Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
AMPLIFY: a randomized, Phase III study evaluating the efficacy and safety of aclidinium/formoterol vs monocomponents and tiotropium in patients with moderate-to-very severe symptomatic COPD
Authors Sethi S ![]() , Kerwin E
, Kerwin E ![]() , Watz H, Ferguson GT
, Watz H, Ferguson GT ![]() , Mroz RM
, Mroz RM ![]() , Segarra R, Molins E, Jarreta D, Garcia Gil E
, Segarra R, Molins E, Jarreta D, Garcia Gil E
Received 2 October 2018
Accepted for publication 31 January 2019
Published 22 March 2019 Volume 2019:14 Pages 667—682
DOI https://doi.org/10.2147/COPD.S189138
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Sanjay Sethi,1 Edward Kerwin,2 Henrik Watz,3 Gary T Ferguson,4 Robert M Mroz,5,6 Rosa Segarra,7 Eduard Molins,7 Diana Jarreta,7 Esther Garcia Gil7
1Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, SUNY, Buffalo, NY, USA; 2Clinical Research Institute, Medford, OR, USA; 3Pulmonary Research Institute at LungenClinic Grosshansdorf, Airway Research Center North, German Center for Lung Research, Grosshansdorf, Germany; 4Pulmonary Research Institute of Southeast Michigan, Farmington Hills, MI, USA; 5Centrum Medycyny Oddechowej, Białystok, Poland; 6Medical University of Białystok, Białystok, Poland; 7AstraZeneca, Barcelona, Spain
Background: AMPLIFY assessed the efficacy and safety of aclidinium bromide/formoterol fumarate (AB/FF) vs its monocomponents and tiotropium (TIO) in patients with moderate-to-very severe symptomatic COPD (NCT02796677).
Methods: In this 24-week, Phase III, double-dummy, active-controlled study, symptomatic patients (COPD Assessment Test score ≥10) were randomized to twice-daily AB/FF 400/12 µg, AB 400 µg, or FF 12 µg, or once-daily TIO 18 µg. Co-primary endpoints were change from baseline at week 24 in 1-hour morning post-dose FEV1 (AB/FF vs AB) and in pre-dose (trough) FEV1 (AB/FF vs FF). Non-inferiority of AB vs TIO in pre-dose FEV1 was also an objective. Normalized area under the curve (AUC)0–3/3 h FEV1 and nighttime and early morning symptoms were also assessed. A subgroup participated in a 24-hour serial spirometry sub-study.
Results: A total of 1,594 patients were randomized; 566 entered the sub-study. At week 24, 1-hour post-dose FEV1 significantly improved with AB/FF vs AB, FF, and TIO (84, 84, and 92 mL; all P<0.0001). AB/FF significantly improved trough FEV1 vs FF (55 mL, P<0.001) and AB was non-inferior to TIO. AB/FF significantly improved AUC0–3/3 h FEV1 vs all comparators (P<0.0001) and provided significant improvements in early morning symptoms vs TIO. The 24-hour spirometry demonstrated significantly greater improvements with AB/FF in AUC12–24/12 h vs all comparators, and in AUC0–24/24 h vs FF or TIO at week 24.
Conclusion: In patients with moderate-to-very severe symptomatic COPD, twice-daily AB/FF significantly improved lung function vs monocomponents and TIO, and early morning symptom control vs TIO.
Keywords: aclidinium bromide, bronchodilators, LAMA, LABA, 24-hour lung function
Introduction
Combination therapy with a long-acting muscarinic antagonist/long-acting β2-agonist (LAMA/LABA) is recommended by the Global initiative for chronic Obstructive Lung Disease as maintenance therapy for patients with stable COPD who experience persistent symptoms and/or a high risk of exacerbations.1
While the efficacy of the LAMA/LABA aclidinium bromide/formoterol fumarate (AB/FF) vs placebo and its monocomponents (AB and FF) has been previously reported,2,3 AMPLIFY (NCT02796677) examined the effect of AB/FF compared with AB, FF, and tiotropium (TIO) on 24-hour lung function, health-related quality of life, and symptoms in a population of patients with moderate-to-very severe symptomatic COPD (COPD Assessment Test [CAT] score ≥10). As the efficacy of AB/FF, AB, and FF vs placebo has been demonstrated previously, only active comparators were included in this study. In addition, a previous study has investigated AB vs TIO over 6 weeks,4 but AMPLIFY aimed to confirm the non-inferior bronchodilation of AB vs TIO over the longer term, and, for the first time, to provide a direct comparison of AB/FF vs TIO.
Materials and methods
AMPLIFY was a Phase III, 24-week, randomized, parallel-group, double-blind, double-dummy, active-controlled, multinational study of current or former smokers aged ≥40 years with stable, moderate-to-very severe symptomatic COPD (post-bronchodilator FEV1/FVC <70% and post-bronchodilator FEV1 <80% of predicted at screening, and a CAT score ≥10 at screening and randomization). Patients were randomized 2:3:2:3 to AB/FF 400/12 μg, AB 400 μg, or FF 12 μg twice daily via the Genuair™/Pressair® multidose dry powder inhaler (registered trademarks of AstraZeneca group of companies; for use within the USA as Pressair® and Genuair® within all other licensed territories), or TIO 18 μg once daily, via HandiHaler® (Figure 1). To maintain the double-blind, double-dummy nature of the study, all patients used both the Genuair/Pressair and the HandiHaler inhalers each morning, and the Genuair/Pressair inhaler only in the evening. Patients were excluded if they had a predominant asthma diagnosis, or a clinically significant respiratory condition other than COPD; had any respiratory tract infection or COPD exacerbation 6 weeks prior to/during screening; were hospitalized due to a COPD exacerbation in the previous 3 months; or were unable to maintain regular waking/sleeping cycles. Prohibited medications included: LABAs, LAMAs, short-acting β2-agonists (except albuterol/salbutamol, which were permitted “as needed” throughout all study periods), short-acting muscarinic antagonists (except ipratropium, during wash-out and screening only), methylxanthines, leukotriene modifiers, phosphodiesterase-IV inhibitors, or non-selective β-blocking agents. During the washout and screening periods, patients were permitted to use ipratropium and albuterol/salbutamol. Patients were permitted to continue the following medication during screening, washout, and the treatment period provided administration was stable for ≥4 weeks: inhaled, oral, or parenteral corticosteroids (dose equivalent to ≤10 mg/day prednisone); oxygen therapy (<15 hours/day); or oral sustained-release theophylline, or selective β-blocking agents (eg, atenolol, metoprolol, nebivolol; stable administration for ≥2 weeks).
 | Figure 1 Study design. |
The study was conducted in 11 countries (Bulgaria, Czech Republic, Germany, Hungary, Israel, Poland, Russia, Spain, Ukraine, UK, and USA) between July 5, 2016 and June 8, 2017 in accordance with the International Conference on Harmonisation/Good Clinical Practice guidelines and the Declaration of Helsinki. The protocol was approved by the Institutional Review Boards/Independent Ethics Committees (Supplementary materials), and all patients gave written informed consent.
Objectives and endpoints
AMPLIFY had two objectives: to assess the bronchodilatory effect of AB/FF vs the monocomponents and to assess the non-inferiority of bronchodilation for AB vs TIO in change from baseline in trough FEV1, at week 24.
For the AB/FF objective, the co-primary efficacy endpoints were changed from baseline in:
- 1-hour morning post-dose FEV1 (AB/FF vs AB) at week 24.
- Morning pre-dose (trough) FEV1 (AB/FF vs FF) at week 24.
There were two secondary endpoints (both at 24 weeks):
- Change from baseline in normalized area under the curve (AUC) from 0 to 3 hours (AUC0–3/3 h) FEV1 (AB/FF vs AB or FF).
- Proportion of St George’s Respiratory Questionnaire (SGRQ) total score responders (≥4-unit improvement, the minimal clinically important difference [MCID]).5
Additional endpoints included change from baseline in FEV1 over 3 hours post-dose on day 1 and week 24, including onset of action (post-dose FEV1 at 5 and 15 minutes) on day 1; change from baseline in morning pre-dose (trough) FVC at weeks 1 and 24, and AUC0–3/3 h FVC on day 1 and at week 24; change from baseline in SGRQ and CAT score, and proportion of CAT responders (≥2-unit improvement, the MCID)6 at week 24; rate of moderate and severe healthcare resource utilization exacerbations; and change from baseline over 24 weeks in nighttime and early-morning symptoms, (assessed using the validated Nighttime and Early-Morning Symptoms of COPD Instruments [NiSCI and EMSCI]),7,8 Evaluating-Respiratory Symptoms in COPD (E-RS™: COPD) total score,9 and rescue medication use.
A subset of patients participated in a 24-hour sub-study; serial spirometry was additionally performed between 4 and 24 hours on day 1 and at week 24. Change from baseline in FEV1, normalized AUC0–12/12 h, AUC12–24/12 h, AUC0–24/24 h FEV1, and FVC, overall nighttime and early morning symptom severity, and rescue medication were evaluated.
Treatment-emergent adverse events (TEAEs), serious adverse events (AEs), and major adverse cardiovascular events were recorded throughout the study.
All efficacy analyses, except exacerbations, were performed on the intent-to-treat (ITT) population (all randomized patients receiving ≥1 dose of study medication with baseline FEV1 measurements). Non-inferiority analyses were performed on the per protocol population (patients in the ITT population meeting all inclusion/exclusion criteria, >70% treatment compliance, and no serious protocol deviations). Non-inferiority of AB vs TIO was defined as the lower bound of the two-sided 95% CI >−50 mL. Exacerbations and safety analyses were performed on the safety population (all randomized patients receiving ≥1 dose of study medication).
All lung function, health-related quality of life, and symptom measures were analyzed using a mixed model of repeated measures, responder analyses by logistic random-effect model, and exacerbation rates using a negative binomial regression model. Details are provided in Supplementary materials. To control for multiplicity, endpoints were tested in a pre-specified hierarchical sequence with the order: 1-hour post-dose FEV1, morning pre-dose (trough) FEV1, AUC0–3/3 h FEV1, SGRQ responder analysis.
Results
Of 1,594 patients randomized, 1,583 were included in the ITT and safety populations, 1,403 in the per protocol population, and 1,356 (85.1%) remained on study treatment and completed the study (Figure 2). Baseline characteristics are presented in Table 1. A total of 566 patients were included in the sub-study (35.5%), of whom 563 were included in the sub-study ITT population and 493 (87.1%) remained on study treatment and completed the study. The baseline characteristics in the sub-study were similar to the total population (Table S1).
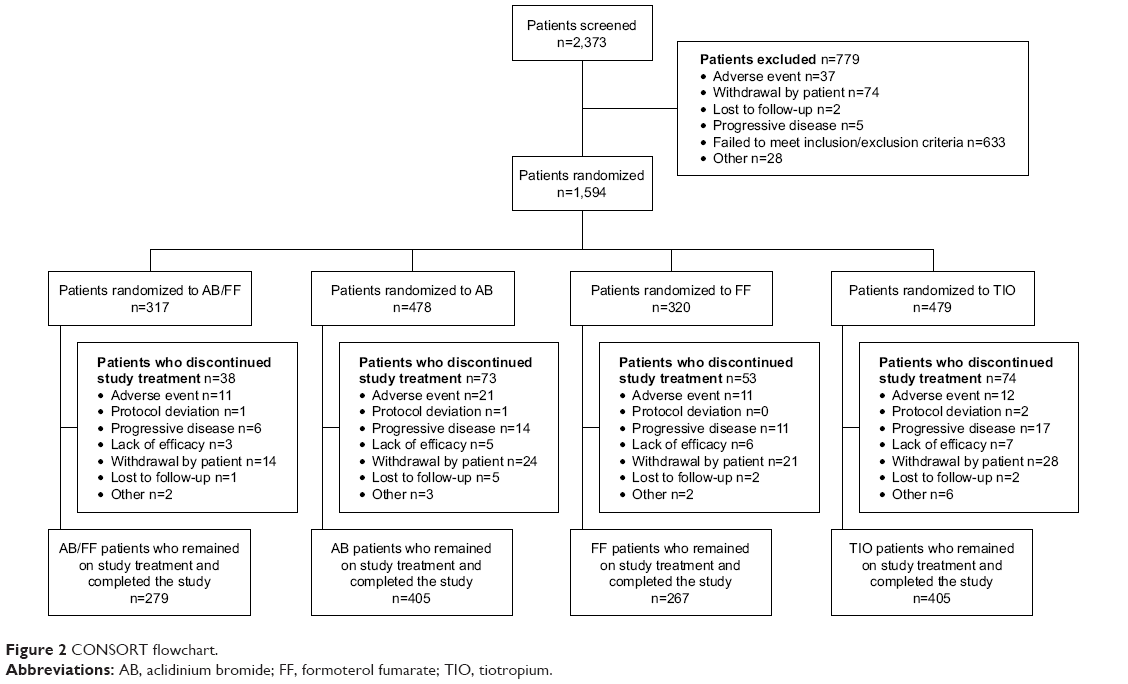 | Figure 2 CONSORT flowchart. |
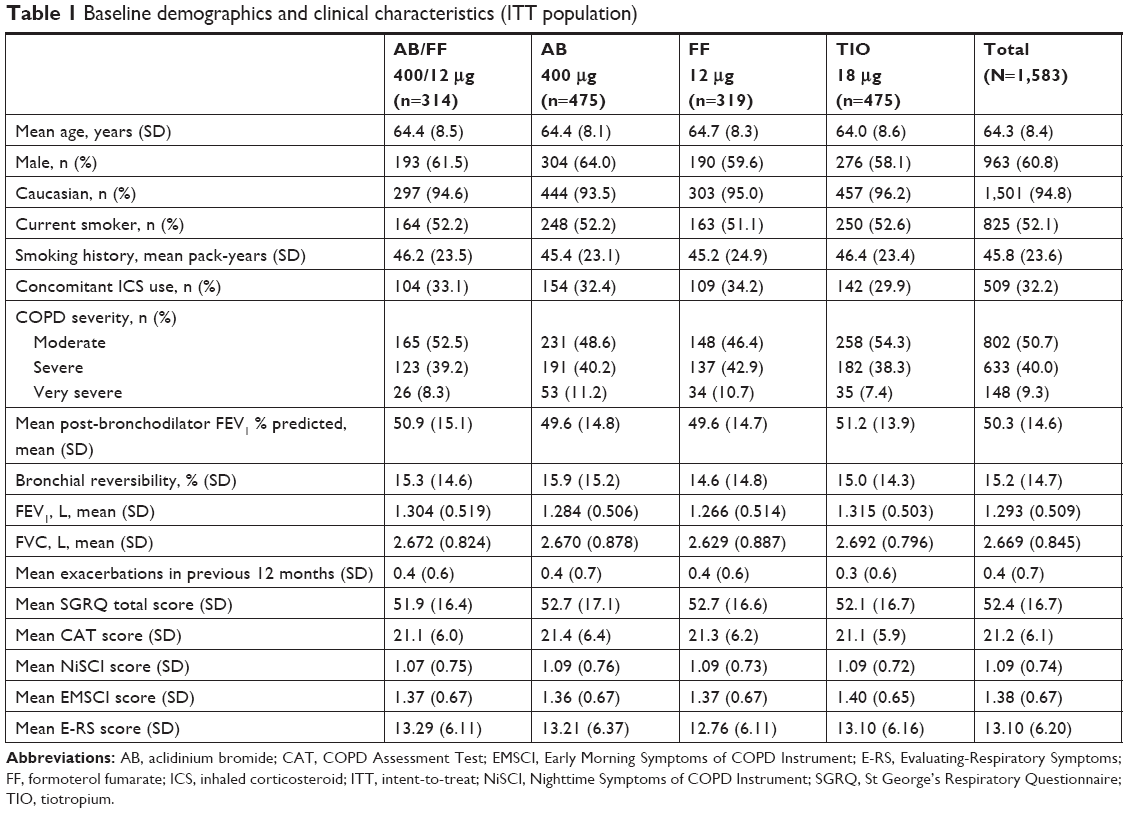 | Table 1 Baseline demographics and clinical characteristics (ITT population) |
Efficacy
At week 24, all active treatments improved 1-hour post-dose FEV1 from baseline (Figure 3). Treatment with AB/FF resulted in significantly greater improvements in 1-hour post-dose FEV1 compared with AB (84 mL, P<0.0001; co-primary endpoint), FF (84 mL, P<0.0001), and TIO (92 mL, P<0.0001).
 | Figure 3 Change from baseline in 1-hour post-dose FEV1 at week 24, ITT population (co-primary endpoint). |
AB/FF led to significantly greater improvements in change from baseline in morning pre-dose (trough) FEV1 vs FF (55 mL, P<0.001; co-primary endpoint); however, the improvements for AB/FF compared with AB (14 mL) and TIO (19 mL) did not reach statistical significance (Figure 4).
 | Figure 4 Change from baseline in trough FEV1 at week 24, ITT population (co-primary endpoint). |
For the AB vs TIO objective, AB was non-inferior to TIO in change from baseline in morning pre-dose (trough) FEV1 at week 24 (least squares [LS] mean difference 7 mL [95% CI: −21 mL, 35 mL; P=0.6377]).
All treatments improved post-dose FEV1 over 3 hours on day 1 and at week 24, and the improvements observed with AB/FF were statistically significant compared with AB, FF, and TIO (Figure 5). On day 1 and at week 24, there were significantly greater improvements from baseline in AUC0–3/3 h FEV1 with AB/FF compared with AB, FF, or TIO (Figure 5). On day 1, all treatments improved post-dose FEV1 from baseline at 5 minutes (143, 52, 122, and 49 mL, for AB/FF, AB, FF, and TIO, respectively) and 15 minutes (173, 101, 148, and 86, for AB/FF, AB, FF, and TIO, respectively). AB/FF treatment led to significantly greater improvements in post-dose FEV1 compared with AB, FF, and TIO at both 5 minutes (91, 21, and 95 mL, respectively, all P<0.05) and 15 minutes (72, 25, and 87 mL, respectively, all P<0.01).
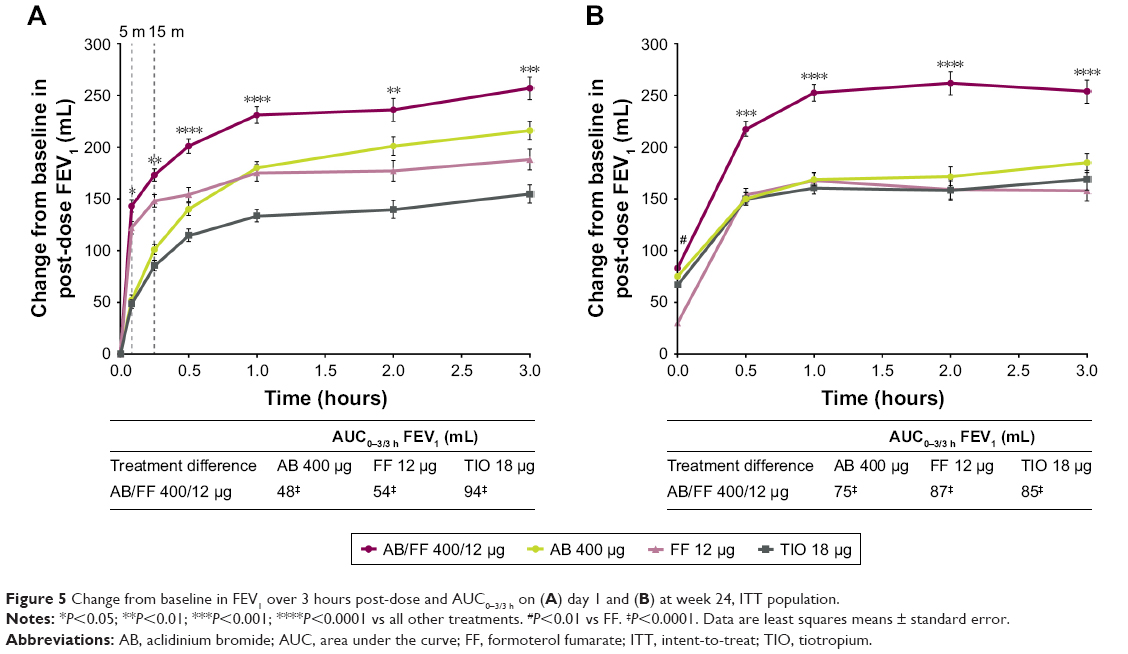 | Figure 5 Change from baseline in FEV1 over 3 hours post-dose and AUC0–3/3 h on (A) day 1 and (B) at week 24, ITT population. |
AB/FF improved trough FVC from baseline at week 1 (152 mL) and week 24 (116 mL). These improvements were significantly greater than FF (difference of 85 and 117 mL, at weeks 1 week 24, respectively, both P<0.001), but there were no significant differences compared with AB, or TIO (Figure S1). Improvements from baseline in normalized AUC0–3/3 h FVC for AB/FF were 340 and 328 mL on day 1 and at week 24, respectively; these improvements were significantly greater compared with AB, FF, and TIO on day 1 (difference of 49, 73, and 134 mL, respectively, all P<0.01) and at week 24 (difference of 105, 139, and 138 mL, respectively, all P<0.0001; Figure S2).
All treatments improved patient-reported health-related quality of life. AB/FF, AB, and TIO improved SGRQ total score vs baseline by more than the MCID at week 24 (4.68, 4.95, and 5.58 units, respectively). FF improved SGRQ by 3.96 units compared with baseline. At week 24, all treatments improved CAT score vs baseline by more than the MCID (2.85, 2.54, 2.78, and 2.53 units for AB/FF, AB, FF, and TIO, respectively). There were no significant differences between treatments for SGRQ total score or CAT score, or in the proportion of SGRQ responders (48.1%, 49.1%, 49.6%, and 50.6% for AB/FF, AB, FF, and TIO, respectively) or CAT responders (58.8%, 59.5%, 57.8%, and 55.4% for AB/FF, AB, FF, and TIO, respectively).
The rate of moderate and severe exacerbations was similar between AB/FF, AB, and TIO (0.46, 0.48, and 0.41 exacerbations per patient per year, respectively) and lower than reported with FF (0.61).
Overall nighttime and overall early morning symptom severity scores showed numerical improvements with AB/FF compared with all treatments over 24 weeks, with significant improvements in overall early morning symptom severity score vs TIO (Figure 6). The change from baseline in E-RS total score over 24 weeks was similar between treatment groups (−1.73, −1.64, −1.25, and −1.38 units for AB/FF, AB, FF, and TIO, respectively).
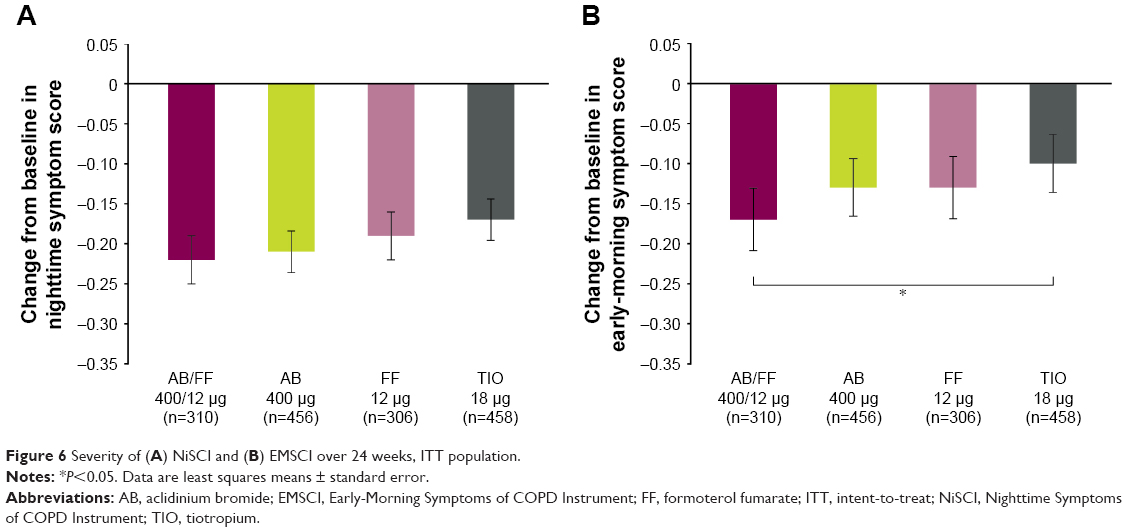 | Figure 6 Severity of (A) NiSCI and (B) EMSCI over 24 weeks, ITT population. |
At week 24, AB/FF significantly decreased the use of rescue medication vs TIO (−0.39 puffs/day, P<0.05). Numerical reductions were seen with AB/FF compared with AB (−0.16 puffs/day, P=0.295) and FF (−0.18 puffs/day, P=0.291).
24-Hour serial spirometry sub-study
The sub-study demonstrated improvements from baseline in FEV1 with AB/FF vs monotherapies across the 24-hour post-dose period on day 1 and at week 24 (Figure 7). On day 1, AB/FF significantly improved FEV1 at all time points vs FF (except 15 minutes) and TIO (except 12 hours). At week 24, AB/FF significantly improved FEV1 at all time points vs FF. At week 24, AB/FF demonstrated significantly greater improvements vs all monotherapies in AUC12–24 h FEV1, and significant improvements in AUC0–12 h/12 h FEV1 vs FF, and in AUC0–24 h/24 h FEV1 vs FF and TIO (Figure 8). AB/FF improved FVC across the 24-hour period (Figure S3); AB/FF improved AUC0–24/24 h FVC from baseline by 314 and 239 mL on day 1 and at week 24, respectively (Figure S3). Compared with all three monotherapies, AB/FF significantly improved AUC12–24/12 h FVC on day 1 and week 24, and AUC0–24/24 h FVC at week 24 (Figure S3).
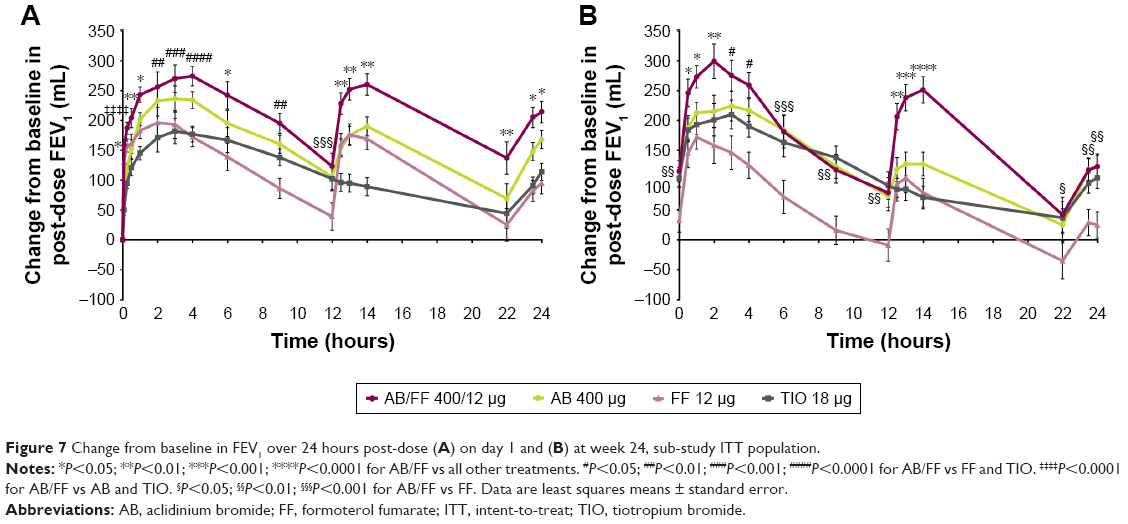 | Figure 7 Change from baseline in FEV1 over 24 hours post-dose (A) on day 1 and (B) at week 24, sub-study ITT population. |
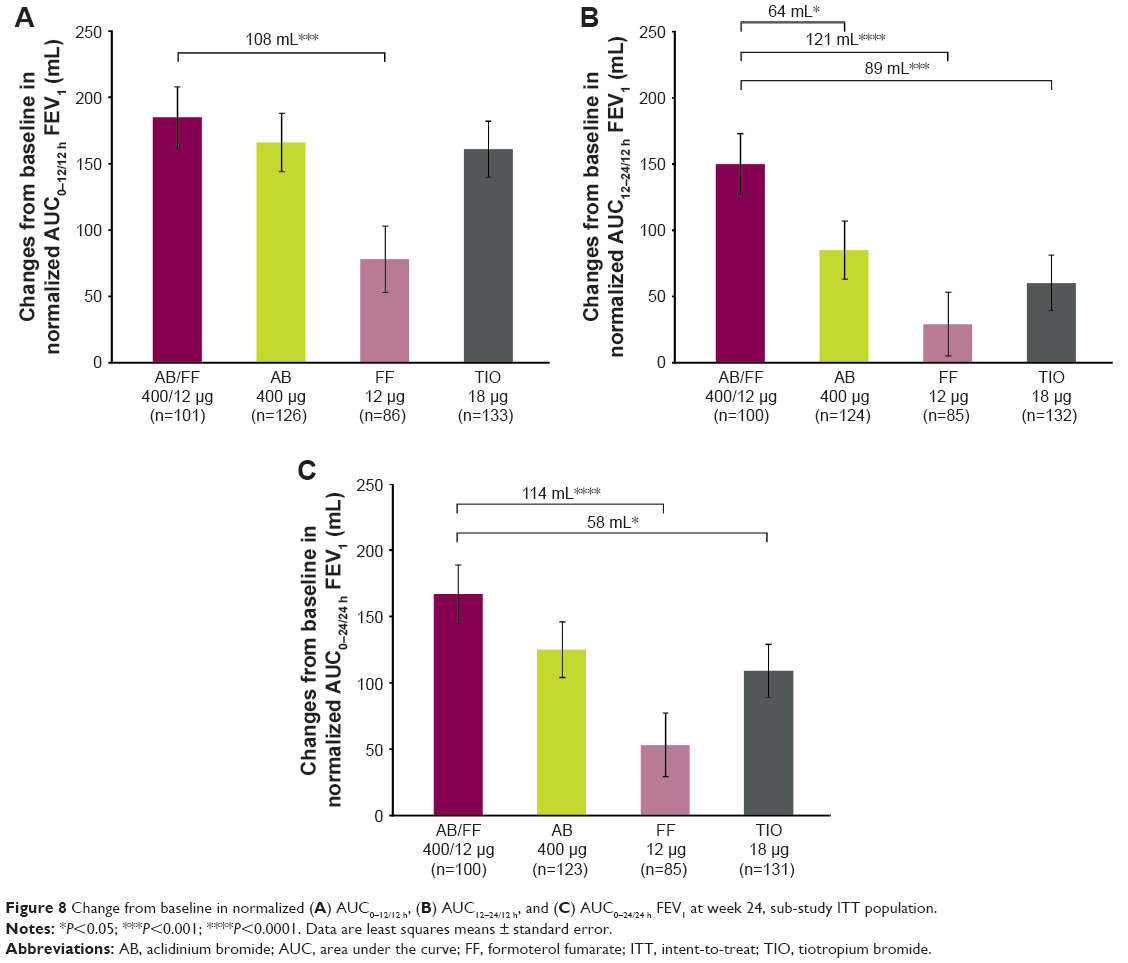 | Figure 8 Change from baseline in normalized (A) AUC0–12/12 h, (B) AUC12–24/12 h, and (C) AUC0–24/24 h FEV1 at week 24, sub-study ITT population. |
Overall nighttime and overall early morning symptom severity score in the sub-study showed numerical improvements with AB/FF vs AB and TIO over 24 weeks. AB/FF demonstrated significant improvements in overall early morning symptom severity vs TIO (Figure 9).
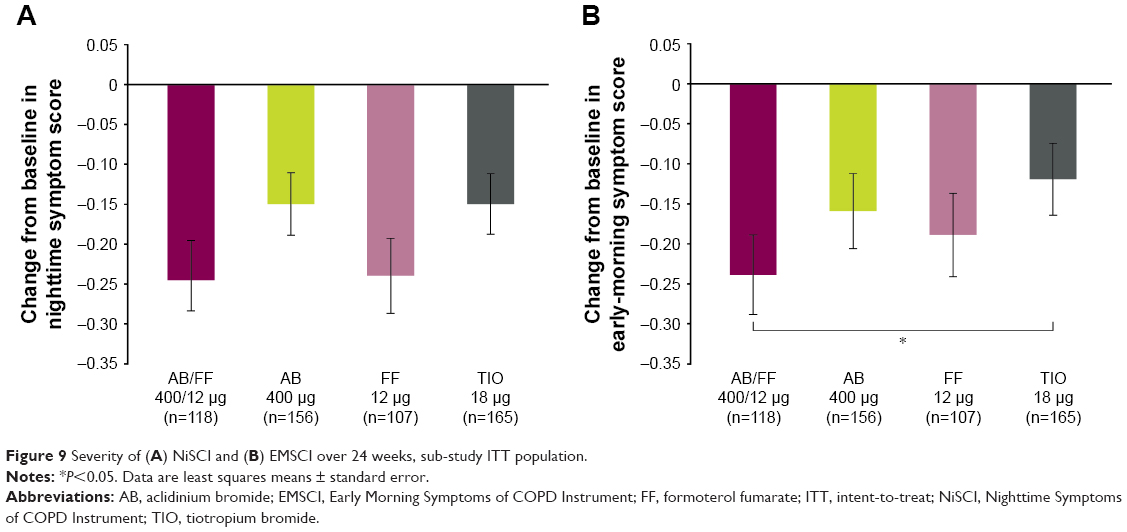 | Figure 9 Severity of (A) NiSCI and (B) EMSCI over 24 weeks, sub-study ITT population. |
At week 24 of the sub-study, all treatments decreased rescue medication use, and AB/FF significantly decreased the use of rescue medication vs AB, FF, and TIO (difference of −0.47, −0.67, and −0.58 puffs/day, respectively, all P<0.05).
Safety
The proportion of patients reporting TEAEs was similar between treatment groups, as was incidence of the most common events (COPD exacerbation, nasopharyngitis, and headache; Table 2). The incidence of serious AEs, major adverse cardiovascular events, and AEs leading to discontinuation or death was low and similar across treatment groups (Table 2).
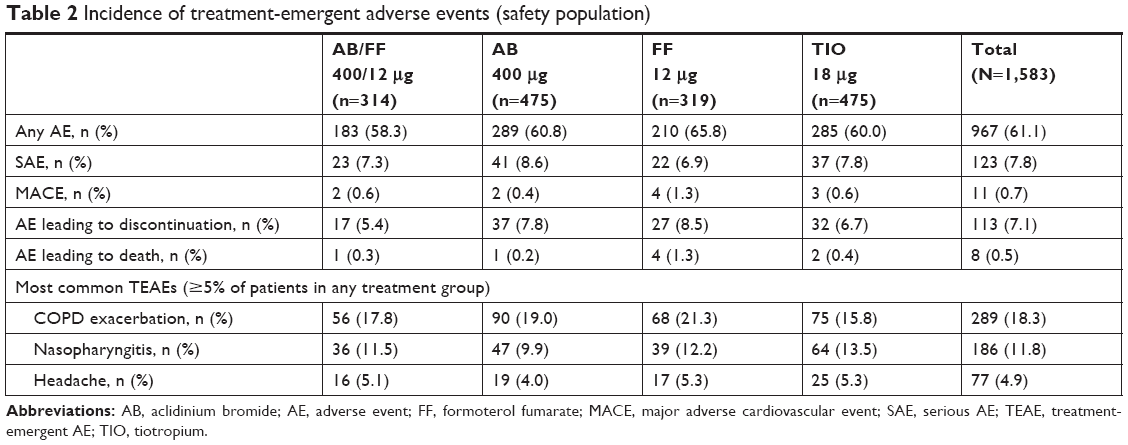 | Table 2 Incidence of treatment-emergent adverse events (safety population) |
Discussion
In this 24-week, Phase III, active-controlled study in patients with moderate-to-very severe symptomatic COPD, treatment with AB/FF 400/12 μg led to rapid improvements in lung function vs monocomponents and TIO that were sustained over 24 hours and reduced overall early morning COPD symptoms vs TIO. The lung function improvements observed in this study were within the range of those reported in previous studies of AB/FF vs monocomponents,2,3,10,11 and the improvements from baseline in 1-hour post-dose FEV1 reported for AB/FF vs AB confirm the contribution of FF to the efficacy of AB/FF. Although this study was not intended to investigate hyperinflation, the post-dose FVC results indicate that AB/FF increased the overall vital capacity of the lung vs monocomponents and TIO.
This was the first study to report the efficacy of AB/FF vs TIO; a previous study of AB demonstrated similar lung function efficacy compared with TIO over 6 weeks,4 and the present study confirmed that AB is statistically non-inferior to TIO over a longer treatment period. This was also the first study to report improvements in AUC0–3/3 h and 24-hour bronchodilation for AB/FF vs both the monocomponents and TIO.
In the sub-study, AB/FF demonstrated increased 24-hour bronchodilation vs comparators with the greatest benefits observed in the first few hours following administration and in the 12–24-hour post-dose period, during nighttime and early morning hours. Interestingly, the attenuated FEV1 response following evening administration, compared with the morning response seen for all three twice-daily treatments (AB/FF, AB, and FF), has been observed previously4 and can potentially be explained by the natural diurnal changes in lung function that have been reported in patients with COPD.12 Although the link between lung function and time of day is not well characterized, particularly given the heterogeneous nature of COPD, 24-hour spirometry has shown circadian variation in FEV1, with the highest values reported mid-morning, a steady decline throughout the day, and the lowest values in the early hours of the morning.4,12 Therefore, twice-daily bronchodilators, such as AB/FF or AB, may be beneficial in counteracting this characteristic dip in airflow during the evening and at nighttime.
For patients receiving AB/FF, significant improvements were observed in overall early morning symptoms vs TIO in the sub-study and total population. Numerical improvements were observed in overall nighttime and early morning symptoms for AB/FF vs all monotherapies in the sub-study and total population, except for nighttime symptoms vs FF in the sub-study population. The magnitude of the improvements observed in both the nighttime and early morning symptom scores with AB/FF was similar to those seen previously.2,3,11 The lung function improvements observed with AB/FF 12–24 hours post-dose illustrate the beneficial effects of dual bronchodilation over monotherapy, but together with the improvements in symptom scores, also suggest the potential benefits of twice-daily administration of dual bronchodilators for some patients.
The proportion of patients achieving clinically important improvements in patient-reported health-related quality of life outcomes was similar across all treatment groups. Nearly half the patients in this study reported clinically significant improvements in SGRQ, which is in line with previous studies,2,3,11 and nearly 60% reported clinically relevant improvements in CAT. The improvements in CAT score observed are consistent with those reported in a previous 24-week study of AB/FF vs salmeterol/fluticasone 50/500 μg. This is noteworthy, as both studies enrolled symptomatic patients13 with baseline CAT scores of ~20 units, so improvements in health status could be of particular significance for this group of patients. It was unexpected that although they achieved the MCID, the improvements in SGRQ were smaller than previously reported,2,3 and AB/FF did not provide significantly greater improvements in SGRQ total score or responder rates vs monotherapies or TIO. Recent improvements in the standard of COPD patient care and effective modern bronchodilator therapy might mean that it is more difficult to achieve large changes in the SGRQ total score, particularly in a patient population with more severe COPD than previous studies. In addition, patient-reported outcome measures give useful insight into how a patient perceives their treatment is progressing, but are, by definition, subjective. As a result, patient-reported outcomes may lack the sensitivity to detect more subtle improvements between active treatments.
When considering possible limitations of this study, it should be noted that the population was not enriched for patients who experienced frequent exacerbations. Exacerbation rates were similar between patients receiving AB/FF, AB, and TIO and were in line with similar studies in patients with moderate-to-severe COPD,10,11,14 where AB/FF demonstrated significant improvements vs placebo in the pooled analysis of the pivotal studies.14 A longer duration study in a population of exacerbation-prone patients may further elucidate any potential differences for AB/FF compared with monotherapy.
With regard to the strength of the symptom score instruments, Mocarski et al and Hareendran et al showed good internal consistency, reliability, and test–retest reliability for the NiSCI and EMSCI scores, and moderate-to-strong correlations with the SGRQ scores and E-RS total score.7,8 Further validation and increasing adoption of the early morning and nighttime COPD symptom instruments would help to strengthen the results of this study.
One of the strengths of this study, however, lies in the selection of a symptomatic population with more severe COPD; patients were required to have a CAT score of ≥10 (mean baseline CAT score: 21.2), and, unlike other AB/FF studies, this study had no lower limit to FEV1, which resulted in a population with more severe and more symptomatic patients than previous studies.2,3,10 Another area of this study that should be considered a strength was the inclusion of the 24-hour serial spirometry sub-study, which coupled with the nighttime and early morning symptom score data allowed evaluation of effects of AB/FF on lung function and symptoms simultaneously over the 24-hour period.
Additional studies of twice-daily AB/FF compared with twice-daily AB, and FF, and once-daily TIO over the longer term and/or in specific sub-groups of patients, would further support the encouraging findings of this study. It would be interesting, for example, to look at AB/FF treatment in patients who specifically report higher levels of nighttime symptoms, and who may derive additional benefit from the twice-daily dosing of AB/FF. Furthermore, as AB/FF has previously demonstrated improvements in physical activity compared with placebo15 within the suggested MCID range for physical activity,16 additional studies with standardized exercise tests or activity measurement could confirm whether the increased daytime and evening bronchodilation reported for AB/FF vs once-daily bronchodilator monotherapy may also provide improved activity tolerance for patients with COPD.
Conclusion
In conclusion, twice-daily AB/FF 400/12 μg treatment demonstrated significantly greater bronchodilation in patients with moderate-to-very severe symptomatic COPD compared with monocomponents and TIO at 24 weeks. AB monotherapy demonstrated non-inferiority to TIO in trough FEV1, and all treatments in this study improved health-related quality of life outcomes at week 24. The 24-hour serial spirometry sub-study demonstrated that the most significant lung function benefits over monotherapies were observed in the 12–24-hour post-dose period, which coincided with significant improvements in overall early-morning symptom severity score compared with TIO. Overall, the findings of AMPLIFY support the sustained efficacy of AB/FF over 24 hours and indicate that AB/FF 400/12 μg may provide improved nighttime and early morning symptom control, compared with monotherapies.
Data availability
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
The protocol and the statistical analysis plan can be found at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/View?id=22775.
Acknowledgments
The authors would like to thank the patients, investigators, and site staff involved in the study. The authors would also like to thank Ferran Chuecos for his role in statistical programming, Alejhandra Lei, Ana Frances, and Marta Pujol for their role in providing medical safety monitoring and for input on the definition and interpretation of the safety analyses, and Joaquim Delgadillo, a former employee of AstraZeneca, for his contribution to the study design.
Medical writing support, under the direction of the authors, was provided by Siobhán Hoy of CMC CONNECT, a division of McCann Health Medical Communications Ltd, Glasgow, UK, funded by AstraZeneca, Cambridge, UK, in accordance with Good Publication Practice (GPP3) guidelines.17
Data of this manuscript have previously been presented at the 2018 meeting of the American Thoracic Society, the 2018 meeting of the European Respiratory Society, the 2018 Aspen Lung Conference, and the 2018 Eastern Pulmonary Conference (Am J Respir Crit Care Med 2018;197:A4235; Am J Respir Crit Care Med 2018;197:A4241).
This study was funded by AstraZeneca with support from Circassia. The sponsor did not place any restriction on authors about the statements made in the final article.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
SS has received grants from AstraZeneca, Dey, and Pearl Therapeutics. He has received personal fees from AstraZeneca, Bayer, Boehringer Ingelheim, Cempra, CSL Behring, Forest, GlaxoSmithKline, Merck, Pearl Therapeutics, Pulmonx, Reckitt Benckiser, Sunovion, and Theravance.
EK has served on advisory boards, speaker panels, or received travel reimbursement from Amphastar, AstraZeneca, GlaxoSmithKline, Mylan, Novartis, Oriel, Pearl Therapeutics, Sunovion, Teva, and Theravance. He has conducted multicenter clinical trials for ~40 pharmaceutical companies.
HW has received honoraria for consultancies, lectures, and travel support to attend scientific congresses from Almirall, AstraZeneca, Berlin-Chemie, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Novartis, and Takeda. His institution received investigator fees for participation in clinical trials from Almirall, AstraZeneca, Bayer Health Care, Berlin-Chemie, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Novartis, Roche, Sanofi Aventis, and Takeda.
GTF reports consulting and advisory board participation for AstraZeneca, Boehringer Ingelheim, Forest Laboratories, Novartis, Pearl Therapeutics, Sunovion, and Verona Pharma; consulting fees from Receptos; speaking engagements for AstraZeneca, Boehringer Ingelheim, Forest Laboratories, GlaxoSmithKline, Pearl Therapeutics, and Sunovion; and research grants from AstraZeneca, Boehringer Ingelheim, Forest Laboratories, GlaxoSmithKline, Novartis, Pearl Therapeutics, Sanofi, Sunovion, and Theravance Biopharma.
RMM has received consulting fees, speaker’s fees, and travel expenses from Boehringer Ingelheim and has also received compensation for participating in advisory boards for Almirall, AstraZeneca, Boehringer Ingelheim, and Novartis. Furthermore, RMM has received compensation for participation in multicenter clinical trials in the past 5 years from several companies including Almirall, AstraZeneca, Boehringer Ingelheim, GSK, Merck Sharp & Dohme, Mundipharma, Novartis, Pearl, Roche, and Takeda.
RS, EM, DJ, and EGG are employees of AstraZeneca PLC, Barcelona, Spain.
The authors report no other conflicts of interest in this work.
References
Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease; 2018. Available from: https://goldcopd.org/wp-content/uploads/2017/11/GOLD-2018-v6.0-FINAL-revised-20-Nov_WMS.pdf. Accessed October 11, 2018. | ||
Singh D, Jones PW, Bateman ED, et al. Efficacy and safety of aclidinium bromide/formoterol fumarate fixed-dose combinations compared with individual components and placebo in patients with COPD (ACLIFORM-COPD): a multicentre, randomised study. BMC Pulm Med. 2014;14(1):178. | ||
D’Urzo AD, Rennard SI, Kerwin EM, et al. Efficacy and safety of fixed-dose combinations of aclidinium bromide/formoterol fumarate: the 24-week, randomized, placebo-controlled AUGMENT COPD study. Respir Res. 2014;15(1):123. | ||
Beier J, Kirsten A-M, Mróz R, et al. Efficacy and safety of aclidinium bromide compared with placebo and tiotropium in patients with moderate-to-severe chronic obstructive pulmonary disease: results from a 6-week, randomized, controlled phase IIIB study. COPD. 2013;10(4):511–522. | ||
Jones PW. St George’s Respiratory Questionnaire: MCID. COPD. 2005;2(1):75–79. | ||
Kon SSC, Canavan JL, Jones SE, et al. Minimum clinically important difference for the COPD Assessment Test: a prospective analysis. Lancet Respir Med. 2014;2(3):195–203. | ||
Mocarski M, Zaiser E, Trundell D, Make BJ, Hareendran A. Evaluation of the psychometric properties of the Nighttime Symptoms of COPD Instrument. Int J Chron Obstruct Pulmon Dis. 2015;10:475–487. | ||
Hareendran A, Make BJ, Zaiser E, Garcia Gil E. Evaluation of the psychometric properties of the Early Morning Symptoms of COPD Instrument (EMSCI). Int J Chron Obstruct Pulmon Dis. 2018;13:1633–1645. | ||
Leidy NK, Murray LT, Monz BU, et al. Measuring respiratory symptoms of COPD: performance of the EXACT-Respiratory Symptoms Tool (E-RS) in three clinical trials. Respir Res. 2014;15(1):124. | ||
Donohue JF, Soong W, Wu X, Shrestha P, Lei A. Long-term safety of aclidinium bromide/formoterol fumarate fixed-dose combination: results of a randomized 1-year trial in patients with COPD. Respir Med. 2016;116:41–48. | ||
D’Urzo A, Rennard S, Kerwin E, et al. A randomised double-blind, placebo-controlled, long-term extension study of the efficacy, safety and tolerability of fixed-dose combinations of aclidinium/formoterol or monotherapy in the treatment of chronic obstructive pulmonary disease. Respir Med. 2017;125:39–48. | ||
Calverley PMA, Lee A, Towse L, van Noord J, Witek TJ, Kelsen S. Effect of tiotropium bromide on circadian variation in airflow limitation in chronic obstructive pulmonary disease. Thorax. 2003;58(10):855–860. | ||
Vogelmeier C, Paggiaro PL, Dorca J, et al. Efficacy and safety of aclidinium/formoterol versus salmeterol/fluticasone: a phase 3 COPD study. Eur Respir J. 2016;48(4):1030–1039. | ||
Bateman ED, Chapman KR, Singh D, et al. Aclidinium bromide and formoterol fumarate as a fixed-dose combination in COPD: pooled analysis of symptoms and exacerbations from two six-month, multicentre, randomised studies (ACLIFORM and AUGMENT). Respir Res. 2015;16(1):92. | ||
Watz H, Troosters T, Beeh KM, et al. ACTIVATE: the effect of aclidinium/formoterol on hyperinflation, exercise capacity, and physical activity in patients with COPD. Int J Chron Obstruct Pulmon Dis. 2017;12:2545–2558. | ||
Demeyer H, Burtin C, Hornikx M, et al. The minimal important difference in physical activity in patients with COPD. PLoS One. 2016;11(4):e0154587. | ||
Battisti WP, Wager E, Baltzer L, et al. Good publication practice for communicating company-sponsored medical research: GPP3. Ann Intern Med. 2015;163(6):461–464. |
Supplementary materials
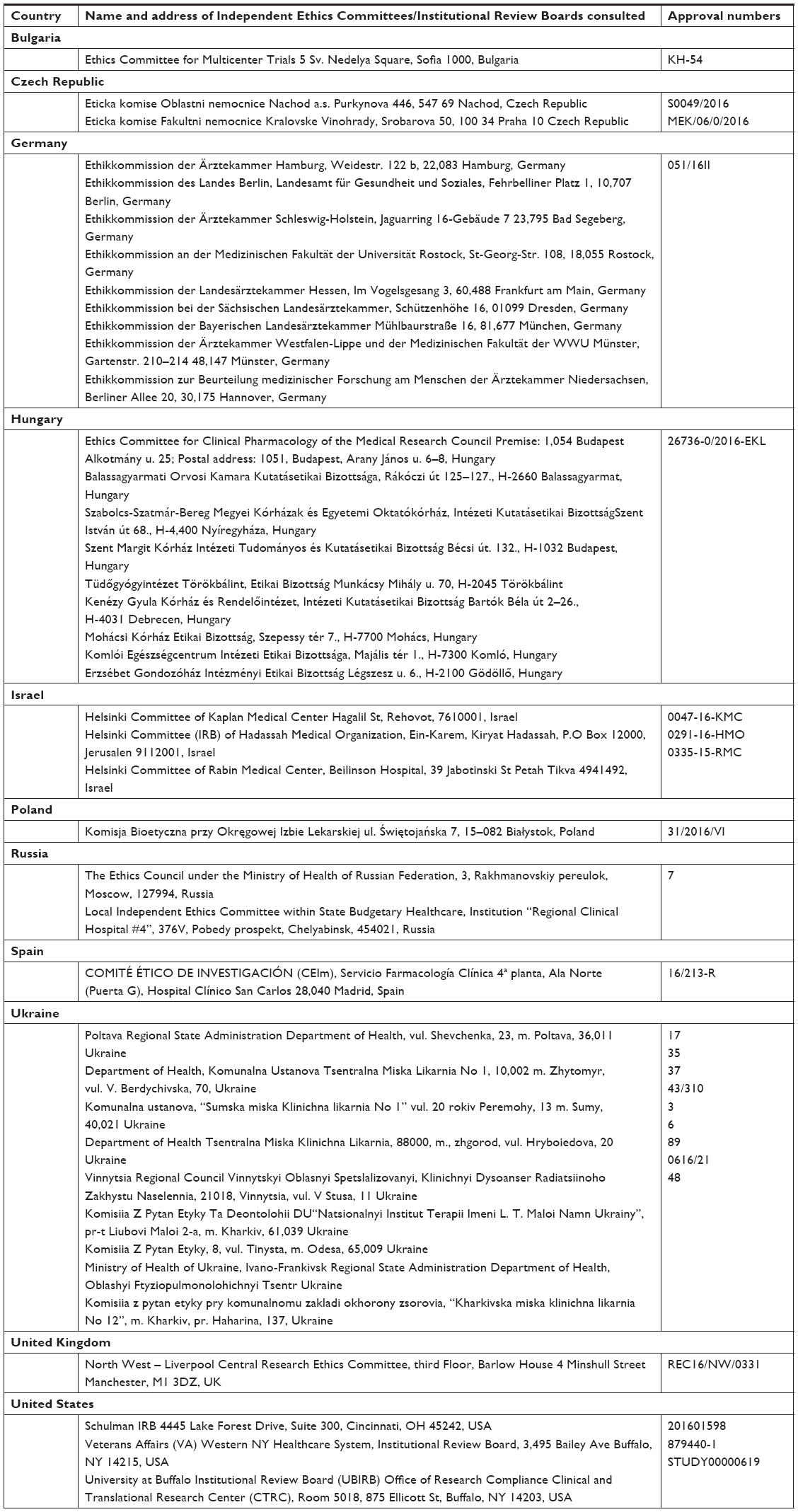 | Independent Ethics Committees/Institutional Review Boards and Approval Numbers |
Statistical analyses
Sample size was calculated to provide ≥90% power to detect a statistically significant difference of 100 mL between AB/FF and AB in change from baseline in 1-hour morning post-dose FEV1, and 65 mL between AB/FF and FF in change from baseline in morning pre-dose (trough) FEV1, at week 24, with an SD of 230 mL based on a two-sided test at a 5% significance level. The sample size also has 90% power to show the lower bound of the two-sided 95% CI for the change from baseline in morning pre-dose (trough) FEV1 at week 24 between AB and TIO of over −50 mL (expected difference: 0 mL; SD: 230 mL).
All lung function, health-related quality of life, and nighttime and early morning symptoms measures were analyzed by means of a mixed model for repeated measures, adjusted for covariates (pre- and post-bronchodilator FEV1 at screening [lung function endpoints only], age, and baseline score/measure), fixed-effect factors (treatment group, sex, smoking status, visit, and treatment group-by-visit interaction), and random intercept (country).
The proportion of SGRQ and CAT responders was analyzed based on a logistic random-effect model including a random intercept to account for the variability between subjects, and adjusting for fixed factors (treatment, sex, smoking status, country, visit, and treatment group-by-visit interaction), and covariates (age and baseline).
The rate of COPD exacerbations was analyzed by negative binomial regression models, adjusting for factors (sex, baseline ICS use, baseline COPD severity, smoking status, country, prior history of exacerbation, treatment group, and offset) and covariate (age).
To control for multiplicity, endpoints were tested in a pre-specified hierarchical sequence with the order: change from baseline in 1-hour post-dose FEV1; change from baseline in trough FEV1; change from baseline in AUC0–3/3 h FEV1; SGRQ responder analysis. The non-inferiority between AB and TIO in change from baseline in morning pre-dose (trough) FEV1 at week 24 will be tested at a significance level of 0.05.
 | Table S1 Baseline demographics and clinical characteristics (sub-study ITT population) |
 | Figure S1 Change from baseline in morning pre-dose (trough) FVC (A) at week 1, and (B) week 24, ITT population. |
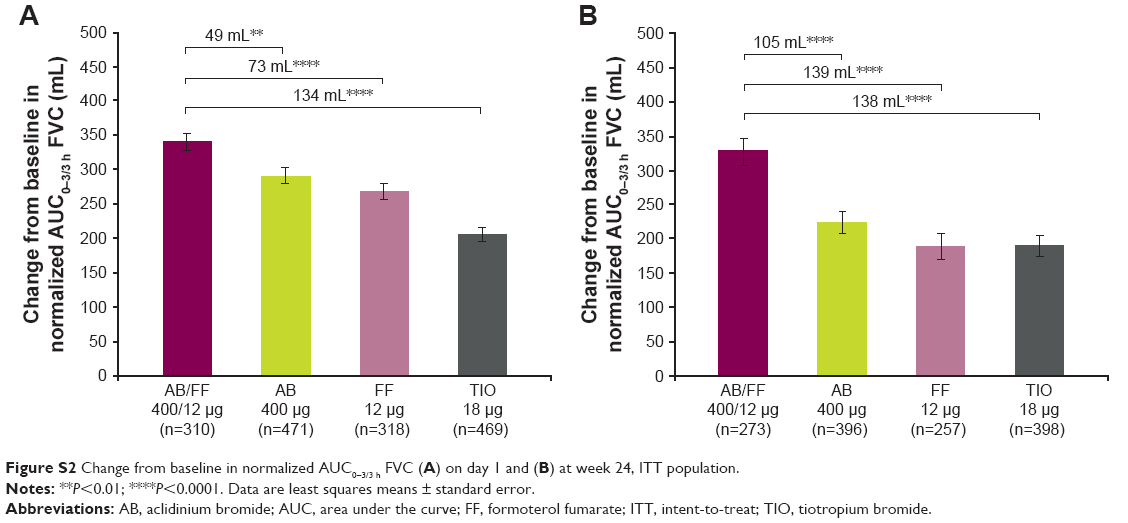 | Figure S2 Change from baseline in normalized AUC0–3/3 h FVC (A) on day 1 and (B) at week 24, ITT population. |
 | Figure S3 Change from baseline in normalized (A and B) AUC0–12/12 h, (C and D) AUC12–24/12 h, and (E and F) AUC0–24/24 h FVC at day 1 and week 24, sub-study ITT population. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.