Back to Journals » Hepatic Medicine: Evidence and Research » Volume 15
Ambiguous Pathogenic Roles of Macrophages in Alcohol-Associated Liver Diseases
Authors Ait Ahmed Y, Lafdil F, Tacke F ![]()
Received 25 May 2023
Accepted for publication 7 September 2023
Published 21 September 2023 Volume 2023:15 Pages 113—127
DOI https://doi.org/10.2147/HMER.S326468
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Gerry Lake-Bakaar
Yeni Ait Ahmed,1 Fouad Lafdil,2– 4 Frank Tacke1
1Department of Hepatology & Gastroenterology, Charité - Universitätsmedizin Berlin, Campus Virchow-Klinikum (CVK) and Campus Charité Mitte (CCM), Berlin, Germany; 2Université Paris-Est, UMR-S955, UPEC, Créteil, France; 3Institut National de la Sante et de la Recherche Medicale (INSERM), U955, Créteil, France; 4Institut Universitaire de France (IUF), Paris, France
Correspondence: Frank Tacke, Charité - Universitätsmedizin Berlin, Department of Hepatology and Gastroenterology, Campus Virchow-Klinikum (CVK) and Campus Charité Mitte (CCM), Augustenburger Platz 1, D-13353, Berlin, Germany, Tel +49 (30) 450 553 022, Fax +49 (30) 450 553 902, Email [email protected]
Abstract: Alcohol-associated liver disease (ALD) represents a major public health issue worldwide and is a leading etiology of liver cirrhosis. Alcohol-related liver injuries include a range of manifestations including alcoholic hepatitis (AH), simple steatosis, steatohepatitis, hepatic fibrosis, cirrhosis and liver cancer. Liver disease occurs from several pathological disturbances such as the metabolism of ethanol, which generates reactive oxygen species (ROS) in hepatocytes, alterations in the gut microbiota, and the immune response to these changes. A common hallmark of these liver affections is the establishment of an inflammatory environment, and some (broad) anti-inflammatory approaches are used to treat AH (eg, corticosteroids). Macrophages, which represent the main innate immune cells in the liver, respond to a wide variety of (pathogenic) stimuli and adopt a large spectrum of phenotypes. This translates to a diversity of functions including pathogen and debris clearance, recruitment of other immune cells, activation of fibroblasts, or tissue repair. Thus, macrophage populations play a crucial role in the course of ALD, but the underlying mechanisms driving macrophage polarization and their functionality in ALD are complex. In this review, we explore the various populations of hepatic macrophages in alcohol-associated liver disease and the underlying mechanisms driving their polarization. Additionally, we summarize the crosstalk between hepatic macrophages and other hepatic cell types in ALD, in order to support the exploration of targeted therapeutics by modulating macrophage polarization.
Keywords: macrophages, alcohol, liver disease, fibrosis, cirrhosis
Alcohol-associated liver disease (ALD) is a major public health concern worldwide and represents the leading etiology of liver cirrhosis in many parts of the world, including Europe, India and South America.1 It is estimated that more than 800 million people in the world suffer from chronic liver disease accounting for 2 million deaths per year, including 1 million from cirrhosis complications.2,3 Since the liver is the main site of alcohol metabolism, it is the most vulnerable tissue to chronic and heavy alcohol consumption. The effects of variable drinking patterns and individual (genetically determined) susceptibility translate into a large range of hepatic injuries ranging from steatosis to advanced conditions such as alcoholic steatohepatitis (ASH), alcohol-associated hepatitis (AH), and alcohol-associated cirrhosis (AC), all bearing the risk of leading to liver failure. People with ALD regularly experience stigma and discrimination, which contributes to inferior healthcare, low likelihood to request help, contributing to disease progression.4 To alleviate the stigma related to ALD, a new nomenclature has been proposed, replacing the term “alcoholic” by “alcohol-associated”, nonetheless maintaining the familiar abbreviations ALD, ASH and AC, with the exception of alcoholic hepatitis due to its long-established usage.5
Alcohol-induced liver damage occurs from different factors, via the alcohol metabolism which generates reactive oxygen species (ROS),6 alterations of gut microbiota called dysbiosis7 and the immune response to these disturbances that contribute to the development and progression of ALD.8 In addition, heavy alcohol consumption can lead to fat accumulation in the liver, in lipid droplets inside hepatocytes, and represents the first reaction to excessive alcohol exposure. This stage is referred to as alcohol-associated fatty liver disease, an early stage of ALD. Fatty liver disease is rarely accompanied by clinical symptoms; however, it represents a warning signal about harmful alcohol drinking pattern. At this stage, fatty liver disease is reversible by adopting a healthier lifestyle and stopping alcohol consumption. Steatosis can evolve to steatohepatitis following a second hit by another toxic agent and is accompanied by inflammation.9
AH is an acute manifestation of ALD and is a life-threatening condition with acute decompensation of the liver and an important risk of liver failure and mortality. AH is associated with necrosis and apoptosis alongside substantial recruitment of immune cells in the liver and accompanied by jaundice and ascites.10
Despite the wide variety of alcohol-induced liver injuries, inflammation remains a common hallmark of all alcohol-associated liver diseases.11 More specifically, macrophages, which constitute the main immune cells in the liver representing approximately 20% of the liver non-parenchymal cells, are key actors of inflammation-associated liver diseases due to their capacity to react to a large range of stimuli.12 Therefore, numerous studies have focused on liver macrophages throughout the last decades, shedding light on their versatility and the complexity of their polarization abilities, contrasting with the old classification mainly based on two subsets, inflammatory (“M1”) and anti-inflammatory (“M2”) macrophages. Technological advances such as single-cell RNA sequencing, spatial transcriptomics and proteo-transcriptomics13 or multiplex immunohistochemistry have broadened our understanding of macrophage heterogeneity, which was particularly useful to understand macrophage functionality in the context of metabolic, non-alcohol-associated fatty liver diseases.14 In this review, we focus on the various populations of hepatic macrophages in alcohol-associated liver disease and the underlying mechanisms driving their polarization. We also review the current knowledge about hepatic macrophage crosstalk with other hepatic cell populations in liver diseases, in order to support the exploration of targeted therapeutics by modulating macrophage polarization.
Macrophage Heterogeneity in the Liver
Kupffer Cells
Macrophages represent the largest immune cell population in the liver. They were initially described as phagocytic cells capable of recognizing, engulfing and degrading pathogens and cellular debris. Every 10 hepatocytes are accompanied by about 3 macrophages.15 In principle, two major macrophage populations exist that are either tissue-resident (ie Kupffer cells) or originate from infiltrating bone marrow-derived monocytes (ie monocyte-derived macrophages).16 Within the liver, the resident macrophages known as Kupffer cells (KCs) are large cells located along the sinusoids tightly attached to endothelial cells. Due to this strategic location, they represent the first cell population meeting gut-derived substances brought through the portal vein. They highly express scavenger, complement and pattern recognition receptors (PRR) such as toll-like receptors (TLR) and represent a major source of cytokines in the liver, making them key actors in the host defense against gut-derived microorganisms, whole-body homeostasis and in hepatic regeneration.17
Indeed, in response to their microenvironment, KC can produce a wide variety of cytokines and can acquire a pro-inflammatory phenotype (traditionally referred to as “M1”) activated by interferon (IFN)-γ, lipopolysaccharide (LPS) and characterized by a great ability to present antigens, produce inflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1, IL-6, IL-12, IL-23, nitric oxide, and are considered to be tumoricidal and microbicidal. Alternatively, KC can acquire an anti-inflammatory phenotype (previously termed “M2”) characterized by their ability to balance the inflammatory responses and facilitate tissue repair through the release of IL-10, IL-4, IL-13 and transforming growth factor (TGF)-β and low production of IL-12, IL-6 and TNF-α. In homeostasis, KCs contribute to maintain a “tolerogenic” immune environment in the liver.18
While this dichotomic view illustrates the spectrum of Kupffer cell responses towards environmental signals, more recent studies highlighted KC heterogeneity in the liver beyond their inflammatory polarization. While KCs are commonly described as CD45+F4/80+CD11bintCLEC4F+ cells in mice (Figure 1),19,20 fatty liver due to obesity provokes the occurrence of two KC populations in mice, a major CD206loESAM− population (KC1) and a minor CD206hiESAM+ population (KC2). The CD206hiESAM+ KC2 population is actively involved in fatty acid metabolism via CD36, demonstrating that KCs actively participate in metabolic processes.21 RNA sequencing analysis further highlighted the existence of a variety of hepatic macrophage subsets. Several genes have been identified to segregate between the different populations including the MAcrophage Receptor with COllagenous structure (MARCO) gene which is only expressed in non-inflammatory KC (Figure 1).
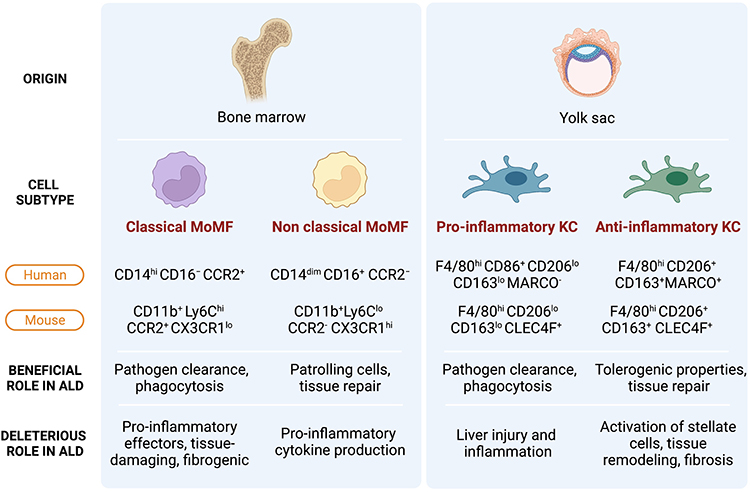 |
Figure 1 Hepatic macrophage populations and their roles in ALD. Hepatic macrophages are mainly composed of two ontogenetically distinct subtypes, monocyte-derived macrophages and liver resident Kupffer cells. Each subtype can further be regarded as heterogeneous populations based on their inflammatory properties, functions. Within Kupffer cells (KCs) and monocyte-derived macrophages (MoMFs), pro- and anti-inflammatory subtypes co-exist. The spectrum of macrophage phenotypes and their roles in ALD is presented in a simplified manner in this figure. |
Monocyte-Derived Macrophages
Although KCs represent the largest subpopulation of hepatic macrophages at steady state, upon hepatic injury circulating monocytes patrolling through the hepatic vascular network infiltrate the liver tissue and differentiate into monocyte-derived macrophages (MoMFs).12,16,22 For example, following alcohol consumption, an accumulation of monocytes in mouse liver has been reported.23,24 Their activation through the stimulation of TLR-2 was shown to contribute to inflammation and alcohol-associated hepatic injury in humans.25
Although KCs and MoMFs share most markers such as the ionized calcium-binding adapter molecule 1 (IBA1) also expressed by macrophages in other organs such as the microglia,26 a few markers have been proposed to distinguish KC from recruited macrophages.
The MoMFs are derived from bone marrow CX3CR1+CD117+ Lin− progenitors. CX3CR1, the G-protein coupled fractalkine receptor, is found on monocytes and capsular macrophages but is not expressed on (adult) KC.27 In humans, blood monocytes are classified into two main subpopulations (as well as an intermediate state) according to the expression of CD14, CD16, CCR2 and CX3CR1. The classical subset, which represents about 90% of circulating monocytes, is characterized by the phenotype CD14hiCD16− and CCR2+/CX3CR1lo.28 The non-classical monocytes are CD14dimCD16+ and CCR2−/CX3CR1hi/CCR5hi (Figure 1).
Similarly, in mice, we find the classical Ly6Chi monocytes comparable to the human CD14hiCD16− monocytes as well as non-classical Ly6Clo monocytes with some similarities to human CD14dimCD16+ monocytes (Figure 1). Ly6Chi monocytes can invade inflamed tissues but may switch to a Ly6Clo phenotype upon phagocytosis of apoptotic hepatocytes.29 Tissue Ly6Clo monocyte-derived macrophages have been associated with tissue repair.30
Macrophage Polarization in Early Stages of Alcohol-Associated Liver Disease
Role of Macrophage Populations in the Development of ALD
In ALD patients, increased macrophage numbers in portal areas have been observed along with increased macrophage-related biomarkers in the circulation.28 Conversely, hepatic macrophage depletion mediated by the administration of gadolinium chloride (GdCl3) protects against alcohol-provoked inflammation in the rat liver,31 indicating a crucial role of liver macrophages in the pathogenesis of alcohol-induced liver disease.
One of the functions of hepatic macrophages is the clearance of alcohol and gut-derived substances coming from the portal vein. Alcohol consumption is associated with an impairment of the intestinal barrier and alteration of the gut microbiota, which augments gut permeability and allows translocation of bacteria and their metabolites (such as LPS) that reach the liver via the portal circulation and activate hepatic macrophages (Figure 2).32 This has been particularly well described in severe alcoholic hepatitis, where enhanced gut permeability and increased portal levels of endotoxins result in the activation of Kupffer cells.33 Interestingly, the depletion of gut microbiota by the use of antibiotics or their direct clearance by KC can relieve ALD.34 Moreover, bacterial translocation appears to be especially pronounced in ALD in comparison to other types of liver injury, as concluded from comparisons of different mouse models.35
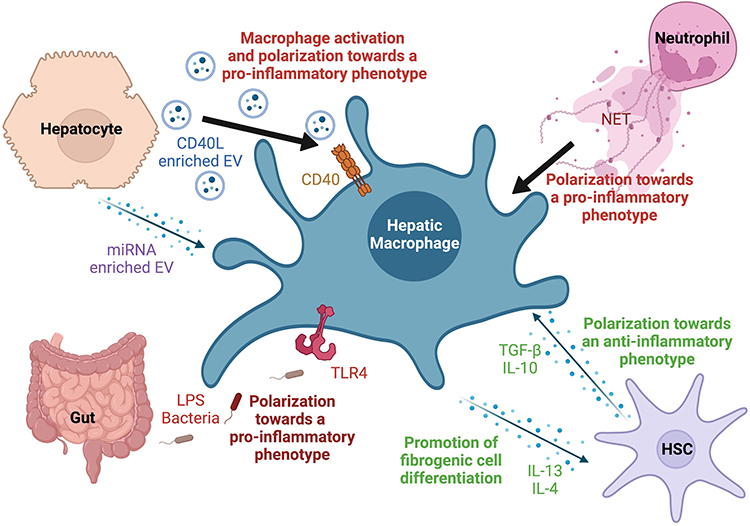 |
Figure 2 Macrophage crosstalk with hepatic cell populations and soluble signals in the context of ALD. Macrophages are sensitive to a wide variety of signals and pathogens and play an essential role in maintaining homeostasis. In the context of ALD, several factors such as extracellular vesicles (EVs) derived from injured hepatocytes, gut derived lipopolysaccharide (LPS) and other mediators can trigger their activation and polarize them towards a pro-inflammatory phenotype. Their interaction and mutual feedback with other non-parenchymal cells such as neutrophils and hepatic stellate cells (HSCs) play a major role in the development or containment of liver injury and fibrosis. |
The exposure to such pathogen-associated molecular patterns (PAMPs) from microbial pathogens (eg, bacteria, parasites, viruses) triggers inflammasome activation. Inflammasomes are important innate immune sensors in the form of cytoplasmic multiprotein complexes involved in maintaining cellular integrity in response to pathogens or stress signals.36 Upon stimulation, the activation of the inflammasome leads to the cleavage of caspase-1 and production of pro-inflammatory cytokines such as IL-1β and IL-18. More specifically, activation of the inflammasome inside hepatic macrophages has been reported in chronic liver diseases, such as ALD and non-alcoholic steatohepatitis (NASH).37 IL-1β, which is released subsequently to the activation of inflammasome in KC, plays a key role in the development of alcohol-mediated steatosis and inflammation, contributing to liver injury.38 In mice, deletion of caspase-1 or caspase-1 adaptor (ASC) impaired IL-1β production, thereby alleviating ALD.38
Alongside liver-resident Kupffer cells, following chronic alcohol consumption in mice, it has been shown that monocyte-derived macrophages accumulate in the liver, consistently with observations from other types of chronic liver injuries.29 A more detailed phenotypic analysis revealed a polarization of monocyte-derived macrophages based on the expression of Ly6C, where Ly6C+ macrophages displayed inflammatory and tissue-damaging effect, while Ly6Clo monocytes showed anti-inflammatory and tissue-protective properties. In this study, using C57BL/6J mice subjected to a 4-week ethanol feeding, the ratio of Ly6Chi/Ly6Clow macrophages is higher in mice chronically fed ethanol and binged, which substantially aggravated liver injury.29
Mechanisms of Macrophage Polarization in Early Stages of Steatosis and Steatohepatitis
Researchers have investigated how alcohol consumption may trigger hepatic macrophage activation in early stages of ALD. One of the main signals involved in the early stages of ALD, and in particular alcohol-associated steatosis, is oxidative stress. In fact, reactive oxygen species (ROS) production may be one of the mechanisms responsible for Kupffer cell sensitization to LPS in alcohol-associated liver disease.39 Prolonged exposure to ethanol induces macrophage-derived ROS production.40 Interestingly, rats pretreated with a nicotinamide adenine dinucleotide phosphate (NADPH) inhibitor to reduce ROS production exhibited reduced phosphorylation of extracellular signal-regulated kinase (ERK)-1/2 and lower production of TNF-α in Kupffer cells, highlighting the key role of ROS production in Kupffer-cell activation.39,40
Additionally, the receptor complex CD14/TLR4 has also been described as an important component in alcohol-mediated macrophage activation. The LPS-induced activation of this receptor triggers downstream signaling kinases in macrophages including the IL-1R associated kinase (IRAK) and IkappaB kinase (IKK), leading to the induction of the proinflammatory cytokines TNF-α, IL-6, and macrophage chemoattractant protein (MCP)-1, also known as chemokine C–C motif ligand 2 (CCL2). Indeed, macrophages from alcohol-fed mice display increased LPS responses associated with higher levels of TNF-α and MCP-1.41,42
Increased levels of TNF-α, along with IL-1β, IL-1α, IL-6 and CCL3 have also been reported in KC isolated from mice with myeloid specific deletion of the cannabinoid receptor 2 (CB2Mye−/−), upon LPS stimulation, indicating a macrophage switch towards a pro-inflammatory phenotype.43 The authors of this study further show that the cannabinoid receptor 2 (CB2) on KC protects mice from ALD in an autophagy-dependent pathway. Indeed, in their murine models, mice with myeloid-specific deletion of CB2 receptor (CB2Mye−/−) or autophagy gene ATG5 (ATG5Mye−/−) displayed exacerbated liver inflammation and alcohol-induced steatosis.43 These data highlight the anti-inflammatory and anti-steatogenic properties of macrophage autophagy mediated by CB2 in ALD, suggesting a rationale for the development of CB2 agonists to stimulate macrophage autophagy and inflammation in the context of ALD. In the same line of evidence, disruption of the LC3-associated phagocytosis pathway (a non-canonical form of autophagy) in monocytes from cirrhotic patients and in genetically modified mice with chronic liver disease enhances inflammation and liver fibrosis.44
Besides changes in macrophage activation state and phenotype, the number of hepatic macrophages significantly increases via MoMF recruitment, upon alcohol-induced liver damage.29 However, the contribution of KCs and MoMFs to liver inflammation and injury depends greatly on the disease and model used. For instance, in a murine model of steatohepatitis induced by high-fat diet and alcohol, proinflammatory MoMFs are activated to produce proinflammatory TNF-α and IL-1β in a Notch1-dependent manner with subsequent inflammatory macrophage polarization, with no significant change in the KC population.45,46 Additionally, Wingless and Int-1 (Wnt) expression was shown to be induced in infiltrating macrophages during steatosis.47 Whether these pro-inflammatory macrophages persist or switch phenotype in the course of ALD remains to be clarified. This process is further compounded by alcohol-induced intestinal gut microbial dysbiosis and impaired gut epithelial integrity favoring translocation of gut-derived microbial products such as LPS.
Observations on current drinkers with mild liver injury and steatosis, anti-inflammatory macrophages were more predominant than in patients with severe liver injury who harbor more pro-inflammatory macrophages,48 indicating that in the early stages of ALD such as steatosis, hepatic macrophages seem to display anti-inflammatory properties and to be protective. Several mechanisms driving macrophage polarization towards an anti-inflammatory phenotype have been described.
It is known that modifications of lipid metabolism in Kupffer cells are associated with pro-inflammatory phenotype. Scavenger receptors like MSR1 can bind saturated fatty acids and oxidized LDL, for example, which in turn polarize Kupffer cell phenotype into a pro-inflammatory state, as shown in msr1 knock-out mice subjected to a high fat diet. These mice display less inflammation and greater resistance to fibrosis.49 Additionally, in steatohepatitis, accumulation of toxic lipids including diacylglycerol and ceramide drives KC towards a pro-inflammatory phenotype. Macrophages can also be activated by cholesterol and phagocytosis of apoptotic steatotic hepatocytes.49
Krüppel‐like factor 4 (KLF4) is a transcription factor, which has been reported to play a role in macrophage polarization in the context of ALD. Indeed, in mice overexpressing KLF4 the expression of ethanol-induced pro-inflammatory mediators TNF‐α and IL‐1β was decreased, and the levels of anti-inflammatory markers Arginase (Arg) 1 and Mannose receptor C-type (Mrc) 1, also known as CD206, was increased, indicating a role of KLF4 in macrophage polarization towards an anti-inflammatory phenotype.50
Lastly, prostacyclin synthase (PTGIS) is an enzyme of the prostaglandin pathway with anti-inflammatory properties. Although its role in ALD is not yet fully understood, a recent study showed that PTGIS is downregulated in ALD and that adenovirus-mediated PTGIS overexpression in vivo alleviated the inflammatory response and suppressed pro-inflammatory macrophage phenotype in mice. Loss- and gain-of function-experiments revealed that PTGIS overexpression inhibited macrophage switch to a pro-inflammatory phenotype in favor of an anti-inflammatory phenotype.51
Macrophage Polarization in Advanced Stages of Alcohol-Associated Liver Disease
Mechanisms of Macrophage Polarization in Advanced Stages of Alcohol-Associated Fibrosis and Cirrhosis
In advanced stages of alcohol-associated fibrosis and cirrhosis, macrophages play a critical role in the progression of the disease. The mechanisms of macrophage polarization in advanced stages of fibrosis and cirrhosis are thought to be mediated by various signaling pathways, including those activated by pro-inflammatory chemokines such as IFN-γ and TNF-α, as well as anti-inflammatory cytokines including IL-4 and IL-13.52
Additionally, patients with cirrhosis often experience complications, such as infections, leading to high morbidity and mortality. Indeed, more than 30% of patients with advanced cirrhosis develop bacterial infections.53 The increased susceptibility to infections is the results of distinctive immune alterations in advanced liver diseases, termed cirrhosis-associated immune dysfunction (CAID).54 Variations of monocyte and macrophage phenotype and functions during cirrhosis critically contribute to cirrhosis-associated immunoparesis.55
Chronic alcohol consumption also leads to the activation of hepatic stellate cells (HSCs), which produce excessive extracellular matrix proteins, leading to liver fibrosis. In the context of chronic alcohol consumption, HSCs release anti-inflammatory cytokines such as IL-10 and TGF-β that polarize macrophages towards an anti-inflammatory phenotype. These macrophages are implicated in tissue repair and fibrosis resolution.56 Anti-inflammatory macrophages can in turn produce cytokines such as IL-13 and IL-4, which promote the differentiation of fibrogenic cells such as myofibroblasts (Figure 2). When activation of anti-inflammatory macrophages persists, this profibrogenic state leads to liver fibrosis and cirrhosis by inducing the production of collagen and other extracellular matrix components. Liver fibrosis is characterized by impairment of the composition of the extracellular matrix. In the healthy liver, the extracellular matrix mainly comprises collagen type IV, laminin, and proteoglycans, which are replaced by collagen type Ι and III upon fibrogenesis leading to the disruption of the lobule architecture.57
In response to alcohol-induced liver tissue damage, Ly-6Chi monocytes infiltrate the liver, differentiate into pro-inflammatory macrophages and produce cytokines and chemokines that attract other immune cells. The monocyte recruitment relies on the CCL2-CCR2 axis. In mice, the disruption of the CCL2-CCR2 axis through genetic deletion or pharmacological inhibition led to reduced liver fibrosis.58 Recently, scRNA-seq data from human cirrhotic livers have revealed the presence of scar-associated macrophages (SAM) derived from MoMF, preferentially located in fibrotic areas.59
Fibrogenic macrophages can switch towards a restorative phenotype with low Ly-6C expression and higher levels of anti-inflammatory mediators (such as IL-10) and matrix metalloproteinases (including MMP-9, MMP-12, MMP-13).29,60 The underlying mechanisms driving this phenotypic plasticity have not been fully elucidated yet. Evidence from murine models indicates that reduction of danger signals and resolution of liver injury promotes a restorative macrophage phenotype.60
Mechanisms of Macrophage Polarization in Alcohol-Associated Hepatitis and Acute-on-Chronic Liver Failure
In chronic alcohol-associated liver disease, liver injury is characterized by chronic inflammation and tissue damage, leading to fibrosis and cirrhosis. Macrophages are involved in the pathogenesis of alcohol-associated chronic hepatitis and can undergo polarization towards different phenotypes depending on the microenvironmental cues present in the liver. The principal relevance of macrophages for alcohol-associated hepatitis is probably best demonstrated by the fact that their accumulation in portal areas was identified as the strongest immunological feature determining disease severity in a recent analysis of human liver biopsies by sequential multiplex immunostaining.61
In a group of heavy alcohol drinkers, the expression of the anti-inflammatory macrophage markers CD206 and CD163 in the liver was lower in patients with advanced liver injury than in those with minimal hepatic damage. Consistently, following ethanol feeding, BALB/c mice, which display higher anti-inflammatory markers compared to alcohol fed C57BL6/J, exhibit significantly less damage and fat accumulation in the liver than their C57BL6/J counterparts. Interestingly, in the BALB/c mice, which displayed a significant resistance to ethanol injury mediated by F4/80+CD206+iNOS− anti-inflammatory macrophages, the authors reported a mechanism driving the apoptosis of the pro-inflammatory subset mediated by anti-inflammatory KC. Mechanistically, IL-10 secreted by anti-inflammatory KC promotes the death of F4/80+CD206−iNOS+ pro-inflammatory macrophages displaying high expression of inducible nitric oxide synthase, through the induction of arginase. In alcohol-fed mice, IL-10 neutralization led to an impairment in pro-inflammatory macrophage apoptosis. BALB/c mice chronically exposed to alcohol displayed a significant reduction in the number of KC, associated with a lower density of pro-inflammatory KC. Remaining KC in alcohol-fed mice predominantly (60% of F4/80+CD206+ macrophages) acquired an anti-inflammatory phenotype.48
Mechanistically, hepatic macrophage polarization towards a pro-inflammatory phenotype is highly regulated by the nuclear regulatory factor κB (NFκB), a key regulator of cellular stress in virtually all hepatic cell types. By forming p65/p50 heterodimers in macrophages, NFκB binds to the promoter region of pro-inflammatory genes leading to their activation. Increased NFκB activation in both liver resident macrophages and monocytes is observed in chronic ALD patients compared to controls.62
One of the mechanisms leading to NFκB activation involves the activation of TLR4 by circulating LPS in chronic alcohol-mediated liver injury. At steady state, the proteins of the NFκB family, which include RelA/p65, RelB, c-Rel and p50, form dimers with inhibitory κB (IκB) molecules in the cytosol. Upon stress signals and injury, danger signals induce IκB phosphorylation leading to its ubiquitination and degradation by the proteosome. The dissociation of IκB exposes NFκB translocation sites. In murine models of chronic alcohol feeding, liver analysis showed increased NFκB nuclear translocation and DNA binding.62
Transcription factor AP-1 is also upregulated following alcohol exposure and has been shown to increase macrophage sensitization leading to higher CD14 mRNA levels and pro-inflammatory cytokine production.63 Indeed, ROS inhibition in the liver abrogates the expression of AP-1 activity and CD14. Additionally, in human monocytes, the alcohol-mediated activation of AP-1 is dependent on Src kinase and has been shown to promote IL-10 production.64
Recent data reported a role of extracellular vesicles (EVs) in ALD and their potential use for diagnostics (as biomarkers for instance) or therapeutic purposes.65 Indeed, AH patients display higher levels of circulating EVs/exosomes, suggesting that they could serve as potential diagnostic markers and may facilitate the development of novel strategies for diagnostics, monitoring, and therapeutics of AH.66 From a macrophage perspective, EVs may be a central component on how alcohol-injured hepatocytes communicate with macrophages and other non-parenchymal cells to initiate inflammatory and fibrogenic activation.
Recently, in a mouse model of alcoholic hepatitis (AH), significant increases in EV levels have been observed following alcohol consumption. Analysis of the content of these EVs revealed that 20 miRNAs were upregulated (including miR-181 and miR-27a) and 40 were downregulated, in mice exposed to alcohol. Transfection of miR-181 and miR-27a in HSCs leads to the inhibition of NR1D2 (repressor of transcription). Moreover, the analysis of EV composition of hepatic macrophages showed that damage-associated molecular patterns (DAMPs), such as mitochondrial DNA and organelle proteins, induce IL17A and IL1β upregulation mediated by TLR9.67
Another study, carried out on mice with alcohol-associated liver disease, reported that heat shock protein 90 of circulating EVs from ALD mice demonstrates a functional role in hepatic macrophage activation.68 In mice who received a transfer of ALD EVs, hepatocytes displayed higher levels of monocyte chemoattractant protein 1, which was not observed in control EV-administered mice, indicating a role of ALD EVs on monocyte recruitment. In addition, mice who received ALD EV displayed significantly more F4/80hi CD11blo KCs and higher percentages of pro-inflammatory TNF-α+IL-12+IL-23+ KCs and F4/80int CD11bhi recruited monocytes and lower percentage of anti-inflammatory CD206+CD163+ KC than control EV-administered mice. In vitro, ALD EV administration induced IL-1β and TNF-α expression in macrophages and inhibited CD163 and CD206 expression. The authors further observed that heat shock protein 90 contained inside ALD EV was a key macrophage activator in ALD.68
Alcohol-associated hepatitis can further evolve to acute-on-chronic liver failure (ACLF) with a high risk of short-term death within 28 days after hospital admission. This life threatening liver condition is characterized by important hepatocellular loss exceeding the liver’s ability to regenerate leading to acute hepatic decompensation, jaundice, ascites, single- or multiple-organ failure with intense systemic inflammation.69 Paradoxically, patients with liver failure are also immunosuppressed with apparent immune dysfunctions making them more vulnerable to infections.70 Patients with ACLF have higher risk of bacterial translocation and infections.71 In this context, monocytes and macrophages play a major role in sensing gut-derived PAMPs. Manifold adaptations have been reported in monocytes and macrophages during ACLF, especially in circulating monocytes, that include their subset distribution, cellular metabolism, expression of sensing receptor molecules, phagocytosis capacity and expression of effector mediators such as cytokines and chemokines.55,72
Understanding Macrophage Plasticity Through Their Crosstalk with Other Hepatic Cells
Hepatocytes
Hepatocyte ballooning is a common sign of cellular injury that histologically translates into swollen hepatocytes, reduced cytoplasm and loss of staining for cytokeratin 8 and 18.73 KCs have been shown to be crucial for hepatocyte regeneration and tissue repair after alcohol-induced liver damage. Indeed, KC depletion by clodronate-loaded liposomes leads to the impairment of proliferating hepatocyte DNA synthesis in mice during liver repair after alcohol-induced injury.74 Conversely, upon ethanol exposure, hepatocytes play a major role in macrophage activation. However, the underlying mechanisms responsible for macrophage activation by hepatocyte exposed to alcohol in ALD are not fully understood.
The development of efficient techniques for the study of EV has shed light on their key functions in cellular communication and crosstalk in the context of ALD. In vitro, EV from alcohol treated hepatocytes stimulated macrophage activation and their polarization towards a pro-inflammatory phenotype with induction of TNF-α, IL-1β and IL-6 production.75 Further analysis revealed that EVs derived from alcohol-exposed hepatocyte (Alc-EV) were enriched in CD40L (TNFSF5), a member of the TNF family which mediates macrophage activation (Figure 2). Neutralization of CD40L from Alc-EV abrogated the induction of pro-inflammatory cytokine production from activated THP-1 macrophages and primary macrophages. In addition, CD40L neutralization attenuated EV-induced ERK activation indicating a role of ERK in pro-inflammatory cytokine production.75 As ethanol-induced EV release requires caspase activation, alcohol-induced injury and macrophage infiltration were significantly reduced in mice receiving a pan-caspase inhibitor or with genetic deletion of CD40 or the caspase-activating TRAIL receptor, in vivo. Consistently, serum from patients with alcoholic hepatitis showed increased levels of CD40L-enriched EV.75
Monocyte chemoattractant protein 1 mRNA levels were also increased in hepatocytes isolated from mice after transfer of ALD EVs.68 Compared to control-EV-recipient mice, ALD-EV administration led to higher number of F4/80hiCD11blo KC and percentages of proinflammatory KC producing TNF-α, IL-12 and IL-23 along with higher rates of F4/80intCD11bhi infiltrating monocytes, and a reduction of anti‐inflammatory CD206+CD163+ KC percentage.68
Another study on EV reported the role of hepatocyte-derived macrophage migration inhibitory factor (MIF) on liver macrophage populations following ethanol exposure. Immunohistochemical analysis performed on liver tissue sections from AH patients showed that macrophage MIF was predominantly expressed within hepatocytes. Interestingly, MIF levels in suprahepatic serum were found to be positively associated with the severity of the disease and mortality.76 The role of MIF on macrophage population and alcohol-associated liver injury was further deciphered in vivo, using chimeric mice. Mice lacking MIF specifically in non-myeloid cells did not develop liver injury after alcohol feeding. Immunohistochemistry analysis of F4/80+ macrophages and Ly6C+ monocytes revealed that both populations were significantly increased after chronic ethanol exposure, while no change was observed in livers of chimeric mice lacking MIF in hepatocytes and non-myeloid cells indicating a role of hepatocyte-derived MIF on the expansion of macrophage populations in the liver following ethanol exposure.76
Neutrophils
Lobular inflammation is often characterized by a major recruitment of neutrophils, surrounding ballooned hepatocytes,73 that interact with macrophages. In liver biopsies of ALD patients, neutrophils, monocytes and monocyte-derived macrophages were mostly observed in peri-central areas with steatosis among the steatotic patients.77 However, it is important to note that different histopathological subtypes seem to exist, particularly for patients with severe AH, including either high neutrophil accumulation (and low CD8 T lymphocytes) or high CD8 T lymphocytes (and low neutrophil numbers), suggesting immunologically distinct drivers of disease progression in patient subgroups.78
Growing evidence suggests that there is an important cellular crosstalk between hepatic macrophage populations and neutrophils.8,79 As the liver constitutes a major site for the clearance of bacteria, viruses, and other foreign substances, both macrophages and neutrophils play essential roles in orchestrating the immune response to these pathogens. Macrophages team up with neutrophils for the clearance of pathogens, dead cells and cellular debris tagged for phagocytosis. Neutrophils, on the other hand, are recruited to the liver upon infection or injury and are involved in the elimination of invading pathogens and tissue repair.80 Studies have shown that macrophages can regulate neutrophil recruitment to the liver during infection. Additionally, macrophages can also regulate neutrophil activity by producing cytokines and chemokines that either activate or suppress neutrophil function.
Chronic alcohol exposure induces phenotypic changes in neutrophils and augments LPS-induced neutrophil-extracellular trap (NET) production.81 Interestingly, in vitro, co-culture of monocytes with NETs or NET-producing neutrophils led to the differentiation of monocytes into a CD14+CD16+ intermediate phenotype, which is known to be increased in ALD. Additionally, the CD86 expression, a marker of pro-inflammatory macrophages, increased after co-culture with NET-producing neutrophils. These findings indicate that NETs are potent macrophage activators and can drive their differentiation towards a pro-inflammatory phenotype that promotes inflammation.81 Furthermore, flow cytometry analysis on blood from AH patients showed that a subset of neutrophils referred to as low-density neutrophils (LDNs) was enriched in the circulation. Whole transcriptome profile revealed that these neutrophils are characterized by an exhausted phenotype, impaired functions and escape from macrophage clearance. Interestingly, no LDNs were found in the circulation of healthy controls, or that of NASH patients indicating the specificity of enriched LDNs to AH.81
Hepatic Stellate Cells
Activated HSCs are known to be major producers of extracellular matrix proteins that contribute to liver fibrosis. Studies have shown that macrophages play a key role in the progression of ALD by modulating HSC activation and promoting inflammation and fibrosis in the liver (Figure 2).82
Indeed, in vitro, non-classical CD14+CD16+ macrophages have been reported to activate stellate cells.83,84 In a cohort of 226 patients with chronic liver disease (CLD) and 184 healthy controls, blood monocytes were significantly higher in CLD-patients than in healthy controls with a shift towards the non-classical CD14+CD16+ phenotype which correlated with proinflammatory cytokine production and disease progression. Higher numbers of CD14+CD16+ macrophages were also observed in patients with cirrhosis. On average, these cells represented half of the total hepatic monocytes/macrophages in cirrhosis patients while accounting for just 10% in the liver of non-cirrhotic patients, which explains the total increase of macrophages in cirrhotic livers.84 Furthermore, experiments where monocytes were HSC co-cultured with CD14+CD16+ monocytes displayed higher collagen expression, while no induction was observed with CD14+CD16− monocytes. The CD14+CD16+ monocyte-mediated HSC activation was partly abolished with anti-TGF-β antibodies. This highlights that in addition to the production of proinflammatory mediators, monocytes can also favor fibrogenic behaviors on HSC.84
Beyond the HSC activation, monocyte-derived macrophages can also induce the transdifferentiation of HSC into collagen-producing myofibroblasts through the secretion of TGF-β. Upon chronic liver injury, CD11b+F4/80+Ly6C+ monocytes differentiate into macrophages characterized by a high production of nitric oxide synthase along with profibrogenic and proinflammatory properties triggering HSC activation. Indeed, impaired monocyte recruitment in Ccr2−/− mice resulted in weaker HSC activation and reduced liver fibrosis.85
Similar to recruited monocytes, hepatic macrophages have been shown to induce HSC activation, in different contexts including NASH as well.86 A microarray analysis revealed that co-culture of HSCs and F4/80+ hepatic macrophages from fibrotic livers strongly induced the overexpression of NF-κB–regulated genes which were inhibited by the administration of adenoviral IκB suppressor or IKK inhibitor.87 In addition, clodronate-induced macrophage depletion significantly repressed the expression of the NF-κB-dependent gene that were upregulated by hepatic macrophages in the co-culture setup. Further investigation revealed that hepatic macrophages induce NF-κB in a TNF-α and IL-1β-dependent manner. Indeed, TNF-α and IL-1β neutralization with antibodies resulted in a significant reduction of NF-κB-mediated luciferase activity.87
Novel Strategies Using Macrophages as Therapeutic Targets in ALD
To date, alcohol abstinence constitutes the most sustainable and efficient care for ALD. Thus, treatment of ALD cannot solely focus on the liver disease itself but needs to address the alcohol use disorder (AUD) as well, so that liver function recovery is not compromised by the patient returns to alcohol consumption.88 There are multiple pharmacologic and non-pharmacologic therapies for AUD, which are discussed elsewhere.89 In addition to abstinence, several liver-directed pharmacotherapy options have been proposed.
The most widely explored treatment for AH is corticosteroids, which are intended to alleviate the hepatic inflammation.10 In a large prospective trial, prednisolone was correlated with a moderate reduction in short-term mortality of AH patients, while long-term benefit was determined by abstinence.90 More recent clinical observations indicate that AH patients with a MELD (model of end-stage liver disease) score between 25 and 39 are most likely to benefit from steroids.91 The major risk of corticosteroids in patients with AH is the increased number of infectious complications.90
Macrophages play crucial functions in liver injury, but also in host defense. Therefore, there is a concern that impairing their functions or their recruitment might be counterproductive (eg, increasing the risk of infections). Instead, a strategic therapeutic approach based on the use of key factors that trigger a switch in macrophage polarization favoring a restorative phenotype could be more efficient and help accelerate injury resolution and support hepatocyte regeneration. One promising approach is to modulate the activity of macrophages by using drugs that modulate the effects of pro-inflammatory cytokines or that promote the activity of anti-inflammatory cytokines. Anakinra, is an anti-IL-1 monoclonal antibody that has been tested in clinical trials (NCT04072822).92 However, in a recently reported randomized controlled trial, anakinra (in combination with pentoxifylline and zinc) was not superior to current standard treatment with prednisolone in patients with AH.93
Since chemokines play an important role in the recruitment of inflammatory monocytes and in the progression of ALD, they may represent a therapeutic target for the control of liver inflammation. For example, data in mice showed that MIF exerts protective properties against ethanol-mediated liver damage.94 Preclinical evidence on mice also showed that cenicriviroc, a CCR2/5 antagonist can prevent and reverse alcohol-mediated inflammation, steatosis and liver injury.95 However, cenicriviroc did not provide sustained histological benefit in clinical trials involving patients with NASH,96 making it unlikely that this compound is tested in patients with AH.
Another strategy for targeting macrophages in ALD is to modulate their polarization to inhibit pro-inflammatory signaling pathways and promote anti-inflammatory signaling pathways to shift the balance of macrophage polarization towards anti-inflammatory macrophages, which are responsible for the resolution of inflammation and tissue repair.97 One example is adiponectin, an anti-inflammatory adipokine. Indeed, globular adiponectin treatment abrogated LPS-mediated TNF-α expression in Kupffer cells in an IL-10/STAT3/heme oxygenase-1 dependent manner and through the induction of an anti-inflammatory phenotype.98,99 Conversely, another potent strategy for the treatment of ALD might aim at promoting IL-10-mediated desensitization of alcohol-exposed macrophages. Indeed, lower levels of IL-10 following alcohol exposure play an important role in sensitizing macrophages, and data obtained in IL-10 knock-out mice reported higher levels of proinflammatory cytokines following alcohol exposure.100 On the contrary, TLR-3 activation was found to alleviate alcohol-induced liver injury by promoting the production of IL-10 from KC and HSC.101
In the same line of evidence, another study showed that TLR2 and TLR3-deficiency ameliorated and accentuated ALD-induced liver injury, respectively. The underlying mechanisms involve Kupffer cell inactivation, a polarization switch from pro- to anti-inflammatory phenotype, and STAT3-mediated IL-10 expression resulting in the TLR3 stimulation and TLR2 inhibition. Furthermore, the authors identified polyphenol epigallocatechin-3-gallate (EGCG), a green tea extract that interacts with TLR2 and TLR3 on macrophages to promote IL-10 production. IL-10 deficiency worsened ALD and abolished EGCG-induced hepatoprotection, while macrophage depletion partially recovered liver injury but abrogated the effect of EGCG.102
In more advance hepatic conditions such as liver failure syndromes, immune dysfunction is common, which translates to a higher risk for the patients to develop infectious complications. In this context, the effects of hematopoietic growth factors aiming at restoring immune functions represent an interesting therapeutic approach.103 In a study conducted with ACLF patients, administration of granulocyte colony-stimulating factor (G-CSF) for 28 days, classically secreted by macrophages and other immune cells, was described to reduce disease gravity, infections and improved survival.104 However, this finding was not replicated in a larger multicenter European trial, which involved mainly patients with AH or ACLF based on alcohol-associated liver cirrhosis. In fact, G-CSF caused more (and partly serious) adverse effects while not providing a substantial survival benefit.105 Clearly, a much more granular understanding of pathomechanisms is needed to tailor a “macrophage-targeted” intervention. In experimental mouse models of ACLF, G-CSF combined with a TLR4 inhibitor reduced inflammation and promoted liver regeneration.106
Conclusions
Alcohol-related liver disease represents a major public health concern worldwide with high morbidity and mortality. Beyond the prior dichotomic classifications of macrophages, hepatic macrophages represent heterogenous cell populations involving circulating monocytes infiltrating the hepatic parenchyma as well as liver resident KC. These cells play various roles in homeostasis and pathology, which correspond to a large spectrum of phenotypes. Data have shown that macrophage polarization is a key element in the early phase and the development of liver diseases. The underlying mechanisms involved in macrophage polarization in ALD are complex and not fully understood. However, studies show that chronic alcohol consumption modulates macrophage polarization through several mechanisms, including gut-derived endotoxemia, which activates TLR4 signaling pathway and leads to macrophage polarization towards a pro-inflammatory phenotype and the production of pro-inflammatory cytokines; conversely, the activation of signaling pathways associated with CB2 tends to favor an anti-inflammatory phenotype. Although anti-inflammatory macrophages are beneficial to alleviate alcohol-associated inflammation, their prolonged activation leads to the stimulation of HSC, which significantly contributes to liver fibrosis and the development of ALD. Given the lack of efficient therapeutic options for advanced stages of ALD, targeting macrophages should be further explored as a promising strategy. Rather than aiming at depleting hepatic macrophages, the focus at early stages should be to suppress inflammation and excessive macrophage activation by inhibiting bacterial translocation with the administration of antibiotics, which have been shown to improve steatohepatitis, and inhibit pathogen recognition receptors using TLR antagonists,107 for example. Additionally, some novel therapeutic strategies developed for later stages of ALD include drugs that inhibit monocyte recruitment, modulate macrophage polarization towards an anti-inflammatory phenotype to promote tissue repair and reduce inflammation in ALD or regulate macrophage-mediated cytokine production. The intensive research aiming at characterizing macrophage populations has improved our knowledge about hepatic macrophages and refined our perception of this widely heterogenous population. This rapid increase in scientific knowledge regarding the underlying mechanisms driving macrophage polarization in ALD is leading to the identification of new potential new therapeutic target and novel promising therapies.
Acknowledgments
The authors sincerely thank all lab members and collaborators for helpful discussions. F.T. is supported by the German Research Foundation (DFG Ta434/8-1, SFB/TRR 296 and CRC1382, Project-ID 403224013) and the German Ministry of Education and Research (BMBF DEEP-HCC consortium, BMBF Immun Avatar consortium).
Disclosure
FT’s lab has received research funding from Allergan, Bristol-Myers Squibb, Gilead and Inventiva. FT has received honoraria for consulting or lectures from Astra Zeneca, Gilead, AbbVie, BMS, Boehringer, Madrigal, Intercept, Falk, Ionis, Inventiva, Merz, Pfizer, Alnylam, NGM, CSL Behring, Novo Nordisk, Novartis. The other authors report no conflicts of interest in this work.
References
1. Huang DQ, Terrault NA, Tacke F, et al. Global epidemiology of cirrhosis - aetiology, trends and predictions. Nat Rev Gastroenterol Hepatol. 2023:1–11. doi:10.1038/s41575-023-00759-2
2. Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol. 2019;70(1):151–171. doi:10.1016/j.jhep.2018.09.014
3. Marcellin P, Kutala BK. Liver diseases: a major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int. 2018;38(Suppl 1):2–6. doi:10.1111/liv.13682
4. Schomerus G, Leonhard A, Manthey J, et al. The stigma of alcohol-related liver disease and its impact on healthcare. J Hepatol. 2022;77(2):516–524. doi:10.1016/j.jhep.2022.04.026
5. Crabb DW, Im GY, Szabo G, Mellinger JL, Lucey MR. Diagnosis and Treatment of Alcohol‐Associated Liver Diseases: 2019 Practice Guidance From the American Association for the Study of Liver Diseases. Hepatology. 2020;71(1):306. doi:10.1002/hep.30866
6. Tan HK, Yates E, Lilly K, Dhanda AD. Oxidative stress in alcohol-related liver disease. World J Hepatol. 2020;12(7):332–349. doi:10.4254/wjh.v12.i7.332
7. Day AW, Kumamoto CA. Gut microbiome dysbiosis in alcoholism: consequences for health and recovery. Front Cell Infect Microbiol. 2022;12. doi:10.3389/fcimb.2022.840164
8. Xu H, Wang H. Immune cells in alcohol-related liver disease. Liver Research. 2022;6(1):1–9. doi:10.1016/j.livres.2022.01.001
9. Singh S, Osna NA, Kharbanda KK. Treatment options for alcoholic and non-alcoholic fatty liver disease: a review. World J Gastroenterol. 2017;23(36):6549–6570. doi:10.3748/wjg.v23.i36.6549
10. Bataller R, Arab JP, Shah VH. Alcohol-Associated Hepatitis. N Engl J Med. 2022;387(26):2436–2448. doi:10.1056/NEJMra2207599
11. Lackner C, Stauber RE, Davies S, et al. Development and prognostic relevance of a histologic grading and staging system for alcohol-related liver disease. J Hepatol. 2021;75(4):810–819. doi:10.1016/j.jhep.2021.05.029
12. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol. 2021;18(1):45–56. doi:10.1038/s41423-020-00558-8
13. Govaere O, Hasoon M, Alexander L, et al. A proteo-transcriptomic map of non-alcoholic fatty liver disease signatures. Nature Metabolism. 2023;5(4):572–578. doi:10.1038/s42255-023-00775-1
14. Vonderlin J, Chavakis T, Sieweke M, Tacke F. The Multifaceted Roles of Macrophages in NAFLD Pathogenesis. Cell Mol Gastroenterol Hepatol. 2023;S2352-345X(23)00039–5. doi:10.1016/j.jcmgh.2023.03.002
15. Lopez BG, Tsai MS, Baratta JL, Longmuir KJ, Robertson RT. Characterization of Kupffer cells in livers of developing mice. Comp Hepatol. 2011;10(1):2. doi:10.1186/1476-5926-10-2
16. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17(5):306–321. doi:10.1038/nri.2017.11
17. Ait Ahmed Y, Fu Y, Rodrigues RM, et al. Kupffer cell restoration after partial hepatectomy is mainly driven by local cell proliferation in IL-6-dependent autocrine and paracrine manners. Cell Mol Immunol. 2021;18(9):2165–2176. doi:10.1038/s41423-021-00731-7
18. Heymann F, Peusquens J, Ludwig-Portugall I, et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology. 2015;62(1):279–291. doi:10.1002/hep.27793
19. Hsieh SLE, Yang CY. CLEC4F, A Kupffer Cells Specific Marker, Is Critical for Presentation of Alfa-Galactoceromide to NKT Cells (78.38). J Immunol. 2009;182(1_Supplement):78. doi:10.4049/jimmunol.182.Supp.78.38
20. Yang CY, Chen JB, Tsai TF, et al. CLEC4F Is an Inducible C-Type Lectin in F4/80-Positive Cells and Is Involved in Alpha-Galactosylceramide Presentation in Liver. PLoS One. 2013;8(6):e65070. doi:10.1371/journal.pone.0065070
21. Blériot C, Barreby E, Dunsmore G, et al. A subset of Kupffer cells regulates metabolism through the expression of CD36. Immunity. 2021;54(9):2101–2116.e6. doi:10.1016/j.immuni.2021.08.006
22. Cai J, Zhang XJ, Li H. The role of innate immune cells in nonalcoholic steatohepatitis. Hepatology. 2019;70(3):1026–1037. doi:10.1002/hep.30506
23. Alharshawi K, Fey H, Vogle A, Klenk T, Kim M, Aloman C. Alcohol Consumption Accumulation of Monocyte Derived Macrophages in Female Mice Liver Is Interferon Alpha Receptor Dependent. Front Immunol. 2021;12. doi:10.3389/fimmu.2021.663548
24. Yin F, Wu M, Wei X, et al. Hepatic NCoR1 deletion exacerbates alcohol-induced liver injury in mice by promoting CCL2-mediated monocyte-derived macrophage infiltration. Acta Pharmacol Sin. 2022;43(9):2351–2361. doi:10.1038/s41401-022-00863-0
25. Maccioni L, Kasavuli J, Leclercq S, et al. Toll-like receptor 2 activation in monocytes contributes to systemic inflammation and alcohol-associated liver disease in humans. Hepatol Commun. 2023;7(5):67.
26. Guillot A, Buch C, Jourdan T. Kupffer Cell and Monocyte-Derived Macrophage Identification by Immunofluorescence on Formalin-Fixed, Paraffin-Embedded (FFPE) Mouse Liver Sections. In: Aouadi M, Azzimato V editors. Kupffer Cells: Methods and Protocols. Springer US; 2020:45–53. doi:10.1007/978-1-0716-0704-6_6
27. Yona S, Kim KW, Wolf Y, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91. doi:10.1016/j.immuni.2012.12.001
28. Ju C, Mandrekar P. Macrophages and alcohol-related liver inflammation. Alcohol Res. 2015;37(2):251–262.
29. Wang M, You Q, Lor K, Chen F, Gao B, Ju C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J Leukoc Biol. 2014;96(4):657–665. doi:10.1189/jlb.6A0114-004RR
30. Peiseler M, Schwabe R, Hampe J, Kubes P, Heikenwälder M, Tacke F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease - novel insights into cellular communication circuits. J Hepatol. 2022;77(4):1136–1160. doi:10.1016/j.jhep.2022.06.012
31. Dou L, Shi X, He X, Gao Y. Macrophage Phenotype and Function in Liver Disorder. Front Immunol. 2019;10:3112. doi:10.3389/fimmu.2019.03112
32. Li F, McClain CJ, Feng W. Microbiome dysbiosis and alcoholic liver disease☆. Liver Res. 2019;3(3–4):218–226. doi:10.1016/j.livres.2019.09.001
33. Suraweera DB, Weeratunga AN, Hu RW, Pandol SJ, Hu R. Alcoholic hepatitis: the pivotal role of Kupffer cells. World J Gastrointest Pathophysiol. 2015;6(4):90–98. doi:10.4291/wjgp.v6.i4.90
34. Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: pathophysiological basis for therapy. J Hepatol. 2020;72(3):558–577. doi:10.1016/j.jhep.2019.10.003
35. Hsu CL, Wang Y, Duan Y, et al. Differences in Bacterial Translocation and Liver Injury in Ethanol Versus Diet-Induced Liver DiseaseDear author, please note that CrossRef and PubMed databases have been used to validate the references and mismatches have been updated in the manuscript. Please check carefully and confirm these have been done correctly. Dig Dis Sci. 2023. doi:10.1007/s10620-023-07860-1
36. Luan J, Ju D. Inflammasome: a double-edged sword in liver diseases. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.02201
37. Knorr J, Wree A, Tacke F, Feldstein AE. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin Liver Dis. 2020;40(3):298–306. doi:10.1055/s-0040-1708540
38. Petrasek J, Bala S, Csak T, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;122(10):3476–3489. doi:10.1172/JCI60777
39. Thakur V, Pritchard MT, McMullen MR, Wang Q, Nagy LE. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J Leukoc Biol. 2006;79(6):1348–1356. doi:10.1189/jlb.1005613
40. Kono H, Rusyn I, Yin M, et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest. 2000;106(7):867–872. doi:10.1172/JCI9020
41. Nagy LE. Recent insights into the role of the innate immune system in the development of alcoholic liver disease. Exp Biol Med (Maywood). 2003;228(8):882–890. doi:10.1177/153537020322800803
42. Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology. 2011;54(6):2185–2197. doi:10.1002/hep.24599
43. Denaës T, Lodder J, Chobert MN, et al. The Cannabinoid Receptor 2 Protects Against Alcoholic Liver Disease Via a Macrophage Autophagy-Dependent Pathway. Sci Rep. 2016;6:28806. doi:10.1038/srep28806
44. Wan J, Weiss E, Ben Mkaddem S, et al. LC3-associated phagocytosis protects against inflammation and liver fibrosis via immunoreceptor inhibitory signaling. Sci Transl Med. 2020;12(539):eaaw8523. doi:10.1126/scitranslmed.aaw8523
45. Gao B, Ahmad MF, Nagy LE, Tsukamoto H. Inflammatory pathways in alcoholic steatohepatitis. J Hepatol. 2019;70(2):249–259. doi:10.1016/j.jhep.2018.10.023
46. Xu J, Chi F, Guo T, et al. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J Clin Invest. 2015;125(4):1579–1590. doi:10.1172/JCI76468
47. Debebe A, Medina V, Chen CY, et al. Wnt/β-catenin activation and macrophage induction during liver cancer development following steatosis. Oncogene. 2017;36(43):6020–6029. doi:10.1038/onc.2017.207
48. Wan J, Benkdane M, Teixeira-Clerc F, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. 2014;59(1):130–142. doi:10.1002/hep.26607
49. Gilgenkrantz H, Mallat A, Moreau R, Lotersztajn S. Targeting cell-intrinsic metabolism for antifibrotic therapy. J Hepatol. 2021;74(6):1442–1454. doi:10.1016/j.jhep.2021.02.012
50. Saha B, Bala S, Hosseini N, Kodys K, Szabo G. Krüppel‐like factor 4 is a transcriptional regulator of M1/M2 macrophage polarization in alcoholic liver disease. J Leukoc Biol. 2015;97(5):963–973. doi:10.1189/jlb.4A1014-485R
51. Pan X, Wang L, You H, et al. Alternative activation of macrophages by prostacyclin synthase ameliorates alcohol induced liver injury. Lab Invest. 2021;101(9):1210–1224. doi:10.1038/s41374-021-00531-7
52. Koda Y, Nakamoto N, Kanai T. Regulation of Progression and Resolution of Liver Fibrosis by Immune Cells. Semin Liver Dis. 2022;42(4):475–488. doi:10.1055/a-1957-6384
53. Singanayagam A, Triantafyllou E. Macrophages in Chronic Liver Failure: diversity, Plasticity and Therapeutic Targeting. Front Immunol. 2021;12. doi:10.3389/fimmu.2021.661182
54. Albillos A, Martin-Mateos R, Van der Merwe S, Wiest R, Jalan R, Álvarez-Mon M. Cirrhosis-associated immune dysfunction. Nat Rev Gastroenterol Hepatol. 2022;19(2):112–134. doi:10.1038/s41575-021-00520-7
55. Geng A, Flint E, Bernsmeier C. Plasticity of monocytes and macrophages in cirrhosis of the liver. Front Netw Physiol. 2022;2:937739. doi:10.3389/fnetp.2022.937739
56. Carter JK, Friedman SL. Hepatic Stellate Cell-Immune Interactions in NASH. Front Endocrinol (Lausanne). 2022;13:867940. doi:10.3389/fendo.2022.867940
57. Karsdal MA, Daniels SJ, Holm Nielsen S, et al. Collagen biology and non-invasive biomarkers of liver fibrosis. Liver Int. 2020;40(4):736–750. doi:10.1111/liv.14390
58. Baeck C, Wei X, Bartneck M, et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C+ macrophage infiltration in mice. Hepatology. 2014;59(3):1060–1072. doi:10.1002/hep.26783
59. Hammerich L, Tacke F. Hepatic inflammatory responses in liver fibrosis. Nat Rev Gastroenterol Hepatol. 2023. doi:10.1038/s41575-023-00807-x
60. Ramachandran P, Pellicoro A, Vernon MA, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A. 2012;109(46):E3186–3195. doi:10.1073/pnas.1119964109
61. Guillot A, Winkler M, Silva Afonso M, et al. Mapping the hepatic immune landscape identifies monocytic macrophages as key drivers of steatohepatitis and cholangiopathy progression. Hepatology. 2023. doi:10.1097/HEP.0000000000000270
62. Mandrekar P, Szabo G. Signalling pathways in alcohol-induced liver inflammation. J Hepatol. 2009;50(6):1258–1266. doi:10.1016/j.jhep.2009.03.007
63. Wheeler MD, Thurman RG. Up-regulation of CD14 in liver caused by acute ethanol involves oxidant-dependent AP-1 pathway. J Biol Chem. 2003;278(10):8435–8441. doi:10.1074/jbc.M212076200
64. Norkina O, Dolganiuc A, Shapiro T, Kodys K, Mandrekar P, Szabo G. Acute alcohol activates STAT3, AP-1, and Sp-1 transcription factors via the family of Src kinases to promote IL-10 production in human monocytes. J Leukoc Biol. 2007;82(3):752–762. doi:10.1189/jlb.0207099
65. Morán L, Cubero FJ. Extracellular vesicles in liver disease and beyond. World J Gastroenterol. 2018;24(40):4519–4526. doi:10.3748/wjg.v24.i40.4519
66. Momen-Heravi F, Saha B, Kodys K, Catalano D, Satishchandran A, Szabo G. Increased number of circulating exosomes and their microRNA cargos are potential novel biomarkers in alcoholic hepatitis. J Transl Med. 2015;13:261. doi:10.1186/s12967-015-0623-9
67. Eguchi A, Yan R, Pan SQ, et al. Comprehensive characterization of hepatocyte-derived extracellular vesicles identifies direct miRNA-based regulation of hepatic stellate cells and DAMP-based hepatic macrophage IL-1β and IL-17 upregulation in alcoholic hepatitis mice. J Mol Med (Berl). 2020;98(7):1021–1034. doi:10.1007/s00109-020-01926-7
68. Saha B, Momen-Heravi F, Furi I, et al. Extracellular vesicles from mice with alcoholic liver disease carry a distinct protein cargo and induce macrophage activation through heat shock protein 90. Hepatology. 2018;67(5):1986–2000. doi:10.1002/hep.29732
69. Arroyo V, Moreau R, Jalan R. Acute-on-Chronic Liver Failure. N Engl J Med. 2020;382(22):2137–2145. doi:10.1056/NEJMra1914900
70. Triantafyllou E, Woollard KJ, McPhail MJW, Antoniades CG, Possamai LA. The Role of Monocytes and Macrophages in Acute and Acute-on-Chronic Liver Failure. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.02948
71. Cai Q, Liu W, Zhu M, Sheng J. Microbial Infections as a Trigger for Acute-on-Chronic Liver Failure: a Review. Med Sci Monit. 2019;25:4773–4783. doi:10.12659/MSM.915637
72. Zhang IW, Curto A, López-Vicario C, et al. Mitochondrial dysfunction governs immunometabolism in leukocytes of patients with acute-on-chronic liver failure. J Hepatol. 2022;76(1):93–106. doi:10.1016/j.jhep.2021.08.009
73. Sakhuja P. Pathology of alcoholic liver disease, can it be differentiated from nonalcoholic steatohepatitis? World J Gastroenterol. 2014;20(44):16474–16479. doi:10.3748/wjg.v20.i44.16474
74. Owumi SE, Corthals SM, Uwaifo AO, Kamendulis LM, Klaunig JE. Depletion of Kupffer cells modulates ethanol-induced hepatocyte DNA synthesis in C57Bl/6 mice. Environ Toxicol. 2014;29(8):867–875. doi:10.1002/tox.21814
75. Verma VK, Li H, Wang R, et al. Alcohol stimulates macrophage activation through caspase-dependent hepatocyte derived release of CD40L containing extracellular vesicles. J Hepatol. 2016;64(3):651–660. doi:10.1016/j.jhep.2015.11.020
76. Marin V, Poulsen K, Odena G, et al. Hepatocyte-derived macrophage migration inhibitory factor mediates alcohol-induced liver injury in mice and patients. J Hepatol. 2017;67(5):1018–1025. doi:10.1016/j.jhep.2017.06.014
77. Guilliams M, Bonnardel J, Haest B, et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell. 2022;185(2):379–396.e38. doi:10.1016/j.cell.2021.12.018
78. Ma J, Guillot A, Yang Z, et al. Distinct histopathological phenotypes of severe alcoholic hepatitis suggest different mechanisms driving liver injury and failure. J Clin Invest. 2022;132(14):e157780. doi:10.1172/JCI157780
79. Watanabe K, Uchida Y, Sugawara K, et al. Sequential therapy consisting of glucocorticoid infusions followed by granulocyte–monocyte absorptive apheresis in patients with severe alcoholic hepatitis. J Gastroenterol. 2017;52(7):830–837. doi:10.1007/s00535-016-1287-9
80. Calvente CJ, Tameda M, Johnson CD, et al. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223. J Clin Invest. 2019;129(10):4091–4109. doi:10.1172/JCI122258
81. Cho Y, Bukong TN, Tornai D, et al. Neutrophil extracellular traps contribute to liver damage and increase defective low-density neutrophils in alcohol-associated hepatitis. J Hepatol. 2023;78(1):28–44. doi:10.1016/j.jhep.2022.08.029
82. Matsuda M, Seki E. Hepatic Stellate Cell-Macrophage Crosstalk in Liver Fibrosis and Carcinogenesis. Semin Liver Dis. 2020;40(3):307–320. doi:10.1055/s-0040-1708876
83. Liaskou E, Zimmermann HW, Li KK, et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology. 2013;57(1):385–398. doi:10.1002/hep.26016
84. Zimmermann HW, Seidler S, Nattermann J, et al. Functional contribution of elevated circulating and hepatic non-classical CD14CD16 monocytes to inflammation and human liver fibrosis. PLoS One. 2010;5(6):e11049. doi:10.1371/journal.pone.0011049
85. Karlmark KR, Weiskirchen R, Zimmermann HW, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50(1):261–274. doi:10.1002/hep.22950
86. Wallace SJ, Tacke F, Schwabe RF, Henderson NC. Understanding the cellular interactome of non-alcoholic fatty liver disease. JHEP Reports. 2022;4(8). doi:10.1016/j.jhepr.2022.100524
87. Pradere JP, Kluwe J, De Minicis S, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58(4):1461–1473. doi:10.1002/hep.26429
88. Arab JP, Addolorato G, Mathurin P, Thursz MR. Alcohol-Associated Liver Disease: integrated Management With Alcohol Use Disorder. Clin Gastroenterol Hepatol. 2023;S1542-3565(23)00159–3. doi:10.1016/j.cgh.2023.02.017
89. Arab JP, Izzy M, Leggio L, Bataller R, Shah VH. Management of alcohol use disorder in patients with cirrhosis in the setting of liver transplantation. Nat Rev Gastroenterol Hepatol. 2022;19(1):45–59. doi:10.1038/s41575-021-00527-0
90. Thursz MR, Richardson P, Allison M, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med. 2015;372(17):1619–1628. doi:10.1056/NEJMoa1412278
91. Arab JP, Díaz LA, Baeza N, et al. Identification of optimal therapeutic window for steroid use in severe alcohol-associated hepatitis: a worldwide study. J Hepatol. 2021;75(5):1026–1033. doi:10.1016/j.jhep.2021.06.019
92. Bruellman R, Llorente C. A Perspective Of Intestinal Immune-Microbiome Interactions In Alcohol-Associated Liver Disease. Int J Biol Sci. 2021;17(1):307–327. doi:10.7150/ijbs.53589
93. Szabo G, Mitchell M, McClain CJ, et al. IL-1 receptor antagonist plus pentoxifylline and zinc for severe alcohol-associated hepatitis. Hepatology. 2022;76(4):1058–1068. doi:10.1002/hep.32478
94. Poulsen KL, McMullen MR, Huang E, et al. Novel Role of Macrophage Migration Inhibitory Factor in Upstream Control of the Unfolded Protein Response After Ethanol Feeding in Mice. Clin Exp Res. 2019;43(7):1439–1451. doi:10.1111/acer.14065
95. Ambade A, Lowe P, Kodys K, et al. Pharmacological Inhibition of CCR2/5 Signaling Prevents and Reverses Alcohol‐Induced Liver Damage, Steatosis, and Inflammation in Mice. Hepatology. 2019;69(3):1105. doi:10.1002/hep.30249
96. Wiering L, Tacke F. Treating inflammation to combat non-alcoholic fatty liver disease. J Endocrinol. 2023;256(1):e220194. doi:10.1530/JOE-22-0194
97. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66(6):1300–1312. doi:10.1016/j.jhep.2017.02.026
98. Mandal P, Pratt BT, Barnes M, McMullen MR, Nagy LE. Molecular mechanism for adiponectin-dependent M2 macrophage polarization: link between the metabolic and innate immune activity of full-length adiponectin. J Biol Chem. 2011;286(15):13460–13469. doi:10.1074/jbc.M110.204644
99. Mandal P, Roychowdhury S, Park PH, Pratt BT, Roger T, Nagy LE. Adiponectin and heme oxygenase-1 suppress TLR4/MyD88-independent signaling in rat Kupffer cells and in mice after chronic ethanol exposure. J Immunol. 2010;185(8):4928–4937. doi:10.4049/jimmunol.1002060
100. Hill DB, D’Souza NB, Lee EY, Burikhanov R, Deaciuc IV. A role for interleukin-10 in alcohol-induced liver sensitization to bacterial lipopolysaccharide. Alcohol Clin Exp Res. 2002;26(1):74–82.
101. Byun JS, Suh YG, Yi HS, Lee YS, Jeong WI. Activation of toll-like receptor 3 attenuates alcoholic liver injury by stimulating Kupffer cells and stellate cells to produce interleukin-10 in mice. J Hepatol. 2013;58(2):342–349. doi:10.1016/j.jhep.2012.09.016
102. Luo P, Wang F, Wong NK, et al. Divergent Roles of Kupffer Cell TLR2/3 Signaling in Alcoholic Liver Disease and the Protective Role of EGCG. Cell Mol Gastroenterol Hepatol. 2020;9(1):145–160. doi:10.1016/j.jcmgh.2019.09.002
103. Kedarisetty CK, Anand L, Bhardwaj A, et al. Combination of granulocyte colony-stimulating factor and erythropoietin improves outcomes of patients with decompensated cirrhosis. Gastroenterology. 2015;148(7):1362–1370.e7. doi:10.1053/j.gastro.2015.02.054
104. Garg V, Garg H, Khan A, et al. Granulocyte colony-stimulating factor mobilizes CD34(+) cells and improves survival of patients with acute-on-chronic liver failure. Gastroenterology. 2012;142(3):505–512.e1. doi:10.1053/j.gastro.2011.11.027
105. Engelmann C, Herber A, Franke A, et al. Granulocyte-colony stimulating factor (G-CSF) to treat acute-on-chronic liver failure: a multicenter randomized trial (GRAFT study). J Hepatol. 2021;75(6):1346–1354. doi:10.1016/j.jhep.2021.07.033
106. Engelmann C, Habtesion A, Hassan M, et al. Combination of G-CSF and a TLR4 inhibitor reduce inflammation and promote regeneration in a mouse model of ACLF. J Hepatol. 2022;77(5):1325–1338. doi:10.1016/j.jhep.2022.07.006
107. Tilg H, Adolph TE, Tacke F. Therapeutic modulation of the liver immune microenvironment. Hepatology. 2023. doi:10.1097/HEP.0000000000000386
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.