model. 2013;53(1):201–209. | ||
Nowakowski J, Cronin CN, McRee DE, et al. Structures of the cancer-related Aurora-A, FAK, and EphA2 protein kinases from nanovolume crystallography. Structure. 2002;10(12):1659–1667. | ||
Ghosh S, Nie A, An J, Huang Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr Opin Chem Biol. 2006;10(3):194–202. | ||
Hann MM. Molecular obesity, potency and other addictions in drug discovery. Med Chem Commun. 2011;2(5):349–355. | ||
Totrov M, Abagyan R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr Opin Struct Biol. 2008;18(2):178–184. | ||
Carlson HA, McCammon JA. Accommodating protein flexibility in computational drug design. Mol Pharmacol. 2000;57(2):213–218. | ||
Meanwell NA. Improving drug design: an update on recent applications of efficiency metrics, strategies for replacing problematic elements, and compounds in nontraditional drug space. Chem Res Toxicol. 2016;29(4):564–616. | ||
Cortes-Ciriano I. Benchmarking the predictive power of ligand efficiency indices in QSAR. J Chem Inf Model. 2016;56(8):1576–1587. | ||
Li J, Bai F, Liu H, Gramatica P. Ligand efficiency outperforms pIC50 on both 2D MLR and 3D CoMFA models: a case study on AR antagonists. Chem Biol Drug Des. 2015;86(6):1501–1517. | ||
Hawkins PC, Warren GL, Skillman AG, Nicholls A. How to do an evaluation: pitfalls and traps. J Comput Aided Mol Des. 2008;22(3–4):179–190. | ||
Michel J, Essex JW. Prediction of protein-ligand binding affinity by free energy simulations: assumptions, pitfalls and expectations. J Comput Aided Mol Des. 2010;24(8):639–658. | ||
Palmer BD, Smaill JB, Rewcastle GW, et al. Structure-activity relationships for 2anilino-6-phenylpyrido[2,3-d]pyrimidin-7(8H)-ones as inhibitors of the cellular checkpoint kinase Wee1. Bioorg Med Chem Lett. 2005;15(7):1931–1935. | ||
McCluskey A, Keane MA, Walkom CC, et al. The first two cantharidin analogues displaying PP1 selectivity. Bioorg Med Chem Lett. 2002;12(3):391–393. | ||
Kolb P, Phan K, Gao ZG, Marko AC, Sali A, Jacobson KA. Limits of ligand selectivity from docking to models: in silico screening for A(1) adenosine receptor antagonists. PLoS One. 2012;7(11):e49910. |
Supplementary materials
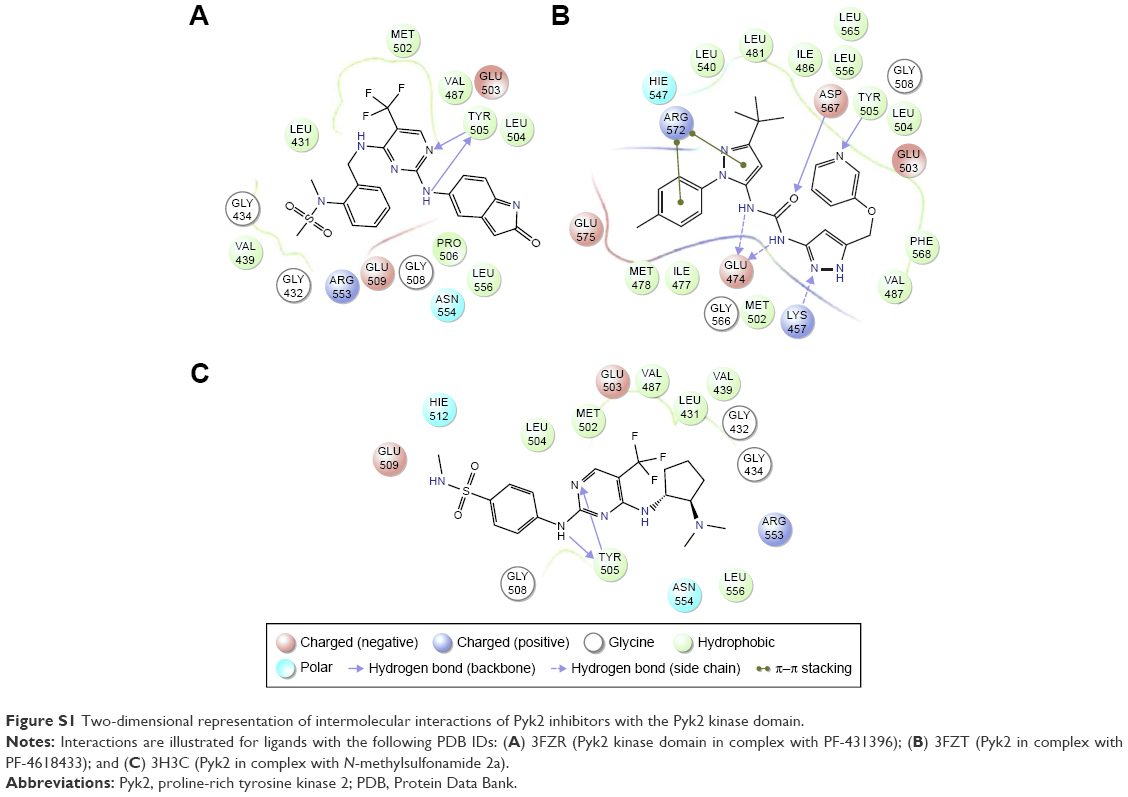 | Figure S1 Two-dimensional representation of intermolecular interactions of Pyk2 inhibitors with the Pyk2 kinase domain. |
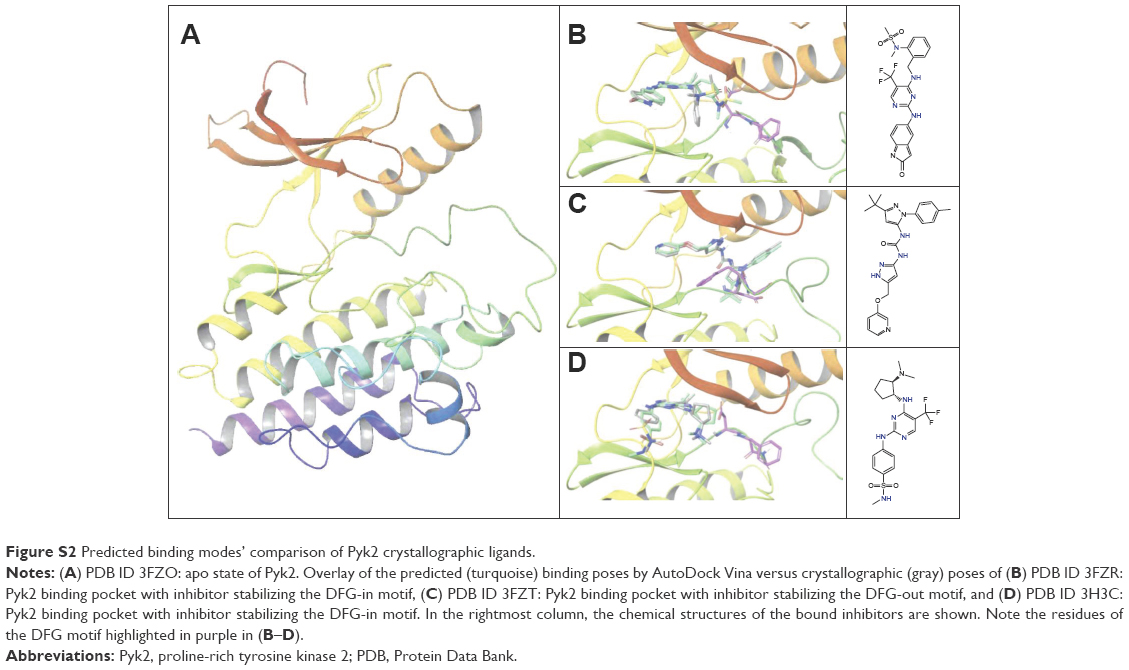 | Figure S2 Predicted binding modes’ comparison of Pyk2 crystallographic ligands. |
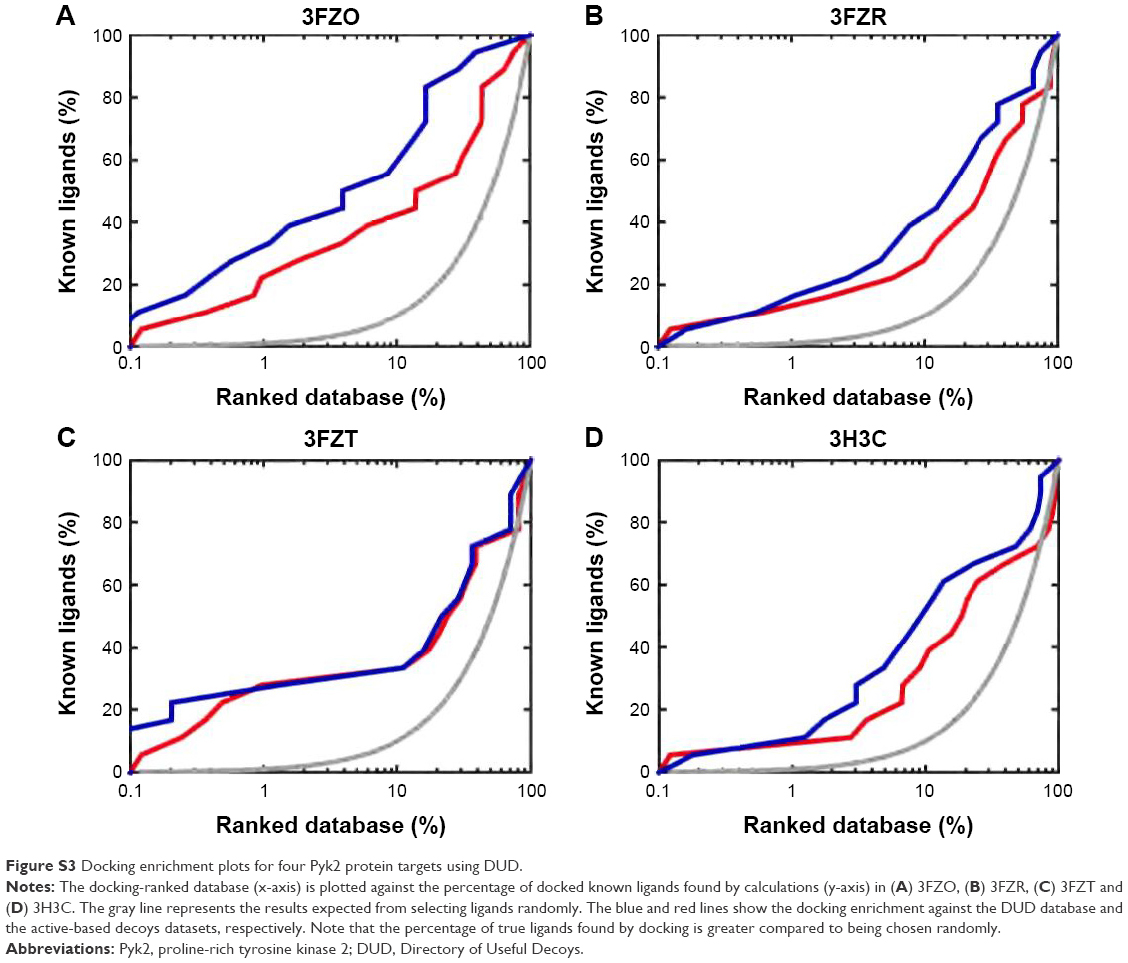 | Figure S3 Docking enrichment plots for four Pyk2 protein targets using DUD. |
 | Table S1 Predicted binding energies of the cognate ligands |
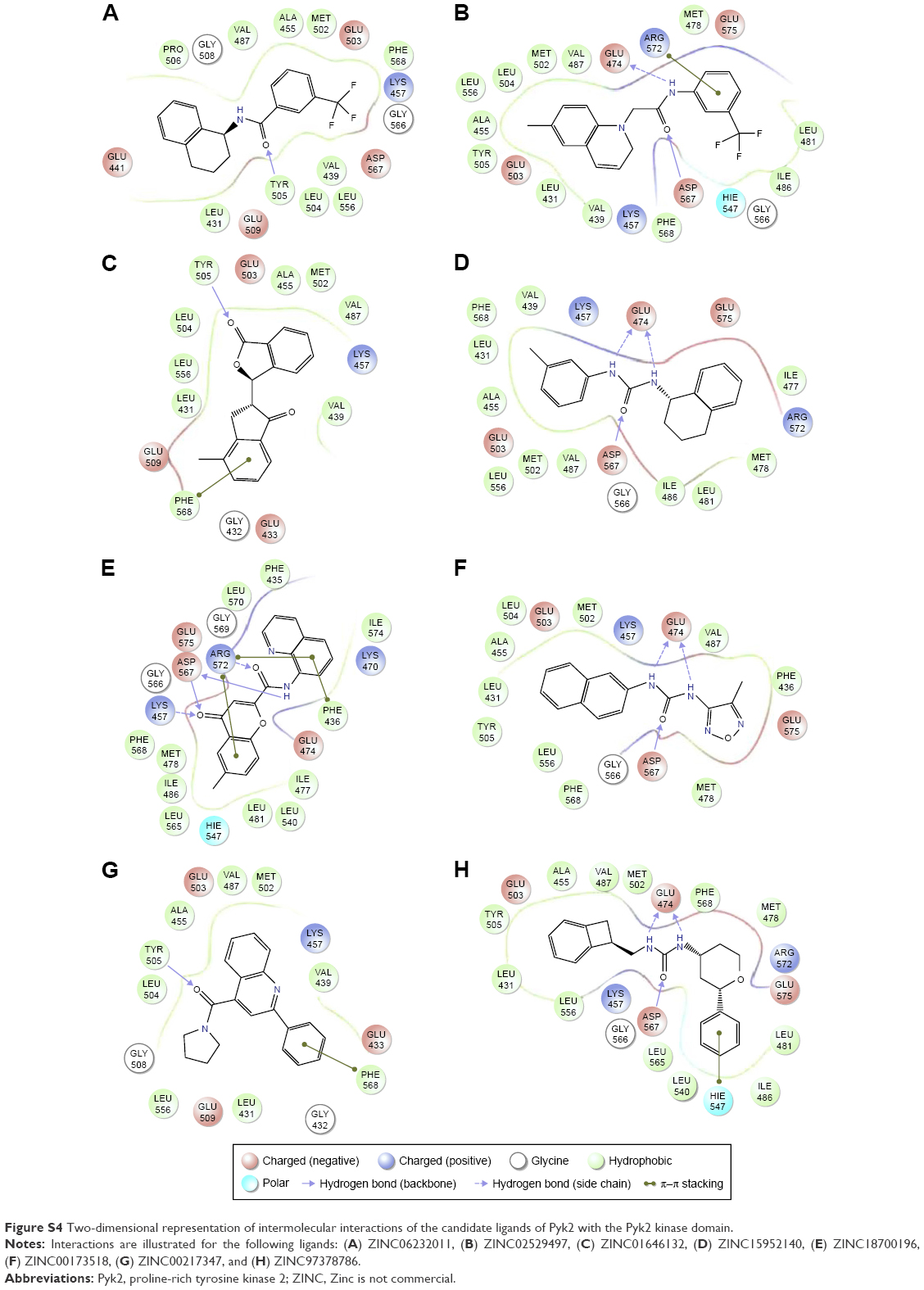 | Figure S4 Two-dimensional representation of intermolecular interactions of the candidate ligands of Pyk2 with the Pyk2 kinase domain. |
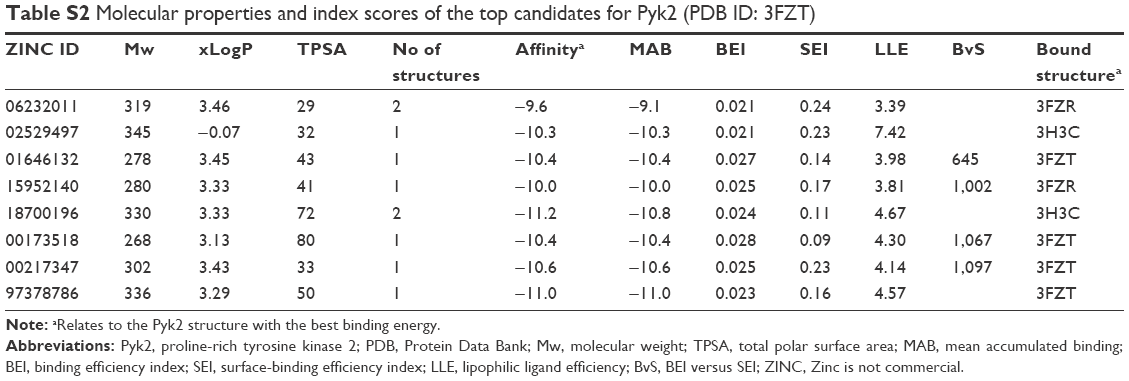 | Table S2 Molecular properties and index scores of the top candidates for Pyk2 (PDB ID: 3FZT) |
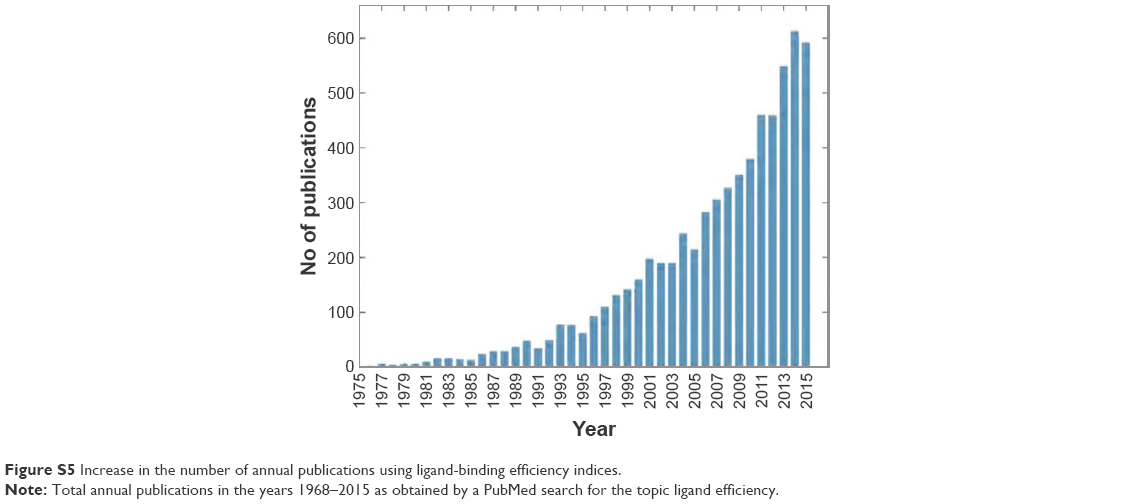 | Figure S5 Increase in the number of annual publications using ligand-binding efficiency indices. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.