Back to Journals » Vascular Health and Risk Management » Volume 13
An evidence-based review on urate-lowering treatments: implications for optimal treatment of chronic hyperuricemia
Authors Bove M, Cicero AFG ![]() , Veronesi M, Borghi C
, Veronesi M, Borghi C
Received 20 July 2016
Accepted for publication 21 October 2016
Published 8 February 2017 Volume 2017:13 Pages 23—28
DOI https://doi.org/10.2147/VHRM.S115080
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Daniel Duprez
Marilisa Bove, Arrigo Francesco Giuseppe Cicero, Maddalena Veronesi, Claudio Borghi
Department of Medical and Surgical Sciences, University of Bologna, Bologna, Italy
Abstract: Several studies suggest that chronic hyperuricemia, the main precursor of gout, is involved in the pathogenesis of different systemic disorders that affect cardiovascular and renal systems, such as hypertension, obesity, hypercholesterolemia, atherosclerosis, metabolic syndrome, chronic heart failure, and chronic kidney disease. Recent epidemiological evidence has shown an increasing trend in the prevalence of hyperuricemia and gout in the Western world: a number of population-based studies estimate a prevalence of up to 21% for hyperuricemia and 1%–4% for gout. As such, early detection and careful management of this pathological condition is required, starting from lifestyle changes (mainly based on a diet low in red meat, sugars, and alcoholic beverages, with increased intake of vegetables, water, and vitamin C sources), adding specific drugs to lead serum uric acid (SUA) levels under the target value of 7 mg/dL. In particular, nonselective and selective XO inhibitors (allopurinol, oxypurinol, febuxostat) reduce SUA levels and the overproduction of reactive oxygen species, mainly related to XO overactivity that often causes inflammatory damage to the vascular endothelium. The effect of lowering SUA levels via XO inhibition includes an attenuation of oxidative stress and related endothelial dysfunction that largely contribute to the pathophysiology of metabolic syndrome and cardiovascular diseases. Therefore, the inhibition of XO overactivation seems to be an excellent therapeutic option to limit the harmful effects of excess UA and reactive oxygen species. In conclusion, rapid diagnosis and correct therapy for hyperuricemia may also improve the prevention and/or treatment of serious and multifactorial diseases. The available evidence supports the importance of promoting new experimental clinical trials to confirm the emerging antioxidant role of XO inhibitors, which could effectively contribute to cardiovascular and chronic kidney disease prevention.
Keywords: hyperuricemia, cardiorenal diseases, therapy, xanthine oxidoreductase inhibitors
Introduction
A chronic increment of serum uric acid (SUA) levels, or hyperuricemia, is the main pathological condition for gout development. According to a revised guideline for the management of hyperuricemia and gout, the normal target value of SUA is 7 mg/dL,1 but the clinically relevant levels of SUA are not entirely clear, and their definition will require new considerations and reflections in the light of recent epidemiological and therapeutic data. For example, the American College of Rheumatology guidelines for management of gout indicate a target value of SUA of 6 mg/dL, probably more suitable, considering the increased prevalence of gout in the general population2 and the already verified association between hyperuricemia, cardiovascular disease (CVD), and chronic kidney disease (CKD). According to these considerations, a scientific goal to achieve as soon as possible is to establish a “normal” value universally agreed upon by researchers and clinicians. In fact, most authors have described an evident increment in SUA in the last few decades, such as Trifirò et al, who reported an increase in the prevalence of hyperuricemia using a cutoff of 6 mg/dL from 2005 (8.5%) to 2009 (11.9%).3 Furthermore, other epidemiological evidence confirms this trend, above all in Western countries: population-based studies have estimated a prevalence of up to 21% for hyperuricemia and 1%–4% for gout.4,5 This has important implications, because hyperuricemia is often included among the diagnostic criteria for metabolic syndrome, a complex disorder of the cardiometabolic system with possible serious systemic and hemodynamic consequences.6 Therefore, careful management of hyperuricemia, either resulting in crystal deposition or not, is crucial to prevent or even treat consequent CVD and CKD. As such, a first approach to the patient with hyperuricemia would surely be based on lifestyle changes (mainly defined as a diet low in red meat, sugars, and alcoholic beverages – in particular beer – with an increased intake of vegetables, some flavonoids, vitamin C sources, and water), but this is often insufficient to lower SUA levels to or below the target value, and drug therapy is required.7 The most common drugs used for the management of hyperuricemia are uricostatic agents (eg, allopurinol, oxypurinol, febuxostat), which reduce the production of UA through competitive inhibition of XO, and uricosuric agents (eg, probenecid, benzbromarone, and the most recent – lesinurad), which favor the urinary excretion of UA, modulating the resorption of urate in the renal tubule.8 The aim of this review is to emphasize the importance of a rapid diagnosis of hyperuricemia, considered as a multifactorial pathological condition closely related to cardiovascular and renal complications. We would like to raise awareness among general practitioners to test SUA levels more often, especially in subjects with one or more risk factors for improving cardiovascular and renal risk global framing. We also summarize the main classes of drugs in use, and particularly the role of XO inhibitors, in the careful management of hyperuricemia in clinical practice.
Management of hyperuricemia
Role of xanthine oxidoreductase in uric acid metabolism
UA is the final product of purine-based (adenine and guanine) catabolism, formed by the liver and excreted primarily by the kidneys and in a lesser amount by the intestine.9 UA is capable of antioxidant activity in the extracellular ambient,10 but intracellularly it has opposite effects, promoting oxidative stress and inflammatory processes.11 Under ischemic conditions or because of tissue damage, the prooxidant effect of UA promotes lipid oxidation, obstructing the protective action of high-density lipoproteins, and favors atheromatous lesion development.12 It is well known that excess UA is strongly related to greater oxidative stress, and XO seems to have a crucial role in this process. XO is the main enzyme complex responsible for UA formation. It catalyzes in the biochemical process of UA synthesis the final two conversions of hypoxanthine to xanthine and xanthine to UA. It also participates in other different biochemical chains contributing to the metabolism of purines, pterins, and aromatic heterocycles; moreover, XO in particular contexts can oxidize nicotinamide adenine dinucleotide (NADH), inducing the reactive oxygen species (ROS) formation that favors vascular inflammation.13
Drug treatment for hyperuricemia
Currently, the drugs of first choice for long-term management of hyperuricemia are XO inhibitors with uricostatic features (eg, allopurinol, oxypurinol, febuxostat), which lower SUA levels through competitive inhibition of XO. Following oral administration, allopurinol is rapidly absorbed in the upper gastrointestinal tract, reaching peak plasma concentrations in approximately 30 minutes, and it has a plasma half-life of 2–3 hours. The main active metabolite of allopurinol is oxypurinol, excreted and partly reabsorbed in the kidneys; it has the same mechanism of action, a longer plasma half-life of 14–30 hours, but lower oral bioavailability than its precursor.14 Febuxostat is a new nonpurine, selective inhibitor of XO, blocks the active pterin–molybdenum center of the enzyme complex, and unlike allo/oxypurinol, does not determine enzyme turnover, avoiding the consequent undesired production of ROS. Following oral administration, it is completely absorbed in the upper gastrointestinal tract, reaching peak plasma concentrations in approximately 1 hour, with a plasma half-life of 1.5–16 hours. Febuxostat is metabolized and excreted predominantly through hepatic conjugation processes. Recent studies have shown that it is more potent and effective than allopurinol in producing anti-inflammatory effects on the endothelium and lowering SUA levels.15,16 Malik et al pointed to febuxostat’s ability of to inhibit glycosaminoglycan-bound and endothelial cell-bound XO in support of its better efficacy.17 The standard dosage of febuxostat is 80–120 mg/day, even though remarkable decreases in SUA levels have been observed with a lower dose of 40 mg/day. In fact, Ernst and Fravel claimed that a greater number of subjects reached target SUA values (6 mg/dL) with febuxostat 40 mg (45%) than with allopurinol 300 mg (42%), and a higher dose of febuxostat (80 mg) resulted in a further increase in this percentage (67%).18 Ye et al showed that a greater number of subjects were able to reach target SUA values with administration of more selective and less toxic febuxostat (40 mg/day) than allopurinol (100–300 mg/day) (odds ratio [OR] 3.14, P<0.01); furthermore, at the final visit 50.9% of the febuxostat group versus 46.5% of the allopurinol group achieved SUA levels ≤6 mg/dL (OR 1.25; 95% confidence interval [CI] 1.05–1.49; P=0.01).19 These data were widely confirmed by Li et al in a recent review, where they suggested with pairwise meta-analysis that febuxostat was the most effective drug to achieve target SUA levels (Figure 1).20
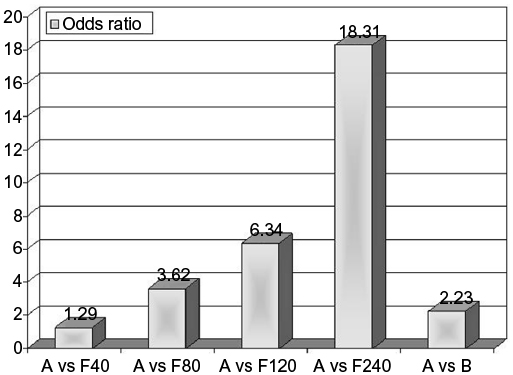 | Figure 1 Results from pairwise meta-analysis on the size effect of urate-lowering therapy with major drugs. Note: Adapted from Clin Ther, 2013;35, Ye P, Yang S, Zhang W, et al, Efficacy and tolerability of febuxostat in hyperuricemic patients with or without gout: a systematic review and meta-analysis, Copyright (2013), with permission from Elsevier.19 Abbreviations: A, allopurinol (100–300 mg/day); F80, febuxostat 80 mg/day; B, benzbromarone (100–200 mg/day); F120, Febuxostat 120 mg/day; F240, Febuxostat 240 mg/day. |
A new and powerful inhibitor of XO that is still being studied is 3,4-dihydroxy-5-nitrobenzaldehyde (DHNB), which has a time-dependent mechanism of action similar to that of allopurinol. Some studies suggest it has low toxicity and enhances therapeutic effectiveness, even at low doses, if coadministered with allopurinol. Moreover, DHNB has a direct antioxidant capacity, reducing free radicals and ROS production.21
In clinical practice, there is an alternative class of drugs often used in subjects intolerant to XO inhibitors – the uricosuric agents (eg, probenecid, benzbromarone, and the most recent – lesinurad) – which result in increased urinary excretion of urate by modulating renal tubule reabsorption.8 Probenecid, the older uricosuric drug, acts at the proximal renal tubule level by inhibiting the main protein transporter of urate (URAT1). After oral administration, it is rapidly absorbed in the upper gastrointestinal tract, reaching peak plasma concentrations in approximately 2–4 hours, with a variable plasma half-life of 4–17 hours. It is not indicated in patients with kidney stones of UA, CKD, or blood dyscrasias. Benzbromarone (URAT1 inhibitor) is rarely used, because of its evident hepatotoxicity.12 Lesinurad, another important URAT1 inhibitor, lowers SUA levels to target value if coadministered with allopurinol or febuxostat in >60% of subjects.22 In some patients refractory to conventional treatments, it is possible to use for a short period polyethylene glycolated uricases (pegloticase and pegadricase) that stimulate UA metabolism,17,18 but at the same time lead to the formation of H2O2 molecules (UA + O2 → allantoin + H2O2), with obvious consequences to be considered also in everyday clinical practice.23,24
The main drugs approved for the treatment of hyperuricemia and their mechanism of action are reported in Table 1. We also cite cases of gout, diagnosed according to recent and more appropriate classification criteria25 and treated following the clinical and therapeutic measures proposed by the last European League Against Rheumatism update.26 Urate-lowering therapy is closely indicated from the first clinical manifestation of gout to achieve target SUA values (<6 mg/dL); also, in patients with severe gout, target values are lower (<5 mg/dL) to facilitate crystal dissolution and resolution. In our clinical practice, the choice of drug/s is related to the severity of clinical conditions, to previous contraindications, and to the presence of kidney impairment and/or other comorbidities, so we fully adhere to the aforementioned guidelines.25
 | Table 1 Main approved drugs to treat hyperuricemia Abbreviations: DHNB, 3,4-dihydroxy-5-nitrobenzaldehyde; PEG, polyethylene glycol. |
Safety and tolerability of XO inhibitors
Allopurinol is considered a safe drug with good overall tolerability; nonetheless, in rare cases (<2%) it can cause hypersensitivity reactions, sometimes even fatal. Moreover, its use has been associated with side effects, such as diarrhea, stomach pain, skin rash, headache, dizziness, and drowsiness. Dose-related and idiosyncratic side effects occur mainly in patients with renal impairment, where the use of lower doses of allopurinol does not adequately control the risk of gout.27 Febuxostat has an excellent safety and tolerability profile, and also in subjects with moderate renal impairment, because of its hepatic metabolism. The most frequent side effects are increased levels of liver transaminases, gastrointestinal disorders, and skin rash. Clinical trials have shown that effectiveness and tolerability of febuxostat are significantly higher than allopurinol in patients with hyperuricemia or gout,13,14 but above all in patients with renal failure, where febuxostat was safer in terms of relative risk (RR) of adverse events (OR 0.85, 95% CI 0.75–0.97).28 These data support previous clinical findings that the SUA-lowering efficacy of febuxostat is greater than that of allopurinol in patients with hyperuricemia with or without CKD.
Cardiovascular and renal effect of XO inhibitors
In the recent past, several studies have suggested that hyperuricemia is involved in the pathogenesis of different systemic disorders that affect cardiovascular and renal systems, such as hypertension, atherosclerosis, chronic heart failure (CHF), CKD, and stroke.7,9,29,30 Some investigators emphasize the role of oxidative stress as a common event promoting the development of the aforementioned pathologies. As described earlier, UA has prooxidant effects in the intracellular context, reducing nitric oxide availability and increasing ROS production, low-density lipoprotein peroxidation,31 and endothelial dysfunction.32 Therefore, high SUA levels, responsible for different inflammatory and prooxidant processes, are strongly related to an elevated cardiovascular risk.33 Moreover, it is well known that an increase in XO activity may induce higher SUA and ROS levels, with a direct impact of XO expression in severe inflammatory and ischemic events.34,35 Therefore, treatment with XO inhibitors has beneficial effects on hyperuricemia, gout, and different forms of ischemic and vascular injuries and inflammatory and metabolic diseases.36,37 The protective role of allopurinol on the cardiovascular system is expressed not only by lowering SUA levels but also by different nonselective actions causing free-radical scavenging and reduced lipid peroxidation.29 As reported in the literature, a decrease in SUA levels is associated not only with an improvement in blood pressure values20,38 but also with some additional benefits, such as better control of the ascertained residual cardiovascular risk, present also in subjects with well-controlled hypertension.39 Most of these favorable effects have been achieved by the antioxidant capacity of XO inhibitors, rather than their lowering effect on SUA levels.28 According to different authors, treatment with allopurinol (at elevated dosage) may cause an improvement in endothelial function in subjects with CHF and type 2 diabetes mellitus with mild hypertension,40,41 but no similar effect was observed after the administration of probenecid, despite a comparable reduction in SUA.33 Doehner et al suggested relevant improvements in forearm and leg blood-flow peak (detected with plethysmography) of 24% and 23%, respectively, in CHF patients after therapy with allopurinol (300 mg/day) compared to the placebo phase. Moreover, these patients showed an important reduction of 20% in allantoin values, a marker of oxygen production due to free radical action, compared to baseline.42 Butler et al41 described a significant decrease in malondialdehyde in subjects with type 2 diabetes mellitus treated with allopurinol (300 mg/day) versus a placebo group (0.3±0.04 versus 0.34±0.05 µmol/L, P<0.05); malondialdehyde is a biomarker of low-density lipoprotein peroxidation due to free radical insult.33
In many epidemiological and clinical studies, a relationship between hyperuricemia and risk of hypertension and CKD development is claimed.31,43 The excess UA could interfere with the renin–angiotensin system, causing reduced availability of nitric oxide and resulting in renal vasoconstriction with an increase in blood pressure; in addition, pro-inflammatory effects of elevated SUA levels promote CKD development or progression.30 Allopurinol treatment may improve the course of CKD,44 as reported by Goicoechea et al, who showed that allopurinol administration reduces CRP values, improves and slows the evolution of renal disorders, reduces the number of hospitalizations (RR 62%), and lowers cardiovascular risk (RR 71%).45
Long-term treatment
Although all possible side effects due to allopurinol treatment have been reported in the literature, including allopurinol-hypersensitivity syndrome, associated with high mortality (20%–25%), most studies mentioned herein did not detect any of them. The daily dose of allopurinol recommended for reaching target values of uricemia is 100–600 mg, although sometimes it must be up to 900 mg in patients without impaired renal function.46 Different studies show a dose–response effectiveness for allopurinol to achieve beneficial effects on blood pressure and endothelial function; in fact, a higher dosage of allopurinol (300 mg twice daily) has resulted in quality improvement of vascular biology and left ventricular hypertrophy.47 In obese or hypertensive adolescents, the dosage of allopurinol administered was 200 mg twice daily,48,49 but in older adults it seems necessary to increase the dosage to lower blood pressure values.29 Beattie et al suggested the use of allopurinol at doses of ≥300 mg daily or the more selective febuxostat at lower dosage (80–120 mg) in future clinical trials, to confirm and clarify their role in CVD and CKD prevention and treatment.38 The main recommendations for optimal management of silent hyperuricemia to prevent gout development and possible cardiorenal sequelae according to current guidelines are resumed in Table 2.1,40,50
 | Table 2 Key recommendations for the management of hyperuricemia Abbreviation: SUA, serum uric acid. |
Conclusion
In the recent past, hyperuricemia was considered a multifactorial pathological condition, implicated in the pathogenesis of several important metabolic disorders and closely related to CVD and renal incident development.6,7,9 It is well known that XO-inhibitor administration is the first choice to treat hyperuricemia and gout. Besides effectiveness in reducing SUA levels, antioxidant effects of allopurinol (at high doses) and febuxostat and their ability to improve endothelial function have been already shown in patients with CHF32 and type 2 diabetes mellitus.33 As mentioned earlier, elevated SUA levels are considered a predisposing condition to hypertension onset and consequently related to CVD and CKD.51 Even in subjects with well-controlled hypertension, residual cardiovascular risk remains, which in light of recent findings and especially after future studies could be attenuated by this class of drugs, particularly by febuxostat, which is more selective and better tolerated than allopurinol. Do XO inhibitors represent a new strategic class of drug for better future management of CVD and CKD? This question remains open, but we hope that in the near future will have a concrete answer based on growing scientific evidence and ongoing clinical trials.
Disclosure
The authors report no conflicts of interest in this work.
References
Yamanaka H. Essence of the revised guideline for the management of hyperuricemia and gout. Japan Med Assoc J. 2012;55:324–329. | ||
Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63:3136–3141. | ||
Trifirò G, Morabito P, Cavagna L, et al. Epidemiology of gout and hyperuricaemia in Italy during the years 2005-2009: a nationwide population-based study. Ann Rheum Dis. 2013;72:694–700. | ||
Roddy E, Doherty M. Epidemiology of gout. Arthritis Res Ther. 2010;12:223. | ||
Kuo CF, Grainge MJ, Mallen C, et al. Rising burden of gout in the UK but continuing suboptimal management: a nationwide population study. Ann Rheum Dis. 2015;74:661–667. | ||
Choi HK, Ford ES. Prevalence of the metabolic syndrome in individuals with hyperuricemia. Am J Med. 2007;120:442–447. | ||
Grassi D, Ferri L, Desideri G, et al. Chronic hyperuricemia, uric acid deposit and cardiovascular risk. Curr Pharm Des. 2013;19:2432–2438. | ||
Suresh E, Das P. Recent advances in management of gout. QJM. 2012;105:407–417. | ||
Kannangara DR, Phipps-Green AJ, Dalbeth N, et al. Hyperuricaemia: contributions of urate transporter ABCG2 and the fractional renal clearance of urate. Ann Rheum Dis. 2016;75:1363–1366. | ||
Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A. 1981;78:6858–6862. | ||
Billiet L, Doaty S, Katz JD, Velasquez MT. Review of hyperuricemia as new marker for metabolic syndrome. ISRN Rheumatol. 2014;2014:852954. | ||
McGillicuddy FC, de la Llera Moya M, Hinkle CC, et al. Inflammation impairs reverse cholesterol transport in vivo. Circulation. 2009;119:1135-45. | ||
Zhang Z, Blake DR, Stevens CR, et al. A reappraisal of xanthine dehydrogenase and oxidase in hypoxic reperfusion injury: the role of NADH as an electron donor. Free Radic Res. 1998;28:151–164. | ||
Pea F. Pharmacology of drugs for hyperuricemia: mechanisms, kinetics and interactions. Contrib Nephrol. 2005;147:35–46. | ||
Becker MA, Schumacher HR Jr, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450–61. | ||
Schumacher HR Jr, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum. 2008;59:1540–1548. | ||
Malik UZ, Hundley NJ, Romero G, et al. Febuxostat inhibition of endothelial-bound XO: implications for targeting vascular ROS production. Free Radic Biol Med. 2011;51:179–184. | ||
Ernst ME, Fravel MA. Febuxostat: a selective xanthine-oxidase/xanthine-dehydrogenase inhibitor for the management of hyperuricemia in adults with gout. Clin Ther. 2009;11:2503–2518. | ||
Ye P, Yang S, Zhang W, et al. Efficacy and tolerability of febuxostat in hyperuricemic patients with or without gout: a systematic review and meta-analysis. Clin Ther. 2013;35:180–189. | ||
Li S, Yang H, Guo Y, et al. Comparative efficacy and safety of urate-lowering therapy for the treatment of hyperuricemia: a systematic review and network meta-analysis. Sci Rep. 2016;6:33082. | ||
Jian-Ming L, Yao Q, Chen C. 3,4-Dihydroxy-5-nitrobenzaldehyde (DHNB) is a potent inhibitor of xanthine oxidase: a potential therapeutic agent for treatment of hyperuricemia and gout. Biochem Pharmacol. 2013;86:1328–1337. | ||
Crittenden DB, Pillinger MH. New therapies for gout. Annu Rev Med. 2013;64:325–337. | ||
Bentley R, Neuberger A. The mechanism of the action of uricase. Biochem J. 1952;52:694–699. | ||
Kelley EE. Comment on Fabbrini et al. Effect of plasma uric acid on antioxidant capacity, oxidative stress, and insulin sensitivity in obese subjects. Diabetes 2014;63:976-981. Diabetes. 2014;63:e18. | ||
Neogi T, Jansen TL, Dalbeth N, et al. 2015 Gout classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis. 2015;74:1789–1798. | ||
Richette P, Doherty M, Pascual E, et al. 2016 Updated EULAR evidence-based recommendations for the management of gout. Ann Rheum Dis. Epub 2016 Jul 25. | ||
Hande KR, Noone RM, Stone WJ. Severe allopurinol toxicity: description and guidelines for prevention in patients with renal insufficiency. Am J Med. 1984;76:47–56. | ||
Borghi C, Perez-Ruiz F. Urate lowering therapies in the treatment of gout: a systematic review and meta-analysis. Eur Rev Med Pharmacol Sci. 2016;20:983–992. | ||
Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med. 2008;359:1811–1821. | ||
Galassi FM, Borghi C. A brief history of uric acid: from gout to cardiovascular risk factor. Eur J Intern Med. 2015;26:373. | ||
Cicero AF, Rosticci M, Cagnati M, et al. Serum uric acid and markers of low-density lipoprotein oxidation in nonsmoking healthy subjects: data from the Brisighella Heart Study. Pol Arch Med Wewn. 2014;124:661–668. | ||
Rablink TJ, Luscher TF. Endothelial nitric oxide synthase: host defense enzyme of the endothelium? Arterioscler Thromb Vasc Biol. 2006;26:267–271. | ||
McGillicuddy FC, de la Llera Moya M, Hinkle CC, et al. Inflammation impairs reverse cholesterol transport in vivo. Circulation. 2009;119:1135–1145. | ||
Harrison R. Structure and function of xanthine oxidoreductase: where are we now? Free Radic Biol Med. 2002;33:774–797. | ||
Harrison R. Physiological roles of xanthine oxidoreductase. Drug Metab Rev. 2004;36:363–375. | ||
Borghi C, Desideri G. Urate-lowering drugs and prevention of cardiovascular disease: the emerging role of xanthine oxidase inhibition. Hypertension. 2016;67:496–498. | ||
Pacher P, Nivorozhkin A, Csaba S. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev. 2006;58:87–114. | ||
Beattie CJ, Fulton RL, Higgins P, et al. Allopurinol initiation and change in blood pressure in older adults with hypertension. Hypertension. 2014;64:1102–1107. | ||
Borghi C, Rosei EA, Bardin T, et al. Serum uric acid and the risk of cardiovascular and renal disease. J Hypertens. 2015;33:1729–1741. | ||
Farquharson CA, Butler R, Hill A, Belch JJ, Struthers AD. Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation. 2002;106:221–226. | ||
Butler R, Morris AD, Belch JJ, Hill A, Struthers AD. Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension. 2000;35:746–751. | ||
Doehner W, Schoene N, Rauchhaus M, et al. Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: results from 2 placebo-controlled studies. Circulation. 2002;105:2619–2624. | ||
Edwards NL. The role of hyperuricemia and gout in kidney and cardiovascular disease. Cleve Clin J Med. 2008;5:13–16. | ||
Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis. 2006;47:51–59. | ||
Goicoechea M, de Vinuesa SG, Verdalles U, et al. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol. 2010;5:1388–1393. | ||
Roddy E, Doherty M. Treatment of hyperuricaemia and gout. Clin Med. 2013;13:400–403. | ||
Rekhraj S, Gandy S, Szweijkowski B, et al. High-dose allopurinol reduces left ventricular mass in patients with ischemic heart disease. Circulation. 2013;106:221–226. | ||
Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA. 2008;300:924–932. | ||
Soletsky B, Feig DI. Uric acid reduction rectifies prehypertension in obese adolescents. Hypertension. 2012;60:1148–1156. | ||
Desideri G, Puig JG, Richette P. The management of hyperuricemia with urate deposition. Curr Med Res Opin. 2015;31:S27–S32. | ||
Edwards NL. The role of hyperuricemia and gout in kidney and cardiovascular disease. Cleve Clin J Med. 2008;75:S13–S16. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.