Back to Journals » Journal of Inflammation Research » Volume 15
An AAV-Based NF-κB-Targeting Gene Therapy (rAAV-DMP-miR533) to Inflammatory Diseases
Authors Luo T, Wang Y, Tang H ![]() , Zhou F, Chen Y, Pei B, Wang J
, Zhou F, Chen Y, Pei B, Wang J ![]()
Received 16 February 2022
Accepted for publication 28 May 2022
Published 14 June 2022 Volume 2022:15 Pages 3447—3466
DOI https://doi.org/10.2147/JIR.S362732
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Zili You
Tao Luo,1 Yile Wang,1 Hailin Tang,1 Fei Zhou,2 Ying Chen,3 Bing Pei,4 Jinke Wang1
1State Key Laboratory of Bioelectronics, Southeast University, Nanjing, 210096, People’s Republic of China; 2School of Food Engineering and Biotechnology, Hanshan Normal University, Chaozhou, 521041, People’s Republic of China; 3School of Medical Technology, Xuzhou Medical University, Xuzhou, 221004, People’s Republic of China; 4Department of Clinical Laboratory, the Affiliated Suqian First People’s Hospital of Nanjing Medical University, Suqian, Jiangsu, 223800, People’s Republic of China
Correspondence: Jinke Wang, State Key Laboratory of Bioelectronics, Southeast University, Nanjing, 210096, People’s Republic of China, Email [email protected]
Background: The inflammatory diseases pose a great threat to human health. Variant anti-inflammatory agents have been therefore developed. However, the current anti-inflammatory drugs are still challenged by low response and side effects. There remain great unmet treatments to inflammatory diseases.
Methods: In this work, we fabricate a recombinant adeno-associated virus (rAAV), rAAV-DMP-miR533, by packaging a DNA molecule DMP-miR533 into AAV, in which DMP is a NF-κB-activatable promoter composed of a NF-κB decoy and a minimal promoter and miR533 codes an artificial microRNA targeting NF-κB RELA. We evaluate the in vitro and in vivo anti-inflammatory effect of the virus with inflammatory cells and the mice of three typical inflammatory diseases including the dextran sulphate sodium-induced acute colitis, imiquimod-induced psoriasis, and collagen-induced arthritis.
Results: We found that rAAV-DMP-miR533 had marked anti-inflammatory effect in both cells and mice. In addition, rAAV-DMP-miR533 showed biosafety in mice.
Conclusion: This study thus provides a promising gene therapy to variant inflammatory diseases by directly targeting NF-κB, an established hub regulator of inflammation.
Keywords: NF-κB, inflammation, therapy, AAV, microRNA
Introduction
Inflammation is the protective reaction for organism in response to infection and body injury. The moderate inflammatory response helps to maintain homeostasis in the body. However, the abnormal inflammatory responses will cause inflammatory diseases. For example, some infectious agents can lead to systematic inflammation, which may result in sepsis, cytokine release syndrome, acute respiratory distress syndrome, and even multiple organs failure.1 Some persistent infections lead to chronic inflammation and form high risk of cancers, such as hepatitis B virus-induced hepatis and hepatoma.2–4 Many chronic inflammations resulted from abnormal changes in adaptive immunity lead to various autoimmune diseases, such as arthritis, inflammatory bowel disease, lupus, psoriasis, dermatitis, asthma, multiple sclerosis, steatohepatitis, and even atherosclerosis, diabetes, neurodegenerative diseases, and inflammaging.5–7 Additionally, many abnormal changes in innate immunity induce various autoinflammatory diseases.8,9 The abnormal inflammation therefore poses wide and serious threats to human health.
To treat inflammatory diseases, the molecular mechanisms underlying these diseases have been extensively investigated. The basic signaling pathways (eg NF-κB and JAK-STAT) and main involved molecules have been identified,10–15 and many potential targets for developing anti-inflammation drugs have been discovered. Based on these findings, many drugs have been developed for treating various inflammatory diseases such as the widely used corticosteroids (eg glucocorticoid).16,17 In recent years, many anti-inflammatory biologicals have been developed, such as monoclonal antibodies to cytokines (eg pro-inflammatory cytokine TNF-α, IL-1α, IL-1β, IL-5, IL-6, IL-12, IL-17A, IL-17F, IL-23 and anti-inflammatory cytokine IL-4, IL-10, IL-11, IL-13, TGFβ), antibodies or antagonists to cytokine receptors (eg IL-6R, IL-5Rα, IL-4Rα), and antibodies to CDs (eg CD4, CD14, CD19, CD20, CD38, CD40).18–25 The small molecules as JAK inhibitors (JAK1, JAK2, JAK3, TYK2) are new promising anti-inflammatory drugs rapidly developed.23,26–28 Undoubtedly, the current anti-inflammatory drugs have already greatly benefitted the patients. However, the current anti-inflammatory drugs are still challenged by several key limitations, such as primary non-response,29 resistance or loss of response,30 recurrence,31,32 and multiple side effects.33–37 Therefore, there are still unmet treatments to inflammatory diseases.
NF-κB,11 a family of sequence-specific DNA-binding transcription factors including RelA/p65, p50, p52, RelB, and c-Rel, plays a key regulatory role in inflammation.24 After induced by various inducers,38 the activated NF-κB (mainly RelA-p50 heterodimer) can induce expression of its target genes by directly binding to the discrete DNA sequence in promoters and enhancers, and thus participates in cell proliferation, apoptosis, innate immune response, and other processes.38,39 NF-κB directly regulates the expression of many inflammatory genes including adhesion factors (eg ICAM-1, VCAM-1), cytokines (eg IL-1α, IL-1β, IL-2, IL-6, IL-8, IL-10, IL-11, IL-12, IL-13, IL-15, IL-17, IL-23, TNF-α, IFNβ, IFN-γ), and chemokines (eg CCL5, CCL17, CCL19, CCL20, CCL22, CCL23, CCL28).38,40,41 Conversely, NF-κB can be activated by some pro-inflammatory cytokines (eg TNF-α) to form a positive feedback loop strengthening inflammation process.38 Nowadays, NF-κB is identified as a primary driver of inflammation in the body. The aberrant activation of NF-κB is widely known to play key roles in all inflammatory diseases and cancers.42–44 Therefore, NF-κB and its signaling pathway are regarded as important targets for developing anti-inflammatory drugs.45 Especially, NF-κB itself is the most attractive target for anti-inflammatory drug development because NF-κB lies at the heart of inflammations.44,46 Many chemicals targeting NF-κB have been therefore developed,38,47–49 but only few of them did even reach clinical trials due to their low specificity and side effects.
The nucleic acids, such as decoy oligonucleotides,50–53 and small interfering RNA (siRNA),54–57 have also ever been explored as candidate NF-κB inhibitor. However, because of uncontrollable activity, difficult delivery and easy degradation, the two kinds of NF-κB inhibitors also failed in clinical trials. To take their advantages but overcome their limitations, we recently developed a new NF-κB inhibitor molecule, a plasmid vector DMP-miR533, by combining NF-κB decoy and miRNA interference.58 DMP is a NF-κB-specific promoter that consists of a NF-κB Decoy and a Minimal Promoter. MiR533 is an artificial miRNA targeting NF-κB RELA. We demonstrated that DMP-miR533 can sense and control the intracellular NF-κB activity in inflammatory cells. Transfection of DMP-miR533 made inflammatory cells apoptosis but exerted little effect on normal cells.58 Comparatively, a typical small molecule as NF-κB inhibitor, BAY 11–7082, induced apoptosis of both inflammatory and normal cells.58
In this work, to address the in vivo delivery of DMP-miR533 and explore its in vivo anti-inflammatory effect, we packaged DMP-miR533 in adeno-associated virus (AAV), a safe gene delivery vector applied to gene therapy.59 We first evaluated the anti-inflammatory effect of the prepared recombinant AAV (rAAV), rAAV-DMP-miR533, with inflammatory cells in vitro. We then treated the mice of three typical inflammatory diseases, including the dextran sulphate sodium (DSS)-induced acute colitis, imiquimod (IMQ)-induced psoriasis, and collagen-induced arthritis.
Methods
Plasmid Construction
The miRNAs targeting human or murine RELA was designed by BLOCK-iT™ RNAi Designer (https://rnaidesigner.thermofisher.com/rnaiexpress/) (Table S1 and S2). The DMP-miR533 fragment was amplified from pDMP-miR533 (our previous work)58, then ligated into pAAV-MCS (VPK-410, Stratagene) by using MluI (upstream) and XbaI (downstream) restriction sites to construct the vector pAAV-DMP-miR533 (Figure S1). The CMV-EGFP fragment was amplified with a pair of primers with the upstream MluI and downstream EocRI sites from the pEGFP-C1 (Clontech). The CMV-EGFP fragment was then cloned into pAAV-MCS to obtain the vector pAAV-CMV-EGFP (Figure S1). The DNA fragments were amplified by PCR using the HieffTM PCR Master Mix (With Dye) (Yeasen). The PCR amplified DNA fragments were run with agarose gel and purified with the AxyPrep DNA Gel Extraction Kit (Axygen). The digestion-ligation reaction contained the proper restriction endonucleases (ThermoFisher Scientific) and T4 DNA Ligase (ThermoFisher Scientific). The plasmid pAAV-DMP-miR533-CMV-EGFP (Figure S1) was constructed by cloning the CMV-EGFP fragment into the pAAV-DMP-miR533. As a negative control vector, the miR-NT fragment was synthesized according to the sequence of plasmid pcDNA™ 6.2-GW/EmGFP-miR-Neg (Thermo Fisher Scientific) and inserted into pDMP-miR, named pDMP-NT. The DMP-NT fragment was cloned from pDMP-NT and inserted into pAAV-MCS to get pAAV-DMP-NT vector. All plasmids including pAAV-MCS, pAAV-DMP-NT, pAAV-CMV-EGFP, pAAV-DMP-miR533, pAAV-DMP-miR533-CMV-EGFP, pAAV-Helper, and pAAV-RC were transfected into the E. coli DH5α (Tiangen) and purified with the EndoFree Plasmid kits (CWBio). All plasmids were verified by DNA sequencing. Oligonucleotides and primers used in this study were synthesized by Sangon Biotech (Shanghai, China) (Table S2 and S3).
Cell Culture
All Cell lines used in this research were acquired from the cell resource center of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, included HEK-293T (human fetal kidney cells), HT-29 (human colon cancer cells), CT-26 (mouse colon cancer cells), HL7702 (human normal hepatocytes), NIH-3T3 (mouse embryonic fibroblast). HT-29, CT-26, HL7702 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco). NIH-3T3, HEK-293T were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco). All media were supplemented with 10% fetal bovine serum (HyClone), 100 units/mL penicillin (Thermo Fisher), and 100 μg/mL streptomycin (Thermo Fisher). Cells were incubated at 37 °C in a humidified incubator containing 5% CO2.
Virus Preparation
HEK-293T cells were seeded into 75 cm2 flasks at a density of 5×106 cells per flask and cultivated for 24 h. Cells were then co-transfected with two helper plasmids (pHelper and pAAV-RC; Stratagene) and one of the pAAV plasmids (pAAV-MCS, pAAV-DMP-NT, pAAV-DMP-miR533, pAAV-DMP-miR533-CMV-EGFP, pAAV-CMV-EGFP) using Lipofectamine 2000 (Thermo Fisher) according to the manufacturer’s instructions. Cells were cultured for another 72 h. The cells and media were collected and kept at −80 °C overnight. The cells and media were then incubated in 37 °C water bath for 2 h. This freeze-thaw process was totally repeated three times. The 1/10 volume of pure chloroform was added to the cell lysate and the mixture was vigorously shaken at 37 °C for 1 h. The mixture was added NaCl to a final concentration of 1 M and shaken until NaCl dissolved. The mixture was centrifuged at 15,000 revolutions per minute (rpm) at 4 °C for 15 min and the supernatant was collected. The supernatant was added PEG8000 at a final concentration of 10% (w/v) and shaken until PEG8000 dissolved. The mixture was centrifuged at 15,000 rpm at 4 °C for 15 min. The supernatant was discarded and the pellet was dissolved into PBS. DNase and RNase were added to a final concentration of 1 µg/mL to the dissolved pellet. The mixture was incubated at room temperature for 30 min. The mixture was extracted once with chloroform (1:1 volume) and the aqueous layer that contained the purified virus was transferred to a new tube. Titers of AAVs were determined by qPCR using the primers AAV-F/R (Table S3). Quantified viruses were aliquoted and kept at −80°C for later use. The obtained viruses were named as rAAV-MCS, rAAV-DMP-NT, rAAV-DMP-miR533, rAAV-DMP-miR533-CMV-EGFP, and rAAV-CMV-EGFP.
Treatment of Cells
Cells (1×105) were seeded into 24-well plates and cultured at 37°C in 5% CO2 overnight. Cells were then transfected with various pAAVs plasmids (500 ng/well) by using Lipofectamine 2000 (Thermo Fisher) according to the manufacturer’s instruction. After transfection, cells were cultured for 24 h, 48 h, 72 h, respectively. If needed, the normal cells were first stimulated with TNF-α (Sigma-Aldrich) at a final concentration of 10 ng/mL for 1 h before transfection. Cells were stained with acridine orange (Solarbio) according to the manufacturer’s instructions, in which live cells appears uniformly green. Cells were imaged with a fluorescence microscope (IX51, Olympus) and counted with the Image-Pro Plus software.
Cell viability was analyzed with the CCK-8 assay by using a Cell Counting Kit-8 (Yeasen) according to the manufacturer’s instructions. Cells (5×103) were seeded into 96-well plate and cultured at 37°C in 5% CO2 overnight. Cells were then transfected with various pAAV plasmids (200 ng/well) by using Lipofectamine 2000 (Thermo Fisher) according to the manufacturer’s instruction. After transfection, cells were cultured for 24 h, 48 h, 72 h, respectively. If needed, the normal cell lines were first stimulated with TNF-α (Sigma-Aldrich) at a final concentration of 10 ng/mL for 1 h before transfection. Cells were added CCK-8 reagent (10 μL/well) and incubated for 1 h. The absorbance was measured at 450 nm using a microplate reader (BioTek).
Cells (5×105) were seeded into 6-well plate and cultured at 37°C in 5% CO2 overnight. Cells were then transfected with various pAAV plasmids (4 μg/well) by using Lipofectamine 2000 (Thermo Fisher) according to the manufacturer’s instruction. After transfection, cells were cultured for 24 h, 48 h, 72 h, respectively. If needed, the normal cells were first stimulated with TNF-α (Sigma-Aldrich) at a final concentration of 10 ng/mL for 1 h before transfection. Cell apoptosis was then detected with Flow Cytometry (Calibur, BD, USA) by using an AnnexinV-FITC/PI cell apoptosis detection kit (Vazyme) according to the manufacturer’s instructions.
Cells (5×103) were seeded into 96-well plate and cultured at 37°C in 5% CO2 overnight. Cells were then infected with various rAAVs (5×107 vg/well). After infection, cells were cultured for 48 h. If needed, the normal cells were first stimulated with TNF-α (Sigma-Aldrich) at a final concentration of 10 ng/mL for 1 h before transfection. The EGFP fluorescence was imaged with a fluorescence microscope (IX51, Olympus) and quantitatively analyzed by Flow Cytometry (Calibur, BD, USA). The cell apoptosis was detected by Flow Cytometry (Calibur, BD, USA) as described above.
Animals
All animal experiments in this study were conducted under a Project License (SYXK (su) 2021–0021) authorized by Jiangsu Provincial Science and Technology Department in accordance with the China laboratory Animal Laws (2017) and approved by the Animal Care and Use Committee of Southeast University (Nanjing, China).
Treatment of Colitis Model
The BALB/c mice purchased from Cavens (China) were randomly divided into 4 groups (n = 6), including Blank, dextran sulphate sodium (DSS), DSS+MCS, and DSS+miR533 groups. The mice of the Blank group drank water while those of other 3 groups drank water containing 3% Dextran sulfate sodium (DSS) (M. W=36000-50,000) (MP). On the 3rd and 5th day after drinking water containing 3% DSS, mice of the DSS+MCS and DSS+miR533 groups were intravenously injected with 100 μL of 1×1010 vg/mL rAAV-MCS and rAAV-DMP-miR533, respectively. The body weight was measured every day. On the 8th day, the mice were sacrificed and the colon from anus to ileocecal region was isolated and blood was collected. The length of the colon was measured. The colon tissue was used to prepare paraffin sections and detect gene expression. The sections were stained with hematoxylin-eosin (H&E), imaged, and scored. The histopathological scoring of colon tissue was blindly performed by other technicians according to four levels: 0 score, no obvious pathological changes; 1 score, focal inflammatory cell infiltration; 2 score, extensive inflammatory cell infiltration; 3 score, diffuse inflammatory cell infiltration; 4 score, inflammatory cell infiltration, tissue degeneration and necrosis, fibrous connective tissue hyperplasia. The level of TNF-α and IL-6 in serum was detected by ELISA using the TNF-α (ab208348, abcam) and IL-6 ELISA kits (ab222503, abcam) according to the manufacturer’s instructions.
Treatment of Psoriasis Model
Two batches of animal experiments were performed in the psoriasis model on male BALB/c mice (8 weeks; Cavens, China). After shaved the back of mice (shaved area was around 2.5 cm × 2.5 cm), they were randomly divided into 3 groups (n = 6), including Blank, MCS, and miR533 groups. The mice of the Blank group were treated with Vaseline cream. The mice of the MCS and miR533 groups were topically applied with commercially available 5% w/w of imiquimod (IMQ) (Sichuan Mingxin, China) at a dose of 62.5 mg lotion on the shaved back.
In the first batch of animal experiment, after 6 days of drug application, three mice of the Blank and MCS groups (n = 3) were sacrificed and the skin and blood samples were collected. The remained mice of the MCS group (n = 3) and miR533 group (n = 6) were intravenously injected with 100 μL of 1×1010 vg/mL rAAV-MCS and rAAV-DMP-miR533, respectively. The mice of the MCS and miR533 groups were remained with 5% w/w of IMQ treatment every day. The mice of the Blank group were similarly treated with Vaseline cream. After another 6 consecutive days, all mice were sacrificed and the skin and blood samples were collected. The level of TNF-α and IL-6 of skin and serum samples were detected by RT-qPCR and ELISA kits, respectively. At the same time, treatments with subcutaneous administration (i.h.) and administration usum externum (for external use) (ad us. ext.) of rAAV-DMP-miR533 were tried on one mouse of psoriasis model (n = 1). The mouse with completed 5% w/w of IMQ treatment was subcutaneously injected or smeared with 100 μL of 1×1010 vg/mL rAAV-DMP-miR533 every day, respectively. After another 6 days, all mice were sacrificed, and the sample collection and detection were the same as the previous intravenous injection experiment.
In the second batch of animal experiment, after 6 consecutive days of 5% w/w of IMQ treatment, the mice of the MCS and miR533 groups (n = 6) were smeared with 100 μL of 1×1010 vg/mL rAAV-MCS and rAAV-DMP-miR533 on shaved area every day (ad us. ext.) for 6 consecutive days, respectively. The 100 µL of virus solution was mixed with 0.5 g Vaseline for topical treatment of each mouse. The mice of the MCS and miR533 groups were remained with 5% w/w of imiquimod treatment every day. The Blank group kept smearing Vaseline cream until euthanasia. All mice were sacrificed and photographed on the 12th day and the skin and blood samples were collected. The body weight of the mice and Psoriasis Area and Severity Index (PASI) were monitored and recorded daily. Erythema, scales and thickness on the skin were scored independently from 0 to 4: 0, none; 1, slight; 2, moderate; 3, marked; 4, very marked. The sum of three indicators indicated the severity of inflammation (score, 0‒12). The level of TNF-α and IL-6 in serum was detected as described above. The skin tissue was used to perform H&E staining analysis and gene expression detection. The histopathological scoring of skin tissue was blindly performed by other technicians as described above.
Treatment of Arthritis Model
Thirty male DBA/1J mice (8 weeks; Cavens, China) were randomly divided into 5 groups, one of which was a healthy group injected with PBS (n = 6) as a control. The collagen-induced arthritis (CIA) mouse model was established by a double immunization. For the first immunization, mice were injected intradermally at the end of the tail with an emulsion of equal volume chick type-II collagen solution (2 mg/mL) (Chondrex, Redmond, WA, USA) and complete Freund’s adjuvant (2 mg/mL) (Chondrex, Redmond, WA, USA). After 21 days of the first immunization, the boost immunization was given to the mice with chick type-II collagen solution emulsified in incomplete Freund’s adjuvant (Chondrex, Redmond, WA, USA). The booster injection was administered at a location different from the first immunization. Disease was assessed by scoring all paws of each mouse as follows: 0, normal; 1, mild swelling and erythema confined to the midfoot and ankle joint; 2, mild swelling and erythema extending to the midfoot and ankle joint; 3, moderate swelling and erythema extending from the metatarsal joints to the ankle; 4, severe swelling and erythema encompassing the foot, ankle and digits. The qualitative clinical score of each mouse was the sum of scores of four paws.
The CIA mice were divided into four experiment groups: (1) CIA group: CIA mice were injected with PBS as a control group (i.v.; n = 6), (2) MTX-treated group: CIA mice were injected with MTX (1 mg/kg) every other day for 2 weeks (i.v.; n = 6), (3) NT-treated group: CIA mice were intravenously administered every other day for three times with rAAV-NT (1×109 vg /mouse; n = 6), and (4) miR533-treated group: CIA mice were intravenously administered every other day for three times with rAAV-DMP-miR533 (1×109 vg/mouse; n = 6). The body weight and clinical score of the mice were monitored every other day. Ankle width, paw thickness and tail thickness were measured with a Vernier caliper. All mice were euthanatized and photographed after MTX or NT or miR533 administration for 4 weeks. Serum samples from each group were collected for detection of biochemical indices. The level of TNF-α and IL-6 in serum was detected as described above. Tissues including heart, liver, spleen, lung, and kidney were harvested for H&E staining analysis. Spleen was weighted. The ankle joint tissue was used to perform H&E staining analysis and gene expression detection. The histopathological score of ankle joint tissue was blindly performed by other technician according to four levels: 0, normal synovium; 1, hypertrophy and cell invasion present in the synovial membrane; 2, pannus and cartilage erosions present; 3, erosions of cartilages and subchondral bone; and 4, dysfunction and rigidity of the whole joint.
Hematoxylin and Eosin Staining
Tissues including heart, liver, spleen, lung, kidney, paw (including ankle joint) were dissected, embedded in paraffin, sectioned, and stained with Hematoxylin and eosin (H&E) using routine methods. Briefly, tissues were resected and fixed overnight in 4% paraformaldehyde solution (Sangon Biotech, China) at room temperature. Subsequently, fixed specimens were decalcified, and embedded in paraffin. Tissue sections were prepared and stained with hematoxylin staining solution (C0107, Beyotime) and eosin staining solution (C0109, Beyotime). The prepared slides were photographed by a microscope (IX51, Olympus). The histopathological score was blindly performed by other researchers.
Micro-CT
All DBA/1J mice were sacrificed and their paws (including ankle joints) were collected for Micro-CT imaging by using an in-vivo micro CT scanner (vivaCT 80, SCANCO Medical AG, Switzerland). High-resolution tomographic image reconstruction and analysis were performed in the SCANCO GPU Accelerated Reconstruction System.
Quantitative PCR
Total RNA was isolated from the cultured cells and mouse tissues using TRIzol™ (Invitrogen) according to the manufacturer’s protocol. The complementary DNA (cDNA) was generated using PrimeScriptTM RT reagent Kit with gDNA Eraser (Takara). Expression of target genes from cDNA was detected with quantitative PCR (qPCR) on an ABI StepOne Plus (Applied Biosystems) by using the Fast SYBR Green Master Mix (Roche). Each sample was detected in three technical replicates. The relative mRNA transcript level was calculated as 2–ΔCt or 2–ΔΔCt, in which, ΔCt = Cttarget ‒ CtGADPH and ΔΔCt = ΔCttreatment – ΔCtcontrol. 2–ΔΔCt was also defined as relative quantity (RQ). The specificity of all qPCR primers (Table S3) was verified using melting curve analysis.
Statistical Analysis
All data are presented as means values ± standard deviation (SD), and statistical analysis and graphs were performed through GraphPad Prism 8.0 software. Statistical differences between two groups were determined using two-tailed Student’s t-test. Comparisons of three or more groups were determined by one-way or two-way analysis of variance (ANOVA) with Tukey’s or Sidak’s multiple comparison test when appropriate. Differences at p < 0.05 were considered statistically significant.
Results
Principle of Treatment
NF-κB is widely over-activated in inflammatory cells.60–62 To inhibit the NF-κB activity, we fabricate a rAAV named as rAAV-DMP-miR533 (Figure 1A), in which DMP is a promoter that consists of a NF-κB decoy and a minimal promoter, and miR533 codes an artificial microRNA targeting NF-κB RELA.58 When the DMP-miR533 was transfected into inflammatory cells such as human colon cancer cell HT-29, mouse colon cancer cell CT-26, and TNF-α-induced human normal hepatocyte HL7702 and mouse embryonic fibroblast NIH-3T3 that have NF-κB over activity (Figure 1B), DMP can be bound by NF-κB and the transcription of miR533 can be activated. The NF-κB is thus inhibited by decoy and RNA interference and the inflammatory cytokines as NF-κB target genes such as TNF-α and IL-6 will be knocked down. The NF-κB inhibition can lead the inflammatory cells to apoptosis. In contrast, in normal cells such as human normal hepatocyte HL7702 and mouse embryonic fibroblast NIH-3T3 that have little NF-κB activity (Figure 1B), DMP-miR533 cannot function due to lack of NF-κB activity (Figure 1A). To fully evaluate the anti-inflammatory effect of DMP-miR533 in cells and in animal, we designed and prepared miR533 targeting both human and mouse NF-κB RELA transcripts, respectively (Table S1). The human cells were treated by DMP-miR533 targeting human NF-κB RELA and the mouse cells and mice were treated by DMP-miR533 targeting mouse NF-κB RELA.
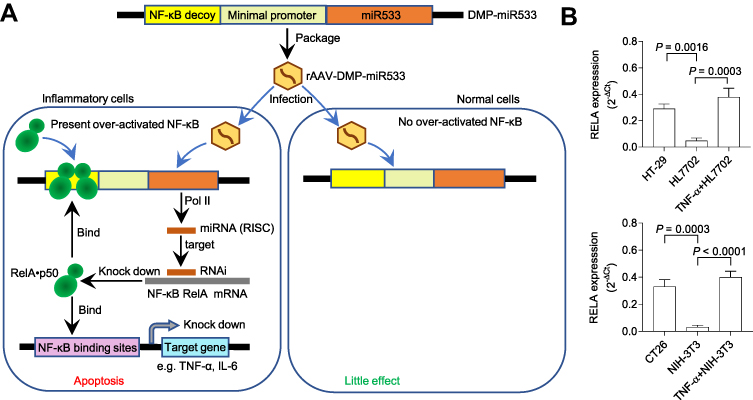 |
Figure 1 Schematic illustration of treatment of inflammation with rAAV-DMP-miR533. (A) Schematic of inflammation treatment with rAAV-DMP-miR533. DMP, Decoy-Minimal Promoter; Pol II, RNA polymerase II; RISC, RNA-induced silencing complex. (B) The NF-κB expression in the inflammatory and normal cells. The cancer cells HT-29 and CT-26 with NF-κB activity are used as natural inflammatory cells. The normal cells HL7702 and NIH-3T3 have little NF-κB activity. However, when stimulated with pro-inflammatory cytokine TNF-α, the two cells become the induced inflammatory cells with NF-κB activity. The NF-κB RELA expression was detected by qPCR (n = 3 wells). |
Treatment of Inflammatory Cells
To evaluate the feasibility of DMP-miR533 system for killing inflammatory cells, HT-29 cell, a human colon cancer cell with NF-κB over activity, was first transfected with pAAV-DMP-miR533 for 24 h to 72 h. The acridine orange (AO) staining of cells revealed that pAAV-DMP-miR533 had significant cytotoxicity to HT-29 cells (Figure 2A and B). The cell growth curve also revealed that the growth of HT-29 cells was significantly inhibited by pAAV-DMP-miR533 (Figure 2C). Meanwhile, the viability and growth of the cells were not significantly affected by a control treatment of pAAV-MCS, an empty vector (Figure 2B and C). To further verify interference efficiency of pAAV-DMP-miR533 in vitro, the expression of NF-κB RELA and its target genes were detected with qPCR. The results indicated that the expression of these genes was significantly knocked down by pAAV-DMP-miR533 but not by Lipofectamine (Blank) and pAAV-DMP-MCS (Figure 2D). These results indicate that pAAV-DMP-miR533 can significantly inhibited the growth of cells with NF-κB activity by knocking down the expression of NF-κB and its target genes.
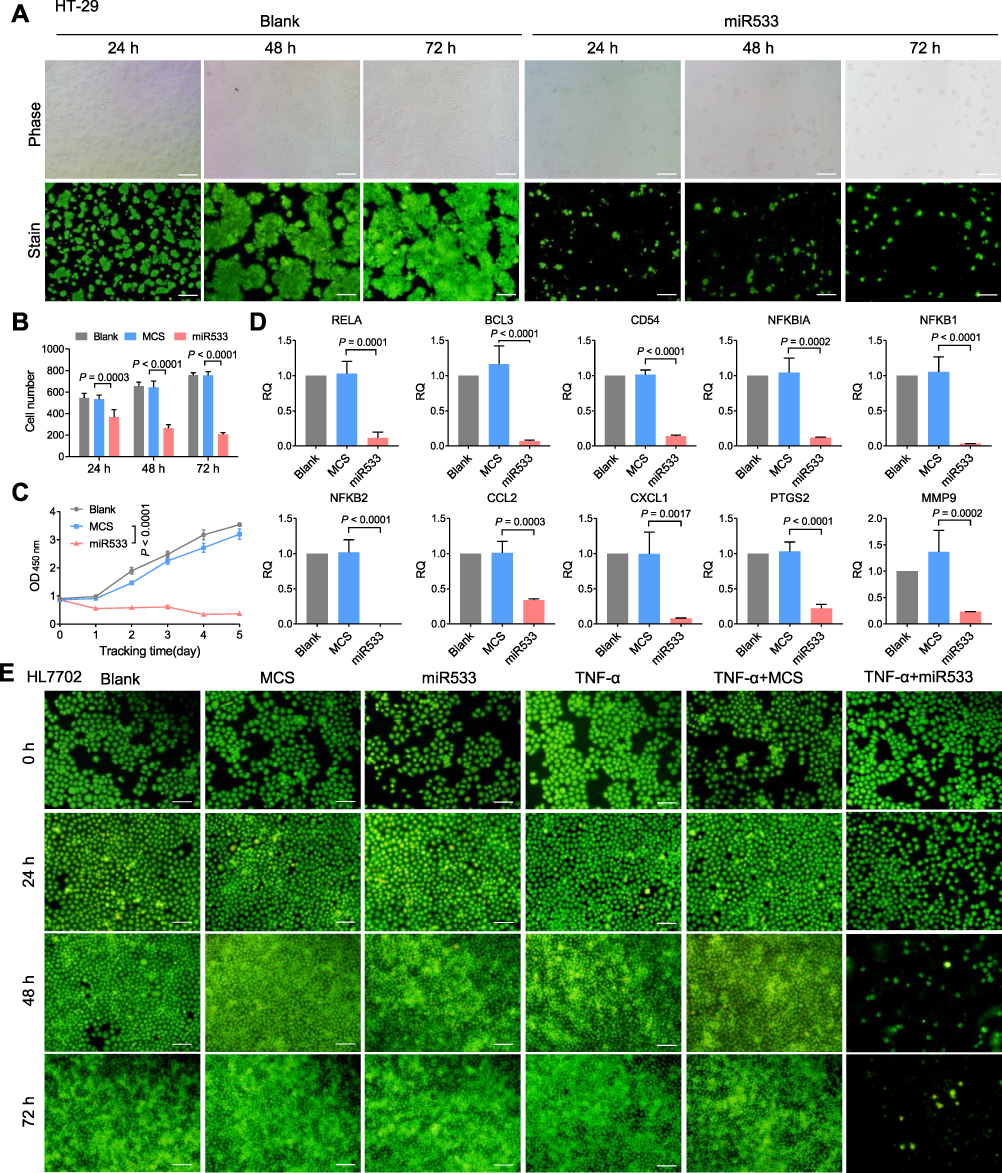 |
Figure 2 Treatment of inflammatory cells with pAAV-DMP-miR533. HT-29 cells were transfected with various plasmids and then cultured for 24, 48, and 72 h, respectively. (A) Representative fluorescent images of acridine Orange (OA)-stained HT-29 cells. Scale bar: 100 μm. (B) Living cell counting at various time points (n = 3 images). Cells were counted from the OA-stained fluorescent images with Image-Pro Plus software. (C) Growth curve of HT-29 cells (n = 3 wells). Cell viability was detected by CCK-8. (D) Relative expression of NF-κB and its target genes in HT-29 cells transfected with various plasmids and cultured for 48 h (n = 3 wells). Gene expression was detected by qPCR. RQ =2–ΔΔCt. RQ, relative quantity. (E) Representative fluorescent images of OA-stained HL7702 cells. Scale bar: 100 μm. HL7702 cells were first stimulated with or without TNF-α and then transfected with various plasmids. The transfected cells were cultured for 24, 48, and 72 h, respectively. Blank, MCS, and miR533: cells transfected with Lipofectamine, pAAV-MCS, and pAAV-DMP-miR533, respectively; TNF-α: TNF-α-stimulated cells (cells stimulated with TNF-α at a final concentration of 10 ng/mL for 1 h); TNF-α+MCS and TNF-α+miR533: TNF-α-stimulated cells transfected with pAAV-MCS and pAAV-DMP-miR533, respectively. |
To further explore whether pAAV-DMP-miR533 has effect on the inflammation, HL7702, a normal human liver cell, was stimulated with a known NF-κB stimulator, TNF-α, to construct a cellular inflammation model. HL7702 cell not stimulated with TNF-α was used as control. The AO-staining of cells revealed that pAAV-DMP-miR533 had no obvious effect on normal HL7702 cell but induced significant death of TNF-α-stimulated HL7702 cell (Figure 2E). These results indicate that pAAV-DMP-miR533 can cause the inflammatory cell death but exerts not obvious effect on non-inflammatory cells.
For the in vivo application, rAAV-MCS, rAAV-CMV-EGFP, rAAV-DMP-miR533 and rAAV-DMP-miR533-CMV-EGFP were then constructed by packaging different DNA fragments into virus (Figure S1). The CMV-EGFP fragment was inserted into rAAV-DMP-miR533 for monitoring the infection of rAAV-DMP-miR533 into cells. A virus rAAV-MCS that contained no inserted gene under the CMV promoter was used as a negative control. The TNF-α-untreated and -treated HL7702 cells were infected by these viruses. The EGFP expression and apoptosis were analyzed by flow cytometry. The results showed that the HL7702 cell infected by rAAV-DMP-miR533-CMV-EGFP had the similar EGFP expression to that infected by rAAV-CMV-EGFP (Figure 3A and B; Figure S2), indicating that the prepared viruses could efficiently infect cells. Moreover, the infection of both rAAV-DMP-miR533 and rAAV-DMP-miR533-CMV-EGFP induced the significant apoptosis in the TNF-α-stimulated HL7702 cell; however, the same infections induced no significant apoptosis in the normal HL7702 cell (Figure 3C and D). The cell viability assay also revealed that only the infections of rAAV-DMP-miR533 and rAAV-DMP-miR533-CMV-EGFP induced significant decrease of cell viability in the TNF-α-induced HL7702 cell (Figure 3E).
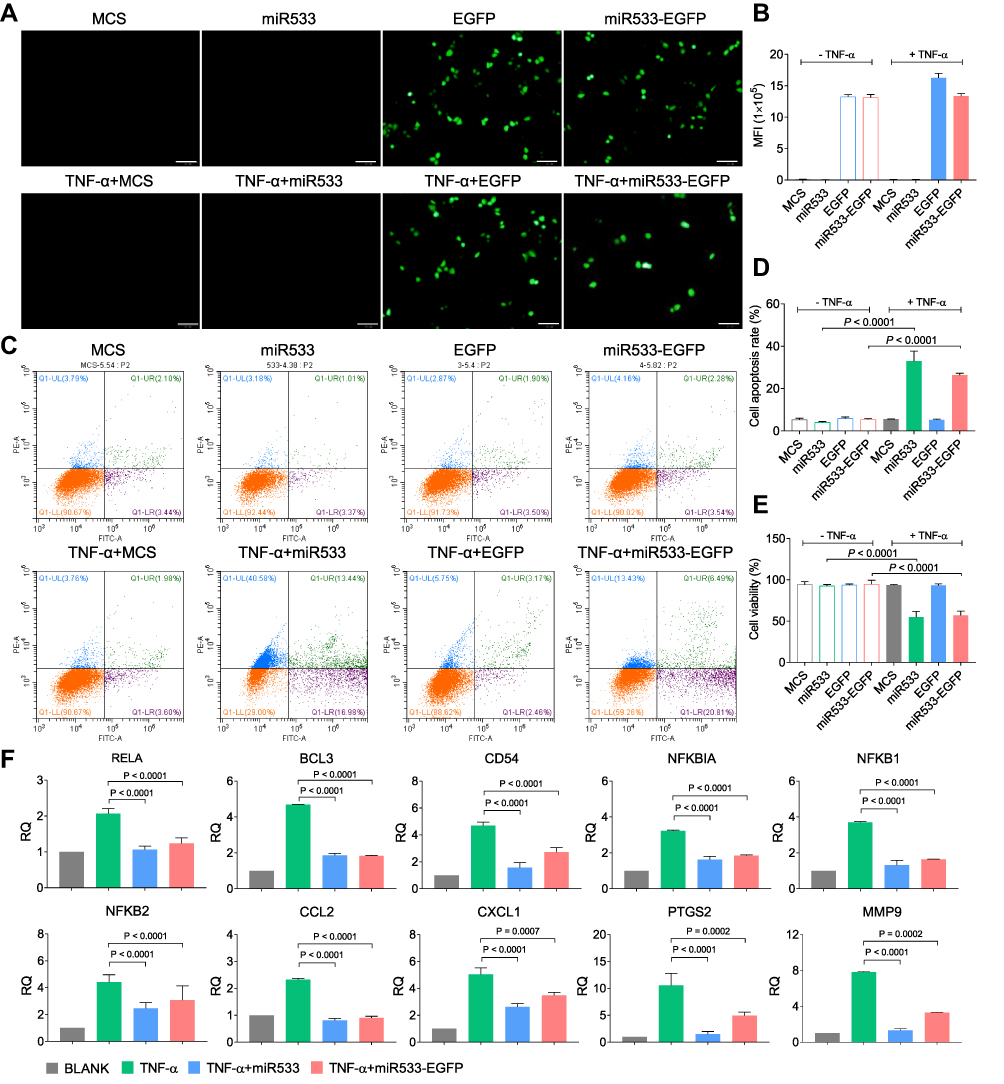 |
Figure 3 Treatment of inflammatory cells with rAAV-DMP-miR533-CMV-EGFP. HL7702 cells were first stimulated with or without TNF-α for 1 h and then infected with various rAAVs for 48 h. (A) Fluorescent images of cells. Scale bar: 100 μm. (B) Cell fluorescence intensity analyzed by Flow Cytometry (n = 3 wells). (C) Representative Flow Cytometry analysis of cell apoptosis. (D) Cell apoptosis analyzed by Flow Cytometry (n = 3 wells). (E) Cell viability detected by CCK-8 (n = 3 wells). (F) QPCR-detected expression of NF-κB and its target genes in cells treated for 48 h (n = 3 wells). Blank, MCS, miR533, EGFP, and miR533-EGFP: cells infected with phosphate buffered saline (PBS), rAAV-MCS, rAAV-DMP-miR533, rAAV-CMV-EGFP, and rAAV-DMP-miR533-CMV-EGFP, respectively; TNF-α: TNF-α-stimulated cells (cells stimulated with TNF-α at a final concentration of 10 ng/mL for 1 h); TNF-α+MCS, TNF-α+miR533, TNF-α+EGFP, TNF-α+miR533-EGFP: TNF-α-stimulated cells infected with rAAV-MCS, rAAV-DMP-miR533, rAAV-CMV-EGFP, and rAAV-DMP-miR533-CMV-EGFP, respectively. |
To further verify the principle of inflammatory cell apoptosis upon rAAV-DMP-miR533 infection, the expression of NF-κB RELA and its target genes in the HL7702 cells were detected with qPCR. The results indicated that the TNF-α stimulation significantly induced the expression of NF-κB RELA and its target genes (Figure 3F). However, the infection of rAAV-DMP-miR533 and rAAV-DMP-miR533-CMV-EGFP significantly reversed their expression (Figure 3F). Altogether, DMP-miR533 could inhibit the expression of NF-κB RELA and lead to cell apoptosis and viability decrease, indicating the in vitro anti-inflammatory effect of rAAV-DMP-miR533.
Treatment of Colitis
To evaluate the in vivo anti-inflammatory effect of DMP-miR533, a colitis mouse model was then constructed by DSS inducement (Figure 4A). By observing the mental state and stools status of the 4 groups of mice at the same time every day, it was found that the mice in the blank group were all normal, the stools were hard and solid, and there was no blood in the stools. However, the DSS-induced mice gradually became sluggish and inactive. On the 3rd day after drinking 3% DSS, the fecal characteristics changed from normal to wet and soft, and the stools showed obvious blood in the 5th day. On the 7th day, the anus remained bloody. These symptoms indicate that DSS-induced acute colitis mice model was successfully established. The mice were then treated with various reagents. After treatment, the DSS-induced mice treated with rAAV-MCS behaved similarly to the DSS-induced mice treated with PBS; their stools became soft and bloody stools appeared. However, the DSS-induced mice treated with rAAV-miR533 showed the change of the stool status and the decreased anus bloody (Figure 4B). The dynamic measurement of body weight revealed that the DSS-induced mice treated with rAAV-MCS and PBS lost body weight but the DSS-induced mice treated with rAAV-DMP-miR533 got body weight after treatment (Figure 4C). The measurement of colon length indicates that the DSS inducement led to the decrease of colon length (Figure 4D and E). Only the treatment of rAAV-DMP-miR533 recovered the length of colon (Figure 4D and E). The H&E-stained colon tissue sections revealed that DSS induced significant inflammatory pathological damages of colon such as disappeared colon crypts in the mucosa, loss of goblet cells, cell degeneration, obvious dense lymphocyte infiltration, and obvious infiltration of neutrophils and plasma cells. However, the treatment of rAAV-DMP-miR533 reversed these damages such as the relatively complete mucosal layer structure, obvious crypt structures, and little infiltration of neutrophils and plasma cells (Figure 4F and G). The measurement of typical pro-inflammatory factors revealed that DSS induced significant increase of TNF-α and IL-6 in serum (Figure 4H). However, the treatment of rAAV-DMP-miR533 significantly decreased levels of the two factors (Figure 4H). In addition, DSS induced the significantly activated expression of NF-κB RELA and its target genes (Figure 4I); however, the treatment of rAAV-DMP-miR533 significantly decreased the expression of these genes (Figure 4I). Similarly, the DSS-induced inflammatory cytokines (TNF-α, IL-6, IL-10, IL-1β, IFN-γ, IL-12A) were inhibited by rAAV-DMP-miR533 (Figure S3A). These favorable anti-inflammatory effects were attributed to the expression of miR533 in the DSS-induced acute colitis mice (Figure S3B). Another independent biological replicate of above animal experiment obtained the similar results (Figure S4). Altogether, these results demonstrated that rAAV-DMP-miR533 had the significant in vivo anti-inflammatory effect in the DSS-induced acute colitis mice.
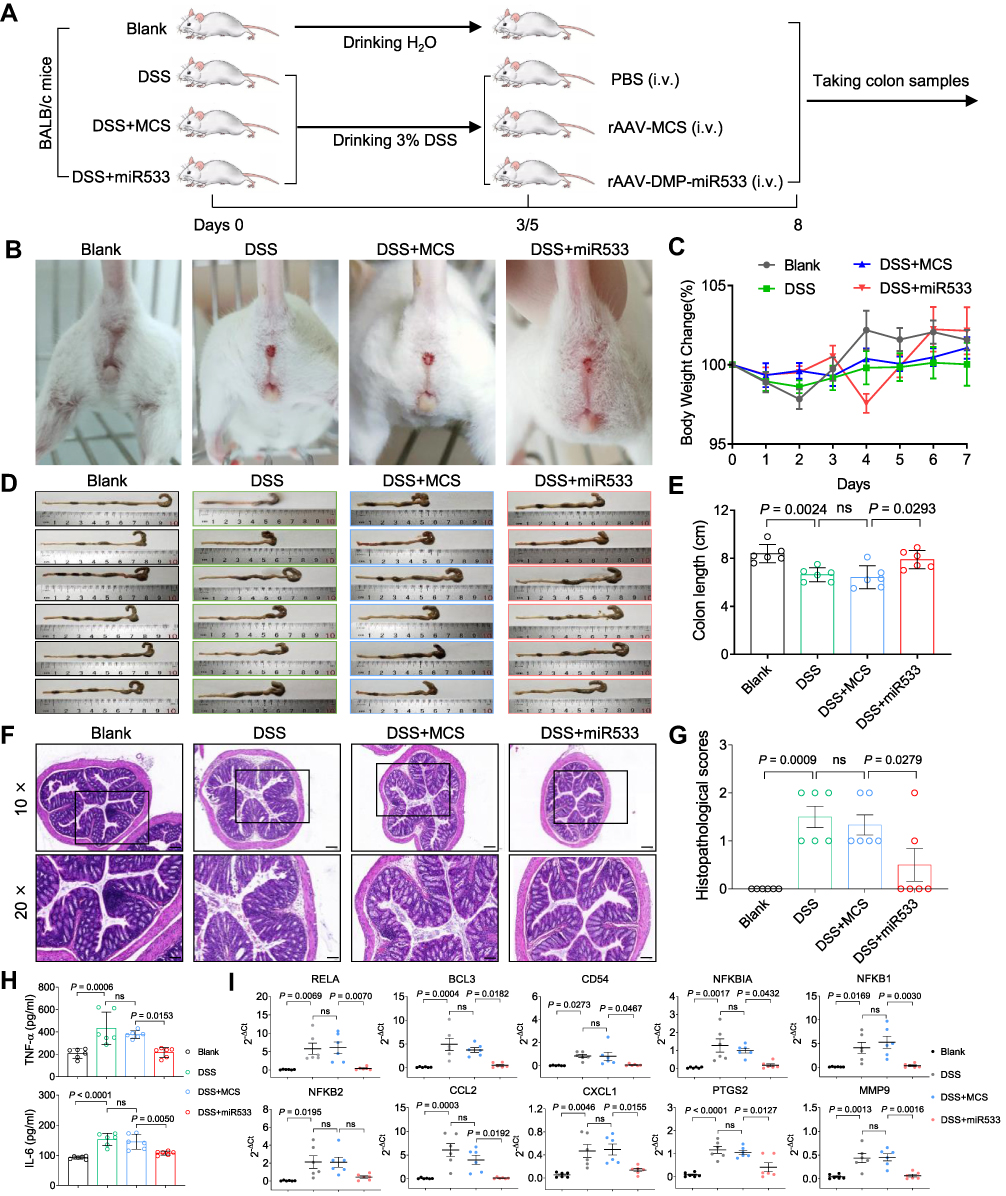 |
Figure 4 Treatment of colitis mice with rAAV-DMP-miR533. The colitis mouse model was established by the dextran sulphate sodium (DSS) inducement and treated with intravenous injection (i.v.) of rAAVs. (A) Schematic diagram of construction of DSS-induced acute colitis mice model and treatment. (B) Bloody traces around mice anus. (C) Body weight of mice. (D) Colons of mice. (E) Colons length of mice (n = 6 mice). (F) Representative H&E-stained sections of colon tissue. The amplified area in black box is showed below. Scale bar: 200 μm (10×) and 100 μm (20×). (G) Histopathological scores of colon tissue (n = 6 mice). (H) TNF-α and IL-6 level in serum detected by ELISA (n = 6 mice). (I) Expression of NF-κB RELA and its target genes in colon tissue detected by qPCR (n = 6 mice). Blank, mice drinking water and treated with PBS; DSS, mice drinking 3% DSS (DSS-induced mice) and treated with phosphate buffered saline (PBS); MCS, DSS-induced mice treated with rAAV-MCS; MiR533, DSS-induced mice treated with rAAV-DMP-miR533. ns, no significance. |
Treatment of Psoriasis
To further confirm the in vivo anti-inflammatory effect of rAAV-DMP-miR533, a second inflammatory mouse model, psoriasis, was established by inducing mice with imiquimod (IMQ) and treated with various reagents (Figure 5A). After six successive days application of IMQ on back skin, the IMQ-induced mice showed red, inflamed, itchy, and thickened skin and silvery scales (Figure 5B). The IMQ-induced mice were then treated with the intravenously injected PBS, rAAV-MCS and rAAV-DMP-miR533. As a result, the rAAV-MCS treatment showed no visible recovery of pathological damages; however, the rAAV-DMP-miR533 treatment obviously recovered the health of IMQ-damaged skins close to the PBS treatment (healthy group) (Figure 5B). The H&E staining of skin tissue sections also revealed that IMQ-induced significant pathological changes of skin such as abscesses, hyper keratosis and infiltration of inflammatory cells; however, only the treatment of rAAV-DMP-miR533 significantly made significant healing of skins (Figure 5C-E; Figure S5A). Detection of pro-inflammatory cytokines in serum indicated that the IMQ induced the increased level of TNF-α and IL-6 (Figure 5F). However, only the treatment of rAAV-DMP-miR533 significantly reversed the increase of two factors in serum (Figure 5F). The detection of gene expression in skins revealed that the expression of TNF-α, IL-6, NF-κB RELA and its target genes was significantly activated by the IMQ inducement (Figures 5G and S5B). However, only the treatment of rAAV-DMP-miR533 significantly reversed the expression of these genes (Figures 5G and S5B). These results indicated that rAAV-DMP-miR533 had a marked curative effect against psoriasis in mouse by intravenous injection.
 |
Figure 5 Treatment of psoriasis mice with rAAV-DMP-miR533. The psoriasis mouse model was established by the Imiquimod (IMQ) inducement and treated by intravenous injection (i.v.) of rAAVs. (A) Schematic diagram of construction of psoriasis mouse model and treatment. (B) Imaging of back skins of mice. (C) Representative H&E-stained sections of skin tissue. Scale bar: 200 μm (10×) and 100 μm (20×). (D) Histopathological scores of skin tissue induced with IMQ for 6 days (n = 3 mice). (E) Histopathological scores of skin tissue induced with IMQ for 12 days (Blank and MCS, n = 3 mice; miR533, n = 6 mice). (F) TNF-α and IL-6 level in serum detected by ELISA (Blank and MCS, n = 3 mice; miR533, n = 6 mice). (G) TNF-α and IL-6 mRNA level in skin tissue detected by qPCR (Blank and MCS, n = 3 mice; miR533, n = 6 mice). Blank, Vaseline-induced mice treated with phosphate buffered saline (PBS); MCS, IMQ-induced mice treated with rAAV-MCS; miR533, IMQ-induced mice treated with rAAV-DMP-miR533. ns, no significance. |
To find other drug administration ways, one IMQ-induced mouse was also tried to be treated by subcutaneous injection and skin application, respectively. As a result, both administrations obtained similar therapeutical effects to intravenous injection (Figure S6), including skin recovery (Figure S6A and S6B), serum pro-inflammatory factors (Figure S6C), and expression of NF-κB RELA and its target genes (Figure S6D). In view of better therapeutical effect and convenience of drug administration, an amplified treatment of skin application was performed that included more mice (n=6) (Figure 6A). The psoriasis mice were treated with skin administration of Vaseline and rAAV-DMP-miR533 mixed in Vaseline for 6 days (Figure 6A). As a result, the fine therapeutical effects were obtained with rAAV-DMP-miR533, including the recovered skin appearance (Figure 6B), low Psoriasis Area and Severity Index (PASI) (Figure 6C), healed skin tissue structure (Figure 6D and 6E; Figure S7A), improved splenomegaly (Figure 6F and G), increased body weight (Figure 6H), lowered serum TNF-α and IL-6 level (Figure 6I), and down-regulated expression of TNF-α and IL-6 (Figure S7B) and NF-κB RELA and its target genes (Figure S7C) in the skin tissues. Similarly, the IMQ-induced other inflammatory cytokines (IL-10, IL-1β, IFN-γ, IL-12A) were inhibited by rAAV-DMP-miR533 (Figure S8A). The favorable therapeutic effects were attributed to the expression of miR533 in IMQ-induced psoriasis mice (Figure S8B). Altogether, these results demonstrate that rAAV-DMP-miR533 had the significant in vivo anti-inflammatory effect in the IMQ-induced psoriasis mice.
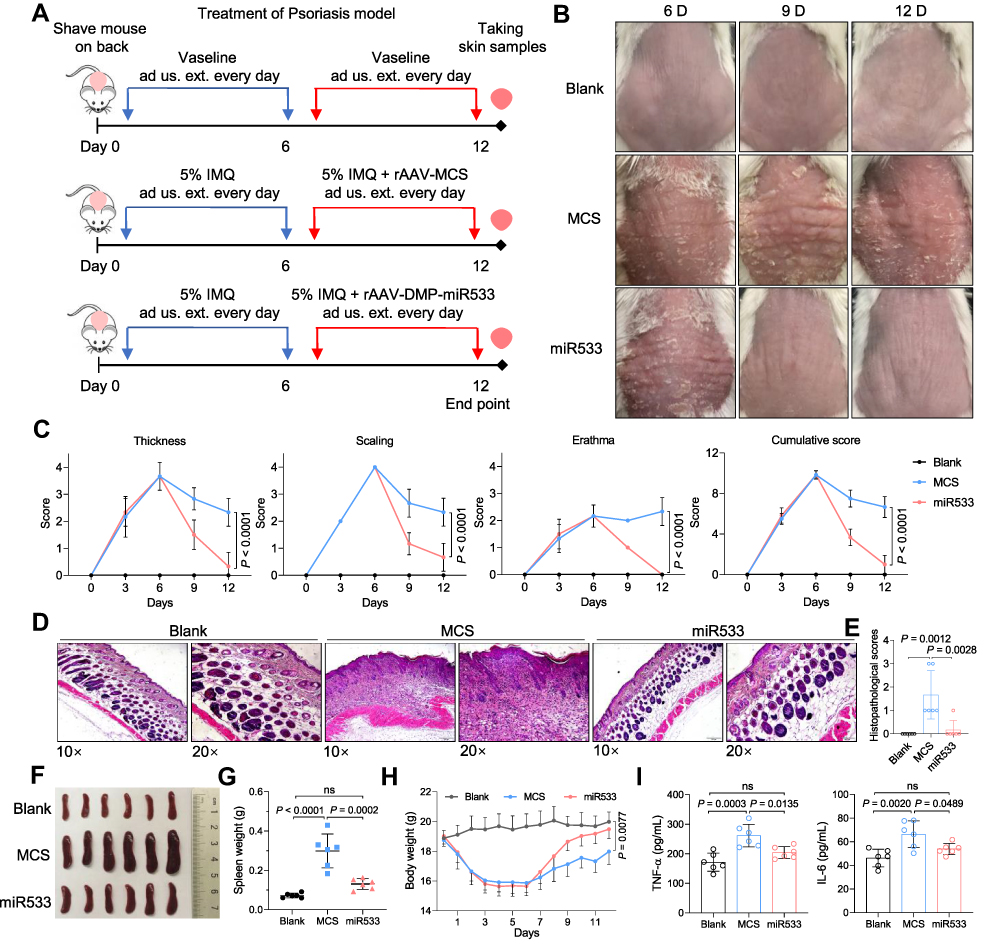 |
Figure 6 Treatment of psoriasis mice with rAAV-DMP-miR533. The psoriasis mice model was established by the Imiquimod (IMQ) inducement and treated by administration usum externum (ad us. ext.) (for external use) of rAAVs. (A) Schematic diagram of construction of psoriasis mouse model and treatment. (B) Imaging of back skins of mice. (C) Psoriasis area and severity index (PASI) score (n = 6 mice). (D) Representative H&E-stained sections of skin tissue. Scale bar: 200 μm (10×) and 100 μm (20×). (E) Histopathological scores of skin tissue (n = 6 mice). (F) Spleen photographs. (G) Spleen weight (n = 6 mice). (H) Average body weight (n = 6 mice). (I) TNF-α and IL-6 level in serum by detected by ELISA (n = 6 mice). Blank, Vaseline-induced mice treated with phosphate buffered saline (PBS); MCS, IMQ-induced mice treated with rAAV-MCS; miR533, IMQ-induced mice treated with rAAV-DMP-miR533. ns, no significance. |
Treatment of Arthritis
In the above cell and mouse experiments, pAAV-MCS and rAAV-MCS were used as negative control to pAAV-DMP-miR533 and rAAV-DMP-miR533, respectively. To provide a more appropriate negative control to DMP-miR533, a new vector, DMP-NT, was constructed that codes a miR targeting no target (NT) in human and mouse genomes. To evaluate this vector, the mouse colon cancer cell CT26 was transfected with pAAV-DMP-miR533 and pAAV-DMP-NT. The results revealed that pAAV-DMP-miR533 induced significant apoptosis and growth inhibition in CT26 (Figure S9A-C); however, pAAV-DMP-NT had no significant effect on the apoptosis and growth of the cell (Figure S9A-C). To further evaluate the two vectors, a normal mouse embryonic fibroblast cell, NIH-3T3, was transfected by two vectors. The results revealed that two vectors induced no significant apoptosis and growth inhibition in the cell (Figure S10A-C). However, when the cell was stimulated by TNF-α, pAAV-DMP-miR533 induced the significant apoptosis and growth inhibition in the cell (Figure S10A-C). But pAAV-DMP-NT still exerted no significant effect on the cell even it was stimulated by TNF-α (Figure S10A-C). The qPCR detection also revealed that pAAV-DMP-miR533 significantly knocked down the expression of NF-κB RELA and its target genes in CT26 and TNF-α-induced NIH-3T3 (Figure S11A and S11B); however, pAAV-DMP-NT showed no effect on gene expression in the two cells (Figure S11A and S11B). Altogether, these results indicate the in vitro anti-inflammatory effect of DMP-miR533 by inhibiting NF-κB activity. In this case, rAAV-DMP-NT were prepared by packaging pAAV-DMP-NT into AAV for further evaluating the in vivo anti-inflammatory effect of rAAV-DMP-miR533 by using rAAV-DMP-NT as a counterpart negative control. The CT26 and NIH-3T3 cells were further treated with rAAV-DMP-miR533 and the newly packaged rAAV-DMP-NT. The expression of miR533 and inflammatory cytokines were evaluated by qPCR detection. As results, the expression of miR533 was time-dependently increased (Figure S12) and that of six inflammatory cytokines (TNF-α, IL-6, IL-10, IL-1β, IFN-γ, IL-12A) were inhibited by rAAV-DMP-miR533 but not rAAV-DMP-NT in CT26 and TNF-α-induced NIH-3T3 cells (Figure S13).
Rheumatoid arthritis (RA) is an insidious autoimmune inflammatory disease of the joints.63 A collagen-induced arthritis (CIA) mouse model was established by a double immunization and used for the treatment of RA (Figure 7A). On the 28th day after primary immunization, the CIA mice were randomly divided into 4 groups (n = 6) and intravenously injected three times with PBS, Methotrexate solution (MTX),64 rAAV-DMP-NT, and rAAV-DMP-miR533, respectively. The healthy mice just intravenously injected with PBS were used as healthy control (n = 6). On the 46th day, all mice were euthanized. Imaging of paw (including ankle joint) revealed that rAAV-DMP-miR533 obtained better therapeutic effect than MTX (Figures 7B and S14). The rAAV-DMP-miR533 treatment recovered the normal volume and weight of spleen, whereas the MTX treatment made more serious splenomegaly than CIA (Figure 7C and D). The rAAV-DMP-miR533 treatment kept the mouse body weight, whereas the MTX treatment made mice losing their body weight (Figure 7E). The dynamic measurement of pathological changes of mice revealed that both rAAV-DMP-miR533 and MTX treatments significantly improved the clinical score (Figure 7F), paw thickness (Figure 7G), and ankle width (Figure 7H). In addition, only the rAAV-DMP-miR533 treatment made the tail thickness close to the healthy mice (Figure 7I). The CIA modeling significantly increased the TNF-α and IL-6 levels in serum; however, the rAAV-DMP-miR533 and MTX treatments significantly lowered the levels of the two factors in serum (Figure 7J). Importantly, rAAV-DMP-miR533 obtained better therapeutical effect than MTX (Figure 7J). These therapeutical effects were also confirmed by the mRNA expression levels of some inflammatory cytokines (TNF-α, IL-6, IL-10, IL-1β, IFN-γ, IL-12A) in paw tissues (Figure 7K and Figure S15A). Furthermore, the rAAV-DMP-miR533 treatment significantly reversed the upregulation of expression of NF-κB RELA and its target genes in paws (Figure S15B) due to the expression of miR533 (Figure S15C). However, the MTX treatment showed no effect on the activated expression of these genes (Figure S15B), indicating inflammatory cells still exist. More convincingly, the Micro-CT imaging of rear paws of mice revealed that the CIA modeling resulted in serious bone erosion in ankle and finger joints (Figure 7L). However, the rAAV-DMP-miR533 treatment greatly improved the bone erosion, which was obviously better than the MTX treatment (Figure 7L). This therapeutical effect was also supported by the results of the H&E staining and histopathological evaluation of joints (Figure 7M and N; Figure S16). The CIA modeling induced extensive pannus formation, severe bone destruction, extensive cartilage damage, inflammatory cell infiltration. However, both the rAAV-DMP-miR533 and MTX treatment significantly improved these pathological changes. In comparison, the rAAV-DMP-miR533 treatment obtained better therapeutical effects than the MTX treatment. Notably, the rAAV-DMP-NT treatment showed no therapeutic effect in above all aspects (Figure 7, Figure S15 and S16). Importantly, the detection of the major organs (heart, liver, spleen, lung, and kidney) by H&E staining of tissue sections indicated that the rAAV-DMP-miR533 treatment significantly recover the tissue damages induced by CIA modeling, especially lung tissue (Figure S17A). Comparatively, the MTX treatment did not significantly recovered lung tissue damage like the rAAV-DMP-miR533 treatment (Figure S17A). The MTX treatment also showed significant damage to liver and some necrotic areas in spleen (Figure S17A). The detection of biochemical indices in serums that were collected on the 46th day revealed that the rAAV-DMP-miR533 treatment showed no effects on these biochemical indices, indicating its in vivo biosafety (Figure S17B and S17C). However, the MTX treatment showed high ALT, AST and ALP, indicating hepatotoxicity (Figure S17B). Altogether, these results demonstrate the significant in vivo anti-inflammatory effect of rAAV-DMP-miR533 in the collagen-induced arthritis mice.
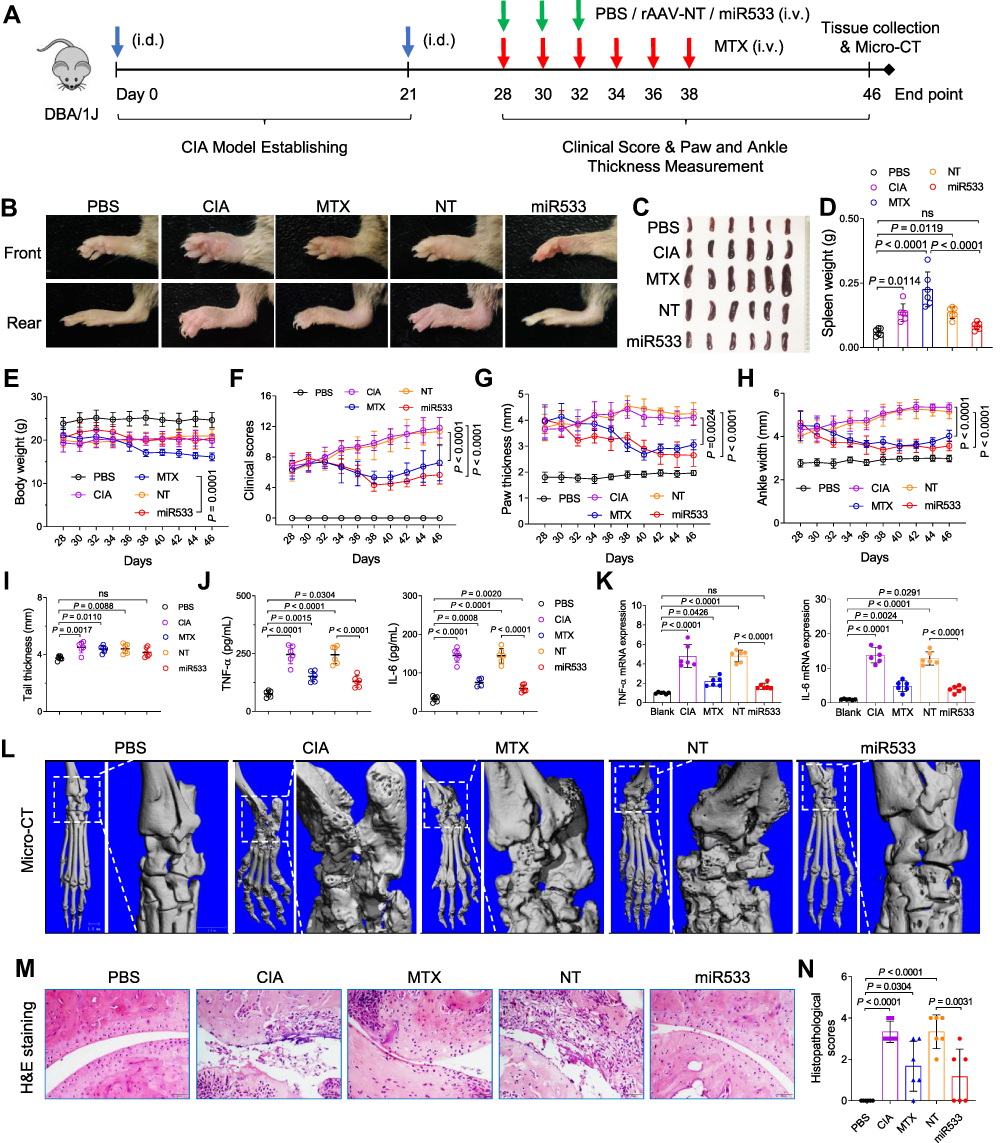 |
Figure 7 Treatment of the arthritis mice with rAAV-DMP-miR533. The collagen-induced arthritis (CIA) mouse model was established by the collagen inducement and treated by intravenous injection (i.v.) of rAAVs. (A) Schematic diagram of construction of CIA mice model and treatment. PBS, a healthy group treated with phosphate buffered saline (PBS). (B) Representative photos of forepaws and bearpaws in different groups. (C) Spleen photographs. (D) Spleen weight (n = 6 mice). (E) Average body weight (n = 6 mice). (F) Clinical score of arthritis severity (n = 6 mice). (G) Paw thickness (n = 6 mice). (H) Ankle width (n = 6 mice). (I) Tail thickness (n = 6 mice). (J) TNF-α and IL-6 level in serum detected by ELISA (n = 6 mice). (K) TNF-α and IL-6 mRNA level in rear paw tissue detected by qPCR (n = 6 mice). (L) Micro-CT imaging of rear paw with ankle joints. The outlined areas are shown at a high resolution. (M) Representative H&E-stained sections of ankle joints. Scale bar: 50 μm. (N) Histopathological scores of ankle joints (n = 6 mice). PBS, normal mice treated with PBS; CIA, CIA mice treated with PBS; MTX, CIA mice treated with Methotrexate (MTX); NT, CIA mice treated with rAAV-NT; miR533, CIA mice treated with rAAV-DMP-miR533. ns, no significance. NT, no target. |
Discussion
In this study, we packaged a DNA fragment, DMP-miR533 that consist of a NF-κB-decoy-minimal prompter and an artificial NF-κB RELA-targeting microRNA, into safe gene delivery vector AAV to fabricate a rAAV named as rAAV-DMP-miR533. We investigated its anti-inflammatory effect at both cell and living body levels. The results indicate that this rAAV showed significant anti-inflammatory effect in vitro and in vivo. Especially, this rAAV showed excellent anti-inflammatory effect in three typical inflammation mouse models, including the dextran sulphate sodium (DSS)-induced mouse of an acute colitis model, imiquimod (IMQ)-mouse of psoriasis model, and collagen-induced mouse of arthritis model. In addition, rAAV-DMP-miR533 showed fine biosafety in these in vivo treatments.
In mechanism, we found that rAAV-DMP-miR533 can make the inflammatory cells apoptosis, which eradicates the inflammatory cells. The inflammatory cells can secrete the pro-inflammatory cytokines and thus intensify and deteriorate the inflammation process. By eradicating the inflammatory cells, the source of inflammation can be removed. This mechanism is different from the current anti-inflammation strategies that depend on antibodies or antagonists of cytokines and their receptors. Most cytokines are direct target genes of NF-κB,41 inhibiting cytokines with their antibodies just neutralizes the transiently produced cytokines, but not eradicates the inflammatory cells producing them. Cytokines can still be persistently produced by the inflammatory cells. This is the reason why inflammatory diseases are easily recurrent with the treatment of the antibodies of cytokines. Additionally, the pleiotropy and redundancy in the actions of many cytokines and their receptors9,65,66 can limit the efficacy of single neutralizing agents. This is the reason why inflammatory diseases can resist to a current treatment. Moreover, the cytokine as key player or pivotal regulator to a certain inflammatory disease may be different in individuals, this is the reason why most the current treatments have low response rate in patients. In contrast, rAAV-DMP-miR533 knocks down all inflammation-related cytokines by eradicating the inflammatory cells producing them. For example, rAAV-DMP-miR533 knocked down two main proinflammation factors TNF-α and IL-6 at both mRNA and protein levels in all three inflammation mice models. Therefore, rAAV-DMP-miR533 provides a new strategy and tool with a more universal anti-inflammation mechanism, which may overcome some key challenges of the current anti-inflammation treatments, such as low response, resistance, recurrence, and side effects.
An advantage of rAAV-DMP-miR533 over the traditional NF-κB inhibitors of small molecules, decoy and siRNA is that it can avoid over inhibition of NF-κB activity in normal cells. This study revealed that rAAV-DMP-miR533 induced significant apoptosis of cancer cells that have NF-κB over activity but had little effect on normal cells. However, when the normal cells were induced with a NF-κB inducer, TNF-α, they became inflammatory cells and were induced to apoptosis by rAAV-DMP-miR533. Therefore, rAAV-DMP-miR533 overcomes the side effect of traditional NF-κB inhibitors and thus has translational potential.
AAV is a safe gene delivery tool and approved to the human clinical gene therapy,67–70 because of its some advantages such as low immunogenicity, no pathogenicity, no genomic insertion, and long-term stable expression.59 At the used dosage, this study showed that rAAV-DMP-miR533 showed no significant toxicity to mice of three inflammatory diseases. Especially, at the highest dosage of three systematic administrations to arthritis mice, rAAV-DMP-miR533 showed no detectable effect on biochemical indices and spleen. In contrast, the widely used anti-arthritis drug, MTX,64,71 showed high toxicities to liver and spleen. Moreover, we for the first time find that rAAV (rAAV-DMP-miR533) could be administered via external use by mixing into Vaseline to treat psoriasis. This is helpful for the convenient treatment of local skin inflammations. Therefore, rAAV-DMP-miR533 provides a new safe anti-inflammation reagent with multiple routes of administration. Anyway, the potential clinical application of rAAV-DMP-miR533 is still challenged by the pre-existing or neutralizing antibodies to AAV vectors blocking administration and readministration of AAV, a current common blockage of AAV-based therapies.72,73 However, the hurdle can be overcome by some new approaches such as eliminating pre-existing anti-AAV antibodies with endopeptidase (eg Imlifidase (IdeS),9 or IgG-degrading enzyme (IdeZ),74 transiently repressing endogenous Myd88 with CRISPR,75 and using engineered AAV with TLR9-inhibitory sequences.76 In addition, the non-viral vectors such as lipid nanoparticles (LNPs)77 can also be used to deliver DMP-miR533.
In this study, we develop an AAV-based gene therapy to inflammation diseases. At present, the AAV-based gene therapies are mainly used to treat human genetic diseases.78 Anyway, several AAV-based gene therapy is now used in clinical trial to treat inflammation diseases, especially autoimmune diseases such as rheumatoid arthritis by expressing IFN-β or TNFR-IgG1 Fc fusion protein.78 However, these treatments still target one inflammatory-related cytokine. In contrast, rAAV-DMP-miR533 directly targets NF-κB itself, an established hub regulator of inflammation. The treatment of three typical inflammation diseases indicates that rAAV-DMP-miR533 provides a more universal AAV-based gene therapy to variant inflammatory diseases.
Conclusions
In this study, we fabricated a rAAV, rAAV-DMP-miR533, by packaging a DNA molecule DMP-miR533 into AAV. We evaluated the anti-inflammatory effect of the virus with inflammatory cells and the mice of three typical inflammatory diseases including colitis, psoriasis, and arthritis. We found that rAAV-DMP-miR533 had both excellent anti-inflammatory effect and biosafety in these inflammatory diseases. This study thus provides a promising universal gene therapy to variant inflammatory diseases by directly targeting NF-κB. Soon, this therapy should be preferentially used to treat psoriasis via skin application in the clinical test. With the safety on patients in this case, the similar approach should be applied to treat other skin inflammatory diseases such as atopic dermatitis. The arthritis can be tried to treat by the intra-articular injection. With the support of increasing safety and efficacy data, psoriasis and arthritis can be tried to treat by systemic injection. The treatment by systemic injection is very important to chronic inflammatory diseases for lowering serum levels of pro-inflammatory cytokines such as TNF-α and IL-6 detected in this study, because the pro-inflammatory cytokines secreted by inflammatory focus lead to systemic damages.
Acknowledgments
We are acknowledged to Prof. Xiaohui Zhang of Nanjing Agricultural University for his blindly scoring of inflammation level of all H&E-stained tissue sections. We are also acknowledged to Prof. Shouhua Luo of Southeast University and Prof. Zhenhua Feng of Gulou Hospital for imaging mouse paws (including ankle joints) with micro CT.
Funding
This work was mainly supported by the National Natural Science Foundation of China (61971122). This research was also partially supported by Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX21-0145).
Disclosure
The authors declare no competing financial and non-financial interests.
References
1. Ye Q, Wang BL, Mao JH. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J Infect. 2020;80(6):607–613. doi:10.1016/j.jinf.2020.03.037
2. Multhoff G, Molls M, Radons J. Chronic inflammation in cancer development. Front Immunol. 2011;2:98. doi:10.3389/fimmu.2011.00098
3. Wu Y, Antony S, Meitzler JL, Doroshow JH. Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett. 2014;345(2):164–173. doi:10.1016/j.canlet.2013.08.014
4. Wang S, Liu Z, Wang L, Zhang X. NF-κB signaling pathway, inflammation and colorectal cancer. Cell Mol Immunol. 2009;6(5):327–334. doi:10.1038/cmi.2009.43
5. Netea MG, Balkwill F, Chonchol M, et al. A guiding map for inflammation. Nat Immunol. 2017;18(8):826–831. doi:10.1038/ni.3790
6. Franceschi C, Campisi J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J Gerontol a-Biol. 2014;69:S4–S9. doi:10.1093/gerona/glu057
7. Sanada F, Taniyama Y, Muratsu J, et al. Source of Chronic Inflammation in Aging. Front Cardiovasc Med. 2018;5:12. doi:10.3389/fcvm.2018.00012
8. Doria A, Zen M, Bettio S, et al. Autoinflammation and autoimmunity: bridging the divide. Autoimmun Rev. 2012;12(1):22–30. doi:10.1016/j.autrev.2012.07.018
9. Auphan N, Didonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-κB activity through induction of IκB synthesis. Science. 1995;270(5234):286–290.
10. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. doi:10.1038/sigtrans.2017.23
11. Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46(5):705–716. doi:10.1016/0092-8674(86
12. Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: current and Future Prospects. Drugs. 2017;77(5):521–546. doi:10.1007/s40265-017-0701-9
13. Salas A, Hernandez-Rocha C, Duijvestein M, et al. JAK-STAT pathway targeting for the treatment of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17(6):323–337. doi:10.1038/s41575-020-0273-0
14. Malemud CJ, Pearlman E. Targeting JAK/STAT Signaling Pathway in Inflammatory Diseases. Curr Signal Transd T. 2009;4(3):201–221. doi:10.2174/157436209789057467
15. Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018;27(12):1984–2009. doi:10.1002/pro.3519
16. Williams DM. Clinical Pharmacology of Corticosteroids. Respir Care. 2018;63(6):655–670. doi:10.4187/respcare.06314
17. Stone S, Malanga GA, Capella T. Corticosteroids: review of the History, the Effectiveness, and Adverse Effects in the Treatment of Joint Pain. Pain Physician. 2021;24(S1):S233–S246.
18. Dinarello CA. Anti-inflammatory Agents: present and Future. Cell. 2010;140(6):935–950. doi:10.1016/j.cell.2010.02.043
19. Lai YP, Dong C. Therapeutic antibodies that target inflammatory cytokines in autoimmune diseases. Int Immunol. 2016;28(4):181–188. doi:10.1093/intimm/dxv063
20. Luchetti MM, Balloni A, Gabrielli A. Biologic Therapy in Inflammatory and Immunomediated Arthritis: safety Profile. Curr Drug Saf. 2016;11(1):22–34. doi:10.2174/1574886310666151014115401
21. Scheinecker C, Redlich K, Smolen JS. Cytokines as therapeutic targets: advances and limitations. Immunity. 2008;28(4):440–444. doi:10.1016/j.immuni.2008.03.005
22. Rider P, Carmi Y, Cohen I. Biologics for Targeting Inflammatory Cytokines, Clinical Uses, and Limitations. Int J Cell Biol. 2016;2016:9259646. doi:10.1155/2016/9259646
23. Zarrin AA, Bao K, Lupardus P, Vucic D. Kinase inhibition in autoimmunity and inflammation. Nat Rev Drug Discov. 2021;20(1):39–63. doi:10.1038/s41573-020-0082-8
24. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-κB: a Blossoming of Relevance to Human Pathobiology. Cell. 2017;168(1–2):37–57. doi:10.1016/j.cell.2016.12.012
25. Scheinfeld N. Adalimumab (HUMIRA): a review. J Drugs Dermatol. 2003;2(4):375–377.
26. O’Shea JJ, Gadina M. Selective Janus kinase inhibitors come of age. Nat Rev Rheumatol. 2019;15(2):74–75. doi:10.1038/s41584-018-0155-9
27. Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;16(12):843–862. doi:10.1038/nrd.2017.201
28. Muller R. JAK inhibitors in 2019, synthetic review in 10 points. Eur J Intern Med. 2019;66:9–17. doi:10.1016/j.ejim.2019.05.022
29. Berkhout LC, I’Ami MJ, Ruwaard J, et al. Dynamics of circulating TNF during Adalimumab treatment using a drug-tolerant TNF assay. Sci Transl Med. 2019;11(477):eaat3356. doi:10.1126/scitranslmed.aat3356
30. Atreya R, Neurath MF. Mechanisms of molecular resistance and predictors of response to biological therapy in inflammatory bowel disease. Lancet Gastroenterol Hepatol. 2018;3(11):790–802. doi:10.1016/S2468-1253(18
31. Ji CC, Takano S. Clinical efficacy of Adalimumab versus infliximab and the factors associated with recurrence or aggravation during treatment of anal fistulas in Crohn’s disease. Intest Res. 2017;15(2):182–186. doi:10.5217/ir.2017.15.2.182
32. Papp K, Crowley J, Ortonne JP, et al. Adalimumab for moderate to severe chronic plaque psoriasis: efficacy and safety of retreatment and disease recurrence following withdrawal from therapy. Br J Dermatol. 2011;164(2):434–441. doi:10.1111/j.1365-2133.2010.10139.x
33. Wongrakpanich S, Wongrakpanich A, Melhado K, Rangaswami J, Comprehensive A. Review of Non-Steroidal Anti-Inflammatory Drug Use in The Elderly. Aging Dis. 2018;9(1):143–150. doi:10.14336/Ad.2017.0306
34. Scheinfeld N. Adalimumab: a review of side effects. Expert Opin Drug Saf. 2005;4(4):637–641. doi:10.1517/14740338.4.4.637
35. Scheinfeld N. A comprehensive review and evaluation of the side effects of the tumor necrosis factor alpha blockers etanercept, infliximab and Adalimumab. J Dermatolog Treat. 2004;15(5):280–294. doi:10.1080/09546630410017275
36. Vonkeman HE, van de Laar MAFJ. Nonsteroidal Anti-Inflammatory Drugs: adverse Effects and Their Prevention. Semin Arthritis Rheum. 2010;39(4):294–312. doi:10.1016/j.semarthrit.2008.08.001
37. Bindu S, Mazumder S, Bandyopadhyay U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: a current perspective. Biochem Pharmacol. 2020;180:114147. doi:10.1016/j.bcp.2020.114147
38. Ahn KS, Aggarwal BB. Transcription factor NF-κB: a sensor for smoke and stress signals. Ann N Y Acad Sci. 2005;1056:218–233. doi:10.1196/annals.1352.026
39. Wan F, Lenardo MJ. The nuclear signaling of NF-κB: current knowledge, new insights, and future perspectives. Cell Res. 2010;20(1):24–33. doi:10.1038/cr.2009.137
40. Manning AM, Anderson DC. Chapter 24. Transcription Factor NF-κB: an Emerging Regulator of Inflammation. Annu Rep Med Chem. 1994;29(08):235–244.
41. Pahl HL. Activators and target genes of Rel/NF-κB transcription factors. Oncogene. 1999;18(49):6853–6866. doi:10.1038/sj.onc.1203239
42. Paolo T, Alberto T, Elena B, et al. Charting the NF-κB Pathway Interactome Map. PLoS One. 2012;7(3):e32678. doi:10.1371/journal.pone.0032678
43. DiDonato JA, Mercurio F, Karin M. NF-κB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400. doi:10.1111/j.1600-065X.2012.01099.x
44. Tak PP, Firestein GS. NF-κB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7–11. doi:10.1172/JCI11830
45. Lewis AJ, Manning AM. New targets for anti-inflammatory drugs. Curr Opin Chem Biol. 1999;3(4):489–494. doi:10.1016/S1367-5931(99
46. D’Acquisto F, May MJ, Ghosh S. Inhibition of nuclear factor κB (NF-κB): an emerging theme in anti-inflammatory therapies. Mol Interv. 2002;2(1):22–35. doi:10.1124/mi.2.1.22
47. Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 2010;1799(10–12):775–787. doi:10.1016/j.bbagrm.2010.05.004
48. Gilmore TD, Herscovitch M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene. 2006;25(51):6887–6899. doi:10.1038/sj.onc.1209982
49. Ramadass V, Vaiyapuri T, Tergaonkar V. Small Molecule NF-κB Pathway Inhibitors in Clinic. Int J Mol Sci. 2020;21(14):5164. doi:10.3390/ijms21145164
50. Finotti A, Borgatti M, Bezzerri V, et al. Effects of decoy molecules targeting NF-κB transcription factors in Cystic fibrosis IB3-1 cells: recruitment of NF-κB to the IL-8 gene promoter and transcription of the IL-8 gene. Artif DNA PNA XNA. 2012;3(2):97–296. doi:10.4161/adna.21061
51. Tomita T, Takeuchi E, Tomita N, et al. Suppressed severity of collagen-induced arthritis by in vivo transfection of nuclear factor κB decoy oligodeoxynucleotides as a gene therapy. Arthritis Rheum. 1999;42(12):2532–2542. doi:10.1002/1529-0131(199912)42:12<2532::Aid-Anr5>3.0.Co;2-2
52. Bezzerri V, Borgatti M, Nicolis E, et al. Transcription factor oligodeoxynucleotides to NF-κB inhibit transcription of IL-8 in bronchial cells. Am J Respir Cell Mol Biol. 2008;39(1):86–96. doi:10.1165/rcmb.2007-0176OC
53. Farahmand L, Darvishi B, Majidzadeh AK. Suppression of chronic inflammation with engineered nanomaterials delivering nuclear factor κB transcription factor decoy oligodeoxynucleotides. Drug Deliv. 2017;24(1):1249–1261. doi:10.1080/10717544.2017.1370511
54. Li N, Song Y, Zhao W, et al. Small interfering RNA targeting NF-κB attenuates lipopolysaccharide-induced acute lung injury in rats. BMC Physiol. 2016;16(1):7. doi:10.1186/s12899-016-0027-y
55. Yan H, Duan X, Pan H, et al. Suppression of NF-κB activity via nanoparticle-based siRNA delivery alters early cartilage responses to injury. Proc Natl Acad Sci U S A. 2016;113(41):E6199–E6208. doi:10.1073/pnas.1608245113
56. Wu C, Zhao J, Zhu G, Huang Y, Jin L. SiRNA directed against NF-κB inhibits mononuclear macrophage cells releasing proinflammatory cytokines in vitro. Mol Med Rep. 2017;16(6):9060–9066. doi:10.3892/mmr.2017.7715
57. Lianxu C, Hongti J, Changlong Y. NF-κBp65-specific siRNA inhibits expression of genes of COX-2, NOS-2 and MMP-9 in rat IL-1beta-induced and TNF-alpha-induced chondrocytes. Osteoarthritis Cartilage. 2006;14(4):367–376. doi:10.1016/j.joca.2005.10.009
58. Wang D, Tang H, Xu X, Dai W, Wu J, Wang J. Control the intracellular NF-κB activity by a sensor consisting of miRNA and decoy. Int J Biochem Cell Biol. 2018;95:43–52.doi:10.1016/j.biocel.2017.12.009
59. Wang D, Tai PWL, Gao GP. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18(5):358–378. doi:10.1038/s41573-019-0012-9
60. Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18(5):309–324. doi:10.1038/nri.2017.142
61. Capece D, Verzella D, Tessitore A, Alesse E, Capalbo C, Zazzeroni F. Cancer secretome and inflammation: the bright and the dark sides of NF-κB. Semin Cell Dev Biol. 2018;78:51–61. doi:10.1016/j.semcdb.2017.08.004
62. Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. doi:10.1186/1476-4598-12-86
63. Chen JQ, Qi J, Chen C, et al. Tocilizumab-Conjugated Polymer Nanoparticles for NIR-II Photoacoustic-Imaging-Guided Therapy of Rheumatoid Arthritis. Adv Mater. 2020;32(37):e2003399. doi:10.1002/adma.202003399
64. Cutolo M, Sulli A, Pizzorni C, Seriolo B, Straub RH. Anti-inflammatory mechanisms of methotrexate in rheumatoid arthritis. Ann Rheum Dis. 2001;60(8):729–735. doi:10.1136/ard.60.8.729
65. Ozaki K, Leonard WJ. Cytokine and cytokine receptor pleiotropy and redundancy. J Biol Chem. 2002;277(33):29355–29358. doi:10.1074/jbc.R200003200
66. Lin JX, Migone TS, Tsang M, et al. The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity. 1995;2(4):331–339. doi:10.1016/1074-7613(95
67. George LA, Sullivan SK, Giermasz A, et al. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N Engl J Med. 2017;377(23):2215–2227. doi:10.1056/NEJMoa1708538
68. Mendell JR, Al-Zaidy S, Shell R, et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med. 2017;377(18):1713–1722. doi:10.1056/NEJMoa1706198
69. Miesbach W, Meijer K, Coppens M, et al. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia B. Blood. 2018;131(9):1022–1031. doi:10.1182/blood-2017-09-804419
70. Rangarajan S, Walsh L, Lester W, et al. AAV5-Factor VIII Gene Transfer in Severe Hemophilia A. N Engl J Med. 2017;377(26):2519–2530. doi:10.1056/NEJMoa1708483
71. Cronstein BN, Aune TM. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat Rev Rheumatol. 2020;16(3):145–154. doi:10.1038/s41584-020-0373-9
72. Weber T. Anti-AAV Antibodies in AAV Gene Therapy: current Challenges and Possible Solutions. Front Immunol. 2021;12:658399. doi:10.3389/fimmu.2021.658399
73. Jeune VL, Joergensen JA, Hajjar RJ, Weber T. Pre-existing Anti-Adeno-Associated Virus Antibodies as a Challenge in AAV Gene Therapy. Hum Gene Ther Method. 2013;24(2):59–67. doi:10.1089/hgtb.2012.243
74. Elmore ZC, Oh DK, Simon KE, Fanous MM, Asokan A. Rescuing AAV gene transfer from neutralizing antibodies with an IgG-degrading enzyme. JCI Insight. 2020;5(19):e139881. doi:10.1172/jci.insight.139881
75. Moghadam F, LeGraw R, Velazquez JJ, et al. Synthetic immunomodulation with a CRISPR super-repressor in vivo. Nat Cell Biol. 2020;22(9):1143–1154. doi:10.1038/s41556-020-0563-3
76. Chan YK, Wang SK, Chu CJ, et al. Engineering adeno-associated viral vectors to evade innate immune and inflammatory responses. Sci Transl Med. 2021;13(580):eabd3438. doi:10.1126/scitranslmed.abd3438
77. Verma P, Srivastava A, Srikanth CV, Bajaj A. Nanoparticle-mediated gene therapy strategies for mitigating inflammatory bowel disease. Biomater Sci-Uk. 2021;9(5):1481–1502. doi:10.1039/d0bm01359e
78. Kuzmin DA, Shutova MV, Johnston NR, et al. The clinical landscape for AAV gene therapies. Nat Rev Drug Discov. 2021;20(3):173–174. doi:10.1038/d41573-021-00017
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.