")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Altered γ-aminobutyric acid neurotransmission in major depressive disorder: a critical review of the supporting evidence and the influence of serotonergic antidepressants
Received 20 September 2014
Accepted for publication 8 November 2014
Published 19 January 2015 Volume 2015:9 Pages 603—624
DOI https://doi.org/10.2147/DDDT.S62912
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Shu-Feng Zhou
Alan L Pehrson, Connie Sanchez
External Sourcing and Scientific Excellence, Lundbeck Research USA, Paramus, NJ, USA
Abstract: Evidence suggesting that central nervous system γ-aminobutyric acid (GABA) concentrations are reduced in patients with major depressive disorder (MDD) has been present since at least 1980, and this idea has recently gained support from more recent magnetic resonance spectroscopy data. These observations have led to the assumption that MDD’s underlying etiology is tied to an overall reduction in GABA-mediated inhibitory neurotransmission. In this paper, we review the mechanisms that govern GABA and glutamate concentrations in the brain, and provide a comprehensive and critical evaluation of the clinical data supporting reduced GABA neurotransmission in MDD. This review includes an evaluation of magnetic resonance spectroscopy data, as well as data on the expression and function of the GABA-synthesizing enzyme glutamic acid decarboxylase, GABA neuron-specific cell markers, such as parvalbumin, calretinin and calbindin, and the GABAA and GABAB receptors in clinical MDD populations. We explore a potential role for glial pathology in MDD-related reductions in GABA concentrations, and evidence of a connection between neurosteroids, GABA neurotransmission, and hormone-related mood disorders. Additionally, we investigate the effects of GABAergic pharmacological agents on mood, and demonstrate that these compounds have complex effects that do not universally support the idea that reduced GABA neurotransmission is at the root of MDD. Finally, we discuss the connections between serotonergic and GABAergic neurotransmission, and show that two serotonin-focused antidepressants – the selective serotonin-reuptake inhibitor fluoxetine and the multimodal antidepressant vortioxetine – modulate GABA neurotransmission in opposing ways, despite both being effective MDD treatments. Altogether, this review demonstrates that there are large gaps in our understanding of the relationship between GABA physiology and MDD, which must be remedied with more data from well-controlled empirical studies. In conclusion, this review suggests that the simplistic notion that MDD is caused by reduced GABA neurotransmission must be discarded in favor of a more nuanced and complex model of the role of inhibitory neurotransmission in MDD.
Keywords: GABA, major depressive disorder, serotonin, 5-HT
Introduction
Major depressive disorder (MDD) is a heterogeneous, highly prevalent, and severe psychiatric disorder that affects more than 350 million people worldwide, and is currently thought to be the leading contributor to the global disease burden.1 Although classically conceived as a disorder of mood, MDD is also associated with a high degree of cognitive2 and functional disability, resulting in high socioeconomic burden due to the direct costs of treatment and indirect costs, such as unemployment or reduced productivity of the patient and their caregivers. Patients with MDD are more likely to be unemployed than healthy controls, and those that are employed are more likely to be absent.3 In fact, recent estimates suggest that MDD costs $24 billion yearly in the US in absenteeism and reduced productivity alone.3
The advancement of pharmacotherapies for MDD over the last several decades has resulted in a broad set of effective options for treating patients with MDD. However, a bourgeoning line of evidence suggests that although currently available monoamine-centered antidepressant treatments, such as selective serotonin-reuptake inhibitors (SSRIs) and serotonin–norepinephrine reuptake inhibitors (SNRIs) induce clinically relevant improvements in mood for most MDD patients, the cognitive and functional disability associated with this disease tend to persist.4 Furthermore, these treatments are altogether ineffective for a large subset of depressed patients, and for many others symptom relief is delayed by several weeks after the start of treatment. These issues highlight the strong need to advance our understanding of MDD’s biological underpinnings, in order to find novel treatments capable of resolving these unmet clinical needs.
MDD has unquestionably a heterogeneous set of biological underpinnings, and this is reflected by the numerous etiological theories about MDD, which include theories implicating altered regulation of monoamine neurotransmission,5 sleep,6 and excitatory glutamatergic neurotransmission.7 Another accreting narrative has recently suggested that dysregulation of the inhibitory amino acid neurotransmitter γ-aminobutyric acid (GABA) is involved in the etiology of MDD, with the broad theme that MDD is related to reduced inhibitory GABA neurotransmission. This review seeks to present a critical evaluation of the clinical evidence supporting dysregulated GABA neurotransmission in MDD, the potential for treating MDD using GABAergic mechanisms, and the possibility that serotonergic antidepressants may act in part via modulation of GABAergic neurotransmission.
Regulation of GABA and glutamate in the central nervous system
Before reviewing the data on GABA’s role in MDD, we first summarize the mechanisms by which GABA and the closely related excitatory amino acid neurotransmitter glutamate are synthesized, inactivated and catabolized in the central nervous system (CNS).
GABA synthesis
GABA is produced in the CNS via decarboxylation of glutamate, in a reaction catalyzed by glutamic acid decarboxylase (GAD; Figure 1). This process is thought to occur overwhelmingly in neurons, with glia producing essentially no GABA via this metabolic pathway.8 In general, GAD’s enzymatic activity is thought to be regulated through two processes: 1) the level of enzyme expression, and 2) the degree of association with its cofactor – pyridoxal phosphate (PLP).9
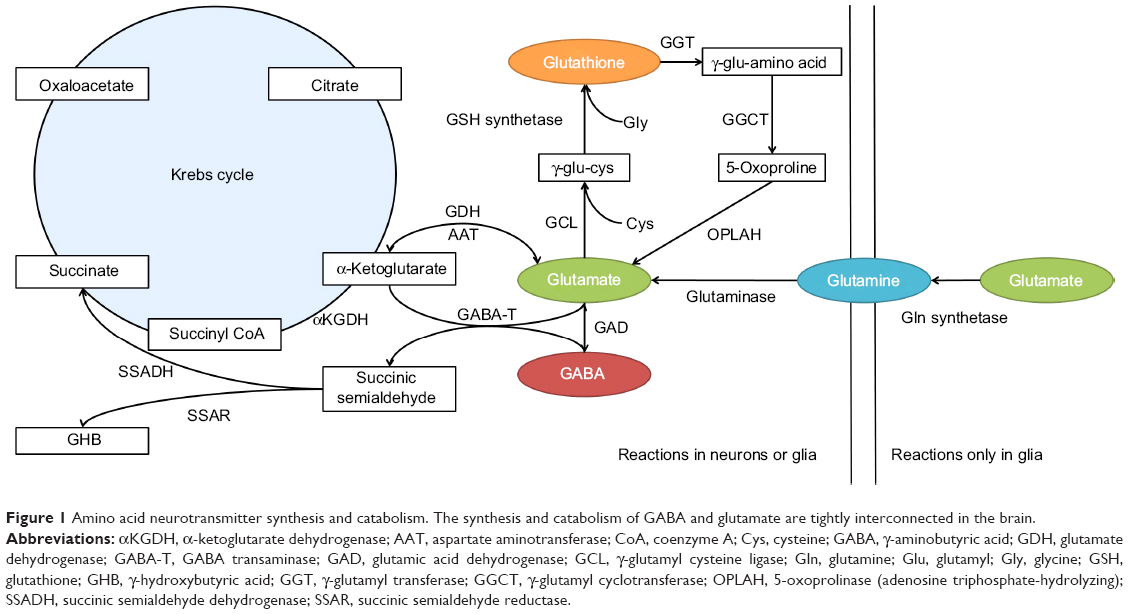 | Figure 1 Amino acid neurotransmitter synthesis and catabolism. The synthesis and catabolism of GABA and glutamate are tightly interconnected in the brain. |
GAD is known to have two separate isoforms in the mammalian CNS – the 65 kDa isoform (GAD65) and the 67 kDa isoform (GAD67) – which are encoded by different genes, have separate patterns of intracellular expression, and are functionally regulated by dissimilar mechanisms. Immunolabeling studies have suggested that GAD65 is located primarily in synaptic terminals, where it is associated with the production of GABA for classical Ca2+-dependent exocytotic release. The majority of GAD65 is dissociated from PLP under normal circumstances, and can markedly increase its activity as demand for synaptic GABA release increases.10 Therefore, the association with its cofactor, rather than the level of expression, appears to be the primary mechanism regulating the metabolic activity of GAD65 in the CNS.9
In contrast, GAD67 is expressed throughout the cytosol, and is primarily associated with PLP under normal conditions. Therefore, rather than being regulated by its association with PLP, GAD67’s enzymatic activity is regulated by its level of expression. Moreover, GAD67’s ubiquitous cytosolic expression and high levels of association with PLP have led to an interpretation that this isoform is primarily involved in cellular metabolic activities via the GABA shunt,8 and is responsible for the majority of the GABA synthesis in the brain.9 However, it has been suggested that cytosolic GABA can be released into the extracellular space via a Ca2+-independent, nonexocytotic mechanism mediated by high-affinity GABA carriers.9 If true, then it is possible that GAD67 plays a role in inhibitory neurotransmission via the stimulation of extrasynaptic GABA receptors. Additionally, it may be that a portion of GAD67-derived GABA is involved in classical exocytotic release, given that GAD65 and GAD67 are both expressed in axon terminals. Therefore, it is not clear based on currently available data whether GABA originating from GAD65 and GAD67 constitutes fully discrete intracellular pools with firmly separated physiological roles.11
GABA inactivation and catabolism
After being released into the extracellular space, GABA is primarily inactivated via reuptake transport, a process that is driven in the CNS by the GABA-selective reuptake transporters GAT-1 and GAT-3.12 GABA transporter proteins can either be expressed on neurons or glial cells. In neurons, the recaptured GABA can either be repackaged for vesicular release or catabolized via the GABA shunt (Figure 1), while GABA captured by glia will largely be catabolized.
Whether it occurs in neurons or glia, GABA catabolism is mediated primarily through the enzyme GABA transaminase (GABA-T). GABA-T is a PLP-dependent enzyme that requires both GABA and α-ketoglutarate as cosubstrates, yielding glutamate and succinic semialdehyde (SSA) as reaction products (Figure 1). SSA can either be metabolized into γ-hydroxybutyric acid (GHB), which can have a role in modulating GABAB receptors, or it can be dehydrogenated into succinate.12 GABA that has been dehydrogenated to succinate via SSA dehydrogenase is then thought to enter the Krebs cycle, where it plays a role in cellular energy metabolism.
Glutamate synthesis
Glutamate may be somewhat unique among central neurotransmitters, in that it has a multiplicity of neurochemical pathways by which it can be produced (Figure 1). Within the CNS, the majority of glutamate is produced from glutamine via the enzyme glutaminase. However, glutamate can also be produced from α-ketoglutarate via several enzymes, including aspartate aminotransferase, or in the GABA shunt via GABA-T. Additionally, central glutamate stores can be derived, at least in part, from the glutathione-cycle metabolite 5-oxoproline.13
Glutamate inactivation and catabolism
As with GABA, neuron-released glutamate is primarily inactivated by uptake from the synapse via high-affinity transport mechanisms. In the case of neurons, this process is mediated by excitatory amino acid transporter (EAAT)-3 in humans (known as excitatory amino acid carrier 1 in rodents), while for glial cells it is mediated via EAAT1/2 (in rodents, glial uptake is mediated by the glutamate aspartate transporter or glutamate transporter 1, respectively). Glutamate that is captured in glial cells is catabolized into glutamine by glutamine synthetase, which is then transported to neurons to be remade into glutamate. This so-called glutamate–glutamine cycle is thought to be the primary source of neuron-released glutamate,14 and may also represent a major source of the glutamate that is used to produce GABA.15 Glutamate also has multiple catabolic pathways, which are depicted in Figure 1.
Evidence for altered GABAergic neurotransmission in MDD: a critical look at the data
GABA concentrations in MDD assessed by magnetic resonance spectroscopy
Neuroscientists have been investigating the potential connection between GABA physiology and MDD for many years, beginning with the observation that GABA concentrations were reduced in the cerebrospinal fluid (CSF) of MDD patients.16 More recently, with the advent of magnetic resonance spectroscopy (MRS) imaging, it has been repeatedly demonstrated that CNS GABA concentrations are reduced in some MDD patients (Table 1). These findings have been most consistent in the occipital cortex (OCC), where several reports have shown reductions compared to healthy controls.17–20 These observations have been replicated in brain regions such as the anterior cingulate cortex18 and in some but not all prefrontal cortex (PFC) areas.21 It is possible that reductions in OCC GABA concentrations are specific to a subpopulation of treatment-resistant patients, as at least one study has shown that treatment-resistant, but not non-treatment-resistant, MDD patients have reduced OCC GABA concentrations.18
 | Table 1 The relationship between major depressive disorder and GABA concentration measured by magnetic resonance spectroscopy |
If major depression is related to reduced GABA concentrations, then a corollary may be the idea that effective treatment for depression should normalize GABA. Some reports have suggested that effective treatment with antidepressants22 or electroconvulsive therapy23 leads to an increase in GABA concentrations in the OCC when assessed compared to the patient’s own baseline. Additionally, some research groups have shown that there are no differences in GABA concentrations between remitted depressed patients and controls in the PFC24,25 and OCC.25 These results alone seem to support the idea that MDD is associated with reduced GABA concentrations. However, Sanacora et al later found that effective cognitive behavioral therapy treatment led to a nonsignificant trend toward a reduction in OCC GABA concentrations,26 while Bhagwagar et al showed that OCC GABA concentrations in recovered MDD patients were still reduced compared to healthy controls.27,28 These differing results may reflect methodological differences in the detection of GABA,25 brain region-specific relationships between GABA concentrations and depression, or differences in the mechanism of antidepressant treatments.
Therefore, the overall consensus appears to be that GABA concentrations are down in the CNS of depressed patients, at least in some brain regions and in treatment-resistant or severely depressed patients. However, there are not enough data available to develop a firm consensus on whether these altered GABA concentrations represent consistent biomarkers for depression states, and in fact there is some evidence both for and against this notion. However, it should be noted that in many cases, depressed patients have cognitive dysfunction that persists even after mood symptoms have been reduced by “successful” antidepressant treatment.29 It is possible that remaining GABA reductions after the retreat of depressed affect are related to a persisting syndrome of cognitive dysfunction. However, none of these studies has evaluated the cognitive state of the patients before and after antidepressant treatment, so this idea is purely speculation at this time. Furthermore, whether the observed reductions in GABA concentrations are circumscribed to specific brain regions or whether there is a broader pattern of reduced CNS GABA concentrations remains unclear. Additionally, given the fact that cellular GABA pools can be loosely separated into metabolic and neurotransmitter subcompartments, with the metabolic compartment representing the lion’s share of cellular GABA,8 it is not possible based on MRS data alone to determine whether the reduced GABA concentrations observed in MDD patients are actually related to changes in GABAergic neurotransmission. These issues can only be clarified with more empirical data. Moreover, as the technology of MRS advances, it will be preferable if future studies focus more heavily on brain regions with greater theoretical relevance to depression-related changes in affect and cognitive function.
GAD expression in MDD patients
If GABA concentrations are reduced in the brain of MDD patients, then it makes sense to look for altered physiology in GAD, the enzyme that produces GABA. Several research groups to date have investigated either the expression or function of GAD in postmortem brains from MDD patients, and these data are summarized in Table 2. Unfortunately, there is some mismatch between the brain regions studied using protein- or gene-expression techniques, which have focused most heavily on the frontal cortex, compared to the MRS studies that have tended to focus on the OCC due to technological limitations. In the frontal cortex, one research group reported that medication-free MDD patients had a significant reduction in the expression of GAD67 compared to healthy control subjects,30 but found that there was no difference in medicated MDD patients versus controls. These data may be in line with MRS data showing reduced GABA concentrations in the dorsal PFC in untreated but not treated depressed patients.21,24 An investigation of GAD67 expression in the cerebellum in a small group of MDD patients compared to controls also found a significant reduction in MDD patients.31 However, other groups that have investigated GAD67 protein expression using either Western blot,32 immunohistochemistry,33 or gene expression using quantitative polymerase chain reaction (PCR),34 have found no significant changes in GAD67 expression in the frontal cortex of MDD patients. Therefore, the evidence that GAD67 expression is altered in the brain of depressed patients is currently equivocal at best. It should be noted, however, that the majority of these studies used tissue samples from a mix of patients who were treated and untreated, and thus may be confounded by the influence of antidepressant medications.
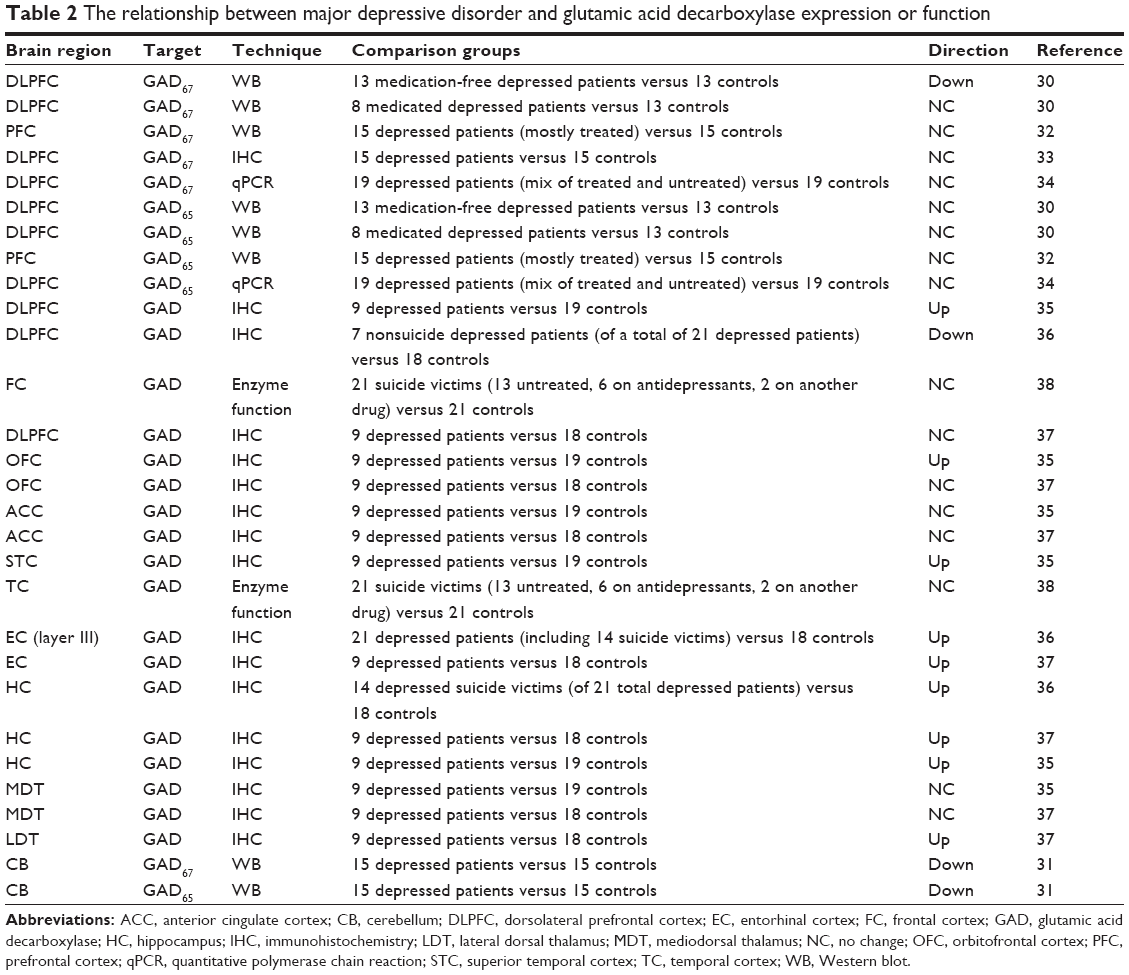 | Table 2 The relationship between major depressive disorder and glutamic acid decarboxylase expression or function |
While GAD67 expression may or may not be reduced in the frontal cortex of MDD patients, research groups that have investigated the expression of GAD65 in the frontal cortex of this patient population have universally shown that there are no changes compared to healthy control patients.30,32,34 Conversely, in the cerebellum of MDD patients, Fatemi et al31 observed a significant reduction in GAD65 expression compared to controls. However, it is important to point out that the primary method of regulating GAD65’s enzymatic activity is its association with PLP, not the degree of its expression, and the vast majority of GAD65 is dissociated from PLP under normal conditions (see earlier discussion under GABA synthesis). Therefore, it is not clear that this reduction in GAD65 expression is from an active enzyme population, and the relevance of this reduction is thus questionable.
In addition to data generated on the expression of specific GAD isoforms, several research groups have investigated the relationship between undifferentiated GAD and MDD in a number of brain regions using either immunohistochemical techniques or enzyme functional assays, and in general the findings have varied by the region studied. In the PFC, the overall results have been equivocal, with one study finding increased density of GAD-immunoreactive (IR) cells,35 another finding significant decreases,36 and a third finding no change.37 Additionally, an investigation of GAD enzymatic activity in the PFC of suicide patients compared to healthy controls found no changes in GAD function.38 Similarly, results in the orbitofrontal cortex, temporal cortex, and thalamus up to this point have had mixed results.35,37,38 However, it appears that there is some agreement that the density of GAD-IR cells is increased in the entorhinal cortex36,37 and hippocampus of MDD patients.35–37
Based on these data, it appears that GAD expression or function is not strongly regulated in either direction in the frontal cortex of depressed patients, including the dorsal PFC, orbitofrontal cortex, and anterior cingulate cortex. Moreover, the seemingly consistent increases in GAD expression observed in the entorhinal cortex and hippocampus may suggest that GABA physiology is not altered in a monolithic, pan-CNS manner, but rather is regionally specific. In any case, at this time the data on GAD expression cannot be considered generally supportive of the idea that GABA concentrations are reduced in MDD due to changes in GAD function. However, there are several significant caveats to consider when evaluating these ideas. A number of these studies were confounded due to the presence of mixed samples of treated and untreated depressed patients, making it difficult to delineate how GAD would be regulated by MDD alone. In addition, these studies have tended to use small patient samples and are probably not numerous enough to confidently make claims about the population of depressed patients. Finally, there is little agreement between the MRS and GAD-expression literature in terms of the brain areas studied. Bearing these significant caveats in mind, it is probably premature to take a firm stance on how GAD expression is regulated in depressed patients, and how this may or may not be relevant to the observed reductions in GABA concentration. Moreover, in order to move this research forward, more studies are required that carefully control for the effects of antidepressant treatments, use larger sample sizes, and study brain regions that have also been studied using MRS.
GABAergic neuron-related Ca2+-binding proteins and the neuropeptide somatostatin
Another potential way to explain the reductions in GABA concentrations seemingly observed in MDD via MRS studies is to examine the density of GABAergic interneurons. To the extent that GABA neurons selectively express GAD in the CNS, the previous section may have already suggested that there are not any consistent changes in GABA neuron density in MDD. However, GABAergic interneurons can be further subdivided based on the expression of the calcium-binding proteins calbindin, calretinin, and parvalbumin (PV),39 and it is possible that changes in GABAergic physiology in MDD are specific for one or more of these cell types. To our knowledge, only a few research groups have investigated the expression of these proteins in depressed patients, and as in studies of GAD have tended to focus on the frontal cortex. Perhaps the most consistent result has suggested that the density of calbindin-IR interneurons is significantly reduced in areas, including the dorsolateral PFC (DLPFC)40 and OCC,41 and trends toward a reduction in the orbitofrontal cortex.40 Reports on the density of calretinin-IR and parvalbumin-IR interneurons in the DLPFC and orbitofrontal cortex have generally found no significant changes.40,42,43 Therefore, it may be the case that there is a reduction in calbindin-IR interneurons in the cortex of depressed patients. However, this observation is based on only two studies in a cumulative sample of 24 depressed patients, and needs further replication before it can be thought of as a consistent biology in MDD.40,41
It should also be noted that several well-controlled studies have demonstrated that the expression of the neuropeptide somatostatin, which is commonly coreleased by GABAergic interneurons, is reduced in postmortem brains of MDD patients in such regions as the amygdala and subgenual cingulate.44,45
A role for glial pathology in disrupted GABA concentrations
It is important to note that there are several lines of histological evidence pointing to glial pathology in the frontal cortex of MDD patients. These observations include reductions in size and density of glia,46 as well as reductions in the gene or protein expression of glial-specific markers, such as glial fibrillary acidic protein, the calcium-binding protein S100B, EAAT1, EAAT2, and glutamine synthetase.47,48 These glial pathologies can be theoretically important for a discussion of GABA neurotransmission from the perspective that glial cells, such as astrocytes, regulate glutamate–glutamine cycling through cellular machinery that includes EAAT1/2 and glutamine synthetase (see earlier discussion on Glutamate inactivation and catabolism). This process is thought to be a major neurochemical source of GABA,14 in part because experimental disruptions of glutamate–glutamine cycling under depolarizing conditions significantly attenuate neuronal GABA pools.15 In fact, there have been some suggestions that glutamate–glutamine cycling is more important for maintenance of GABA neurotransmitter pools than for glutamate stores.49
Current thinking on the consequences of reduced glial density on neurotransmission has imagined a complex set of effects that include not only a reduction in the pool of glutamate available for synaptic neurotransmission but also an increase in the stimulation of extrasynaptic receptors due to the reduced glial uptake, leading to an altered ratio of synaptic to extrasynaptic signaling.47,48 Given that GABA metabolism and inactivation is regulated via glial mechanisms that are very similar to glutamate, the same ideas could be used for GABA neurotransmission in MDD. This theory could plausibly explain the reduced concentrations of GABA observed in MRS studies of MDD, as well as the observations of reduced concentrations of GLX, a combined measure of glutamate and glutamine.50
Although the idea of a compartmentalized effect of glial dysfunction on synaptic and extrasynaptic amino acid neurotransmission is certainly plausible, it is not entirely supported by the data in nonclinical model systems. The idea that reduced postsynaptic GABA neurotransmission can result from disrupted glutamate–glutamine cycling has been observed in nonclinical models.51 However, Paulsen and Fonnum49 investigated the effects of both a selective glial lesion and pharmacological inhibition of glutamine synthetase on basal and potassium-stimulated extracellular GABA and glutamate concentrations. These authors found that within the first 2–3 hours of either treatment, extracellular concentrations of glutamate and GABA were indeed higher, but after 6–7 hours the extracellular concentrations of both amino acid transmitters fell well below those of the control condition. These data do not support the idea that glial pathology would drive compartmentalized effects on synaptic versus extrasynaptic amino acid neurotransmission over a long time frame, although it is possible that the translatability of the models used in this study may be questioned. However, glial pathology is still a plausible explanation for the reduced cortical GABA concentrations observed in MDD.
Cortical silent period and short-interval cortical inhibition
It can be argued that electrophysiological measures associated with transcranial magnetic stimulation, such as cortical silent period and short-interval cortical inhibition, provide evidence of disrupted GABA neurotransmission in depressed patients.52 While it is true that manipulating GABAergic neurotransmission can induce changes in these measures, this can also be said of other neurotransmitter systems.53 Therefore, it seems that there is a complicated physiology underlying cortical silent period and short-interval cortical inhibition that make it difficult to clearly interpret the causes of any changes observed in MDD patients. Therefore, data on these electrophysiological measures have not been considered in this review.
GABA receptors in MDD
GABAA receptors
GABAA receptors are pentameric transmembrane ion channels that have heterogeneous subunit composition and anatomical expression patterns, and are thought to mediate the majority of fast inhibitory neurotransmission in the CNS. The subunit composition of GABAA receptors determines a number of functional factors, including subcellular localization (eg, synaptic versus extrasynaptic localization), channel open probabilities, desensitization rate, and modulatory ligand affinity.
Unfortunately, although there has been evidence that central GABA physiology is altered in MDD since at least 1980, there is a relative paucity of research that has investigated whether altered composition or function of GABAA receptors is present in the brain tissue of clinical MDD populations. Recently, however, a few studies have shown changes in GABAA receptor-gene expression in depressed patients compared to controls. For example, Choudary et al54 found upregulation of the genes governing several GABAA receptor subunits, including GABAAβ3, GABAAδ, and GABAAγ2, in the frontal cortex. Similarly, a larger analysis of multiple brain regions using PCR in depressed suicide victims demonstrated altered expression of numerous GABA receptor-subunit genes, most of which were related to the GABAA receptor (19 of the 27 differentially regulated GABAergic genes), and were mostly upregulated. This notably included increased expression of GABRA5 (coding for the GABAAα5 receptor subunit) and GABRG2 (GABAAγ2 receptor subunit) in the DLPFC and the inferior temporal cortex,55 and reduced expression of GABRG1 in the DLPFC. However, the expression pattern was found to be region-specific. On the basis of gene-expression data alone, it is tempting to conclude that the GABAA receptor is implicated in the pathophysiology of MDD, at least in those patients who committed suicide.
Studies using protein-expression techniques have also found some evidence of changes in GABAA receptor in tissue from depressed patients. For example, Fatemi et al56 found significant increases in GABAA-receptor α1, α2, α6, γ3, and ε subunits in the lateral cerebellum of depressed patients compared to controls, as well as a reduction in the β1 subunit, although significance for the α2, γ3, and β1 subunits was lost when the authors statistically controlled for the effects of antidepressant treatments. Unfortunately, at the time of writing, we were not able to identify any studies that looked at the expression of GABAA-receptor subunits in the frontal cortex, and thus it is difficult to compare these results to the gene-expression data discussed, or to arrive at a consensus on how GABAA receptors are regulated in depression.
Given that the benzodiazepine-binding site is localized between the α- and γ-receptor subunits, the altered expression of the messenger ribonucleic acid (mRNA) governing expression of these subunits may make it plausible to expect that the binding of benzodiazepine-site ligands will be different in depressed patients compared to controls. Unfortunately, there is not much literature on benzodiazepine binding in depressed patients. A recent study by Klumpers et al57 found significant reductions in benzodiazepine-site binding in the parahippocampal gyrus and right lateral superior temporal gyrus of a small sample of MDD patients. Another study found no significant changes in benzodiazepine-site binding in the temporal cortex or frontal cortex in untreated depressed suicide patients, but found significant frontal cortex binding increases only in cases where patients were receiving antidepressant treatment.38 Three additional studies failed to find any differences in benzodiazepine-binding sites in depressed patients in the locus coeruleus,58 the OCC in patients where low GABA concentrations were found via MRS,17 or in the amygdala and hippocampus of suicide victims.59 Therefore, although these results are largely negative, there has been very little overlap in the brain regions studied by these binding studies, leaving open the possibility that benzodiazepine-receptor expression is regulated in a regionally specific manner in MDD. Moreover, the lack of agreement on the brain regions that have been evaluated for benzodiazepine-site binding and altered expression of the genes governing GABAA-receptor expression make it difficult to evaluate whether the altered gene-expression pattern has led to changes in the function of the GABAA receptor. Once again, this issue can only be resolved with more empirical data. To our knowledge, there have been no attempts to investigate binding at the GABAA-receptor orthosteric site in MDD patients at this time.
GABAB receptors
The role of GABAB receptors in MDD has received little attention from the research community in comparison to the GABAA receptor. Fatemi et al60 investigated the expression of GABAB receptors in the lateral cerebellum of depressed patients, and found reduced expression of the GABBR1 and GABBR2 subunits in a small sample of depressed patients compared to controls. However, studies that have investigated GABAB binding in the frontal cortex61,62 and in the temporal cortex and hippocampus62 found no evidence of altered GABAB-receptor binding, with the exception of an increased affinity in the temporal cortex of drug-free suicide victims. Of course, studies in suicide victims can be criticized from the perspective that only a small population of depressed patients actually go through with suicide, and thus it may be argued that a different set of CNS biologies are at play in these patients compared to the broader MDD population. In any case, the available data do not convincingly point to an altered GABAB-receptor biology in MDD patients.
Neurosteroids, GABA neurotransmission, and mood dysfunction
Premenstrual syndrome (PMS) and the more severe premenstrual dysphoric disorder (PMDD), which are associated with a cyclical onset and retreat of negative mood symptoms, may represent clinically relevant examples of a connection between GABA neurotransmission and mood dysfunction. Although the mechanisms underlying these symptoms are not entirely clear, there is a line of evidence implicating abnormal responses to neuroactive steroids that are metabolically derived from ovarian hormones, such as the progesterone metabolite allopregnanolone, in the etiology of these disorders. The highest probability of negative mood onset in these disorders occurs during the late luteal phase, when levels of the ovarian hormone progesterone are falling. Although it should be noted that in PMDD, symptoms can also increase in the early luteal phase when progesterone levels are rising.63 Schmidt et al64 demonstrated that mood symptoms associated with PMS could be blocked by pharmacological inhibition of the menstrual cycle, perhaps helping to implicate hormonal fluctuations. However, these authors found that direct administration of estradiol or progesterone in women with PMS caused increases in negative mood that did not occur in healthy women. Similarly, Smith et al65 used transcranial magnetic stimulation of the motor cortex to demonstrate that women with PMS had an increase in motor-cortex excitability during the luteal versus follicular phase, when within-subject progesterone levels were relatively higher. By comparison, cortical excitability during the luteal phase of control patients was reduced, and importantly these controls did not have different plasma progesterone or estradiol concentrations compared to patients during either phase. Given that the progesterone metabolite allopregnanolone modulates GABAA-receptor function, it seems plausible that the mood dysfunction associated with these disorders is related to abnormal neurosteroid regulation of GABA neurotransmission.
The weight of the available nonclinical data suggests that acute allopregnanolone administration positively modulates most GABAA-receptor conformations, resulting in enhanced GABA-mediated inhibitory neurotransmission.66 Acute allopregnanolone administration also has well-recognized behavioral effects that are consistent with a positive modulatory effect on GABA neurotransmission, including anxiolytic67 and seizure-reducing68 properties. However, allopregnanolone’s effects on GABA neurotransmission are complex and acute administration of this steroid can have paradoxical effects under some conditions in nonclinical model systems,69,70 and in the clinic,64 which can include reductions in GABA-mediated inhibition and anxiogenic properties.
Recent papers suggest that in nonclinical models, these paradoxical effects can be attributed to allopregnanolone’s negative modulatory effects on GABAA receptors containing the α4 and δ subunits.69 The α4 and δ subunit-containing GABAA receptors are normally expressed at low levels in most brain regions, with notable exceptions, such as the dentate gyrus and thalamus.71 However, during periods of fluctuating progesterone levels, eg, in progesterone-withdrawal models, GABAA-receptor composition shifts by sharply increasing the expression of the α4 and δ subunits in the CA1 subregion of the hippocampus,72,73 while at the same time reducing the expression of the α1 subunit.63 In addition to producing paradoxical effects of allopregnanolone, this shift in GABAA receptor-subtype expression causes a complex set of changes to basal inhibitory neurotransmission. For example, the shift from α1 subunit-containing GABAA receptors to α4 subunit-containing GABAA receptors increases the decay kinetics of GABAA receptor-mediated inhibitory currents,74 leading to reduced phasic inhibitory neurotransmission.75 However the increase in δ-containing GABAA receptors, which are localized extrasynaptically and have a low desensitization rate,74 induces an increase in tonic inhibitory neurotransmission.69,76 Therefore, the effects of progesterone withdrawal in nonclinical models can be conceptualized as a shift in the balance of phasic versus tonic GABA neurotransmission.
Moreover, there is some evidence suggesting that these changes in GABAA-receptor conformation are relevant to clinical disorders, such as PMDD. The shift from α1-containing GABAA receptors to α4-containing GABAA receptors in nonclinical model systems is associated with a reduction in sensitivity to positive allosteric modulation at the benzodiazepine site,72 and a similar reduction in sensitivity to benzodiazepines has been observed in human patients with PMS. Therefore, it is plausible to suggest that the mood dysfunction associated with PMS and PMDD are related to increased expression of α4- and perhaps δ-containing GABAA receptors, suggesting a dysregulation of GABAergic neurotransmission. Moreover, data showing increased motor-cortex excitability in women with PMS65 may suggest that inhibitory neurotransmission is overall reduced in this disorder. However, this idea may be complicated by evidence from an MRS study of GABA concentrations, which found that women with PMDD experienced increases in cortical GABA concentrations from the follicular to the luteal phase, when negative mood symptoms increased,77 while control subjects had significantly reduced GABA concentrations across these menstrual phases. Therefore, while it is clear from the accumulated evidence that PMS and PMDD are related to dysregulated GABAergic neurotransmission, the precise nature of this relationship remains unclear at this time.
A relationship between neurosteroids and mood has also been proposed in MDD. Several studies have demonstrated that antidepressant treatments, such as the SSRI fluoxetine, can enhance the production of such neurosteroids as allopregnanolone. For example, preclinical models have demonstrated that acute intraperitoneal administration of 20 mg/kg fluoxetine significantly elevates the brain concentration of allopregnanolone in adrenalectomized, castrated male rats 30 minutes after injection,78 which may suggest that fluoxetine treatment can indirectly modulate GABAA receptor-mediated neurotransmission. Interestingly, the same research group later suggested that intraperitoneal administration of 1.45 μmol/kg fluoxetine (equivalent to approximately 0.45 mg/kg of the base) was able to reverse reductions in brain allopregnanolone concentrations in mice induced by social isolation at 30 minutes after injection.79 Given that the fluoxetine dose required to reach 80% occupancy at the mouse 5-hydroxytryptamine (HT) transporter protein (SERT) is approximately 8 mg/kg,80 these data suggest that fluoxetine may have effects on neurosteroid concentrations in extremely low dose ranges. However, these data may also lead one to question the relevance of this mechanism to treatment response, based on clinical data suggesting that approximately 80% SERT occupancy is present at minimally effective SSRI doses in MDD patients.81
We have identified a small number of studies to date that have measured changes in neurosteroid levels in MDD patients in response to treatment with antidepressants. Uzunova et al82 measured CSF concentrations of such neurosteroids as allopregnanolone in a small patient sample (n=15), and found that at baseline, patients had substantially lower allopregnanolone concentrations than were found in a sample of three control patients. Furthermore, Uzunova et al found that allopregnanolone levels increased significantly with successful treatment with fluoxetine or fluvoxamine over a period of about 8 weeks. These data imply that normalization of CSF neurosteroid concentrations are related to the antidepressant effects of these drugs, which could have ramifications for GABAA receptor-mediated inhibitory neurotransmission. However, there are some caveats to consider when interpreting this study. Despite the fact that the majority of the subjects in this study were women (nine of 15) and allopregnanolone concentrations are known to fluctuate with the menstrual cycle, the authors failed to account for the menstrual status of the women in this study, and in fact most of the apparent variance in treatment effects on allopregnanolone in this study were focused in the female subjects. Therefore, the primary dependent measure of this study was confounded.
However, a separate research group presented another small but better-controlled study,83 which found that a small sample of treatment-naïve outpatients with MDD had altered plasma neurosteroid concentrations compared to healthy controls. Specifically, allopregnanolone and 3α,5β-tetrahydroprogesterone were significantly reduced in patients compared to healthy control subjects, while 3β,5α-tetrahydroprogesterone was significantly increased. These differences were reversed by chronic treatment with fluoxetine. In another experiment presented in the same paper, the authors found similar but much smaller responses in neurosteroid concentrations to successful antidepressant treatments in a more severely depressed sample of patients. However, these authors found no correlation between plasma neurosteroid concentrations and therapeutic response in these experiments, again making the relevance of this neurosteroid response to MDD symptoms questionable. Interestingly, a third clinical study84 found that the antidepressant mirtazapine could significantly increase plasma allopregnanolone concentrations in a small group of patients where only about 40% responded to treatment by the end of the study. However, patients receiving mirtazapine with lithium as an adjuvant had a higher response rate (approximately 75%), but had no changes in plasma allopregnanolone concentrations, thus suggesting that elevated neurosteroid concentrations are not necessary for successful antidepressant treatment. Taken together, these data do suggest that some pharmacological antidepressant treatments are able to modulate neurosteroid concentrations in clinical MDD populations; however, the evidence supporting their relevance for clinical antidepressant efficacy is currently not strong.
GABA pharmacology and depressive symptoms
GABAergic interneurons are likely to exert a powerful regulatory influence on the neural networks responsible for controlling mood and cognitive function. Therefore, it may be possible to normalize the networks responsible for these symptoms in MDD by modulating GABA neurotransmission whether or not there is a primary GABAergic dysregulation at the root of MDD. However, if reduced GABA concentrations are an etiological factor in MDD, then it is logical to expect that increasing GABA neurotransmission should relieve depression-related symptoms, while treatments that reduce GABA neurotransmission should induce depressive symptoms.
Perhaps the best-characterized set of GABA-related pharmacological treatments in MDD is the benzodiazepine class of GABAA receptor-positive allosteric modulators (PAMs). Two review papers that examined the effects of benzodiazepines generally found that classical benzodiazepines, such as chlordiazepoxide and diazepam, are not effective at treating patients with MDD.85,86 However, both studies concluded that treatment with the triazolobenzodiazepine alprazolam was able to reliably reduce MDD scores compared to placebo, and one found that alprazolam treatment was as effective as tricyclic antidepressant treatment,85 at least in cases of mild-to-moderate depression. However, these reviews disagreed about whether these depression-score reductions were mediated by changes in “core symptoms”, such as anhedonia. One study concluded that alprazolam was able to reduce anhedonia,86 while the other concluded that most of alprazolam’s effects on depression scores were due to changes in psychomotor retardation, anxiety, and sleep disturbances, and not due to changes in anhedonia or anergia.85 It is important to note that the doses used for the classical benzodiazepines in these studies were at the recommended doses for treatment of anxiety, whereas alprazolam doses were generally much higher. Additionally, recent clinical data have suggested that coadministration of the benzodiazepine-site PAM eszopiclone with SSRIs can lead to significantly greater treatment response, as well as faster onset of efficacy.87,88 Therefore, treatment with the benzodiazepine-site PAMs, such as alprazolam or eszopiclone as an adjuvant may be effective in treating at least some aspects of MDD, although it remains unclear whether alprazolam is effective in attenuating the core mood symptoms. Additionally, it is unclear whether classical benzodiazepines would be effective if used at higher doses. However, a recent nonclinical experiment demonstrated that diazepam, zolpidem, and low-dose alprazolam induced a depression-like phenotype in rodents, while a high dose of alprazolam induced an antidepressant-like response,89 suggesting that alprazolam has specialized effects in its high-dose range that do not generalize to other benzodiazepine-site PAMs. Whether these effects are due to modulation of GABAA receptors or some other off-target effect is unclear.
Another line of inquiry may be drugs that act as agonists at the GABAA-receptor orthosteric binding site. Although preclinical data suggested that the combination of gaboxadol and escitalopram had synergistic antidepressant-like effects in nonclinical models,90 in a clinical trial 5 and 10 mg gaboxadol did not add any benefit over escitalopram treatment alone.91 Taken together with data on benzodiazepines, it does not seem that increasing activity at GABAA receptors uniformly improves depressive phenotypes. Instead, positive modulation of GABAA-receptor function only had positive effects in the case of some drugs, which may suggest a greater understanding of GABAA-receptor subtypes and their action in MDD is needed.
Systematic studies of the utility of other GABAergic treatments in MDD have been difficult to identify. However, there is evidence tying GABAergic treatments for epilepsy to mood dysregulation. For example, a review by Lambert and Robertson92 found that depressive symptoms are more common in epilepsy patients treated with barbiturates, an idea that is echoed in a report showing that phenobarbital treatment in epileptic children was associated with as much as a 40% prevalence of MDD and a 47% prevalence of suicidal ideation.93 Additionally, a number of papers have suggested that the antiepileptic drug vigabatrin, which acts to increase GABA concentrations in the brain by irreversibly inhibiting the GABA-catabolizing enzyme GABA-T, increases the risk of depressive episodes. Estimates of the rate of this adverse event ranged from 4% in one study (compared to 2.6% in placebo controls)94 to 12% in another (compared to 3% in placebo controls).95 An anecdotal report found that major depressive episodes presented soon after either starting vigabatrin treatment or increasing dose in a small epilepsy patient group, and in seven of ten cases the patient had a prior history of MDD.96 These data are further supported by nonclinical data showing that vigabatrin induced a depression-like phenotype in rodents.89
Interestingly, a recent review of nonclinical data has suggested that pharmacological blockade or genetic knockout of GABAB receptors induces antidepressant-like behavioral effects in a variety of nonclinical rodent MDD models, including chronic mild stress, learned helplessness, and forced swim, while GABAB-receptor activation either blocks the effects of known antidepressants or induces a depressive-like phenotype.97 Although there are no clinical investigations of GABAB-receptor antagonists in MDD that could be identified at this time, a clinical experiment demonstrated that GABAB-receptor activation using baclofen induced symptom exacerbation in the majority of a very small MDD patient sample that reversed upon baclofen withdrawal.98 It should be noted that another GABAB-receptor agonist, GHB, which has been approved for use in narcolepsy, has been suggested for use as an antidepressant, primarily based on its beneficial effects on sleep quality,99 although to our knowledge, there are no clinical data on GHB’s effects in MDD patients.
Taken together, these data suggest that increased activity at GABAergic receptors is beneficial in a few cases, eg, with alprazolam as a standalone treatment and eszopiclone as an adjuvant. However, increased neurotransmission at GABAA and GABAB receptors does not generally have positive effects on MDD symptoms, and in some cases can lead to an exacerbation of symptoms. In fact, it may be beneficial in some cases to reduce rather than increase GABA neurotransmission, particularly at GABAB receptors. These observations are not altogether supportive of the idea that reduced GABA neurotransmission drives MDD symptoms, but instead suggest that if GABA dysregulation is an etiological cause of MDD, then it is more complex than a simple reduction in function. In any case, our understanding of GABA’s role in MDD is still in its infancy, and more investigation is required to clarify whether GABAergic mechanisms can be effectively leveraged to treat MDD-related symptoms.
Serotonin–GABA interactions
It is important to note that although in many cases GABA receptors are sparsely expressed in subcortical brain structures, GABAergic interneurons are nearly ubiquitous, and tend to have broadly branching networks that powerfully modulate the activation state of many CNS circuits. Therefore, treatment strategies that feature direct modulation of GABAergic neurotransmission may open up for broad-ranging side effects that can include cognitive impairment100,101 and abuse liability, as well as tolerance and withdrawal syndromes,100 although it must be noted that some GABAergic treatments, eg, eszopiclone, are not associated with tolerance/withdrawal issues, and are considered relatively benign from a safety and tolerability perspective.
An alternative strategy may be to indirectly modulate GABA neurotransmission using targeted pharmacological actions on serotonergic receptors. Serotonin receptors are known to exist as heteroreceptors on GABAergic interneurons, where they can act to excite or inhibit interneuron function. Additionally, serotonin receptor-expression patterns are relatively circumscribed, and are often present in limbic brain regions that are relevant for the mood and cognitive symptoms associated with MDD. An in-depth discussion of the ways that serotonin signaling can modulate GABAergic neurotransmission is beyond the scope of this review. However, given the ascendant position serotonin modulation has in pharmacotherapeutic strategies for MDD, and the evidence that GABA pharmacology can influence mood and cognitive function, it seems important to highlight some of the mechanisms by which serotonin-receptor modulation can influence GABA neurotransmission. We begin with a review of the well-known serotonergic heteroreceptors that influence GABA neurotransmission, and this will be followed by a discussion of receptors for which there are fewer accumulated data. Figure 2 depicts the relationships between serotonergic heteroreceptors and limbic system GABA and glutamate cells.
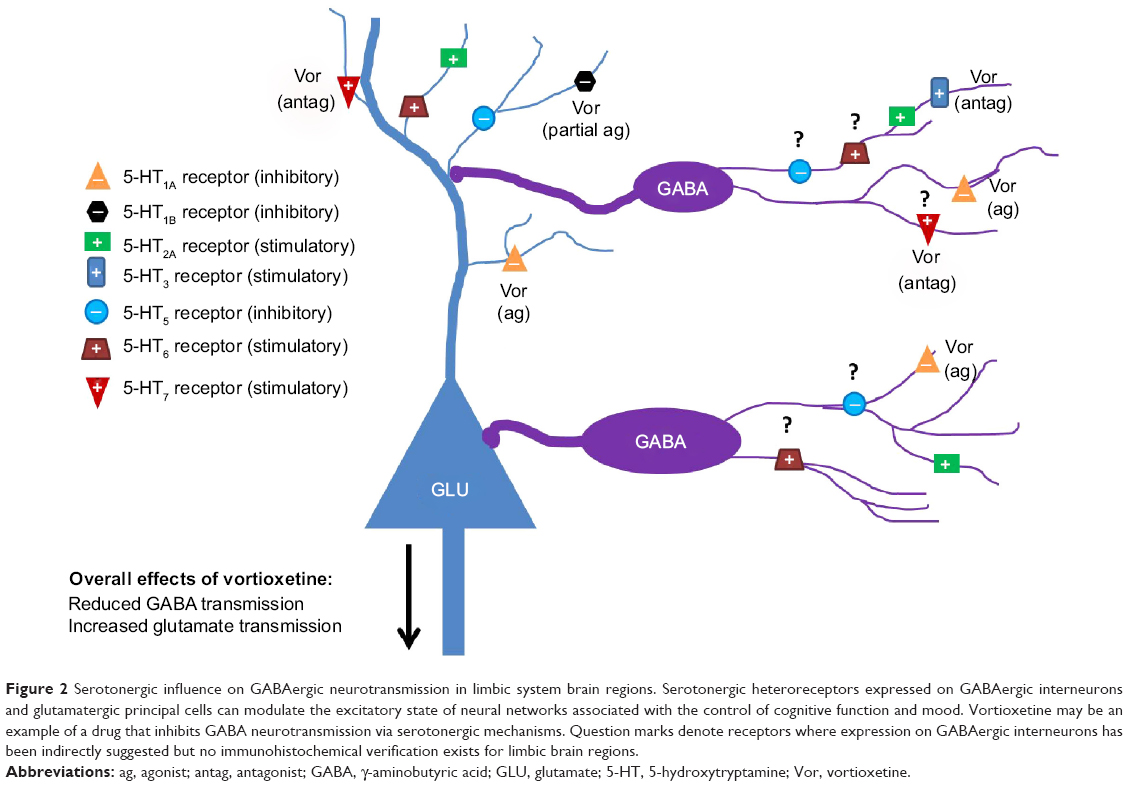 | Figure 2 Serotonergic influence on GABAergic neurotransmission in limbic system brain regions. Serotonergic heteroreceptors expressed on GABAergic interneurons and glutamatergic principal cells can modulate the excitatory state of neural networks associated with the control of cognitive function and mood. Vortioxetine may be an example of a drug that inhibits GABA neurotransmission via serotonergic mechanisms. Question marks denote receptors where expression on GABAergic interneurons has been indirectly suggested but no immunohistochemical verification exists for limbic brain regions. |
Well-known relationships between serotonergic targets and GABA neurotransmission
5-HT1A receptors
As heteroreceptors, 5-HT1A receptors are G-protein-coupled receptors whose activation leads to hyperpolarization of neuronal membranes via the actions of inwardly rectifying potassium channels.102 In rodents, 5-HT1A receptors are expressed in MDD-relevant regions, such as the medial frontal cortex, the anterior cingulate cortex, septum, and hippocampus, and are thought to be present on calbindin- and PV-IR GABAergic interneurons.103 Therefore, treatments that increase activity at 5-HT1A heteroreceptors expressed on GABA cells should lead to a reduction in GABAergic tone, and a disinhibition of downstream circuits. Electrophysiological studies do lend some support to this idea. 5-HT1A-receptor activation causes an increase in cortical pyramidal neuron firing that is thought to be driven by reduced GABAergic inhibitory tone.104–106 However, it should be noted that 5-HT1A receptors are also thought to be expressed on pyramidal neurons, which may be responsible for the fact that in most pyramidal neurons recorded in one study, increased firing was followed by inhibition.106
5-HT2A receptors
5-HT2A receptors are G-protein-coupled receptors that serve to depolarize neuronal membranes via the stimulation of Gq/11 and the resulting increases in intracellular inositol phosphate and Ca2+ concentrations.107 This receptor is thought to be expressed as a postsynaptic receptor, and is strongly localized in MDD-relevant regions, including the frontal cortex, the medial septum, the hippocampus, and the amygdala complex.108 Studies in rodents have found that 5-HT2A heteroreceptors are expressed on GABAergic interneurons, as well as glutamatergic principal cells in numerous brain regions.107,109,110 The degree of expression of the 5-HT2A receptor on inhibitory and principal cells appears to vary by brain region. For example, in the rodent prelimbic cortex, approximately 50% of pyramidal cells express mRNA for the 5-HT2A receptor, while about 30% of GABA cells express the 5-HT2A receptor mRNA. In the infralimbic cortex, only 10% of pyramidal cells and 25% of GABA cells express 5-HT2A receptors.109 However, in the medial septum, hippocampus, and amygdala, nearly all principal excitatory cells and the vast majority of GABAergic interneurons express this receptor.107,110 Therefore, 5-HT2A-receptor activation should have a stimulatory influence on GABAergic interneurons; however, its stimulatory effects on principal cells will again lead to complex actions on local circuits that will likely differ in each brain area.
In general, our understanding of the actions of 5-HT2A receptors in vivo is somewhat limited by the availability of centrally active, selective agonists. In vivo extracellular recordings of neuronal firing have found that activation of 5-HT2A/2C receptors using the nonselective agonist 2,5-dimethoxy-4-iodoamphetamine (DOI) both increased and decreased activity in subsets of normal spiking cells,111,112 which could conceivably include both principal excitatory cells, such as pyramidal neurons, and regular spiking interneurons, such as those expressing the Ca2+-binding proteins calbindin and calretinin. However, the overall effect on population spiking seems to differ based on the region from which the recordings were taken. Puig et al111 found that DOI administration led to an overall 2.4-fold increase in spiking activity in deep layers of the medial frontal cortex, while recordings from the orbitofrontal cortex and anterior cingulate cortex showed that DOI reduced population activity.112,113 Moreover, given the multiplicity of 5-HT2A receptor-expressing GABAergic interneurons on principal cells, eg, in the amygdala and hippocampus,107 it seems likely that the effects of pharmacological treatments that either activate or inhibit 5-HT2A receptors are overwhelmingly mediated by downstream effects of GABA neurotransmission. This may explain evidence that 5-HT2A-receptor antagonism using M100907 leads to seemingly paradoxical increases in excitatory postsynaptic currents and enhancements in hippocampal long-term potentiation (LTP).114
5-HT3 receptors
The 5-HT3 receptor is unique among the 14 known serotonergic receptors, in that it is the only stimulatory ligand-gated ion channel. It is expressed in MDD-relevant brain regions, such as the frontal cortex, anterior cingulate cortex, hippocampus, and related cortical regions, such as the entorhinal and perirhinal cortices and the amygdala complex.115 The 5-HT3 receptor is also seemingly unique in that its expression, at least in the rodent brain, is largely circumscribed to GABAergic interneurons,116,117 where it is thought to provide a serotonin-mediated fast excitatory drive. More specifically, cortical interneurons expressing 5-HT3 receptor mRNA are at their highest density in the shallow cortical layers, and are thought to express the calcium-binding proteins calbindin and calretinin, but not parvalbumin.116,117 Consistent with this idea, 5-HT3-receptor agonists suppress the firing of medial prefrontal cortical pyramidal neurons in rodents, while 5-HT3-receptor antagonists suppress the firing of GABAergic interneurons and concomitantly increase the firing rate of cortical pyramidal cells.117–119 Additionally, 5-HT3-receptor antagonists increase measures of synaptic potentiation, such as theta-burst LTP and drive increases in cortical theta oscillatory rhythms.120
Less well-known relationships between serotonergic targets and GABA neurotransmission
5-HT4 receptors
5-HT4 receptors are G-protein-coupled receptors that are associated with Gs, and thus act to depolarize cellular membranes by stimulation of adenylyl cyclase and the resulting increase in cyclic adenosine monophosphate (cAMP) production.121 Based on autoradiographic mapping, these receptors are thought to be expressed in limbic regions, such as the olfactory tubercle, islands of Calleja, septum, hippocampus, and amygdala;122 however, at this time we have not been able to identify any studies that have investigated the cell types that express this receptor using histological techniques.
Based on evidence from electrophysiological recordings, it is likely that 5-HT4 receptors are expressed on pyramidal neurons in the CA1 region of the hippocampus, given that at least one group has found a 5-HT4 receptor-mediated depolarizing current in these cells.121 However, evidence tying 5-HT4 receptors to GABAergic interneurons is relatively sparse. There is some electrophysiological and neurochemical evidence that stimulation of 5-HT4 receptors can have an effect on GABA-mediated neurotransmission; however, it is not clear that these effects are mediated via direct effects of 5-HT4 receptors on GABAergic cells. For example, Bianchi et al123 found that 5-HT4-receptor stimulation using the selective agonist BIMU-8 in hippocampal slices from guinea pigs had no effect on GABA outflow under basal conditions, but during electrical stimulation low BIMU-8 concentrations elicited GABA release, while higher concentrations inhibited GABA release. These effects could be blocked by a selective 5-HT4-receptor antagonist, or by antagonists selective for muscarinic receptors, making it possible that these effects are mediated indirectly via cholinergic transmission rather than by direct effects on GABAergic interneurons. Similarly, Cai et al124 found that GABAA receptor-mediated currents measured from cortical pyramidal neurons could be either increased or decreased by 5-HT4-receptor stimulation, depending on the activation state of protein kinase A. However, these data only demonstrate that 5-HT4 receptors can modulate GABAA receptor-mediated currents, and do not necessarily suggest that these effects are mediated by direct effects of 5-HT4 receptors on GABAergic cells. Moreover, without histological data on the presence or absence of 5-HT4 receptors on GABAergic interneurons, it is probably premature to draw conclusions on the nature of the relationship between 5-HT4 receptors and GABAergic neurotransmission.
5-HT5 receptors
Studies of the 5-HT5 receptor in stably transfected cell lines have suggested that this receptor is coupled to Gi/Go and is thus negatively coupled to adenylyl cyclase.125 Additionally, there is some evidence suggesting that 5-HT5 receptors can be coupled to inwardly rectifying K+ channels in the rodent PFC.126 Immunohistochemical evidence demonstrates that the 5-HT5 receptor is expressed throughout the CNS in rodents, with its most intense immunoreactivity being noted in the suprachiasmatic nucleus and hypothalamic regions.127 In addition to these regions, 5-HT5 receptors were also observed at weak or moderately weak levels of immunoreactivity in cognition- and mood-relevant regions, such as the frontal cortex, anterior cingulate, and hippocampus.127 Moreover, the evidence linking 5-HT5 receptors to GABAergic interneurons or principal excitatory cells is relatively sparse at this time. Oliver et al127 observed relatively weak 5-HT5-receptor immunoreactivity in striatal medium spiny neurons, and thus the presence of 5-HT5 receptors on GABAergic neurons has been confirmed in at least one brain region. This group also observed 5-HT5-receptor immunoreactivity on principal cells in some regions, including hippocampal pyramidal neurons in regions CA1–3, as well as granule cells in the dentate gyrus. Finally, it is likely that 5-HT5 receptors are also expressed on cortical pyramidal neurons, given evidence of a 5-HT5-mediated inhibitory current in layer V cortical pyramidal neurons.126 Again, based on this sparse evidence, we believe it is premature to make any firm conclusions on the relationship between 5-HT5 receptors and GABAergic neurotransmission at this time.
5-HT6 receptors
5-HT6 receptors are G-protein-coupled receptors that are positively linked to adenylyl cyclase activity when transiently expressed in cell lines via Gs.128 Investigation of the anatomical pattern of 5-HT6-receptor expression found the highest expression in the striatum, olfactory tubercle, hippocampus, and frontal cortex, while more moderate expression was found in the thalamus and substantia nigra, among other regions.128,129
The expression pattern is relatively similar when investigated via in situ hybridization128 or immunohistochemical techniques,129 which may suggest that 5-HT6 receptors tend to have a somatodendritic localization.
Once again, evidence tying 5-HT6 receptors to GABAergic neurotransmission is relatively sparse at this time, and theories about a connection between the two are largely driven by indirect evidence from electrophysiological or neurochemical studies. One notable electrophysiological study by West et al130 found that despite being coupled to Gs, stimulation of 5-HT6 receptors using the 5-HT6-preferring agonist WAY-181187 attenuates theta-burst stimulation-induced LTP in hippocampal slice recordings. Additionally, West et al found that WAY-181187 increased the frequency of inhibitory postsynaptic currents (IPSCs) recorded from pyramidal neurons. Both of these effects were blocked by the selective 5-HT6-receptor antagonist SB-399885. Another study131 found that WAY-181187 significantly increased the concentration of extracellular GABA in the PFC, striatum, and dorsal hippocampus when evaluated in vivo, and further that this effect was blocked by the 5-HT6-receptor antagonist SB-217046. The authors also demonstrated that potassium-stimulated glutamate release in the slice was attenuated by WAY-181187 at 30 and 100 μM concentrations, but not at 10 μM. However, given that this agonist only has about a 60-fold selectivity for the 5-HT6 receptor over the 5-HT2C receptor (ie, 2 nM versus 120 nM, respectively), and is not active at the 10 μM concentration, it is not entirely clear that it is the 5-HT6-receptor mechanism that is driving this effect on glutamate release, particularly given that no attempt was made to antagonize this effect with SB-217046.
These data make it tempting to conclude that 5-HT6 receptors are expressed on GABAergic interneurons and act to increase their activity in such regions as the hippocampus. There is some evidence to support the idea of 5-HT6-receptor colocalization with GABAergic cells; however, it is relatively thin at this time. One study found that 5-HT6 receptors were expressed on nigrostriatal neurons that are known to coexpress GABA, dynorphin, and substance P.129 In another paper, double immunofluorescence was used to demonstrate in an example cell that 5-HT6-IR processes were closely apposed to a GAD67-IR cell.132 Finally, a study in postmortem human striatal tissue found that 5-HT6 immunoreactivity was present in PV-IR cells.133
However, the most comprehensive anatomical study of 5-HT6 receptors to date found substantial heterogeneity in the types of cells that express 5-HT6 receptors in human limbic brain regions, such as the frontal cortex and hippocampus. Marazziti et al134 demonstrated that the highest density of 5-HT6 receptor-IR cells in the frontal cortex was present in layer I, and that those cells were astrocytes rather than neurons. These authors also found that 5-HT6 receptor-IR cells were present in deeper cortical layers, and that those cells had a pyramidal morphology and were positive for the neuronal nuclear label NeuN. Finally, Marazziti et al showed that 5-HT6 receptor-IR cells in human hippocampal regions CA1–4 were pyramidal cells, and did not report colocalization with GABAergic cells in this region. Therefore, while it seems clear from the rodent electrophysiology and neurochemistry data that 5-HT6-receptor activation can modulate GABAergic neurotransmission, it is not clear that this modulation is due to direct effects of 5-HT6 receptors on GABAergic cells.
5-HT7 receptors
The 5-HT7 receptor is a relatively novel G-protein-coupled receptor that is expressed in several MDD-relevant brain regions, such as the frontal cortex, septum, thalamus, hippocampus, and amygdala.135,136 It is coupled to Gs, and thus 5-HT7-receptor stimulation can be expected to increase adenylyl cyclase activity and intracellular cAMP, leading to depolarization of neuronal membranes. The availability of data on the cellular localization of 5-HT7 heteroreceptors is still somewhat limited; however, at this time 5-HT7-receptor expression has been confirmed on some types of GABAergic neurons, eg, on cerebellar Purkinje cells,137 and the presence of 5-HT7 receptors on GABAergic interneurons in other regions has been inferred based on electrophysiology data showing that 5-HT7-receptor stimulation increases the frequency of spontaneous IPSCs.138 Therefore, as with the 5-HT2A and 5-HT3 receptors, stimulation of the 5-HT7 receptor is thought to have an excitatory effect on GABAergic neurotransmission.
However, 5-HT7 receptors are also thought to be expressed on pyramidal cells in the frontal cortex and hippocampus on the basis of immunohistochemical136 and electrophysiological evidence.138–140 Therefore, stimulation of 5-HT7 receptors is likely to have mixed effects on the overall excitatory state of the circuits in which it is present, although on average it seems to lead to an overall excitation of firing.140 This idea is overall supported by observations that repeated administration of the 5-HT7 receptor-selective antagonist SB269970 reduces the frequency and amplitude of spontaneous excitatory postsynaptic potentials from cortical pyramidal neurons.138
In summary, when the direct effects of these serotonergic heteroreceptors on GABAergic cells are considered, it appears that serotonin signaling has generally excitatory effects on GABAergic neurotransmission that are mediated through 5-HT2A, 5-HT3, and perhaps 5-HT7 receptors, although it should be noted that 5-HT1A receptors appear to be a 5-HT-mediated mechanism for GABA inhibition. The direct effects of 5-HT4–7 receptors on GABAergic interneurons are unclear at this time. Importantly, the overall effect of 5-HT stimulation on GABA neurotransmission in vivo will likely vary by region on the basis of which heteroreceptors are expressed locally.
Examples of serotonergic antidepressants modulating GABAergic neurotransmission
Fluoxetine
Fluoxetine is an SSRI that was approved in the US in the late 1980s, and has become a standard treatment for patients with MDD. Its action is characterized in vivo by a combination of its own inhibitory effects at the SERT (Dissociation constant [Ki] =0.9 nM)141 and the SERT-inhibitory effects of its long-acting metabolite norfluoxetine (Ki =2.3 nM),141 and thus fluoxetine is thought to bring about its antidepressant actions by elevating extracellular 5-HT concentrations. Based on the conclusion that serotonin heteroreceptors have a generally excitatory effect on GABAergic neurotransmission, it can be expected that the increases in 5-HT tone induced by fluoxetine treatment will lead to overall increases in GABAergic neurotransmission.
Although in general there is not a great deal of information available on the actions of serotonergic antidepressants on GABA neurotransmission, there is a fair amount of data available on fluoxetine’s effects. Studies of fluoxetine’s effects on interneuron firing using electrophysiological recordings in the slice suggest that bath application of fluoxetine in the range of 10–100 μM leads to significant increases in PFC fast-spiking (FS) interneuron-firing rates, curiously without significantly affecting pyramidal neuron firing.142 A separate research group using slice electrophysiology demonstrated that application of fluoxetine at 10–30 μM concentrations led to significant increases in the frequency but not amplitude of spontaneous IPSCs.143 In addition, recordings from brain slices taken from animals treated with a dose of 10 mg/kg/day fluoxetine for 21 days demonstrated that the intrinsic excitability of FS interneurons (measured as the number of spikes elicited by increasing current injections) was enhanced compared to FS cells recorded from vehicle-treated slices.142 Finally, Gören et al144 found that acute or repeated fluoxetine administration at 5 mg/kg/day significantly increased CSF GABA concentrations in rodents. Each of these observations suggests that fluoxetine administration can induce significant increases in GABA neurotransmission, consistent with the idea that serotonin neurotransmission has a generally excitatory action on GABA.
In addition to these observations, there are several studies using high concentrations of fluoxetine that may suggest it can potentiate GABA neurotransmission independently of its serotonergic mechanisms. For example, Robinson et al145 used human cell lines expressing recombinant GABAA receptors to show that fluoxetine has a positive allosteric modulatory effect on GABAA-receptor responses in the range of 130 μM concentrations, while the active metabolite norfluoxetine had a similar effect in the range of 600 nM concentrations. This effect was found for GABA receptors expressing all α subunits except α5, and was later replicated in a separate set of experiments using a 300 μM fluoxetine concentration.146 Furthermore, a point mutation in the α5 subunit conferred sensitivity to this allosteric modulatory effect, while a separate point mutation in the α1 subunit reduced the sensitivity of GABAA receptors to fluoxetine.146 A separate group using electrophysiological recordings in cultured hippocampal neurons from rodents found that fluoxetine (at concentrations of 10–30 μM) was able to increase the frequency and amplitude of spontaneous IPSCs in a manner that was insensitive to ritanserin or tetrodotoxin, suggesting a postsynaptic action that is at least independent of 5-HT2-receptor actions.147 Therefore, it is possible that fluoxetine has the ability to positively modulate GABA neurotransmission independently of serotonergic signaling. However, it should be pointed out that the clinical relevance of these observations can be questioned from the perspective that they represent very high biophase concentrations of fluoxetine.
Clinical observations of whole-brain exposure suggest that fluoxetine can reach concentrations in the tens of micromoles during treatment for MDD,148 which by itself may suggest that allosteric modulatory effects beginning in the hundreds of micromoles are probably irrelevant in MDD patients. However, it should be noted that whole-brain exposure is not necessarily the best exposure measurement to predict a drug’s CNS actions, as it represents the free brain drug concentration plus drug that is bound to lipids and proteins, and is thus not free to interact with neurotransmitter receptors. Fortunately, for drugs like fluoxetine, the CSF concentration can be a good heuristic of the brain free concentration.149 Measurements of CSF concentrations in humans after 6 weeks of clinically relevant dosing have found that fluoxetine concentrations are at approximately 25 nmol/L, while norfluoxetine concentrations are at about 17 nmol/L.150 Based on this observation, concentrations of 10–30 μM fluoxetine in cultured neurons are probably also at an irrelevantly high concentration, given that cultured neurons are thought to be much more available for drug interactions compared to neurons in brain-slice preparations due to the relative absence of glial cells. Based on this line of evidence, it is possible, although unlikely, that fluoxetine has direct positive allosteric modulatory effects on GABA neurotransmission in the clinic.
It is important to note, however, that fluoxetine does not appear to have universally GABA neurotransmission-enhancing effects. For example, Méndez et al143 demonstrated that 10–20 μM fluoxetine in hippocampal brain slices significantly decreased IPSC amplitudes, and further that this effect was likely due to a change in feed-forward inhibition originating from perisomatic FS interneurons. These authors went on to show that for chronic fluoxetine administration at 20 mg/kg/day, which is a dose that is well above the clinically relevant range,80 the strength of the electrical stimulus required to elicit firing was significantly higher than in slices from saline-treated animals, and further that those IPSCs were smaller than in control slices. However, it is important to note that this group did not observe changes in basal firing frequency, resting membrane potentials, or membrane resistances. Another group used slice electrophysiological recordings from rats chronically treated with the more reasonable dose of 10 mg/kg/day to show that 5-HT2 receptor-mediated IPSCs desensitize faster than in control conditions,151 and further that chronic fluoxetine treatment led to a significant decrease in the effects of 5-HT application on FS interneuron-firing rates.142 Therefore, at the very least, long-term fluoxetine treatment can lead to a desensitization of 5-HT2 receptor-mediated effects on GABA neurotransmission, and may also decrease the excitability of FS interneurons in some hippocampal circuits.
Given that fluoxetine requires chronic administration in the clinic in order to effectively treat MDD, it is reasonable to assume that changes in GABA neurotransmission resulting from chronic fluoxetine treatment are more relevant to fluoxetine’s antidepressant efficacy. However, given that chronic fluoxetine use can increase intrinsic excitability in some interneurons142 and increase CSF GABA concentrations,144 while at the same time reducing serotonin’s effect on phasic GABA activation,142,151 it is unclear whether fluoxetine’s GABA-enhancing effects are beneficial for mood. However, behavioral data from nonclinical model systems have suggested that pharmacological antagonism of GABAA receptors using bicuculline can partially block fluoxetine’s antidepressant-like effects,152 and thus it is at least plausible that fluoxetine’s antidepressant effects are partially mediated by enhancing GABA neurotransmission.
Vortioxetine
Vortioxetine is a multimodal-acting antidepressant that was recently approved in North America and Europe for the treatment of MDD. Vortioxetine is an inhibitor at the SERT (Ki =1.6 nM in human receptors), an antagonist at 5-HT1D (Ki =54 nM), 5-HT3 (Ki =3.7 nM), and 5-HT7 receptors (Ki =19 nM), an agonist at 5-HT1A receptors (Ki =15 nM), and a partial agonist at 5-HT1B receptors (Ki =33 nM).153,154 Based on the earlier discussion, it is expected that vortioxetine’s direct pharmacological effects on GABAergic neurotransmission will be inhibitory overall, based on 5-HT1A-receptor activation, antagonism of 5-HT3, and perhaps also 5-HT7 receptors. Vortioxetine may have some indirect excitatory effects on GABA neurotransmission mediated via increased activation of 5-HT2A receptors due to 5-HT-reuptake inhibition. However, we have hypothesized that on average, vortioxetine will inhibit GABAergic neurotransmission.
Slice electrophysiological recordings have demonstrated that vortioxetine blocks 5-HT-induced increases in spontaneous inhibitory postsynaptic potentials recorded from hippocampal pyramidal neurons and increases hippocampal theta-burst LTP.155 In vivo recordings of putative frontal cortical pyramidal cells also found that acute vortioxetine significantly increased firing rates.156 In each case, the hypothesized mechanism involved 5-HT3-receptor antagonism. Additionally, in vivo electroencephalography has demonstrated that vortioxetine increases frontocortical theta and gamma power during active awake periods.155,157 By comparison, the SSRI escitalopram had no effects on 5-HT-induced spontaneous inhibitory postsynaptic potentials, theta-burst LTP, frontal cortex pyramidal firing, or theta/gamma oscillatory power.155–157 These data fit with the hypothesis that vortioxetine indirectly reduces GABAergic neurotransmission via its serotonergic pharmacological actions. However, more studies are required to further substantiate vortioxetine’s effects on GABA neurotransmission, to understand the mechanisms underlying these effects, and to demonstrate that vortioxetine’s effects on GABA neurotransmission translate into clinical MDD populations.
Importantly, there is a large set of accumulated clinical data demonstrating that vortioxetine is an effective antidepressant.158 Therefore, despite the fact that vortioxetine and fluoxetine have apparently opposing effects on GABAergic neurotransmission, both are effective antidepressants. Moreover, this idea may further call into question the hypothesis that a simple reduction in GABA neurotransmission is at the etiological root of MDD.
Conclusion
In summary, there has been evidence of reduced CNS GABA concentrations in MDD going back to at least 1980, and more recent MRS data have generally supported this idea. However, it is not clear based on current clinical data whether these reductions are circumscribed to certain brain regions, whether the reduced GABA concentrations are from an intracellular pool that is used for neurotransmission, or whether the reduced GABA concentrations are a consistent biomarker for depression. Moreover, the MDD-related reductions in GABA are probably not related to impaired function in GABA synthesis via GAD, but instead it is more likely that these changes in GABA concentrations are related to pathology in calbindin-IR interneurons or in glia, although more data are required to confirm these ideas. Gene-expression data suggest that GABAA-receptor physiology may be altered in MDD, but a relative paucity of receptor-binding, protein-expression, and functional assay data has made it difficult to assess whether these changes have translated into altered receptor function, or what the consequences of these changes might be for GABA neurotransmission. There are GABA pharmacology data that could support the reduced GABA neurotransmission hypothesis in MDD. However, there are also data suggesting that increasing GABA neurotransmission actually induces negative mood (eg, mood dysfunction induced by vigabatrin, barbiturates, or baclofen). Nonclinical data have suggested that reducing some aspects of GABA neurotransmission is actually beneficial for mood, eg, in the case of direct GABAB-receptor antagonists. Finally, two serotonergic antidepressants – fluoxetine and vortioxetine – have apparently opposing effects on GABA neurotransmission, despite both being effective antidepressants. Moreover, it may be important to consider the idea that treatments capable of reducing some aspects of GABA neurotransmission may be more in line with theories claiming that synaptic potentiation is important for the fast onset of antidepressant efficacy seen in ketamine.7 Taken together, these data suggest that the notion that MDD is related to simple reductions in GABAergic neurotransmission must be reevaluated and discarded in favor of a more nuanced and complex model.
Disclosure
Alan L Pehrson and Connie Sanchez are employees of Lundbeck Research USA. The authors report no other conflicts of interest in this work.
References
World Health Organization. Depression. 2012. Available from: http://www.who.int/mediacentre/factsheets/fs369/en. Accessed September 11, 2014. | ||
McIntyre RS, Cha DS, Soczynska JK, et al. Cognitive deficits and functional outcomes in major depressive disorder: determinants, substrates, and treatment interventions. Depress Anxiety. 2013;30(6):515–527. | ||
Birnbaum HG, Kessler RC, Kelley D, Ben-Hamadi R, Joish VN, Greenberg PE. Employer burden of mild, moderate, and severe major depressive disorder: mental health services utilization and costs, and work performance. Depress Anxiety. 2010;27(1):78–89. | ||
Jaeger J, Berns S, Uzelac S, Davis-Conway S. Neurocognitive deficits and disability in major depressive disorder. Psychiatry Res. 2006;145(1):39–48. | ||
Charney DS. Monoamine dysfunction and the pathophysiology and treatment of depression. J Clin Psychiatry. 1998;59 Suppl 14:11–14. | ||
Carotenuto M, Esposito M, Parisi L, et al. Depressive symptoms and childhood sleep apnea syndrome. Neuropsychiatric Dis Treat. 2012;8:369–373. | ||
Zarate C, Duman RS, Liu G, Sartori S, Quiroz J, Murck H. New paradigms for treatment-resistant depression. Ann N Y Acad Sci. 2013;1292:21–31. | ||
Martin DL, Rimvall K. Regulation of gamma-aminobutyric acid synthesis in the brain. J Neurochem. 1993;60(2):395–407. | ||
Soghomonian JJ, Martin DL. Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci. 1998;19(12):500–505. | ||
Pinal CS, Tobin AJ. Uniqueness and redundancy in GABA production. Perspect Dev Neurobiol. 1998;5(2–3):109–118. | ||
Martin DL, Barke KE. Are GAD65 and GAD67 associated with specific pools of GABA in brain? Perspect Dev Neurobiol. 1998;5(2–3):119–129. | ||
Rowley NM, Madsen KK, Schousboe A, White HS. Glutamate and GABA synthesis, release, transport and metabolism as targets for seizure control. Neurochem Int. 2012;61(4):546–558. | ||
Koga M, Serritella AV, Messmer MM, et al. Glutathione is a physiologic reservoir of neuronal glutamate. Biochem Biophys Res Commun. 2011;409(4):596–602. | ||
Behar KL, Rothman DL. In vivo nuclear magnetic resonance studies of glutamate-gamma-aminobutyric acid-glutamine cycling in rodent and human cortex: the central role of glutamine. J Nutr. 2001;131(9 Suppl):2498S–2504S; discussion 2523S–2524S. | ||
Rae C, Hare N, Bubb WA, et al. Inhibition of glutamine transport depletes glutamate and GABA neurotransmitter pools: further evidence for metabolic compartmentation. J Neurochem. 2003;85(2):503–514. | ||
Gold BI, Bowers MB Jr, Roth RH, Sweeney DW. GABA levels in CSF of patients with psychiatric disorders. Am J Psychiatry. 1980;137(3):362–364. | ||
Kugaya A, Sanacora G, Verhoeff NP, et al. Cerebral benzodiazepine receptors in depressed patients measured with [123I]iomazenil SPECT. Biol Psychiatry. 2003;54(8):792–799. | ||
Price RB, Shungu DC, Mao X, et al. Amino acid neurotransmitters assessed by proton magnetic resonance spectroscopy: relationship to treatment resistance in major depressive disorder. Biol Psychiatry. 2009;65(9):792–800. | ||
Sanacora G, Gueorguieva R, Epperson CN, et al. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61(7):705–713. | ||
Sanacora G, Mason GF, Rothman DL, et al. Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 1999;56(11):1043–1047. | ||
Hasler G, van der Veen JW, Tumonis T, Meyers N, Shen J, Drevets WC. Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 2007;64(2):193–200. | ||
Sanacora G, Mason GF, Rothman DL, Krystal JH. Increased occipital cortex GABA concentrations in depressed patients after therapy with selective serotonin reuptake inhibitors. Am J Psychiatry. 2002;159(4):663–665. | ||
Sanacora G, Mason GF, Rothman DL, et al. Increased cortical GABA concentrations in depressed patients receiving ECT. Am J Psychiatry. 2003;160(3):577–579. | ||
Hasler G, Neumeister A, van der Veen JW, et al. Normal prefrontal gamma-aminobutyric acid levels in remitted depressed subjects determined by proton magnetic resonance spectroscopy. Biol Psychiatry. 2005;58(12):969–973. | ||
Shaw A, Brealy J, Richardson H, et al. Marked reductions in visual evoked responses but not gamma-aminobutyric acid concentrations or gamma-band measures in remitted depression. Biol Psychiatry. 2013;73(7):691–698. | ||
Sanacora G, Fenton LR, Fasula MK, et al. Cortical gamma-aminobutyric acid concentrations in depressed patients receiving cognitive behavioral therapy. Biol Psychiatry. 2006;59(3):284–286. | ||
Bhagwagar Z, Wylezinska M, Jezzard P, et al. Reduction in occipital cortex gamma-aminobutyric acid concentrations in medication-free recovered unipolar depressed and bipolar subjects. Biol Psychiatry. 2007;61(6):806–812. | ||
Bhagwagar Z, Wylezinska M, Jezzard P, et al. Low GABA concentrations in occipital cortex and anterior cingulate cortex in medication-free, recovered depressed patients. Int J Neuropsychopharmacol. 2008;11(2):255–260. | ||
Herrera-Guzmán I, Gudayol-Ferré E, Herrera-Guzmán D, Guàrdia-Olmos J, Hinojosa-Calvo E, Herrera-Abarca JE. Effects of selective serotonin reuptake and dual serotonergic-noradrenergic reuptake treatments on memory and mental processing speed in patients with major depressive disorder. J Psychiatr Res. 2009;43(9):855–863. | ||
Karolewicz B, Maciag D, O’Dwyer G, Stockmeier CA, Feyissa AM, Rajkowska G. Reduced level of glutamic acid decarboxylase-67 kDa in the prefrontal cortex in major depression. Int J Neuropsychopharmacol. 2010;13(4):411–420. | ||
Fatemi SH, Stary JM, Earle JA, Araghi-Niknam M, Eagan E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res. 2005;72(2–3):109–122. | ||
Guidotti A, Auta J, Davis JM, et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57(11):1061–1069. | ||
Gilabert-Juan J, Varea E, Guirado R, Blasco-Ibáñez JM, Crespo C, Nácher J. Alterations in the expression of PSA-NCAM and synaptic proteins in the dorsolateral prefrontal cortex of psychiatric disorder patients. Neurosci Lett. 2012;530(1):97–102. | ||
Sibille E, Morris HM, Kota RS, Lewis DA. GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. Int J Neuropsychopharmacol. 2011;14(6):721–734. | ||
Bielau H, Steiner J, Mawrin C, et al. Dysregulation of GABAergic neurotransmission in mood disorders: a postmortem study. Ann N Y Acad Sci. 2007;1096:157–169. | ||
Gos T, Günther K, Bielau H, et al. Suicide and depression in the quantitative analysis of glutamic acid decarboxylase-immunoreactive neuropil. J Affect Disord. 2009;113(1–2):45–55. | ||
Gos T, Steiner J, Bielau H, et al. Differences between unipolar and bipolar I depression in the quantitative analysis of glutamic acid decarboxylase-immunoreactive neuropil. Eur Arch Psychiatry Clin Neurosci. 2012;262(8):647–655. | ||
Cheetham SC, Crompton MR, Katona CL, Parker SJ, Horton RW. Brain GABAA/benzodiazepine binding sites and glutamic acid decarboxylase activity in depressed suicide victims. Brain Res. 1988;460(1):114–123. | ||
Lund JS, Lewis DA. Local circuit neurons of developing and mature macaque prefrontal cortex: Golgi and immunocytochemical characteristics. J Comp Neurol. 1993;328(2):282–312. | ||
Rajkowska G, O’Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology. 2007;32(2):471–482. | ||
Maciag D, Hughes J, O’Dwyer G, et al. Reduced density of calbindin immunoreactive GABAergic neurons in the occipital cortex in major depression: relevance to neuroimaging studies. Biol Psychiatry. 2010;67(5):465–470. | ||
Beasley CL, Zhang ZJ, Patten I, Reynolds GP. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry. 2002;52(7):708–715. | ||
Reynolds GP, Beasley CL, Zhang ZJ. Understanding the neurotransmitter pathology of schizophrenia: selective deficits of subtypes of cortical GABAergic neurons. J Neural Transm. 2002;109(5–6):881–889. | ||
Guilloux JP, Douillard-Guilloux G, Kota R, et al. Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry. 2012;17(11):1130–1142. | ||
Tripp A, Kota RS, Lewis DA, Sibille E. Reduced somatostatin in subgenual anterior cingulate cortex in major depression. Neurobiol Dis. 2011;42(1):116–124. | ||
Rajkowska G, Miguel-Hidalgo JJ, Wei J, et al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45(9):1085–1098. | ||
Rajkowska G, Stockmeier CA. Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr Drug Targets. 2013;14(11):1225–1236. | ||
Sanacora G, Banasr M. From pathophysiology to novel antidepressant drugs: glial contributions to the pathology and treatment of mood disorders. Biol Psychiatry. 2013;73(12):1172–1179. | ||
Paulsen RE, Fonnum F. Role of glial cells for the basal and Ca2+-dependent K+-evoked release of transmitter amino acids investigated by microdialysis. J Neurochem. 1989;52(6):1823–1829. | ||
Yüksel C, Öngür D. Magnetic resonance spectroscopy studies of glutamate-related abnormalities in mood disorders. Biol Psychiatry. 2010;68(9):785–794. | ||
Liang SL, Carlson GC, Coulter DA. Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. J Neurosci. 2006;26(33):8537–8548. | ||
Levinson AJ, Fitzgerald PB, Favalli G, Blumberger DM, Daigle M, Daskalakis ZJ. Evidence of cortical inhibitory deficits in major depressive disorder. Biol Psychiatry. 2010;67(5):458–464. | ||
Paulus W, Classen J, Cohen LG, et al. State of the art: pharmacologic effects on cortical excitability measures tested by transcranial magnetic stimulation. Brain Stimul. 2008;1(3):151–163. | ||
Choudary PV, Molnar M, Evans SJ, et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci U S A. 2005;102(43):15653–15658. | ||
Sequeira A, Mamdani F, Ernst C, et al. Global brain gene expression analysis links glutamatergic and GABAergic alterations to suicide and major depression. PloS One. 2009;4(8):e6585. | ||
Fatemi SH, Folsom TD, Rooney RJ, Thuras PD. Expression of GABAA α2-, β1- and ε-receptors are altered significantly in the lateral cerebellum of subjects with schizophrenia, major depression and bipolar disorder. Transl Psychiatry. 2013;3:e303. | ||
Klumpers UM, Veltman DJ, Drent ML, et al. Reduced parahippocampal and lateral temporal GABAA-[11C]flumazenil binding in major depression: preliminary results. Eur J Nucl Med Mol Imaging. 2010;37(3):565–574. | ||
Zhu H, Karolewicz B, Nail E, Stockmeier CA, Szebeni K, Ordway GA. Normal [3H]flunitrazepam binding to GABAA receptors in the locus coeruleus in major depression and suicide. Brain Res. 2006;1125(1):138–146. | ||
Stocks GM, Cheetham SC, Crompton MR, Katona CL, Horton RW. Benzodiazepine binding sites in amygdala and hippocampus of depressed suicide victims. J Affect Disord. 1990;18(1):11–15. | ||
Fatemi SH, Folsom TD, Thuras PD. Deficits in GABA(B) receptor system in schizophrenia and mood disorders: a postmortem study. Schizophr Res. 2011;128(1–3):37–43. | ||
Arranz B, Cowburn R, Eriksson A, Vestling M, Marcusson J. Gamma-aminobutyric acid-B (GABAB) binding sites in postmortem suicide brains. Neuropsychobiology. 1992;26(1–2):33–36. | ||
Cross JA, Cheetham SC, Crompton MR, Katona CL, Horton RW. Brain GABAB binding sites in depressed suicide victims. Psychiatry Res. 1988;26(2):119–129. | ||
Smith SS, Shen H, Gong QH, Zhou X. Neurosteroid regulation of GABA(A) receptors: focus on the α4 and δ subunits. Pharmacol Ther. 2007;116(1):58–76. | ||
Schmidt PJ, Nieman LK, Danaceau MA, Adams LF, Rubinow DR. Differential behavioral effects of gonadal steroids in women with and in those without premenstrual syndrome. N Engl J Med. 1998;338(4):209–216. | ||
Smith MJ, Adams LF, Schmidt PJ, Rubinow DR, Wassermann EM. Abnormal luteal phase excitability of the motor cortex in women with premenstrual syndrome. Biol Psychiatry. 2003;54(7):757–762. | ||
Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM. Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science. 1986;232(4753):1004–1007. | ||
Carboni E, Wieland S, Lan NC, Gee KW. Anxiolytic properties of endogenously occurring pregnanediols in two rodent models of anxiety. Psychopharmacology. 1996;126(2):173–178. | ||
Devaud LL, Purdy RH, Morrow AL. The neurosteroid, 3α-hydroxy-5α-pregnan-20-one, protects against bicuculline-induced seizures during ethanol withdrawal in rats. Alcohol Clin Exp Res. 1995;19(2):350–355. | ||
Shen H, Gong QH, Aoki C, et al. Reversal of neurosteroid effects at α4β2δ GABAA receptors triggers anxiety at puberty. Nat Neurosci. 2007;10(4):469–477. | ||
Smith SS, Ruderman Y, Frye C, Homanics G, Yuan M. Steroid withdrawal in the mouse results in anxiogenic effects of 3α,5β-THP: a possible model of premenstrual dysphoric disorder. Psychopharmacology. 2006;186(3):323–333. | ||
Wisden W, Laurie DJ, Monyer H, Seeburg PH. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J Neurosci. 1992;12(3):1040–1062. | ||
Gulinello M, Gong QH, Li X, Smith SS. Short-term exposure to a neuroactive steroid increases α4 GABA(A) receptor subunit levels in association with increased anxiety in the female rat. Brain Res. 2001; 910(1–2):55–66. | ||
Sundstrom-Poromaa I, Smith DH, Gong QH, et al. Hormonally regulated α(4)β(2)δ GABA(A) receptors are a target for alcohol. Nat Neurosci. 2002;5(8):721–722. | ||
Smith SS, Gong QH. Neurosteroid administration and withdrawal alter GABAA receptor kinetics in CA1 hippocampus of female rats. J Physiol. 2005;564(Pt 2):421–436. | ||
Smith SS, Gong QH, Li X, et al. Withdrawal from 3α-OH-5α-pregnan-20-one using a pseudopregnancy model alters the kinetics of hippocampal GABAA-gated current and increases the GABAA receptor α4 subunit in association with increased anxiety. J Neurosci. 1998;18(14):5275–5284. | ||
Maguire JL, Stell BM, Rafizadeh M, Mody I. Ovarian cycle-linked changes in GABA(A) receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat Neurosci. 2005;8(6):797–804. | ||
Epperson CN, Haga K, Mason GF, et al. Cortical gamma-aminobutyric acid levels across the menstrual cycle in healthy women and those with premenstrual dysphoric disorder: a proton magnetic resonance spectroscopy study. Arch Gen Psychiatry. 2002;59(9):851–858. | ||
Uzunov DP, Cooper TB, Costa E, Guidotti A. Fluoxetine-elicited changes in brain neurosteroid content measured by negative ion mass fragmentography. Proc Natl Acad Sci U S A. 1996;93(22):12599–12604. | ||
Matsumoto K, Uzunova V, Pinna G, et al. Permissive role of brain allopregnanolone content in the regulation of pentobarbital-induced righting reflex loss. Neuropharmacology. 1999;38(7):955–963. | ||
Pehrson AL, Leiser SC, Gulinello M, et al. Treatment of cognitive dysfunction in major depressive disorder – a review of the preclinical evidence for efficacy of selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors and the multimodal-acting antidepressant vortioxetine. Eur J Pharmacol. Epub 2014 Aug 5. | ||
Meyer JH, Wilson AA, Sagrati S, et al. Serotonin transporter occupancy of five selective serotonin reuptake inhibitors at different doses: an [11C]DASB positron emission tomography study. Am J Psychiatry. 2004;161(5):826–835. | ||
Uzunova V, Sheline Y, Davis JM, et al. Increase in the cerebrospinal fluid content of neurosteroids in patients with unipolar major depression who are receiving fluoxetine or fluvoxamine. Proc Natl Acad Sci U S A. 1998;95(6):3239–3244. | ||
Romeo E, Ströhle A, Spalletta G, et al. Effects of antidepressant treatment on neuroactive steroids in major depression. Am J Psychiatry. 1998;155(7):910–913. | ||
Schule C, Baghai TC, Eser D, et al. Mirtazapine monotherapy versus combination therapy with mirtazapine and aripiprazole in depressed patients without psychotic features: a 4-week open-label parallel-group study. World J Biol Psychiatry. 2007;8(2):112–122. | ||
Birkenhäger TK, Moleman P, Nolen WA. Benzodiazepines for depression? A review of the literature. Int Clin Psychopharmacol. 1995;10(3): 181–195. | ||
Petty F, Trivedi MH, Fulton M, Rush AJ. Benzodiazepines as antidepressants: does GABA play a role in depression? Biol Psychiatry. 1995; 38(9):578–591. | ||
Fava M, McCall WV, Krystal A, et al. Eszopiclone co-administered with fluoxetine in patients with insomnia coexisting with major depressive disorder. Biol Psychiatry. 2006;59(11):1052–1060. | ||
Fava M, Schaefer K, Huang H, et al. A post hoc analysis of the effect of nightly administration of eszopiclone and a selective serotonin reuptake inhibitor in patients with insomnia and anxious depression. J Clin Psychiatry. 2011;72(4):473–479. | ||
El Zahaf NA, Salem Elhwuegi A. The effect of GABAmimetics on the duration of immobility in the forced swim test in albino mice. Libyan J Med. 2014;9:23480. | ||
Christensen T, Bétry C, Mnie-Filali O, et al. Synergistic antidepressant-like action of gaboxadol and escitalopram. Eur Neuropsychopharmacol. 2012;22(10):751–760. | ||
Kasper S, Ebert B, Larsen K, Tonnoir B. Combining escitalopram with gaboxadol provides no additional benefit in the treatment of patients with severe major depressive disorder. Int J Neuropsychopharmacol. 2012;15(6):715–725. | ||
Lambert MV, Robertson MM. Depression in epilepsy: etiology, phenomenology, and treatment. Epilepsia. 1999;40 Suppl 10:S21–S47. | ||
Brent DA, Crumrine PK, Varma RR, Allan M, Allman C. Phenobarbital treatment and major depressive disorder in children with epilepsy. Pediatrics. 1987;80(6):909–917. | ||
Mumford JP, Dam M. Meta-analysis of European placebo controlled studies of vigabatrin in drug resistant epilepsy. Br J Clin Pharmacol. 1989;27 Suppl 1:101S–107S. | ||
Levinson DF, Devinsky O. Psychiatric adverse events during vigabatrin therapy. Neurology. 1999;53(7):1503–1511. | ||
Ring HA, Crellin R, Kirker S, Reynolds EH. Vigabatrin and depression. J Neurol Neurosurg Psychiatry. 1993;56(8):925–928. | ||
Cryan JF, Slattery DA. GABAB receptors and depression. Current status. Adv Pharmacol. 2010;58:427–451. | ||
Post RM, Ketter TA, Joffe RT, Kramlinger KL. Lack of beneficial effects of l-baclofen in affective disorder. Int Clin Psychopharmacol. 1991;6(4):197–207. | ||
Mamelak M. Narcolepsy and depression and the neurobiology of gammahydroxybutyrate. Prog Neurobiol. 2009; 89(2):193–219. | ||
Chouinard G. Issues in the clinical use of benzodiazepines: potency, withdrawal, and rebound. J Clin Psychiatry. 2004;65 Suppl 5:7–12. | ||
Pietrzak RH, Scott JC, Harel BT, Lim YY, Snyder PJ, Maruff P. A process-based approach to characterizing the effect of acute alprazolam challenge on visual paired associate learning and memory in healthy older adults. Hum Psychopharmacol. 2012;27(6):549–558. | ||
Lüscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron. 1997;19(3):687–695. | ||
Aznar S, Qian Z, Shah R, Rahbek B, Knudsen GM. The 5-HT1A serotonin receptor is located on calbindin- and parvalbumin-containing neurons in the rat brain. Brain Res. 2003;959(1):58–67. | ||
Levkovitz Y, Segal M. Serotonin 5-HT1A receptors modulate hippocampal reactivity to afferent stimulation. J Neurosci. 1997;17(14):5591–5598. | ||
Lladó-Pelfort L, Assié MB, Newman-Tancredi A, Artigas F, Celada P. In vivo electrophysiological and neurochemical effects of the selective 5-HT1A receptor agonist, F13640, at pre- and postsynaptic 5-HT1A receptors in the rat. Psychopharmacology. 2012;221(2):261–272. | ||
Lladó-Pelfort L, Santana N, Ghisi V, Artigas F, Celada P. 5-HT1A receptor agonists enhance pyramidal cell firing in prefrontal cortex through a preferential action on GABA interneurons. Cereb Cortex. 2012;22(7):1487–1497. | ||
Bombardi C, Di Giovanni G. Functional anatomy of 5-HT2A receptors in the amygdala and hippocampal complex: relevance to memory functions. Exp Brain Res. 2013;230(4):427–439. | ||
López-Giménez JF, Vilaró MT, Palacios JM, Mengod G. Mapping of 5-HT2A receptors and their mRNA in monkey brain: [3H]MDL100,907 autoradiography and in situ hybridization studies. J Comp Neurol. 2001;429(4):571–589. | ||
Celada P, Puig MV, Artigas F. Serotonin modulation of cortical neurons and networks. Front Integr Neurosci. 2013;7:25. | ||
Lüttgen M, Ove Ogren S, Meister B. Chemical identity of 5-HT2A receptor immunoreactive neurons of the rat septal complex and dorsal hippocampus. Brain Res. 2004;1010(1–2):156–165. | ||
Puig MV, Celada P, Diaz-Mataix L, Artigas F. In vivo modulation of the activity of pyramidal neurons in the rat medial prefrontal cortex by 5-HT2A receptors: relationship to thalamocortical afferents. Cereb Cortex. 2003;13(8):870–882. | ||
Wood J, Kim Y, Moghaddam B. Disruption of prefrontal cortex large scale neuronal activity by different classes of psychotomimetic drugs. J Neurosci. 2012;32(9):3022–3031. | ||
Ashby CR Jr, Jiang LH, Kasser RJ, Wang RY. Electrophysiological characterization of 5-hydroxytryptamine2 receptors in the rat medial prefrontal cortex. J Pharmacol Exp Ther. 1990;252(1):171–178. | ||
Wang RY, Arvanov VL. M100907, a highly selective 5-HT2A receptor antagonist and a potential atypical antipsychotic drug, facilitates induction of long-term potentiation in area CA1 of the rat hippocampal slice. Brain Res. 1998;779(1–2):309–313. | ||
Bloom FE, Morales M. The central 5-HT3 receptor in CNS disorders. Neurochem Res. 1998;23(5):653–659. | ||
Morales M, Bloom FE. The 5-HT3 receptor is present in different subpopulations of GABAergic neurons in the rat telencephalon. J Neurosci. 1997;17(9):3157–3167. | ||
Puig MV, Santana N, Celada P, Mengod G, Artigas F. In vivo excitation of GABA interneurons in the medial prefrontal cortex through 5-HT3 receptors. Cereb Cortex. 2004;14(12):1365–1375. | ||
Ashby CR Jr, Minabe Y, Edwards E, Wang RY. 5-HT3-like receptors in the rat medial prefrontal cortex: an electrophysiological study. Brain Res. 1991;550(2):181–191. | ||
Reznic J, Staubli U. Effects of 5-HT3 receptor antagonism on hippocampal cellular activity in the freely moving rat. J Neurophysiol. 1997;77(1):517–521. | ||
Stäubli U, Xu FB. Effects of 5-HT3 receptor antagonism on hippocampal theta rhythm, memory, and LTP induction in the freely moving rat. J Neurosci. 1995;15(3 Pt 2):2445–2452. | ||
Chapin EM, Haj-Dahmane S, Torres G, Andrade R. The 5-HT(4) receptor-induced depolarization in rat hippocampal neurons is mediated by cAMP but is independent of I(h). Neurosci Lett. 2002;324(1):1–4. | ||
Waeber C, Sebben M, Nieoullon A, Bockaert J, Dumuis A. Regional distribution and ontogeny of 5-HT4 binding sites in rodent brain. Neuropharmacology. 1994;33(3–4):527–541. | ||
Bianchi C, Rodi D, Marino S, Beani L, Siniscalchi A. Dual effects of 5-HT4 receptor activation on GABA release from guinea pig hippocampal slices. Neuroreport. 2002;13(17):2177–2180. | ||
Cai X, Flores-Hernandez J, Feng J, Yan Z. Activity-dependent bidirectional regulation of GABA(A) receptor channels by the 5-HT(4) receptor-mediated signalling in rat prefrontal cortical pyramidal neurons. J Physiol. 2002;540(Pt 3):743–759. | ||
Francken BJ, Jurzak M, Vanhauwe JF, Luyten WH, Leysen JE. The human 5-HT5A receptor couples to Gi/Go proteins and inhibits adenylate cyclase in HEK 293 cells. Eur J Pharmacol. 1998;361(2–3):299–309. | ||
Goodfellow NM, Bailey CD, Lambe EK. The native serotonin 5-HT(5A) receptor: electrophysiological characterization in rodent cortex and 5-HT(1A)-mediated compensatory plasticity in the knock-out mouse. J Neurosci. 2012;32(17):5804–5809. | ||
Oliver KR, Kinsey AM, Wainwright A, Sirinathsinghji DJ. Localization of 5-HT(5A) receptor-like immunoreactivity in the rat brain. Brain Res. 2000;867(1–2):131–142. | ||
Ruat M, Traiffort E, Arrang JM, et al. A novel rat serotonin (5-HT6) receptor: molecular cloning, localization and stimulation of cAMP accumulation. Biochem Biophys Res Commun. 1993;193(1):268–276. | ||
Gerard C, Martres MP, Lefevre K, et al. Immuno-localization of serotonin 5-HT6 receptor-like material in the rat central nervous system. Brain Res. 1997;746(1–2):207–219. | ||
West PJ, Marcy VR, Marino MJ, Schaffhauser H. Activation of the 5-HT(6) receptor attenuates long-term potentiation and facilitates GABAergic neurotransmission in rat hippocampus. Neuroscience. 2009;164(2):692–701. | ||
Schechter LE, Lin Q, Smith DL, et al. Neuropharmacological profile of novel and selective 5-HT6 receptor agonists: WAY-181187 and WAY-208466. Neuropsychopharmacology. 2008;33(6):1323–1335. | ||
Woolley ML, Marsden CA, Fone KC. 5-HT6 receptors. Curr Drug Targets CNS Neurol Disord. 2004;3(1):59–79. | ||
Marazziti D, Baroni S, Pirone A, et al. Distribution of serotonin receptor of type 6 (5-HT(6)) in human brain post-mortem. A pharmacology, autoradiography and immunohistochemistry study. Neurochem Res. 2012;37(5):920–927. | ||
Marazziti D, Baroni S, Pirone A, et al. Serotonin receptor of type 6 (5-HT6) in human prefrontal cortex and hippocampus post-mortem: an immunohistochemical and immunofluorescence study. Neurochem Int. 2013;62(2):182–188. | ||
Hedlund PB, Sutcliffe JG. Functional, molecular and pharmacological advances in 5-HT7 receptor research. Trends Pharmacol Sci. 2004;25(9):481–486. | ||
Neumaier JF, Sexton TJ, Yracheta J, Diaz AM, Brownfield M. Localization of 5-HT(7) receptors in rat brain by immunocytochemistry, in situ hybridization, and agonist stimulated cFos expression. J Chem Neuroanat. 2001;21(1):63–73. | ||
Geurts FJ, De Schutter E, Timmermans JP. Localization of 5-HT2A, 5-HT3, 5-HT5A and 5-HT7 receptor-like immunoreactivity in the rat cerebellum. J Chem Neuroanat. 2002;24(1):65–74. | ||
Tokarski K, Kusek M, Hess G. 5-HT7 receptors modulate GABAergic transmission in rat hippocampal CA1 area. J Physiol Pharmacol. 2011;62(5):535–540. | ||
Bacon WL, Beck SG. 5-Hydroxytryptamine(7) receptor activation decreases slow afterhyperpolarization amplitude in CA3 hippocampal pyramidal cells. J Pharmacol Exp Ther. 2000;294(2):672–679. | ||
Fan LL, Zhang QJ, Liu J, et al. In vivo effect of 5-HT(7) receptor agonist on pyramidal neurons in medial frontal cortex of normal and 6-hydroxydopamine-lesioned rats: an electrophysiological study. Neuroscience. 2011;190:328–338. | ||
Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J Pharmacol Exp Ther. 1997;283(3):1305–1322. | ||
Zhong P, Yan Z. Differential regulation of the excitability of prefrontal cortical fast-spiking interneurons and pyramidal neurons by serotonin and fluoxetine. PloS One. 2011;6(2):e16970. | ||
Méndez P, Pazienti A, Szabó G, Bacci A. Direct alteration of a specific inhibitory circuit of the hippocampus by antidepressants. J Neurosci. 2012;32(47):16616–16628. | ||
Gören MZ, Küçükibrahimoglu E, Berkman K, Terzioglu B. Fluoxetine partly exerts its actions through GABA: a neurochemical evidence. Neurochem Res. 2007;32(9):1559–1565. | ||
Robinson RT, Drafts BC, Fisher JL. Fluoxetine increases GABA(A) receptor activity through a novel modulatory site. J Pharmacol Exp Ther. 2003;304(3):978–984. | ||
Derry JM, Paulsen IM, Davies M, Dunn SM. A single point mutation of the GABA(A) receptor α5-subunit confers fluoxetine sensitivity. Neuropharmacology. 2007;52(2):497–505. | ||
Ye ZY, Zhou KQ, Xu TL, Zhou JN. Fluoxetine potentiates GABAergic IPSCs in rat hippocampal neurons. Neurosci Lett. 2008;442(1):24–29. | ||
Henry ME, Schmidt ME, Hennen J, et al. A comparison of brain and serum pharmacokinetics of R-fluoxetine and racemic fluoxetine: a 19-F MRS study. Neuropsychopharmacology. 2005;30(8):1576–1583. | ||
Liu X, Smith BJ, Chen C, et al. Evaluation of cerebrospinal fluid concentration and plasma free concentration as a surrogate measurement for brain free concentration. Drug Metab Dispos. 2006;34(9):1443–1447. | ||
Mårtensson B, Nyberg S, Toresson G, Brodin E, Bertilsson L. Fluoxetine treatment of depression. Clinical effects, drug concentrations and monoamine metabolites and N-terminally extended substance P in cerebrospinal fluid. Acta Psychiatr Scand. 1989;79(6):586–596. | ||
Zhong P, Yan Z. Chronic antidepressant treatment alters serotonergic regulation of GABA transmission in prefrontal cortical pyramidal neurons. Neuroscience. 2004;129(1):65–73. | ||
Khisti RT, Chopde CT, Jain SP. Antidepressant-like effect of the neurosteroid 3α-hydroxy-5α-pregnan-20-one in mice forced swim test. Pharmacol Biochem Behav. 2000;67(1):137–143. | ||
Pehrson AL, Sanchez C. Serotonergic modulation of glutamate neurotransmission as a strategy for treating depression and cognitive dysfunction. CNS Spectr. 2014;19(2):121–133. | ||
Westrich L, Pehrson A, Zhong H, et al. In vitro and in vivo effects of the multimodal antidepressant vortioxetine (Lu AA21004) at human and rat targets. Int J Psychiatry Clin Pract. 2012;16 Suppl 1:47. | ||
Dale E, Zhang H, Leiser SC, et al. Vortioxetine disinhibits pyramidal cell function and enhances synaptic plasticity in the rat hippocampus. J Psychopharmacol. 2014;28(10):891–902. | ||
Riga MS, Celada P, Sanchez C, Artigas F. Role of 5-HT3 receptors in the mechanism of action of the investigational antidepressant vortioxetine. Eur Neuropsychopharmacol. 2013;23 Suppl 2:S393–S394. | ||
Leiser SC, Pehrson AL, Robichaud PJ, Sanchez C. The multimodal antidepressant vortioxetine increases frontal cortical oscillations unlike escitalopram and duloxetine – a quantitative electroencephalographic study in the rat. Br J Pharmacol. Epub 2014 May 21. | ||
Alvarez E, Perez V, Artigas F. Pharmacology and clinical potential of vortioxetine in the treatment of major depressive disorder. Neuropsychiatric Dis Treat. 2014;10:1297–1307. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.