Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 7
An exploratory, randomized, parallel-group, open-label, relative bioavailability study with an additional two-period crossover food-effect study exploring the pharmacokinetics of two novel formulations of pexmetinib (ARRY-614)
Authors Wollenberg L, Corson D, Nugent C, Peterson F, Ptaszynski A, Arrigo A, Mannila C, Litwiler K, Bell S
Received 3 March 2015
Accepted for publication 30 April 2015
Published 30 September 2015 Volume 2015:7 Pages 87—95
DOI https://doi.org/10.2147/CPAA.S83871
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Arthur E. Frankel
Lance A Wollenberg,1 Donald T Corson,2,3 Courtney A Nugent,1 Farran L Peterson,1 Ann M Ptaszynski,1 Alisha Arrigo,2,3 Coralee G Mannila,2,3 Kevin S Litwiler,1 Stacie J Bell1,4
1Array BioPharma, Boulder, 2Array BioPharma, Longmont, CO, 3Avista Pharma Solutions, Longmont, CO, 4Mallinckrodt Pharmaceuticals, Ellicott City, MD, USA
Background: Pexmetinib (ARRY-614) is a dual inhibitor of p38 mitogen-activated protein kinase and Tie2 signaling pathways implicated in the pathogenesis of myelodysplastic syndromes. Previous clinical experience in a Phase I dose-escalation study of myelodysplastic syndrome patients using pexmetinib administered as neat powder-in-capsule (PIC) exhibited high variability in pharmacokinetics and excessive pill burden, prompting an effort to improve the formulation of pexmetinib.
Methods: A relative bioavailability assessment encompassed three parallel treatment cohorts of unique subjects comparing the two new formulations (12 subjects per cohort), a liquid oral suspension (LOS) and liquid-filled capsule (LFC) and the current clinical PIC formulation (six subjects) in a fasted state. The food-effect assessment was conducted as a crossover of the LOS and LFC formulations administered under fed and fasted conditions. Subjects were divided into two groups of equal size to evaluate potential period effects on the food-effect assessment.
Results: The geometric mean values of the total plasma exposures based upon area-under-the-curve to the last quantifiable sample (AUClast) of pexmetinib were approximately four- and twofold higher after administration of the LFC and LOS formulations, respectively, than after the PIC formulation, when the formulations were administered in the fasted state. When the LFC formulation was administered in the fed state, pexmetinib AUClast decreased by <5% compared with the fasted state. After administration of the LOS formulation in the fed state, pexmetinib AUClast was 34% greater than observed in the fasted state.
Conclusion: These results suggest that the LFC formulation of pexmetinib may achieve greater exposures with lower doses due to the greater bioavailability compared to the PIC, and remain unaffected by coadministration with food.
Keywords: pexmetinib, myelodysplastic syndromes, liquid-filled capsule, bioavailability, food-effect, formulation
Introduction
Pexmetinib (ARRY-614) is an orally bioavailable inhibitor of p38 mitogen-activated protein kinases with additional activity against kinases that are suspected of having a role in malignant disease, such as the endothelial tyrosine kinase receptor, Tie2 (TEK). Currently, pexmetinib is in clinical development for the treatment of patients with myelodysplastic syndromes (MDS), a heterogeneous group of clonal stem cell disorders of the bone marrow.1 MDS are generally characterized by ineffective hematopoiesis that leads to hematologic cell dysfunction, abnormal development of blood and bone marrow, and an increased propensity for progression to acute myeloid leukemia.2 The activity of a p38 inhibitor in the treatment of MDS is thought to occur through two distinct mechanisms: inhibition of myelosuppressive cytokine production in the bone marrow microenvironment3 and inhibition of apoptotic signaling in hematopoietic progenitor cells.4 Inhibition of the p38 pathway in patients with MDS has previously demonstrated clinical activity in a Phase I clinical setting,5 further supporting the validity of this approach in the treatment of MDS. The role of Tie2 in MDS is less well defined; however, increased expression of the endogenous Tie2 ligand, angiopoietin-1, has been shown to be correlated with poor prognosis in MDS.6 Additionally, decreased levels of angiopoietin-2, a natural antagonist of Tie2, have been associated with a decreased survival benefit for patients with acute myeloid leukemia,7 a condition closely related to MDS. As a result, inhibition of the Tie2 signaling pathway is hypothesized to provide additional clinical benefit for this patient population.
A first-in-human study conducted in healthy subjects evaluated the pharmacokinetics (PK), safety, and pharmacodynamics of powder-in-capsule (PIC) freebase pexmetinib delivered as either a single dose (25–400 mg) or multiple doses over 14 days (50–400 mg) administered once daily (QD).8 Ex vivo stimulation of cytokine production in this population was inhibited following administration of pexmetinib PIC in both a time- and dose-dependent manner.8 Based on these findings, a Phase I dose-escalation trial was initiated using a 3+3 design in patients with MDS. Dose cohorts evaluating QD doses ranging from 400 mg to 1,200 mg and BID doses of 200 mg and 300 mg were included.
Neat PIC pexmetinib used in the Phase I dose-escalation trial was associated with high variability in the PK parameters between patients, largely attributed to variability in absorption.8 High lipophilicity (6.8 ClogP) and low aqueous solubility across the physiological pH range of 1–8 (<10 μg/mL) likely contribute to the observed variability in exposure. Additionally, PIC pexmetinib was manufactured at a dose strength of 100 mg, resulting in a substantial pill burden as the dose increased and impacted the dose that could be administered in the Phase I dose-escalation clinical trial. The high pill burden abrogated the dose-escalation sequence, with 1,200 mg QD being the maximum administered dose, resulting in a failure to reach a maximum tolerated dose. Manufacture of higher dose strengths of PIC pexmetinib was not possible due to a low bulk density of neat pexmetinib powder, further spurring the desire to explore new formulation strategies. Additionally, a food-effect arm built into the dose-escalation clinical trial in MDS patients exploring the effect of food on the administration of 400 mg PIC pexmetinib indicated a decrease in pexmetinib exposure when co-administered with food, warranting investigation of potential food-effect with the exploratory formulations.
In an effort to minimize the variability in the disposition of pexmetinib, improve manufacturability, explore the effect of food on disposition, and reduce pill burden, two novel formulations, a liquid oral suspension (LOS) and a liquid-filled capsule (LFC), both containing micronized hydrochloric acid (HCl) salt drug substance, were manufactured. Here, the results of a relative bioavailability study conducted in healthy subjects to compare the PK of the PIC with the novel formulations are reported. Additionally, a food-effect component was included in the study to evaluate the administration of exploratory formulations of pexmetinib under fed and fasting conditions.
Methods
Study design
An exploratory, open-label study evaluated PK, relative bioavailability, potential food-effect, and safety of single oral doses of 400 mg pexmetinib administered as two novel formulations in the fasted and fed state to healthy adult subjects. The new formulations were a LFC and a LOS that were compounded at the clinical research unit (CRU). The study was designed with three parallel treatment cohorts of unique subjects for the evaluation of the two new formulations (12 subjects per cohort) in fed and fasted states. For subjects in the fed and fasted arms, the washout period between fed and fasted treatments was 7 days. A third cohort of PIC pexmetinib, administered under fasted conditions (six subjects, 400 mg), was included as a reference. A tabular description of the design of the study is provided in Table 1.
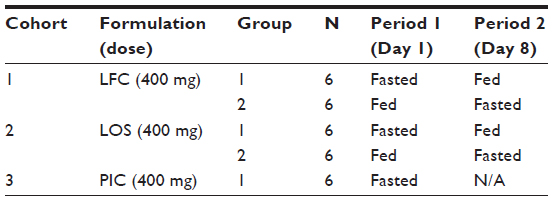 | Table 1 Treatment cohorts and periods |
Prior to study start-up, the study protocol, informed consent form, and printed subject information materials were reviewed and approved by the IntegReview Ethical Review Board (Austin, TX), the CRU’s institutional review board. All subjects were required to provide the written informed consent form prior to study screening. The single-center study described here was conducted at the Pharmaceutical Product Development Phase I clinic (Austin, TX).
Key inclusion criteria for participation in the study included healthy males or females between the ages of 18 years and 60 years, body mass index between 18 kg/m2 and 32 kg/m2, and total body weight between 50 kg and 113.6 kg. Females had to be of nonchildbearing potential or have documented bilateral oophorectomy, and/or hysterectomy. Subjects were excluded from the study for any clinically significant gastrointestinal diseases or other conditions that could affect drug absorption, as well as for screening liver function tests, serum creatinine, and hemoglobin or hematocrit levels outside the normal range. Subjects were also excluded from the study if they were treated with an investigational drug within the past 30 days; prescription medication within the past 28 days; or over-the-counter medication, vitamins, antacids, and dietary and herbal supplements within the past 14 days prior to the first date of the study drug administration. Subjects were also excluded for consumption of grapefruit or grapefruit juice within the past 7 days or tobacco or nicotine-containing products within the past 14 days, or if they had tested positive for alcohol or illicit substances during screening. Subjects abstained from strenuous exercise (eg, heavy lifting, weight training, and aerobic activity) for 48 hours before each blood collection for clinical laboratory tests.
Compounding
Pexmetinib was supplied to the CRU as both a manufactured drug product (PIC) and a micronized HCl salt drug substance. The manufactured PIC drug product contained neat, amorphous pexmetinib freebase at a dose strength of 100 mg contained in a white, opaque, size “0”, hard gelatin capsule. No additional formulating ingredients were used. The HCl salt drug substance was provided in bulk by Array BioPharma (Boulder, CO, USA). The on-site pharmacy compounded the LFC and LOS formulations. The compounded LFC formulation consisted of pexmetinib HCl salt drug substance (200 mg active) suspended in vitamin E d-alpha tocopheryl polyethylene glycol 1000 succinate (TPGS), Labrafac WL Lipophile 1349, and butylated hydroxytoluene contained in a white, opaque, size “00” hard gelatin capsule. The compounded LOS formulation consisted of the pexmetinib HCl salt drug substance suspended in SyrSpend® SF (Fagron Group BV, Rotterdam, the Netherlands) and water for sterile irrigation.
Dose administration
An oral dose of 400 mg was selected for evaluation in this clinical study, based upon the PK and safety data observed to that point in time for pexmetinib in healthy subjects and the MDS patient population in Phase I studies. Additional data from nonclinical toxicology, nonclinical PK, and in vitro formulation data were also considered. For both the PIC and LFC, on Day 1 (Cohorts 1 and 3) and Day 8 (Cohort 1 only), subjects received LFC (Cohort 1) or PIC (Cohort 3) of pexmetinib with 240 mL (±10 mL) water (ambient temperature). Subjects swallowed the study drug whole and did not chew the capsules (four PICs or two LFCs) before swallowing. For the LOS, on Days 1 and 8 (Cohort 2), subjects consumed 60 mL of LOS, followed by two rinse-and-drink cycles with 30 mL (±5 mL) water, followed by consumption of 120 mL (±10 mL) water (ambient temperature). The study drug was administered under the supervision of site personnel, and the oral cavity of each subject was examined following drug administration to ensure that the study drug was swallowed.
Food-effect conditions
For treatments administered in a fasted state, subjects received the study drug after an overnight fast of at least 8 hours. Water was permitted until 1 hour before the study drug administration. Food and water were not permitted for 4 hours following the study drug administration.
For treatments administered in a fed state, subjects fasted overnight for at least 8 hours, after which a standard high-fat meal, which was slightly modified from Food and Drug Administration guidance,9 consisting of 985 calories (153 calories from protein, 257 calories from carbohydrates, and 571 calories from fat) was provided. The high-fat meal consisted of three eggs scrambled in two teaspoons of butter, 1½ pork sausage links, 4 oz of hash brown potatoes, two slices of white toast spread with two teaspoons of butter, and 8 fl oz of whole milk. The entire meal was consumed within 30 minutes. The study drug was administered 30 minutes after completion of the meal. After completion of the study meal, food and water were not permitted for 4 hours following the study drug administration.
PK samples
Blood samples for the determination of plasma concentrations of pexmetinib were collected before dosing and at 0.5 hours, 1 hour, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, 12 hours, 16 hours, 24 hours, and 48 hours after dosing on Day 1 (all cohorts) and Day 8 (LFC and LOS cohorts). PK blood samples (3 mL) were collected into Vacutainers® (Beckton, Dickinson and Company, Franklin Lakes, NJ, USA) containing dipotassium salt of ethylenediaminetetraacetic acid. Within 30 minutes of collection, blood samples were centrifuged and plasma was transferred to cryovials and stored at −20°C until analysis.
Bioanalytical methodology
Plasma concentrations of pexmetinib were determined using a validated liquid chromatography method with tandem mass spectrometric detection. Mobile phase was delivered by a Shimadzu SCL-10A controller with LC-10AD pump (Shimadzu, Columbia, MD, USA). Analysis of the eluent was conducted by an Applied Biosciences Sciex API 5000 (Applied Biosystems Sciex, Ontario, CA, USA) mass spectrometer operating in the positive ion model. Ions were monitored by selected reaction monitoring observing the mass transitions (m/z) of 557.2–256.1 for pexmetinib. Internal standards used in the analysis were stable labeled analogs (d4) of the parent compound and were synthesized at Array BioPharma (Boulder, CO, USA). Mass transitions (m/z) monitored for the internal standard were 561.2–230.2.
Stationary phase for the chromatographic separation was an YMC octadecylsilane column (2.1×100 mm, 5 μm particle size) (Waters, Milford, MA, USA). Mobile phase for the chromatographic separation was a gradient combination consisting of a 0.1% formic acid in water (v/v) and an acetonitrile:methanol (50:50) (v/v) (organic phase) mixture. Upon analyte injection, mobile phase consisted of 60% organic phase and was increased to 70% over a period of 2 minutes. Mobile phase containing 70% organic phase was flowed through the column for a total of 1 minute and was immediately decreased to 60% organic phase over the course of 6 seconds beginning 3 minutes after analyte injection. The 60% organic phase containing mobile phase was held until the end of the run, 4 minutes after analyte injection.
The range of quantitation was 0.5–500 ng/mL for pexmetinib. Quality control standards used in method validation were used to determine assay accuracy and precision. Assay validation indicated a range of intra-assay accuracy from −2.3% to 8.0% and a precision from 1.2% to 4.2% and interassay accuracy ranging from 2.5% to 7.3% and precision from 0.0% to 4.7% for pexmetinib. Acquisition of liquid chromatography method with tandem mass spectrometry data and integration of chromatographic peaks of pexmetinib and its respective internal standards were conducted using Analyst® software (version 1.4.2; Applied Biosystems Sciex, Ontario, CA, USA).
PK data analysis
The plasma concentration–time data for pexmetinib were analyzed by noncompartmental analysis using Phoenix® WinNonlin® software (version 6.1; Certara, Cary, NC, USA). Actual sampling times and nominal dose were used for PK calculations. For the calculation of AUC, a linear-up, log-down method was utilized. For calculation of PK parameters, values below the limit of quantitation before the first quantifiable concentration were set to zero. All below the limit of quantitation values occurring after the first quantifiable concentration were considered as missing and were excluded from the analysis. For the estimation of PK parameters requiring terminal phase extrapolation (ie, the area under the curve extrapolated to infinity [AUCinf] and elimination half-life [t½]), the following criteria were used to determine if extrapolation of terminal PK concentration–time points was appropriate: at least three terminal PK time points exclusive of peak concentration (Cmax) and a coefficient of determination (R2) of those terminal points used in the extrapolation ≥0.8. If these criteria were not met, these subject PK parameter values were not included in the calculation of the appropriate parameter statistics.
Statistical analysis
To determine the relative bioavailability of the novel formulations compared to PIC pexmetinib, plasma Cmax and the area under the curve to the last quantifiable time point (AUClast) values for pexmetinib were log transformed and evaluated with an analysis of variance model with treatment serving as the fixed effect and subject as a random effect. The PIC formulation of pexmetinib served as the reference for the other test formulations. Model outputs were used to calculate geometric mean Cmax and AUClast values for each formulation. Point estimates and 90% confidence intervals (CIs) for differences on the log scale were exponentiated to obtain estimates for ratios of geometric means and 90% CI of the ratios on the original scale. Contrast statements were performed to compare mean values of Cmax and AUClast between each of the test formulations and the reference formulation. A likelihood ratio test was performed to test the homogeneity of variance between the treatment formulations in the fasted state for Cmax and AUClast parameters. Comparison of time-to-peak exposure (Tmax) values was based on the nonparametric methods. The Wilcoxon signed rank (pexmetinib LFC and LOS fed compared with fasted) or Wilcoxon rank sum test (pexmetinib LFC fasted and LOS fasted compared with PIC formulation) was performed on the median Tmax parameter values for this analysis.
To determine the effect of food on the exposure of pexmetinib, 90% CIs were computed on the ratio of the geometric least squares means of log-transformed Cmax and AUClast values for pexmetinib for the LOS and LFC study cohorts using an analysis of variance model with treatment, fed status, treatment by fed status interaction, sequence and period as fixed effects and subject as a random effect (SAS PROC MIXED) (SAS Institute Inc., Cary, NC, USA). The geometric mean ratios for Cmax, and AUClast 90% CI are presented in the later sections, where appropriate.
Safety assessments
Adverse events, physical examination findings, vital sign measurements, electrocardiograph (ECG) readings, and clinical laboratory data were reviewed and summarized to evaluate the safety profile of pexmetinib in healthy subjects.
Results
Subject demographics and disposition
A total of 30 healthy subjects were enrolled in this relative bioavailability study of pexmetinib. The majority of subjects were white males with a mean age of 39.2 years. A broad age range was investigated to encompass older subjects, which would better reflect the elderly MDS population. Summary demographics for all the 30 subjects are listed in Table 2. All subjects completed the study procedures with the exception of one subject in the LOS cohort (Cohort 2) who discontinued due to a family emergency. This subject received the LOS under fasting conditions but did not progress to the fed arm of the study, reducing the number of evaluable subjects in that cohort.
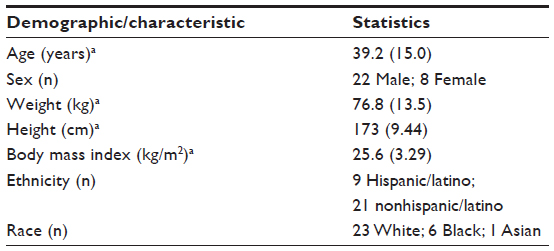 | Table 2 Summary of demographics and baseline characteristics |
Relative bioavailability of three formulations under fasted conditions
After dose administration, plasma concentrations of pexmetinib peaked at a median of 2–4 hours for all treatments in the fasted state. Overall, the extent and rate of absorption were different among the three different formulations, with the LFC formulation appearing to have a greater rate and extent of absorption than the LOS and PIC in the fasted state. The LFC exhibited the shortest median Tmax (2 hours) implying a more rapid absorption of pexmetinib when compared to the median Tmax of the LOS (3 hours) and PIC (3.5 hours). In general, mean plasma concentrations of pexmetinib decreased to <10% of the mean peak plasma concentrations by 48 hours after dosing. Geometric mean concentration–time profiles for pexmetinib for each formulation administered are shown in Figure 1.
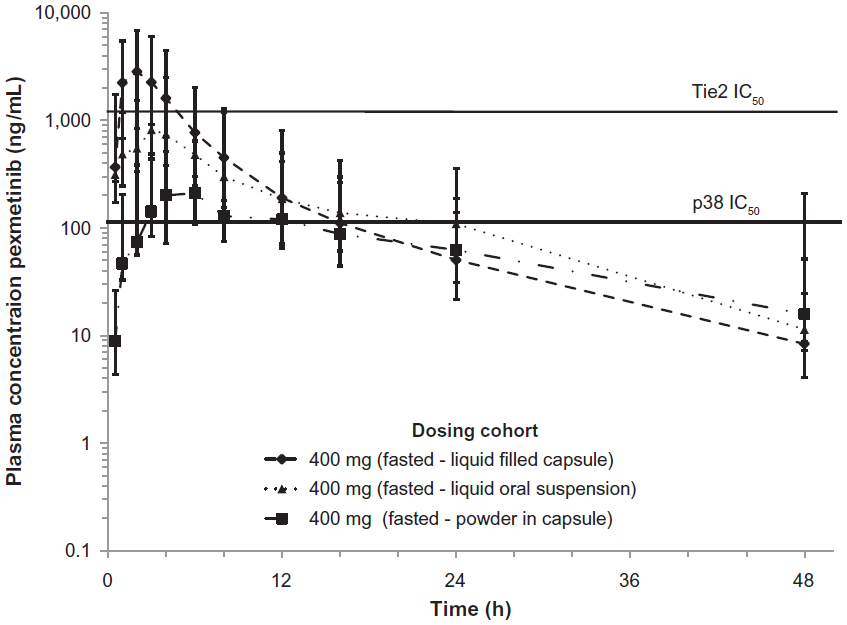 | Figure 1 Fasted concentration-time profile of pexmetinib represented in semi-logarithmic scale. The in vitro half maximal inhibitory concentration (IC50) of ARRY-614 for both p38 and Tie2 is included for reference (solid line). Points represent geometric mean values and error bars ± 1 geometric standard deviation. |
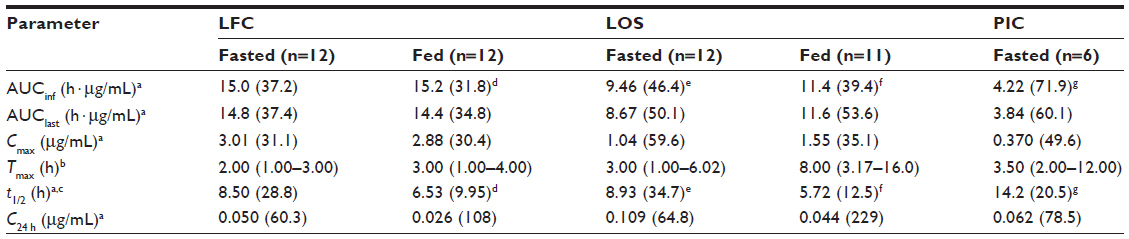
In the fasted state, pexmetinib t½ values were similar between the LOS and LFC, with mean observed t½ values at ~9 hours. However, the mean t½ of the PIC formulation was >50% greater (14.2 hours) than that of either the LOS or the LFC. The ratio of average Cmax to the estimated QD regimen average trough concentration (C24 h) for pexmetinib was 60.2, 9.54, and 5.97 in the fasted state for LFC formulation, LOS formulation, and the PIC, respectively. A summary of the PK parameters calculated during this study is provided in Table 3.
 | Table 3 Summary of fed and fasted plasma PK parameters of ARRY-614 |
The relative bioavailability based on AUClast revealed that the systemic exposure of LFC and LOS formulations were greater than the PIC formulation. The ratios of the geometric means and associated 90% CI values of AUClast for pexmetinib, when administered as the LFC and LOS formulations versus the PIC formulation, were 3.85 (2.63–5.66) and 2.26 (1.54–3.31), respectively. The ratios of the geometric means and associated 90% CI values of Cmax were 8.12 (5.76–11.45) and 2.80 (1.99–3.95), respectively.
An intrasubject variability test could not be performed for the parallel design portion of the current study. Therefore, a likelihood ratio test was performed to evaluate the homogeneity of variance between the treatment formulations. The test results for PK parameters (AUClast and Cmax) showed that the individual residual variances were not sufficiently different between the two test formulations as compared with the PIC formulation in the fasted state (LFC versus PIC formulation and LOS versus PIC formulation) to reject the homoscedastic model (P>0.05). A reduction in the geometric percent coefficient of variation (CV%) values of exposure metrics (AUClast and Cmax) for the novel formulations compared to the PIC was observed. Geometric CV% of AUClast and Cmax for the LFC formulations in the fasted state were 37.4% and 31.1%, respectively, compared with 50.1% and 59.6%, respectively, for the LOS in the fasted state and 60.1% and 49.6%, respectively, for the PIC formulation (in the fasted state).
Food-effect assessment of the LFC and LOS
With the novel formulations of pexmetinib, a less rapid increase in pexmetinib plasma concentration was observed in the fed state, as compared to the fasted state. Overall, the extent and rate of absorption were different among the novel formulations, with the LFC formulation appearing to have a greater rate and extent of absorption than the LOS formulation under a fed state. The median Tmax was 3 hours after administration of the LFC formulation and 8 hours after administration of the LOS formulation in the fed state. The ratio of average Cmax/C24 h values was 110 and 35.2 in the fed state for the LFC and LOS formulations, respectively. The geometric mean concentration–time profiles for each formulation administered with and without food for comparison are shown in Figure 1, for both the LFC (Figure 2A) and LOS (Figure 2B).
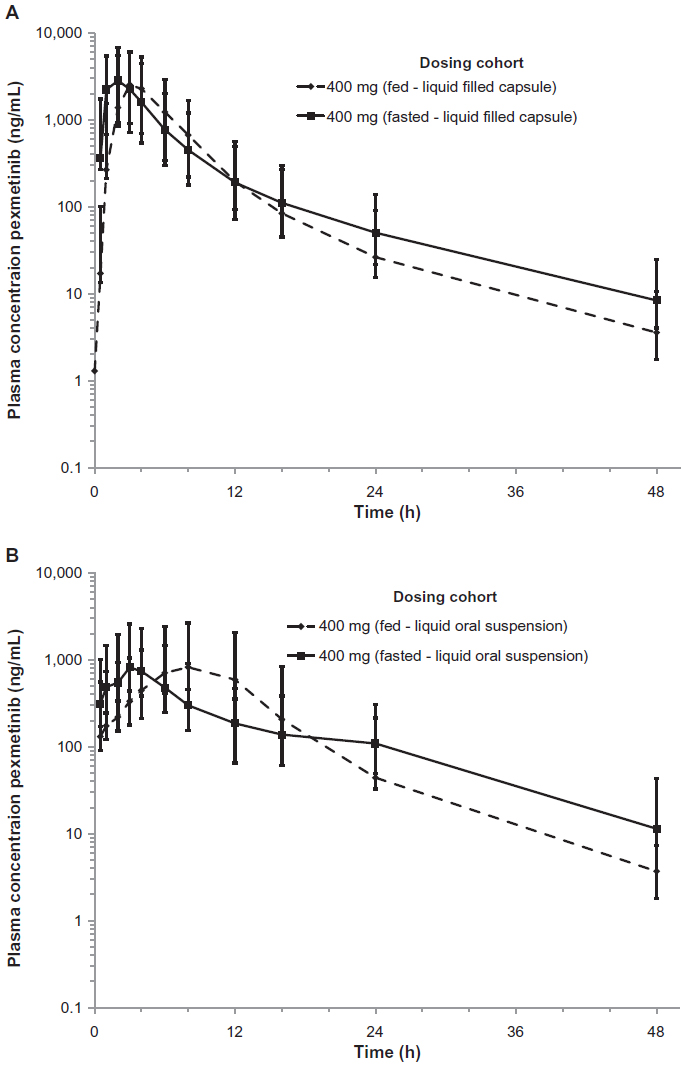 | Figure 2 Concentration-time profile of pexmetinib comparing exposure in both fed and fasted states for both the LFC (A) and LOS (B) represented in semi-logarithmic scale. Points represent geometric mean values and error bars ± 1 geometric standard deviation. |
A slight delay in Tmax after administration of the LFC with food was noted. Comparison of the median Tmax under fed and fasted conditions was statistically different (P<0.05) when pexmetinib was administered as a LFC and LOS, when coadministered with food. The shift in median Tmax observed when the LOS (5-hour shift for pexmetinib) was coadministered with food was greater than the shift in median Tmax observed (1 hour shift for pexmetinib) for the LFC when coadministered with food.
In the fed state, both the Cmax and AUClast of pexmetinib after administration of the LFC formulation decreased <~5% compared with the fasted state, as depicted by the fed/fasted ratios of the geometric means and associated 90% CIs for AUClast and Cmax (0.97 [0.90–1.06] and 0.96 [0.83–1.11]), respectively. However, after administration of the LOS in the fed state, there was a statistically significant increase in AUClast (34%) and Cmax (50%) compared with the fasted state, as depicted by the ratios of the geometric means and associated 90% CI for AUClast and Cmax (1.34 [1.23–1.47] and 1.50 [1.29–1.74]), respectively.
Safety
No clinically significant abnormalities were reported for clinical laboratory test results, vital sign measurements, physical examination findings, or ECG results. With the exception of decreases in the mean heart rate after dosing in the fasted state, no other apparent treatment-related trends were observed in ECG results. No apparent treatment-related trends were observed in clinical laboratory test results, vital sign measurements, and physical examination findings.
Discussion
The dual p38/Tie2 inhibitor pexmetinib is in clinical development as a potential treatment for patients with MDS. Initial Phase I clinical trials of pexmetinib were conducted with PIC and have been characterized by high inter- and intrasubject variability.8 A maximum tolerated dose was not established in a dose-escalation study in patients with MDS due to high pill burden. As a result of these two observations, a change in the oral dosage form from the initial clinical PIC formulation was explored in order to improve the manufacturability of the drug product, reduce pill burden, and potentially reduce the intersubject variability in pexmetinib exposure. The LOS formulation was designed to mimic a well-performing immediate release tablet that effectively wetted and dispersed pexmetinib in vivo. The LFC formulation was designed to increase the aqueous solubility of pexmetinib in vivo through the use of surfactants. The present study was designed to evaluate single-dose plasma PK properties in healthy subjects of these two novel formulations of pexmetinib and their relative bioavailability compared to PIC. A food-effect study arm built into the Phase I clinical study of patients with MDS indicated a significant reduction in exposure when pexmetinib PIC was administered with food (unpublished data), thus the effect of food on exposure of the novel formulations as well as safety and tolerability were assessed.
The increase in exposure of the LFC, compared to the PIC administered at the same dose, suggests that this formulation would improve the bioavailability for engagement of both with p38 as well as Tie2 in vitro IC50 values, compared to the PIC (Figure 1). The LFC demonstrated an approximate fourfold increase and twofold increase in AUClast and Cmax, respectively, when compared to that of the PIC administered at the same dose. A reduction in the geometric CV% from 60.1% to 37.4% was observed when comparing the variability of AUClast of the PIC to the LFC, respectively. A similar reduction in geometric CV% from 49.6% to 31.1% was observed when comparing the variability of Cmax between the PIC and LFC, respectively. Conclusions regarding these differences were limited by the low number of subjects (n=6) in the PIC control arm, relative to the LFC experimental arm (N=12). The reduction in variability was largely attributed to the rapid, consistent absorption facilitated by the LFC formulation, where in vitro dissolution testing indicated the solubility of the drug substance in an aqueous environment was greatly increased in the liquid capsule formulation versus the PIC (unpublished data). The PIC free base drug substance was characterized by poor solubility, which manifested in a protracted absorption phase and likely resulted in the greater variability associated with the PIC administration. Moreover, the decreased in vitro dissolution of the PIC formulation relative to the LFC formulations would suggest that the absorption kinetics were slower, relative to the elimination kinetics. As such, this situation would produce flip-flop kinetics and explain the greater half-life observed during administration of the PIC formulation (14.2 h), relative to the values observed when administering the LFC formulation under similar conditions (8.50 h).
The sum of the preclinical in vitro and in vivo data suggested a high probability of a positive food-effect on exposure of pexmetinib, despite the negative food-effect that was observed in patients for the PIC. In vitro experiments with simulated biofluid suggested differences in solubility between the simulated fasted gastric and intestinal fluids versus the simulated fed gastric and intestinal fluids, with fed gastric and intestinal fluids exhibiting 45-fold and sixfold greater solubility, respectively. In a cynomolgus monkey PK study (unpublished data), an enabled formulation containing surfactants to solubilize the compound, thus simulating a potential food-effect, showed an increase in Cmax and AUClast of two- and fivefold respectively, relative to the PIC formulation (unpublished data). However, administration with food did not significantly affect exposure (Cmax or AUClast) following a single dose of 400 mg pexmetinib administered as a LFC. A slight shift in median Tmax was noted; however, this result may be expected due to slower gastric emptying in the presence of food. It is hypothesized that administration of the LFC results in complete absorption of the dose administered at 400 mg, thus explaining why no food-effect was observed when administering the LFC in the clinical study.
The results presented here suggest that the LFC may be an optimal choice for future clinical development of pexmetinib for the treatment of patients with MDS. The improved bioavailability of the LFC formulation compared to PIC administered at the same dose would be expected to reduce pill burden, as less drug product would be required to achieve plasma concentrations comparable to the PIC. The LFC exhibits manufacturing advantages over the PIC formulation, suggesting the production of pexmetinib drug product for Phase III clinical trials and commercialization is more readily achievable with the LFC. This formulation can be taken without regard to prandial state, making it an optimal choice for administration in patients where chronic, daily administration is necessary for consistent target engagement. Additionally, the improved bioavailability would suggest better engagement of both kinase pathways, p38 and Tie2, known to be inhibited by pexmetinib based on in vitro IC50 values. Moreover, the reduction in intersubject variability of PK parameters for subjects administered the LFC formulation suggests greater consistency in exposure. In addition, the safety profile of a single 400 mg dose in healthy subjects was deemed acceptable. When considered together, the LFC formulation may render important benefits and represents and optimal choice for further exploration in clinical trials for patients with clinical benefits for patients with MDS.
Disclosure
Lance A Wollenberg, Ann M Ptaszynski, and Kevin S Litwiler own shares of Array BioPharma and are current Array BioPharma employees. Donald T Corson, Alisha Arrigo, Coralee G Mannila, Courtney A Nugent, Farran Peterson, and Stacie J Bell own shares of Array BioPharma stock and are former Array BioPharma employees. The authors report no other conflicts of interest in this work.
References
Steensma DP, Tefferi A. The myelodysplastic syndrome(s): a perspective and review highlighting current controversies. Leuk Res. 2003;27(2):95–120. | |
Nimer SD. Myelodysplastic syndromes. Blood. 2008;111(10):4841–4851. | |
Navas T, Zhou L, Estes M, et al. Inhibition of p38alpha MAPK disrupts the pathological loop of proinflammatory factor production in the myelodysplastic syndrome bone marrow microenvironment. Leuk Lymphoma. 2008;49(10):1963–1975. | |
Navas TA, Mohindru M, Estes M, et al. Inhibition of overactivated p38 MAPK can restore hematopoiesis in myelodysplastic syndrome progenitors. Blood. 2006;108(13):4170–4177. | |
Sokol L, Cripe L, Kantarjian H, Sekeres MA, Parmar S, Greenberg P. Randomized, dose-escalation study of the p38alpha MAPK inhibitor SCIO-469 in patients with myelodysplastic syndrome. Leukemia. 2013; 27(4):977–980. | |
Cheng CL, Hou HA, Jhuang JY, et al. High bone marrow angiopoietin-1 expression is an independent poor prognostic factor for survival in patients with myelodysplastic syndromes. Br J Cancer. 2011;105(7):975–982. | |
Loges S, Heil G, Bruweleit M, et al. Analysis of concerted expression of angiogenic growth factors in acute myeloid leukemia: expression of angiopoietin-2 represents an independent prognostic factor for overall survival. J Clin Oncol. 2005;23(6):1109–1117. | |
Winski SL, Freeman B, Remmers AE, Carter L, et al. ARRY-614, a potent P38 MAP kinase inhibitor, is efficacious in preclinical models of hematologic malignancies and significantly reduces ex vivo cytokine production in humans. Ann Oncol. 2009;20(Suppl 3):iii20. | |
United States Food and Drug Administration Center for Drug Evaluation and Research. Food-Effect Bioavailability and Fed Bioequivalence Studies; 2002. Available from: http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM126833.pdf. Accessed: November 6, 2014. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.