Back to Journals » International Journal of Nanomedicine » Volume 14
An efficient and long-acting local anesthetic: ropivacaine-loaded lipid-polymer hybrid nanoparticles for the control of pain
Authors Li A, Yang F, Xin J, Bai X
Received 8 October 2018
Accepted for publication 7 January 2019
Published 31 January 2019 Volume 2019:14 Pages 913—920
DOI https://doi.org/10.2147/IJN.S190164
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Aimei Li,1 Feng Yang,2 Jiaying Xin,1 Xuebo Bai1
1Department of Anesthesiology, Affiliated Hospital of Jining Medical University, Jining, Shandong, China; 2Department of Gastroenterology, Affiliated Hospital of Jining Medical University, Jining, Shandong, China
Purpose: Local anesthetics are used clinically for the control of pain following operation (including gastrointestinal surgery) or for the management of other acute and chronic pain. This study aimed to develop a kind of lipid-polymer hybrid nanoparticles (LPNs), which were constructed using poly(ethylene glycol)-distearoylphosphatidylethanolamine (PEG-DSPE) as the hydrophilic lipid shell and poly-ε-caprolactone (PCL) as the hydrophobic polymeric core.
Methods: Ropivacaine (RPV) was entrapped in the LPNs (RPV-LPNs) and the physicochemical and biochemical properties such as size, zeta potential, drug release, and cytotoxicity were studied. The long-lasting effects and safety aspects of the LPNs were evaluated in vitro and in vivo.
Results: The particle size and zeta potential of RPV-LPNs were 112.3±2.6 nm and -33.2±3.2 mV, with an entrapment efficiency (EE) of 90.2%±3.7%. Ex vivo permeation efficiency of LPNs was better than the drug solution. The RPV-LPNs exhibited a long-lasting in vivo anesthesia effect in both rats and mice.
Conclusion: Considering the low cytotoxicity, the LPNs prepared here could be used as an efficient local anesthetic for the control of pain.
Keywords: postoperative pain, gastrointestinal surgery, poly(ethylene glycol), poly-ε-caprolactone, ropivacaine
Introduction
Postoperative pain management remains one of the most common challenges which have been largely unmet.1,2 Local anesthetics are used clinically for the control of pain following operation (including gastrointestinal surgery), or for the management of other acute and chronic pain.3–5 However, the major limitation of local anesthetics is their short duration of action (<8 hours).6 Another limitation of the clinical application of local anesthetics is their systemic toxicity, including cardiac and neurological toxicity.7 Therefore, more and more attention has been paid to the design and engineering of sustained release systems to prolong the analgesic action for days and to reduce side effects.
Nanotechnology-based drug delivery system (DDS) potentially represents such a disruptive technology for the controlled release of an anesthetic drug and reducing systemic toxicity.8,9 Among various nanocarriers, highly successful results have been recorded for liposomes in pain control and quick patient recovery, such as liposomal bupivacaine in form of DepoFoam bupivacaine.10,11 Liposomes have superior biocompatibility and favorable pharmacokinetics profile. However, liposomes suffer from the drawbacks, such as lower drug loading, fast drug release, and instability.12 Recently, a new generation of nanoparticles (NPs) known as lipid-polymer hybrid nanoparticles (LPNs) has emerged to overcome the shortcomings of both liposomes and polymeric nanoparticles. LPNs consist of two major parts: the polymer and the lipid components.13 Specifically, LPNs comprise polymer cores and lipid shells, which combine the advantages of polymeric nanoparticles like higher drug loading and prolonged drug release, and those of liposomes like suitable biocompatibility.14,15
Poly(ethylene glycol)-distearoylphosphatidylethanolamine (PEG-DSPE) block copolymers have been approved by the Food and Drug Administration (FDA) for medical applications. PEG-DSPE copolymers have been widely used in the preparation of LPNs.16 The amphiphilic polymers (PEG-DSPE) are engineered in LPNs composed of a polymeric core, a lipid layer (DSPE, and/or other lipid), and a hydrophilic shell (PEG). Various researches have verified that PEG could prolong the circulation time and release drugs at a sustained rate in an optimal range of drug concentrations thus reducing toxicity because of drastic plasma drug concentration fluctuation.17,18 In this study, ropivacaine-loaded PEG modified LPNs were designed to prolong its release and reduce side effects.
Ropivacaine (RPV), the pure S-(-)-enantiomer, is a member of the amino amide class of local anesthetics.19 It has been approved by FDA for the production of local or regional anesthesia for surgery and acute pain management. Compared with other local anesthetics, RPV has a longer analgesic effect (3–6 hours) and lower CNS toxicity and cardiotoxicity.20,21 However, the analgesic effect of RPV at present is limited to <6 hours (subcutaneous injection) and <20 hours for a typical femoral nerve block.18 Therefore, it is urgent to develop a long-acting and safe local anesthetic for clinical single use.
In this study, poly-ε-caprolactone (PCL) was used as a hydrophobic polymer to form the polymeric core, and PEG-DSPE as a hydrophilic lipid to form the shell of LPNs. RPV was entrapped in the LPNs and the physicochemical and biochemical properties such as size, zeta potential, drug release, and cytotoxicity were studied. The long-lasting effects and safety aspects of the LPNs were evaluated in vitro and in vivo.
Material and methods
Chemicals and reagents
Ropivacaine hydrochloride monohydrate (RPV, ≥98%, HPLC), L-α-lecithin (from soybean, ≥99%, thin layer chromatography), and Pluronic® F-127 were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). PCL (molecular weight 20 kDa) was provided by Xi’an Ruixi Biological Technology Co., Ltd (Xi’an, Shaanxi, China). PEG2000-DSPE (PEG-DSPE) was purchased from Peng Sheng Biological Co., Ltd (Shanghai, PRC). FBS, DMEM, and MTT were purchased from Invitrogen (Thermo Fisher Scientific, Waltham, MA, USA). Other chemicals and reagents were of analytical grade or HPLC grade.
LPNs preparation
RPV-loaded LPNs (RPV-LPNs, Figure 1) were prepared by the w/o/w double emulsification method.22,23 Briefly, PCL (100 mg) as polymer, PEG-DSPE (100 mg) as lipid, and lecithin (20 mg) as lipophilic surfactant were dissolved in acetone (5 mL) as an organic solvent to form the oil phase (1). RPV (20 mg) was dissolved in sodium dodecyl sulfate (0.5 mL, 5%, w/v) solution to form the internal aqueous phase (2). Next, solution (1) was sonicated (1 minute) in an ice-bathed solution (2) for emulsification. The resultant nano-emulsion was poured into a Pluronic® F-127 solution (20 mL, 1%, w/v) and was sonicated again for 5 minutes in an ice bath. Then the mixture was stirred overnight at room temperature to evaporate off organic solvents, and the resultant RPV-LPNs were collected by centrifugation at 50,000 rpm, washed with ultrapure water (MilliQ water, Merck Millipore, Burlington, MA) and lyophilized to obtain free flowing powder.
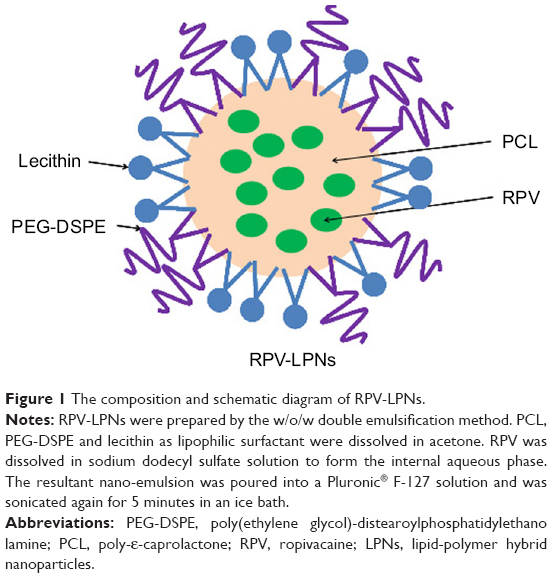 | Figure 1 The composition and schematic diagram of RPV-LPNs. |
Empty LPNs (E-LPNs) was prepared by the same method without the presence of RPV.
Particle size and zeta potential
LPNs samples were diluted in distilled water and the particle size and polydispersity (PDI) were measured using a Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK) at 25°C.24 The zeta potential of the LPNs was measured using the same instrument after dilution in the buffer solution.
Drug loading and in vitro release
RPV content in the LPNs was analyzed by reverse-phase HPLC (Agilent 1,100 Series, Agilent Technologies, Santa Clara, CA, USA).25 Specifically, 10 mL of sample was injected into a mobile phase of 50% PBS (pH 7.4) and 50% acetonitrile (0.1% TFA) at a flow rate of 1 mL/min and passed over a 5 μm reverse-phase column (Zorbax Eclipse SDB-C18, Agilent Technologies). RPV elution was detected at 262 nm.
Drug loading of RPV into the LPNs was evaluated by filtrating the LPN samples through a membrane filter (pore size: 0.1 μm).26 The incorporation of RPV into LPNs was expressed in terms of RPV entrapment efficiency (EE, %) = (amount of encapsulated RPV/total amount of RPV in the LPNs ×100), and RPV loading capacity (LC, %) = (amount of encapsulated RPV/amount of carrier ×100).
Release of RPV from the LPNs was studied using the dialysis bag method. The dialysis bag (cut-off of 2 kD) retained the LPNs but allowed the free drugs to diffuse into the dissolution medium. RPV-LPNs and RPV solution (2 mL) respectively, were sealed into the dialysis bags. The bags were then placed in a glass beaker containing 0.2 L of PBS (pH 7.4) at 37.0°C±0.5°C and stirred at 100 rpm. At pre-set time intervals (0.25, 0.5, 1, 2, 3, 6, 9, 12, 24, 48, 72, and 96 hours), 1 mL of the medium was collected and analyzed for the RPV content by HPLC. An equal volume of PBS (1 mL) maintained at the same temperature was added at every time point to ensure sink conditions.
Cells and animals
Mouse embryonic fibroblasts cells (BALB/c-3T3 cells) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Female BALB/c nude mice (6–8 weeks old) and female Sprague Dawley rats (9–11 weeks old) were purchased from Shandong University Laboratory Animal Center (Jinan, People’s Republic of China). The study protocol followed the National Institutes of Health guide for the care and use of laboratory animals. All animal experiments were approved by the Medical Ethics Committee of Jining Medical University (No. JNMU20180646-001).
In vitro cytotoxicity
Cytotoxicity of RPV-LPNs, E-LPNs, and RPV solution on BALB/c-3T3 cells was measured by MTT assay.27 Cells were seeded in a 48-well plate at a seeding density of 104 cells per well, then incubated in DMEM supplemented with 10% FBS and antibiotics under 5% CO2 at 37°C for 24 hours. The medium was then replaced with various doses of RPV-LPNs, E-LPNs, and RPV solution. MTT solution (5 mg/mL) was added to each well after 48 hours of incubation and incubated for another 4 hours. Dimethyl sulfoxide (DMSO) (200 mL) was added to each well and the absorption of the product formed was measured at 570 nm using a microplate reader (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Ex vivo permeation study
Ex vivo skin permeation study was carried out in Vertical Franz Diffusion cells (Permegear Inc., Bethlehem, PA, USA) with a cross-sectional area of 2.25 cm2 and 8.0 mL cell volume.28 Rats were anesthetized and the abdominal full-thickness skin excised. After removal of hair, muscle, and fat on the skin, the skin was immersed in 15% w/v glycerin containing saline solution for 30 minutes to retain the integrity of the skin and mounted between the donor and receptor compartments with the epidermal side facing upward.29 Physiological saline (20 mL) containing 1% TWEEN 80 which was maintained at 37°C±0.5°C using a water bath was used as a receptor medium to meet the sink condition. RPV-LPNs and RPV solution (0.5 mL) were applied to the epidermal surface of the skin. At pre-set time intervals (0.5, 1, 2, 3, 6, 9, 12, 24, and 48 hours), samples (0.3 mL) were collected from the receiver and replaced with 0.3 mL of fresh receptor medium. The samples were analyzed for the permeate amount of RPV by HPLC method the same as in “Drug loading and in vitro release” section. The cumulative amount of RPV penetrated (Pn) as a function of time was calculated using the equation:
|
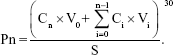
Cn and Ci refer to the RPV concentration of the receptor medium at each sampling time and the RPV concentration of sample i, respectively. V0 and Vi correspond to the volumes of the receptor compartment and the collected sample, respectively. S means the effective diffusion area.
In vivo anesthesia analgesic effect in rats
Radiant heat tail-flick latency (TFL) test was first employed for evaluating the in vivo anesthetic effect of LPNs.31 RPV-LPNs, RPV solution (both containing 20 mg of RPV), E-LPNs, and 0.9% normal saline samples were each applied in turn on the tail of the rats, which were placed in a plastic box, the ventral surface of the distal 5–6 cm of the tail was placed over a 0.5 cm hole, beneath which an infrared radiant bulb was placed. In order to avoid tissue damage, 10 seconds of cutoff time was set. The maximum possible effects (MPEs) were calculated according to the formula: MPE (%) = (test latency − baseline latency)/(cutoff time − baseline latency) × 100.32 The baseline latency was calculated as the mean of three different measurements taken at 10-minute intervals.
In vivo analgesia duration in mice
In vivo analgesia duration test was applied using electrical stimulation testing, which has long been used as a means of determining analgesia in animals and humans.33,34 Testing for analgesia was done by a vocal response to electrical stimulation (beginning at 1 mA and increasing to a maximum of 8 mA) at the skin directly overlying the abdomen at the site of injection using a current generator. The mices’ belly hair was shaved off, and they were screened prior to injection to determine their vocalization threshold, the current required to produce a vocalization response. RPV-LPNs, RPV solution (both containing 2 mg of RPV), E-LPNs, and 0.9% normal saline samples were injected subcutaneous injection into the abdomen of the mice, followed by determination of analgesia at the desired time points. If all the mice in one group (eight each group) did not vocalize to electrical stimulation 2 mA above threshold, it indicates 100% analgesia; if half of the mice in one group vocalized to electrical stimulation, it means 50% analgesia; and if all of the mice in one group vocalized to electrical stimulation, it indicates 0% analgesia (no analgesic effect).
Statistical analysis
All the results were expressed as mean ± SD from at least three independent runs. Comparisons between groups were made using Student’s t-test and with >2 groups, ANOVA was used to compare results. Statistical significance was set at P<0.05. All statistical analysis was performed using SPSS, version 21.0 (IBM Corporation, Armonk, NY, USA).
Results
Particle size, zeta potential, and drug loading
The particle size and PDI of RPV-LPNs were 112.3±2.6 nm and 0.16±0.02, with a zeta potential of −33.2±3.2 mV (Table 1). The similar size and surface charge was found on E-LPNs, which indicated that the encapsulation of RPV had negligible effect on the system. The EE and LC of RPV-LPNs were 90.2%±3.7% and 5.1%±0.4%, respectively.
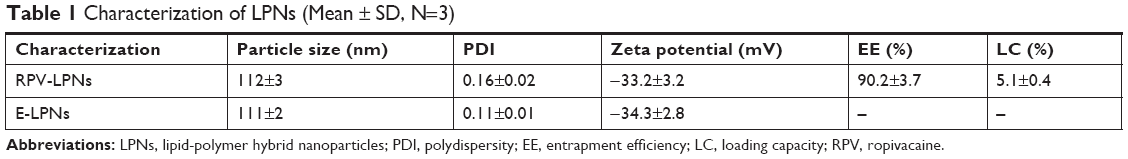 | Table 1 Characterization of LPNs (Mean ± SD, N=3) |
In vitro drug release
In vitro drug release behaviors of LPNs and the solution are depicted as the cumulative RPV release (%) vs time (Figure 2). The results showed that the release of RPV from drug solution was fast and reached over 80% of drug release within 0.5 hour. RPV-LPNs exhibited sustained release behavior and after 24 hours only 50% of drug release was achieved. The time point for RPV-LPNs to reach over 80% drug release was 72 hours.
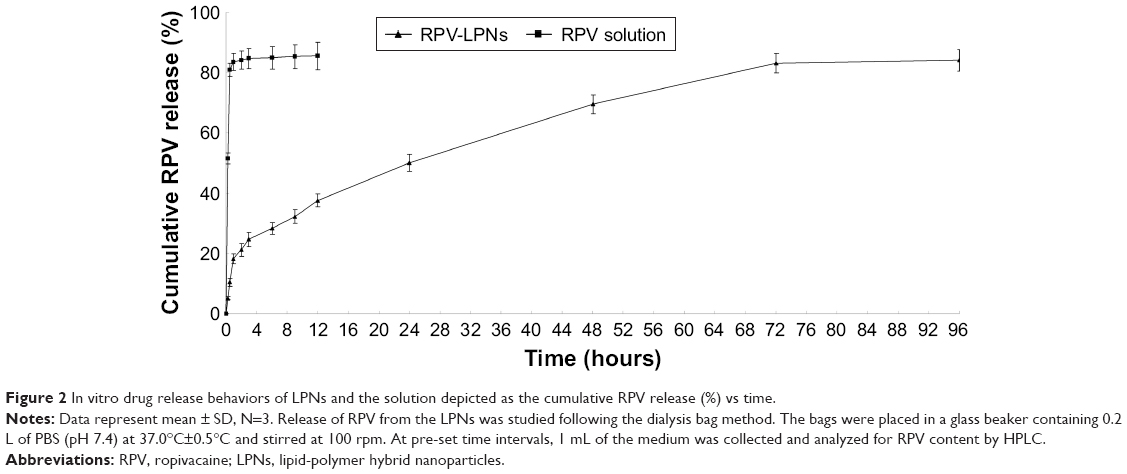 | Figure 2 In vitro drug release behaviors of LPNs and the solution depicted as the cumulative RPV release (%) vs time. |
In vitro cytotoxicity
In vitro cytotoxicity of RPV-LPNs, E-LPNs, and RPV solution was evaluated by assaying the BALB/c-3T3 fibroblast viability (Figure 3). At all the concentrations from 1 to 100 μM, RPV-LPNs, E-LPNs had a moderate effect on cell viability; no obvious changes were found. Decrease in viability of cells occurred in RPV solution at high concentrations (50 and 100 μM), which are shown in Figure 3 (P<0.05).
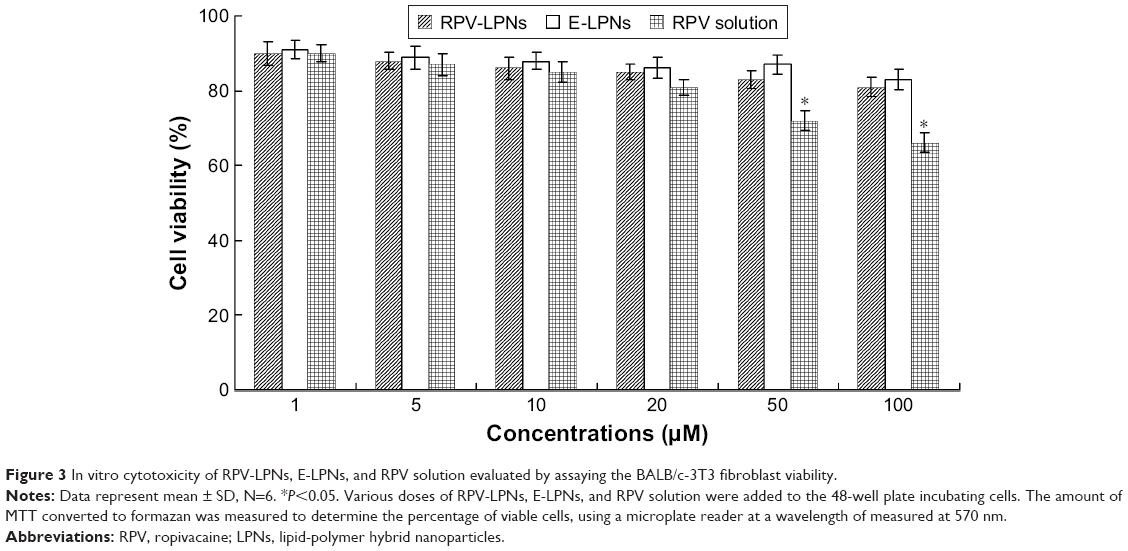 | Figure 3 In vitro cytotoxicity of RPV-LPNs, E-LPNs, and RPV solution evaluated by assaying the BALB/c-3T3 fibroblast viability. |
Ex vivo permeation study
Ex vivo permeation profiles of RPV-LPNs and RPV solution were summarized in Figure 4. RPV solution had completed permeation through the skin in the first few hours, while the permeation of RPV-LPNs was different. First, more RPV was permeated through the skin by RPV-LPNs than RVC injection (P<0.05): the cumulative RPV penetration of RPV-LPNs was 907±35 μg/cm2 at the end of the study (48 hours), which was 2.6-fold higher than that of RVC injection (344±23 μg/cm2).
 | Figure 4 Ex vivo permeation of profiles RPV-LPNs and RPV solution. |
In vivo anesthesia a effect in rats
In vivo anesthesia effect of LPNs was first investigated in rats by TFL test. The MPE curves were presented in Figure 5. E-LPNs showed no significant efficiency compared with the 0.9% saline control group. RVC solution showed a rapid but short time anesthetic effect, with a peak effect seen within 1 hour after the application. RPV-LPNs showed pronounced anesthetic effect last for 10 hours of application, which indicates a more long-lasting effect than the solution formulation (P<0.05).
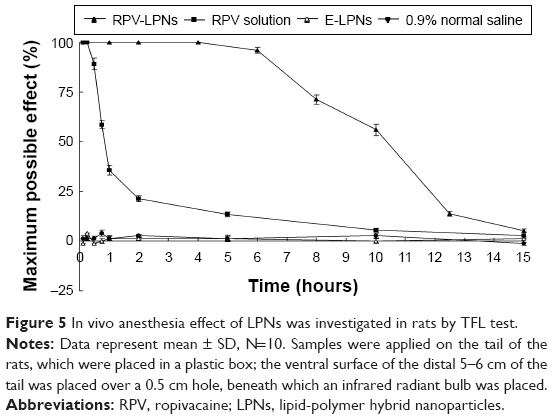 | Figure 5 In vivo anesthesia effect of LPNs was investigated in rats by TFL test. |
In vivo analgesia duration in mice
In vivo analgesia duration results in mice are illustrated in Figure 6. The figure indicates that a single administration of RPV-LPNs to mice provides significantly prolonged analgesia compared with RPV solution. The median duration of analgesia after mice were administered with RPV-LPNs and RPV solution was 36 hours and 0.5 hour, respectively. These results were in accordance with the in vivo anesthesia analgesic effect in rats and the ex vivo permeation results, which could be evidence of the long-lasting and more efficient analgesic effect of the LPNs.
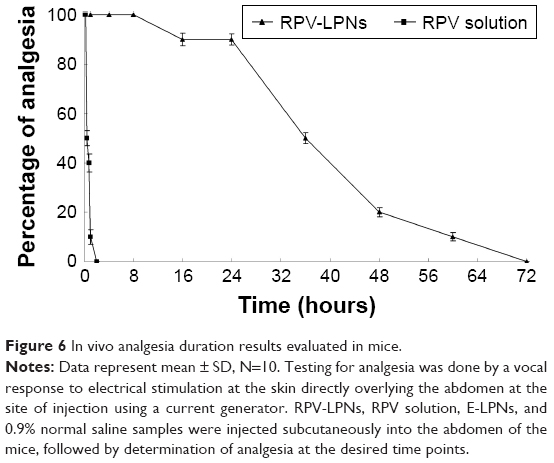 | Figure 6 In vivo analgesia duration results evaluated in mice. |
Discussion
Slight physicochemical differences of NPs may have significant biological implications in the cellular uptake and biological processes of the systems.35 The in vitro and in vivo fate of NPs was influenced by the uniformity of particle size and surface charge. Zeta potential, which can be either positive or negative in polarity depending upon the chemistry of the particles, is an electric potential created by the presence of a charge on the particle surface.36 Zeta potential is an indicator of the degree of repulsion between similarly charged particles in the formulation, thus it can probably indicate the physical stability of a formulation. Generally, a large positive or negative zeta potential is favorable for obtaining particles with better stability. Size and PDI of the NPs are important factors for the nanocarriers, which can influence the distribution of carriers.37 Particle sizes lower than 200 nm (with a PDI lower than 0.2) could decrease the uptake by the liver, prolong circulation time in the blood, and improve bioavailability. The particle size and PDI of RPV-LPNs were 112.3±2.6 nm and 0.16±0.02; the similar size and PDI found on E-LPNs indicated the encapsulation of RPV had negligible effect on the system.
RPV release from the LPNs followed a biphasic process with an initial faster drug release (over 30% in 9 hours) and the remaining 5-fluorouracil (5-FU) being released in a sustained manner over a further period of 72 hours. The initial fast drug release is attributed to the leakage of the lipid on the surface of LPNs which may let the RPV in the outer layer of the particles rapidly diffused into the incubation medium. RPV-LPNs exhibited a sustained-release behavior and complete the drug release until 72 hours of the study. This characteristic may help with the long-lasting action of the system for the local anesthesia. Evaluation of the cytotoxicity of the NPs is essential for the polymers or lipids may bring about a cytotoxic effect. In vitro cytotoxicity of RPV-LPNs and E-LPNs are negligible at all the studied concentrations, which may be used safely in vivo.
Ex vivo permeation study results could be the confirmation of the release behavior. RPV permeation was sustained and much more through the skin using RPV-LPNs compared with RPV solution. The results illustrated that LPNs have better and sustained skin permeation capacity, which may be due to the nano-size, and indicating the biocompatibility for long-term skin administration.38 The results also indicated that LPNs may have the potential to help the RPV to cross the skin layers more efficiently than the free drug and may have good efficiency of action when delivered in vivo.
In vivo local anesthetic effects of RPV-LPNs were evaluated by TFL test in rats and analgesia duration experiments in mice. The MPE results of the TFL test exhibited that RPV-LPNs have a longer lasting effect than the RPV solution, which showed a rapid anesthetic effect with a peak effect seen within 1 hour from the application. RPV-LPNs induce a pronounced anesthetic effect that was a rapid reaction in the first few minutes and also displayed sustained activity compared with the drug solution.39 In vivo analgesia duration results in mice also indicated that a single administration of RPV-LPNs to mice provides significantly prolonged analgesia compared with RPV solution. The RPV-LPNs developed in this study showed a much-prolonged analgesia when compared with free RPV, which was in accordance with the in vivo anesthesia analgesic effect in rats and the ex vivo permeation results.
Conclusion
In the present research, RPV was entrapped in LPNs, which were constructed using PEG-DSPE as hydrophilic lipid shell and PCL as hydrophobic polymeric core. RPV-LPNs had a small size, narrow PDI, high drug encapsulation efficiency, and low cytotoxicity. Ex vivo permeation efficiency of LPNs was better than the drug solution. The sustained release behavior of LPNs was in accordance with the long-lasting in vivo anesthesia effect both in rats and mice. In conclusion, the LPNs prepared in this study could be used as an efficient and long-acting local anesthetic.
Disclosure
The authors report no conflicts of interest in this work.
References
American Society of Anesthesiologists Task Force on acute pain management. Practice guidelines for acute pain management in the perioperative setting: an updated report by the American Society of Anesthesiologists Task Force on acute pain management. Anesthesiology. 2012;116(2):248–273. | ||
Mitra S, Carlyle D, Kodumudi G, Kodumudi V, Vadivelu N. New advances in acute postoperative pain management. Curr Pain Headache Rep. 2018;22(5):35. | ||
Bagshaw KR, Hanenbaum CL, Carbone EJ, et al. Pain management via local anesthetics and responsive hydrogels. Ther Deliv. 2015;6(2):165–176. | ||
Hu D, Onel E, Singla N, Kramer WG, Hadzic A. Pharmacokinetic profile of liposome bupivacaine injection following a single administration at the surgical site. Clin Drug Investig. 2013;33(2):109–115. | ||
Weiniger CF, Golovanevski M, Sokolsky-Papkov M, Domb AJ. Review of prolonged local anesthetic action. Expert Opin Drug Deliv. 2010;7(6):737–752. | ||
Møiniche S, Kehlet H, Dahl JB. A qualitative and quantitative systematic review of preemptive analgesia for postoperative pain relief: the role of timing of analgesia. Anesthesiology. 2002;96(3):725–741. | ||
Harmatz A. Local anesthetics: uses and toxicities. Surg Clin North Am. 2009;89(3):587–598. | ||
Vahabi S, Eatemadi A. Nanoliposome encapsulated anesthetics for local anesthesia application. Biomed Pharmacother. 2017;86:1–7. | ||
Moradkhani MR, Karimi A, Negahdari B. Nanotechnology application to local anaesthesia (LA). Artif Cells Nanomed Biotechnol. 2018;46(2):355–360. | ||
Mcalvin JB, Padera RF, Shankarappa SA, et al. Multivesicular liposomal bupivacaine at the sciatic nerve. Biomaterials. 2014;35(15):4557–4564. | ||
Beiranvand S, Eatemadi A, Karimi A. New updates pertaining to drug delivery of local anesthetics in particular bupivacaine using lipid nanoparticles. Nanoscale Res Lett. 2016;11(1):307. | ||
Mandal B, Bhattacharjee H, Mittal N, et al. Core-shell-type lipid-polymer hybrid nanoparticles as a drug delivery platform. Nanomedicine. 2013;9(4):474–491. | ||
Wakaskar RR. General overview of lipid–polymer hybrid nanoparticles, dendrimers, micelles, liposomes, spongosomes and cubosomes. J Drug Target. 2018;26(4):311–318. | ||
Ma P, Li T, Xing H, et al. Local anesthetic effects of bupivacaine loaded lipid-polymer hybrid nanoparticles: in vitro and in vivo evaluation. Biomed Pharmacother. 2017;89:689–695. | ||
Hadinoto K, Sundaresan A, Cheow WS. Lipid–polymer hybrid nanoparticles as a new generation therapeutic delivery platform: a review. Eur J Pharm Biopharm. 2013;85(3):427–443. | ||
Wang R, Xiao R, Zeng Z, Xu L, Wang J. Application of poly(ethylene glycol)-distearoylphosphatidylethanolamine (PEG-DSPE) block copolymers and their derivatives as nanomaterials in drug delivery. Int J Nanomedicine. 2012;7:4185–4198. | ||
Zhang L, Chan JM, Gu FX, et al. Self-assembled lipid–polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano. 2008;2(8):1696–1702. | ||
Wang Z, Huang H, Yang S, et al. Long-term effect of ropivacaine nanoparticles for sciatic nerve block on postoperative pain in rats. Int J Nanomedicine. 2016;11:2081–2090. | ||
Mcclellan KJ, Faulds D. Ropivacaine: an update of its use in regional anaesthesia. Drugs. 2000;60(5):1065–1093. | ||
Foley PL, Ulery BD, Kan HM, et al. A chitosan thermogel for delivery of ropivacaine in regional musculoskeletal anesthesia. Biomaterials. 2013;34(10):2539–2546. | ||
Zhang Y, Yue Y, Chang M. Local anaesthetic pain relief therapy: in vitro and in vivo evaluation of a nanotechnological formulation co-loaded with ropivacaine and dexamethasone. Biomed Pharmacother. 2017;96:443–449. | ||
Zhao Y, Lin D, Wu F, et al. Discovery and in vivo evaluation of novel RGD-modified lipid-polymer hybrid nanoparticles for targeted drug delivery. Int J Mol Sci. 2014;15(10):17565–17576. | ||
Devrim B, Kara A, Vural İ, Bozkır A. Lysozyme-loaded lipid-polymer hybrid nanoparticles: preparation, characterization and colloidal stability evaluation. Drug Dev Ind Pharm. 2016;42(11):1865–1876. | ||
Ribeiro AF, de Oliveira Rezende RL, Cabral LM, de Sousa VP. Poly ε-caprolactone nanoparticles loaded with Uncaria tomentosa extract: preparation, characterization, and optimization using the Box–Behnken design. Int J Nanomedicine. 2013;8:431–442. | ||
Foley PL, Ulery BD, Kan HM, et al. A chitosan thermogel for delivery of ropivacaine in regional musculoskeletal anesthesia. Biomaterials. 2013;34(10):2539–2546. | ||
Ortiz R, Prados J, Melguizo C, et al. 5-Fluorouracil-loaded poly(ε-caprolactone) nanoparticles combined with phage E gene therapy as a new strategy against colon cancer. Int J Nanomedicine. 2012;7:95–107. | ||
Shao Z, Shao J, Tan B, et al. Targeted lung cancer therapy: preparation and optimization of transferrin-decorated nanostructured lipid carriers as novel nanomedicine for co-delivery of anticancer drugs and DNA. Int J Nanomedicine. 2015;10:1223–1233. | ||
Li H, Liu T, Zhu Y, et al. An in situ-forming phospholipid-based phase transition gel prolongs the duration of local anesthesia for ropivacaine with minimal toxicity. Acta Biomater. 2017;58:136–145. | ||
Shah PP, Desai PR, Channer D, Singh M. Enhanced skin permeation using polyarginine modified nanostructured lipid carriers. J Control Release. 2012;161(3):735–745. | ||
Vitorino C, Almeida A, Sousa J, et al. Passive and active strategies for transdermal delivery using co-encapsulating nanostructured lipid carriers: in vitro vs in vivo studies. Eur J Pharm Biopharm. 2014;86(2):133–144. | ||
You P, Yuan R, Chen C. Design and evaluation of lidocaine- and prilocaine-coloaded nanoparticulate drug delivery systems for topical anesthetic analgesic therapy: a comparison between solid lipid nanoparticles and nanostructured lipid carriers. Drug Des Devel Ther. 2017;11:2743–2752. | ||
Yue Y, Zhao D, Yin Q. Hyaluronic acid modified nanostructured lipid carriers for transdermal bupivacaine delivery: in vitro and in vivo anesthesia evaluation. Biomed Pharmacother. 2018;98:813–820. | ||
Cohen R, Kanaan H, Grant GJ, Barenholz Y. Prolonged analgesia from Bupisome and Bupigel formulations: from design and fabrication to improved stability. J Control Release. 2012;160(2):346–352. | ||
Grant GJ, Piskoun B, Lin A, Bansinath M. An in vivo method for the quantitative evaluation of local anesthetics. J Pharmacol Toxicol Methods. 2000;43(1):69–72. | ||
He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666. | ||
Cho EJ, Holback H, Liu KC, Abouelmagd SA, Park J, Yeo Y. Nanoparticle characterization: state of the art, challenges, and emerging technologies. Mol Pharm. 2013;10(6):2093–2110. | ||
Wang Y, Wang S, Shi P. Transcriptional transactivator peptide modified lidocaine-loaded nanoparticulate drug delivery system for topical anesthetic therapy. Drug Deliv. 2016;23(9):3193–3199. | ||
Chen C, You P. A novel local anesthetic system: transcriptional transactivator peptide-decorated nanocarriers for skin delivery of ropivacaine. Drug Des Devel Ther. 2017;11:1941–1949. | ||
Wang J, Zhang L, Chi H, Wang S. An alternative choice of lidocaine-loaded liposomes: lidocaine-loaded lipid-polymer hybrid nanoparticles for local anesthetic therapy. Drug Deliv. 2016;23(4):1254–1260. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.