")
Back to Journals » Journal of Asthma and Allergy » Volume 16
Alterations in the Gut Microbiome of Young Children with Airway Allergic Disease Revealed by Next-Generation Sequencing
Authors Wan J, Song J , Lv Q, Zhang H, Xiang Q, Dai H, Zheng H, Lin X, Zhang W
Received 23 May 2023
Accepted for publication 17 August 2023
Published 7 September 2023 Volume 2023:16 Pages 961—972
DOI https://doi.org/10.2147/JAA.S422537
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Amrita Dosanjh
Jinyi Wan,1,2,* Jingjing Song,1,* Qingqing Lv,2 Hui Zhang,1 Qiangwei Xiang,1 Huan Dai,1 Hang Zheng,1 Xixi Lin,3 Weixi Zhang1
1Department of Pediatric Allergy and Immunology, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, 325027, People’s Republic of China; 2Department of Pediatric Internal Medicine, Taizhou Hospital of Zhejiang Province affiliated to Wenzhou Medical University, Taizhou, 317000, People’s Republic of China; 3Department of Pharmacy, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, 325027, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xixi Lin; Weixi Zhang, Email [email protected]; [email protected]
Purpose: Recent studies had shown that gut microbiota played a significant role in the development of the immune system and may affect the course of airway allergic disease. We conducted this study to determine unique gut microbial associated with allergic disease in children by shotgun gene sequencing.
Methods: We collected fecal samples from children with allergic asthma (n = 23) and allergic rhinitis (n = 18), and healthy control (n = 19). The gut microbiota of specimens was analyzed by high-throughput metagenomic shotgun gene sequencing.
Results: The intestinal microbiota of children with allergic asthma and allergic rhinitis was characterized by increased microbial richness and diversity. Simpson and Shannon were significantly elevated in children with allergic asthma. Principal coordinates analysis (PCoA) showed that the gut microbial communities cluster patterns of children with asthma or rhinitis were significantly different from those of healthy controls. However, no significant difference was found between asthma group and rhinitis group At the phylum level, higher relative abundance of Firmicutes was found in the allergic rhinitis group and allergic asthma group, while the level of Bacteroidetes was significantly lower. At the genus level, Corynebacterium, Streptococcus, Dorea, Actinomyces, Bifidobacterium, Blautia, and Rothia were significantly enriched in the allergic asthma group. Finally, a random forest classifier model selected 16 general signatures to discriminate the allergic asthma group from the healthy control group.
Conclusion: In conclusion, children in the allergic rhinitis group and allergic asthma group had altered gut microbiomes in comparison with the healthy control group. Compared to healthy children, the gut microbiome in children with allergic diseases has higher pro-inflammatory potential and increased production of pro-inflammatory molecules.
Keywords: childhood, allergic asthma, allergic rhinitis, gut microbiome, metagenomic sequencing
Introduction
Allergic airway diseases affect many people worldwide, and incidences are still increasing gradually. Allergic rhinitis and allergic asthma, are typical chronic nasopharyngitis respiratory diseases in pediatric, causing an enormous burden of illness and high economic costs to the community.1 Allergic rhinitis and allergic asthma are immunoglobulin E (IgE)-mediated inflammatory diseases that occur after exposure to allergens and involve multiple immune active cells and cytokines.2
A growing amount of evidence has pointed to the important roles of the microbiota in human health, including mucosal immunity regulation,3–5 metabolism,6,7 and immune responses.8 With the development of DNA and RNA sequencing technologies, using 16S rRNA sequencing technology, metagenomic shotgun sequencing technology, and other culture-independent methods can conduct detailed assessments of the microbiome diversity, taxonomic composition, and functional characteristics.9 The human gastrointestinal tract is rich in microorganisms including Firmicutes, Bacteroidetes, Actinomyces, and Proteobacteria.10 The gastrointestinal microbiota is a key factor influencing host immune responses, metabolism, and allergic disease progression.11 Microecological disorders, especially in the intestinal tract, usually disrupt intestinal homeostasis and increase intestinal permeability, which in turn influence the function and maturity of the immunology system and induces a variety of immune-induced diseases such as allergic rhinitis and asthma.12
Several longitudinal and prospective studies have revealed that changes in the earlier-life gut microbiota are connected to allergic disease development later in life. The period from gestation to 2 years of age is the window of opportunity for the development of the gut microbiota. During this period, the infant intestinal tract microbiota constitution is susceptible to interference by various factors, including gestational period delivery method, feeding method, drug use, environmental factors, genetic factors, and the colonization and abnormality of the gut microbiota in early infancy are closely related to the development of allergic diseases in later stages.13–15 However, even if the microbiota changes in the near life are neglected, we should pay attention to the possibility of modifying the allergic disease conditions through modulation of the microbiota. In addition, the research on the new concept of the “gut-lung axis” will be a hot topic for future research. We should pay attention to the complex correlation between gut microbiota and lung microbiota and reveal the complex mechanism between them. Meanwhile, the particular bacterial strains associated with the etiology of rhinitis and asthma remain confusing. Current studies have largely been based on 16s rRNA sequencing,16 which has limited capacity to describe non-bacterial modules and functional characteristics of the gut microbiome. Recently, many studies have investigated the role of probiotics in counteracting microbiome dysregulation and improving the patient’s quality of life in allergy and asthma.17,18 However, the specific bacterial strains associated with the pathogenesis of allergic rhinitis and asthma remain elusive. The thorough comprehension of the mechanisms by which the gut microbiome regulates allergic diseases may be valuable for the selection of suitable treatment methods.
In this study, we performed a shotgun metagenomic sequencing of 18 allergic rhinitis children, 23 allergic asthma children, and 19 healthy control subjects. Accordingly, the functional and compositional features of intestinal between allergic patients and healthy individuals were compared. The aim of present study was to identify the specific gut microbiota and potential pathways of gut microbiota that were closely related to allergic rhinitis as well as allergic asthma.
Materials and Methods
Study Participants
We conducted a cross-section survey to investigate the intestinal microflora of children aged 6–14 years diagnosed with allergic rhinitis alone, allergic asthma alone, and healthy controls. This study enrolled children who presented at the Departments of Pediatric Allergy and Immunology of the Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University for allergic rhinitis treatment or allergic asthma treatment from April 2020 to December 2021. In addition, healthy subjects with age and gender-matched and without a history of asthma or allergic rhinitis were recruited from the local community.
The inclusion criteria of allergic asthma were as follows: (i) having the generation of noninfectious wheeze in the past one year, or asthma medical treatment currently in use, according to the guidelines of the Global Initiative for Asthma, (ii) results of positive serum-specific IgE and (iii) positive skin prick tests. People who were diagnosed with allergic rhinitis according to their diagnostic such as sternutation, stuffy nose itching, pale and edematous nasal mucosa, and results of positive serum-specific IgE and/or positive skin prick tests. Healthy children without an allergic history or infections were enrolled as healthy control subjects. The inclusion criteria of healthy children were as follows: (i) no history of the allergic condition; (ii) no respiratory infections; and (iii) voluntary for this research. The exclusion standards of the study were as follows: (i) antibiotics, prebiotics, probiotics, or synbiotics use within 2 months or (ii) suffered an upper respiratory tract infection or chronic viral infection, or (iii) severe gastrointestinal abnormalities, neurodevelopmental disorders and (iv) any severe diseases.
Ethical Permission
The ethical confirmation for this research was approved by the Ethics Committee of the Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University (Permit Number ID: 2021-K-92-01). This research abides by the mission of the Declaration of Helsinki. All of the experimental research were constructed by current guidelines and statutes and all guardians provide their informed consent before they participated in this study.
Sample Collection and Processing
Stool sampling was performed in subjects without receiving probiotics or antibiotics therapy for at least 2 month weeks prior to the sampling. Fresh fecal samples of children used in this study were collected by the nurses of our hospital with instructions on proper method of collection at hospital using a standard stool collection specimen jar. Researchers quickly freeze and transport stools samples on ice to laboratory −80°C freezers until further processing within 2 hours after sampling.
Metagenomic Shotgun Sequencing
Genomic DNA extraction from these fecal samples was multiplexed and paired-end sequenced on the NovaSeq 6000 platform at the Novogene Bioinformatics Technology Co., Ltd by metagenomic shotgun sequencing (150 ×150 bp). The Fastq tool was used to cut adapters and filter out low-quality reads.19 We use metawrap to map trimmed reads to the human reference genome.20 Then, MetaPhlan2 was next used to assess the taxonomic compositional profile of the remaining reads,21 which provided a library of distinct clade-specific markers to explore the taxonomic clades present in each sample and calculate estimates of relative abundance at the species level. Functional profiling and quantification of processed sequence data were profiled using HUMAnN2 with the Uniref50 protein database.22 The output files generated by HUMAnN2 contain pathway abundance, pathway coverage, and gene family abundance. And pathway abundance was chosen for further analysis. The different relative abundance of the pathway between different groups was evaluated by LEfSe.23
Statistical Analysis
All parametric clinical data were analyzed using one-way analysis of variance (ANOVA) and nonparametric data were analyzed using the Kruskal-Wallis test. Analysis of microbiota data was evaluated using R packages (version 4.0.4). Alpha diversity was evaluated using two kinds of different indices: the Simpson index and the Shannon index. We used t-test to evaluate the dissimilarity in alpha diversity between different groups. The Bray-Curtis difference and Jaccard index derived from principal coordinate analysis (PCOA) were used to visualize diversity in a two-dimensional microbial community composition. The Adonis function was used for Permutational multivariate ANOVA (PERMANOVA), which is in the R package Vegan. The linear discriminant analysis (LDA) effect size (LEfSe) algorithm was used to detect the differential abundance of species between groups and further find a biomarker. And taxon with an LDA score>5 and P<0.05 (Kruskal–Wallis test) was considered a significant biomarker. For the significantly different species profile, the R package “RandomForest” was performed to obtain the taxonomic contributions of microbiome communities. The average area under the curve (AUC) was used to evaluate the predictive performance of the random forest model. Mean decrease accuracy (MDA) and mean decrease Gini (MDG) are used for variable ordering and variable selection.
Results
Population Characteristics
In this study, 60 children including 18 children with allergic rhinitis, 23 children with allergic asthma, and 19 healthy controls were recruited. Their age, gender and body mass index were matched. Table 1 showed the demographics and clinical characteristics of children with allergic rhinitis, allergic asthma, and healthy controls. Difference was not statistically significant in sex, height, weight, maternal atopy and BMI between groups. The flow of this study is shown in Figure 1.
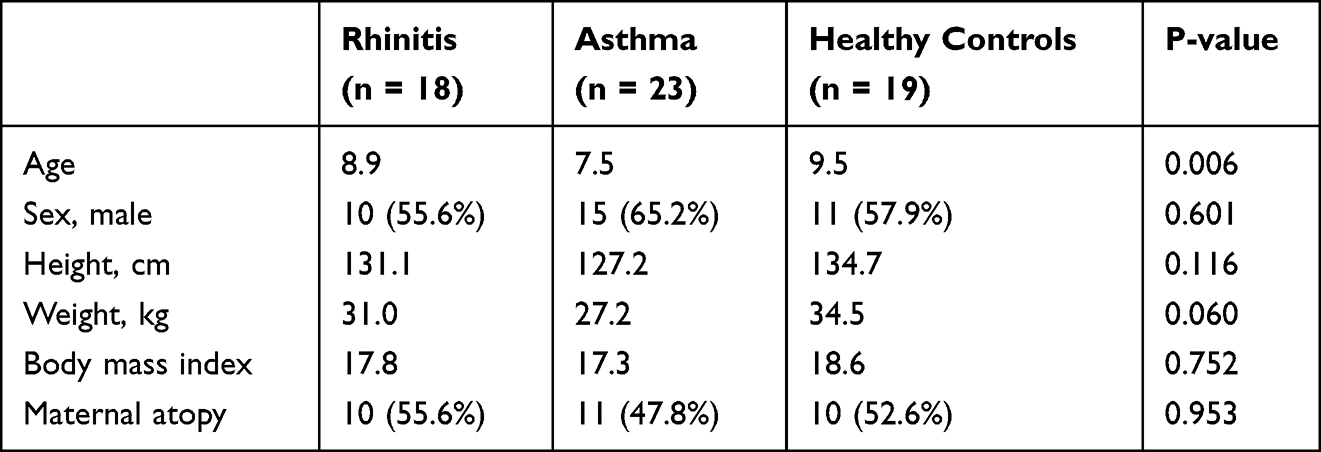 |
Table 1 Demographics and Clinical Characteristics of Participants in This Study |
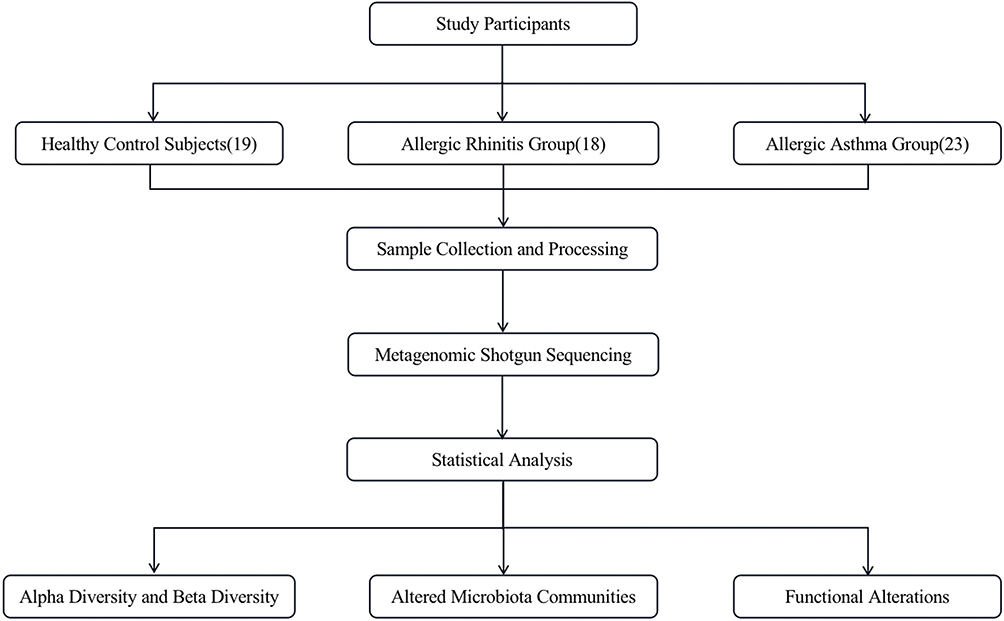 |
Figure 1 The flow of this study. |
Alpha Diversity and Beta Diversity of the Gut Microbiome
Shannon diversity index and Simpson diversity index were used to evaluate Alpha diversity. Significantly higher Shannon’s diversity index and Simpson’s diversity index were found in asthmatic children than those of healthy control group (Kruskal–Wallis test; P=0.0029 for Shannon index and P=0.0016 for Simpson index; Figure 2A). PCOA and PERMANOVA analysis showed that the gut microbial communities cluster patterns of children with asthma or rhinitis were significantly different from that of healthy controls based on Bray-Curtis dissimilarities. However, no significant difference was found between asthma group and rhinitis group (Controls vs asthma, P=0.001; controls vs rhinitis, P=0.001; asthma vs rhinitis, P=0.703; Figure 2B).
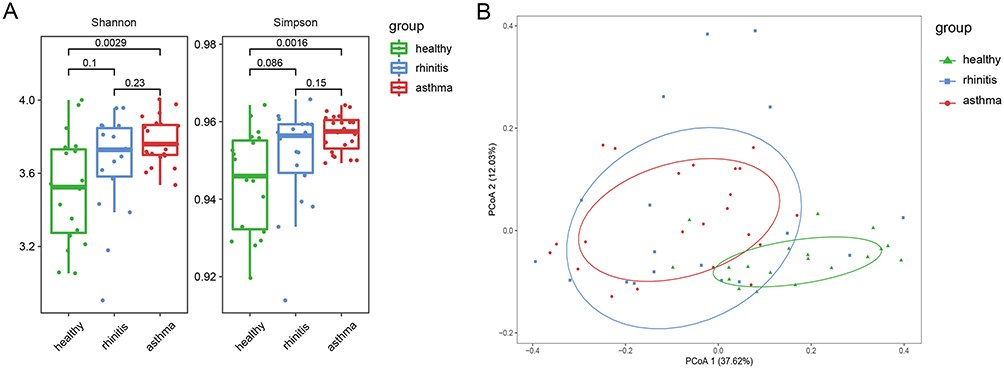 |
Figure 2 Alpha and beta diversity of microbiota. (A) Alpha diversity is based the Shannon diversity index and Simpson index. (B) Beta diversity is on measured by Bray-Curtis dissimilarities. Abbreviation: PCoA, Principal coordinates analysis. |
Altered Microbiota Communities in the Gut Microbiome
To explore the taxonomic composition of allergic asthma and allergic rhinitis groups, we further analyzed the relative abundance of the microbial community in three groups. At the phylum level, we found that the gut microbiome of healthy controls was dominated by Bacteroidetes, followed by Firmicutes (Figure 3A). Compared with controls, asthma, and rhinitis individuals presented a significant increase in the phylum Firmicutes and Proteobacteria in the gut (Figure 3B). On the contrary, asthma and rhinitis group contained significantly lower levels of Bacteroidetes. At the genus level, we find a total of 134 genera, 11 genera with >1% average relative abundance, including Bacteroides, bifidobacterium, Faecalibacterium, Ruminococcus, Alistipes, Blautia, Roseburia, Megamonas, Eubacterium, Parabacteroides, and Subdoligranulum. Out of the 11 genera, Bacteroides, bifidobacterium, Faecalibacterium, Ruminococcus, and Alistipes were the top five genera (P < 0.05, Wilcoxon rank-sum test, Figure 3C). Notably, there was a total of 26 bacteria were discovered to be differentially abundant between any two of the three groups. Seven bacterial populations including Corynebacterium, Streptococcus, Dorea, Actinomyces, Bifidobacterium, Blautia, and Rothia were significantly enriched in the asthma group, whereas three bacterial populations including Bacteroides, Alistipes, and Acidaminococcus were significantly reduced in asthma versus those in the healthy controls (P<0.01, Wilcoxon rank-sum test, Figure 3D and E). The phylogenetic profile of the gut microbiome of asthma patients and rhinitis patients were depicted in a cladogram (Figure 4A). Linear discriminant analysis effect size (LEfSe) was usually used to demonstrate the significant difference of microbial taxa in each group. At the species level, we used LEfSe to further explore the specific gut microbiota taxa associated with asthma groups or rhinitis groups versus healthy controls. Based on the linear discriminant analysis (LDA) selection, 15 discriminative features were identified to be differentially relatively abundant between asthma individuals and controls through LEfSe (LDA score>3, Kruskal–Wallis test, Benjamini-Hochberg FDR, q<0.05; Figure 4B), and 15 specific bacterial were screened to be different between rhinitis and healthy controls (LDA score>3, Kruskal–Wallis test, Benjamini-Hochberg FDR, q<0.05; Figure 4C).
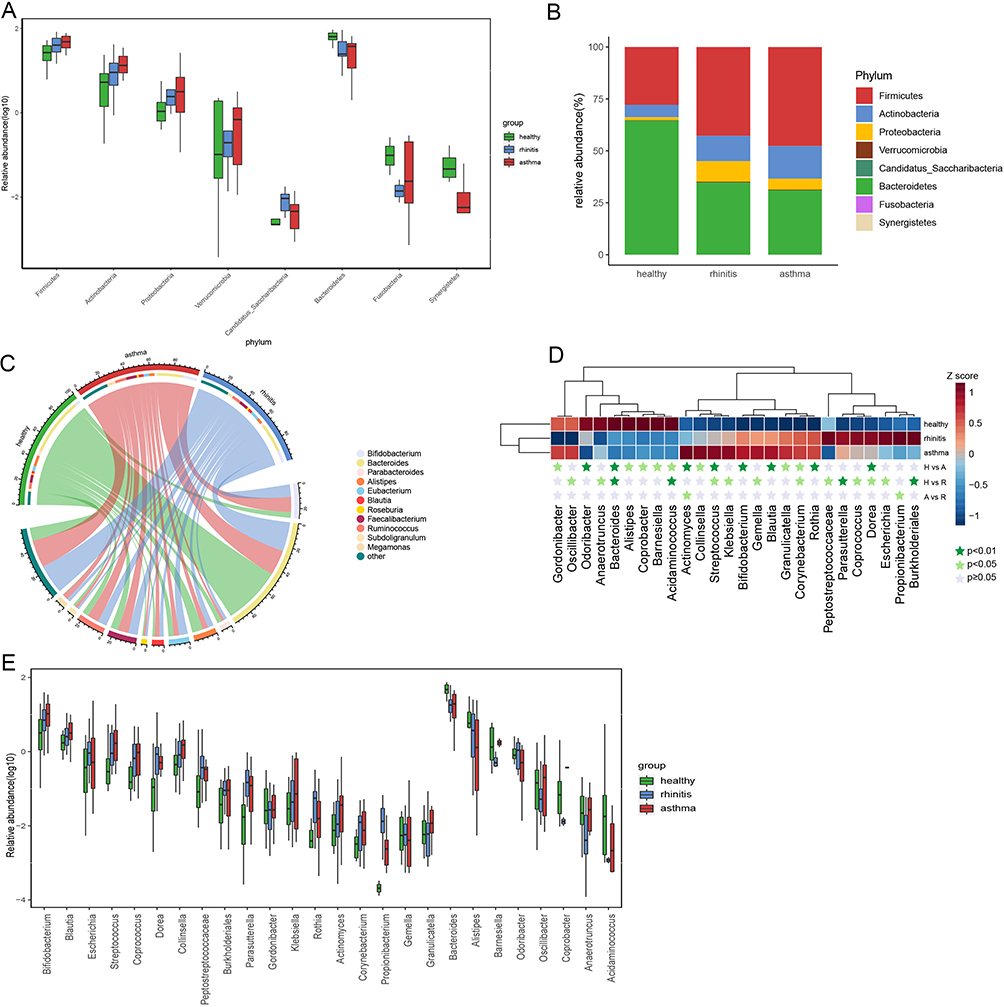 |
Figure 3 Fecal microbial abundance at the major phylum level and major genera in the gut microbiome of patients with allergic rhinitis, allergic asthma, and healthy subjects. (A) Bacterial composition and relative abundance at the phylum level. (B) Differences at the phylum level. (C) Major genera of each group, less abundant (<1%), and unclassified taxa are grouped as “other” (D) The 26 different genus profiles of average relative abundance across three groups. The dark green star indicates P<0.01, the light green star indicates P<0.05, and the gray star indicates P≥0.05. (E) The relative abundance of 26 genera abundant in three groups was exhibited with a box plot. Abbreviations: H, control; A, asthma; R, rhinitis. |
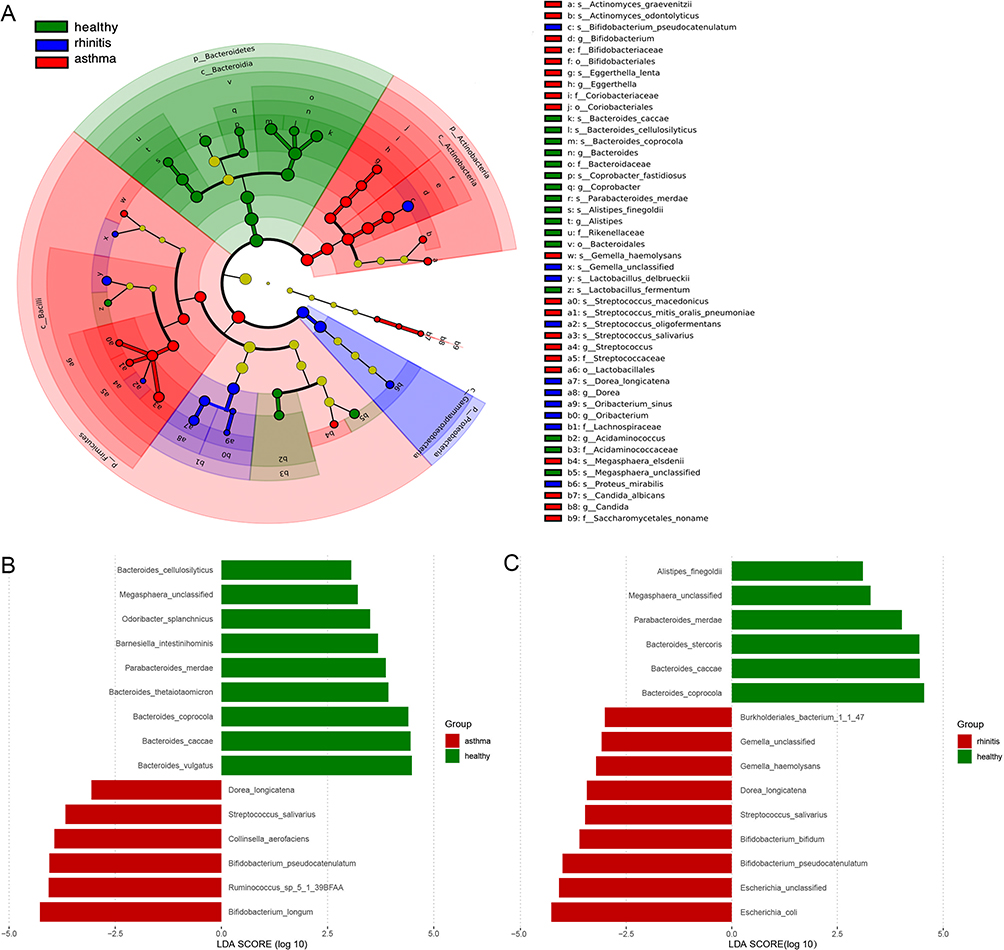 |
Figure 4 Taxonomic changes in the intestinal microbial. (A) Cladogram of the gut microbiome taxa associated with allergic asthma and allergic rhinitis LEfSe analysis. (B) Histogram of the linear discriminant analysis (LDA) scores for crucial bacteria classification with different abundances between allergic asthma and healthy groups. The Significance was calculated from LDA effect size (LEfSe) at P<0.05 (Kruskal–Wallis test) and LDA score>3. (C) Histogram of the linear discriminant analysis (LDA) scores for crucial bacteria taxonomic with different abundance between allergic rhinitis and healthy groups. Significance was obtained by LDA effect size (LEfSe) at P<0.05 (Kruskal–Wallis test) and LDA score>3. |
Classification of Disease Status Using Bacterial Genus-Level Biomarkers
To assess the capacity of intestinal microflora biomarker as a noninvasive diagnostic instrument. We use random forest classifier model to discriminate asthma group from healthy control group. We only choose 34 significant bacterial communities that were significantly different between the two groups for further analysis to build the random forest model. At last, 16 general signatures were selected as the optimal biomarkers set to differentiate the asthma group from the control group. The capability of the best predictive model was evaluated by 100 random ROC analyses, and the average AUC value achieved 92.29% (Figure 5A). We calculated the average MDA value after each characteristic variable was deleted, based on the fact that removing characteristic variables from the prediction model increases the error. The average MDA for the random 100 times of these optimal markers is shown in Figure 5B.
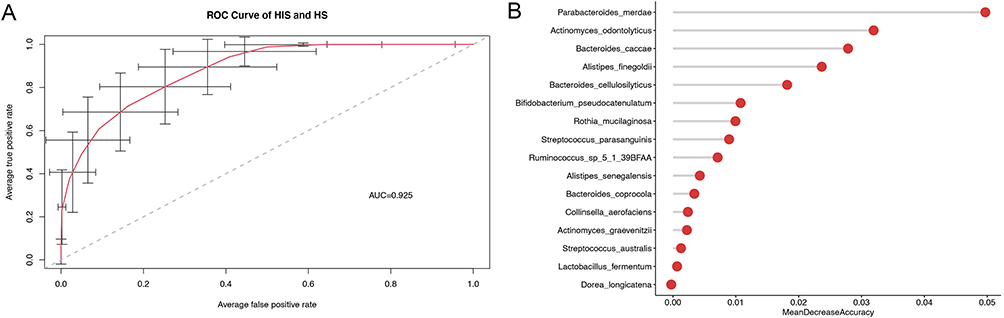 |
Figure 5 Taxonomic changes in the gut microbiome. (A) Species-level biomarkers for the classification of allergic asthma based on random forest model. The average AUC value between allergic asthma and the control group was calculated 100 times. (B) The histogram represents the Mean Decrease Accuracy of all differential species which was calculated by the random forest algorithm. Abbreviations: ROC, Receiver operating characteristics; AUC, Area under roc curve. |
Functional Alterations in the Gut Microbiome
Recently, studies on the relationship between functional composition and gut microbiome are still lacking. In this study, the metagenomic shotgun sequencing results were mapped onto MetaPhlAn2 and ChocoPhlAn pangenome databases using HUMAnN2. The whole diversity in the pathways between groups were visualized in PCoA plots based on an assessment of Bray-Curtis dissimilarity (PERMANOVA; R2=0.198, P=0.025; Figure 6A). Compared with the control group, several functional pathways were identified with significant alterations between the patient and healthy group through LEfSe. The functional gene pathways with significant differences were shown by the heatmap in Figure 6B. We found that the functional pathways related to nucleotides degradation, phylloquinol biosynthesis, and L−arginine biosynthesis were significantly enriched in children with allergic asthma. In contrast the pathway related to peptidoglycan maturation were significantly decreased.
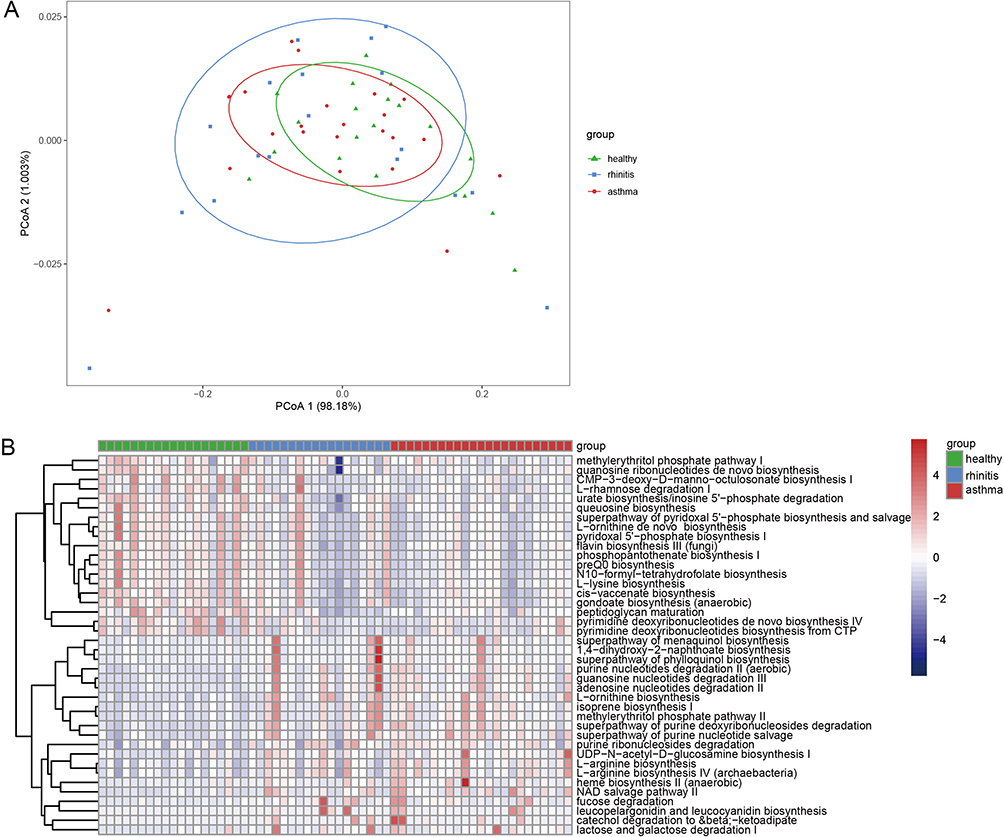 |
Figure 6 Functional alterations in the gut microbiome. (A) The whole diversity in the pathways between different groups. (B) Differentially abundant functional gene pathway heatmap between allergic children and healthy controls; Colored by the relative abundance of each pathway. Identified by LEfSe at P<0.05 (Kruskal-Wallis test) and LDA score>2. P-values are adjusted by the false discovery rate (Benjamini-Hochberg FDR, q<0.05) method. |
Discussion
In this study, we used metagenomic shotgun sequencing and subsequent bioinformatics analysis to compare the species composition and functional characteristics of the intestinal microbiota in child with allergies, including asthmatic child, children with allergic rhinitis, and healthy individuals. The aim was to identify certain microorganisms that differentiate allergic patients from healthy individuals. We systematically compared the bacterial diversity and abundance at different levels. We found that children in the allergic rhinitis group and allergic asthma group had altered gut microbiomes in comparison with the healthy control group. Compared to healthy children, the gut microbiome in children with allergic diseases has higher pro-inflammatory potential and increased production of pro-inflammatory molecules.
The relationship between intestinal microbiome and atopic disease has been investigated in a large number of studies, and microbiota structural signatures for diverse allergic responses have been identified, but the results have not been generally consistent.24,25 Most of the previous studies were based on 16s rRNA sequencing. In recent years, there have been many researches on the new concept of the “gut-lung axis”. We have found that different lung diseases can be affected by the intestinal microenvironment. The mechanism regulating the communication between the intestine and the lungs is still obscure, but it has been suggested that the signals released by endothelial cells can be absorbed by epithelial cells and immune cells, forming a local cytokine microenvironment that leads to changes in the immune response at the distal site.10 Intestinal flora has an important influence on immune cell maturation and resistance to pathogens. The bacterial biodiversity of the intestinal microbiota in the earlier period of life played a very important role in microorganism-host interactions that contribute to the development of the immunology system and allergic disease. Different from previous studies, we found obviously increased alpha-diversity in the allergy group. Many studies showed that the microbiological diversity of the allergic rhinitis group was lower than healthy controls.26–28 However, some studies have found that the bacterial diversity between the allergic rhinitis group and the healthy controls was not significantly different.29 A recent study based on 16s sequencing found that bacterial diversity was significantly higher in the adult patient with allergic rhinitis than in the healthy control group, which corroborates our results.30 Furthermore, based on the Sankey chart of the intestinal microbiome in this research, it was observed that the increased microbiome diversity in individuals with allergic rhinitis because of the bacterial community in Firmicutes. In the analysis of beta diversity, the allergic asthma group and the allergic rhinitis group were significantly separated from the healthy controls, indicating that the gut microbial community structure was significantly altered in the disease group.
Our research found that the relative abundance of Firmicutes was increased and the relative abundance of Bacteroidetes was decrease in the gut microbiota of allergic children compared with healthy controls, which is consistent with previous studies.30,31 The proportions of Bacteroidetes were lower in the allergic asthma group as compared with healthy controls, and Bacteroidetes have been discovered to increase the overall amount of Tregs.32–34 At the same time, increased relative abundance of Proteobacteria in the allergic group, which is inconsistent with the outcomes of a previous research on atopic dermatitis in children, although this dissimilarity mainly ascribed to inequality in participant age.35 All Proteobacteria have an outside membrane mainly consists of lipopolysaccharide, which are gram-negative and the elevated levels of Proteobacteria may result in increased levels of lipopolysaccharide and increase the risk of inflammation and immune abnormalities.36 We found an increase in Proteobacteria in the disease group, further confirming the pathogenesis of phytoconstituent-regulated allergic diseases.
At the genus level, we observed that Corynebacterium, Streptococcus, Dorea, Actinomyces, Bifidobacterium, Blautia, and Rothia were significantly enriched in the allergic asthma group, while Bacteroides, Alistipes, and Acidamcoccus were significantly reduced. In addition, the allergic rhinitis group had significant enrichment of Parasutterella and Burkholderia, while the relative abundance of Bacteroides and Acidaminococcus decreased significantly. Bacteroides spp. showed a significant reduction in relative abundance in children who developed IgE-related allergic disease. Bacteroidetes are intestinal symbionts that stimulate epithelial mucin production and degradation and are important for intestinal barrier function.37 A previous study found that a lower relative abundance of Bacteroides spp. was related to a higher danger of subsequent atopic dermatitis.36 Studies also have found that the genus Parasutterell could affect the flora and host metabolism.38 Together, these studies and our results suggested an important role for changes in bacterial population genera in the course of allergic disease and in host metabolism.
At the species level, compared with the control group, the current study found that many species were differentially enriched in the disease group. Among these species, several were identified as associated with atopies, such as Escherichia coli and Bifidobacterium longum. We chose the best predictors to build a random forests model to discriminate the allergic asthma group from the healthy control group. The integration of the best predictive classification units for identifying the allergic asthma group and the healthy group had high reliability of 92.5%. It indicates that these different microbiome community structure have an identification capability.
As the composition of the microbial altered, the functional profiles were also changed. Short-chain fatty acids (SCFAs) are key signaling molecules in the intestinal that limit inflammation and have been identified as critical signals in the regulation of the immunoregulation system. SCFAs levels are highly rely on the fermented fiber content of the diet and the microorganism community.39 Similarity, dietary fiber can lead to the growth of Bacteroidetes with enhanced SCFAs fermentation by means of a change in the proportion of Firmicutes to bacteroides.40,41 The intestinal microbiota of kids with allergic diseases displayed more lipopolysaccharide biosynthesis-related genes. Lipopolysaccharide can stimulate the cascade inflammation reaction by activating the generation of proinflammatory factor,42,43 and is participated in the pathogenetic mechanism of allergic rhinitis.44 Conversely, the allergic intestinal microbiome showed a reduced likelihood of complex fiber degradation, which explains why lower propionate and butyrate concentrations were found in allergic patients compared to healthy control group.45 Thus, compared with healthy child, intestinal flora in allergic diseases has increased pro-inflammatory capacity, higher generation of pro-inflammatory molecules, and reduced tolerogenic SCFAs and biosynthesis of anti-inflammatory. Because of their protection effects against intestinal and respiratory inflammatory diseases, SCFAs and diets represent a hopeful treatment method, and a better comprehension of the molecular and cellular effects of SCFAs will enhance the potentiality for treatment prevention.
There are some shortcomings in this study. Firstly, the microbiota is modulated by many factors such as age, food, and region, so we still need to expand our sample size to study the role of gut microbiota in childhood airway allergic disease. Secondly, we need large-scale prospective studies and experimental verification to explore the microecological composition and clinical information of patients before and after the disease to understand the disease course and clinical phenotype, the relationship between susceptibility to disease progression, and response to treatment. Future studies can further use multi-omics integrative analysis, including the combined use of 16s, metagenomics, and metabolomics to explore the mechanism of allergic disease development and reveal the multidirectional connections between specific microbiota strains, metabolites, host immune system, and allergens.
Conclusion
We found altered gut microbiomes in the allergic rhinitis group and allergic asthma group in comparison with the healthy control group. Compared with healthy children, the gut microbiota in allergic diseases can increase the production of pro-inflammatory molecules and the reduction of anti-inflammatory molecules and tolerogenic short-chain fatty acids, thus having higher pro-inflammatory potential. As realized that microbial metabolites also play a crucial role, we will continue to be focused on early-life microbiota in the gut and lungs.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hofmaier S. Allergic airway diseases in childhood: an update. Pediatr Allergy Immunol. 2014;25(8):810–816. doi:10.1111/pai.12322
2. Ruby P, Van Cauwenberge P, Khaltaev N. Allergic rhinitis and its impacts on asthma: an evidence based treatment strategy for allergic rhinitis. J Allergy Clin Immunol. 2001;108(5):S147–S334. doi:10.1067/mai.2001.118891
3. Martens EC, Neumann M, Desai MS. Interactions of commensal and pathogenic microorganisms with the intestinal mucosal barrier. Nat Rev Microbiol. 2018;16(8):457–470. doi:10.1038/s41579-018-0036-x
4. Kuhn KA, Pedraza I, Demoruelle MK. Mucosal immune responses to microbiota in the development of autoimmune disease. Rheum Dis Clin North Am. 2014;40(4):711–725. doi:10.1016/j.rdc.2014.07.013
5. Wang L, Zhu L, Qin S. Gut microbiota modulation on intestinal mucosal adaptive immunity. J Immunol Res. 2019;2019:4735040. doi:10.1155/2019/4735040
6. Nieuwdorp M, Gilijamse PW, Pai N, et al. Role of the microbiome in energy regulation and metabolism. Gastroenterology. 2014;146(6):1525–1533. doi:10.1053/j.gastro.2014.02.008
7. Joyce SA, Gahan CG. The gut microbiota and the metabolic health of the host. Curr Opin Gastroenterol. 2014;30(2):120–127. doi:10.1097/MOG.0000000000000039
8. Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10(3):159–169. doi:10.1038/nri2710
9. Weinstock GM. Genomic approaches to studying the human microbiota. Nature. 2012;489(7415):250–256. doi:10.1038/nature11553
10. Clavel T, Desmarchelier C, Haller D, et al. Intestinal microbiota in metabolic diseases: from bacterial community structure and functions to species of pathophysiological relevance. Gut Microbes. 2014;5(4):544–551. doi:10.4161/gmic.29331
11. Yoon SS, Kim EK, Lee WJ. Functional genomic and metagenomic approaches to understanding gut microbiota-animal mutualism. Curr Opin Microbiol. 2015;24:38–46. doi:10.1016/j.mib.2015.01.007
12. Bridgman SL, Kozyrskyj AL, Scott JA, et al. Gut microbiota and allergic disease in children. Ann Allergy Asthma Immunol. 2016;116(2):99–105. doi:10.1016/j.anai.2015.10.001
13. Penders J, Gerhold K, Stobberingh EE, et al. Establishment of the intestinal microbiota and its role for atopic dermatitis in early childhood. J Allergy Clin Immunol. 2013;132(3):601–607 e608. doi:10.1016/j.jaci.2013.05.043
14. van Neerven RJ, Knol EF, Heck JM, et al. Which factors in raw cow’s milk contribute to protection against allergies? J Allergy Clin Immunol. 2012;130(4):853–858. doi:10.1016/j.jaci.2012.06.050
15. Lynch SV, Wood RA, Boushey H, et al. Effects of early-life exposure to allergens and bacteria on recurrent wheeze and atopy in urban children. J Allergy Clin Immunol. 2014;134(3):593–601 e512. doi:10.1016/j.jaci.2014.04.018
16. Chiu CY, Cheng ML, Chiang MH, et al. Gut microbial-derived butyrate is inversely associated with IgE responses to allergens in childhood asthma. Pediatr Allergy Immunol. 2019;30(7):689–697. doi:10.1111/pai.13096
17. Zajac AE, Adams AS, Turner JH. A systematic review and meta-analysis of probiotics for the treatment of allergic rhinitis. Int Forum Allergy Rhinol. 2015;5(6):524–532. doi:10.1002/alr.21492
18. Turner JH, Adams AS, Zajac A, Qin G. Probiotics in prevention and treatment of allergic rhinitis. Am J Rhinol Allergy. 2015;29(6):e224. doi:10.2500/ajra.2015.29.4257
19. Chen S, Zhou Y, Chen Y, et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–i890. doi:10.1093/bioinformatics/bty560
20. Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6(1):158. doi:10.1186/s40168-018-0541-1
21. Truong DT, Franzosa EA, Tickle TL, et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12(10):902–903. doi:10.1038/nmeth.3589
22. Abubucker S, Segata N, Goll J, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol. 2012;8(6):e1002358. doi:10.1371/journal.pcbi.1002358
23. Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. doi:10.1186/gb-2011-12-6-r60
24. Lee KH, Song Y, Wu W, et al. The gut microbiota, environmental factors, and links to the development of food allergy. Clin Mol Allergy. 2020;18:5. doi:10.1186/s12948-020-00120-x
25. Berni Canani R, De Filippis F, Nocerino R, et al. Gut microbiota composition and butyrate production in children affected by non-IgE-mediated cow’s milk allergy. Sci Rep. 2018;8(1):12500. doi:10.1038/s41598-018-30428-3
26. Galazzo G, van Best N, Bervoets L, et al. Development of the microbiota and associations with birth mode, diet, and atopic disorders in a longitudinal analysis of stool samples, collected from infancy through early childhood. Gastroenterology. 2020;158(6):1584–1596. doi:10.1053/j.gastro.2020.01.024
27. Abrahamsson TR, Jakobsson HE, Andersson AF, et al. Low gut microbiota diversity in early infancy precedes asthma at school age. Clin Exp Allergy. 2014;44(6):842–850. doi:10.1111/cea.12253
28. Petersen C, Round JL. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol. 2014;16(7):1024–1033. doi:10.1111/cmi.12308
29. Chiu CY, Chan YL, Tsai MH, et al. Gut microbial dysbiosis is associated with allergen-specific IgE responses in young children with airway allergies. World Allergy Organ J. 2019;12(3):100021. doi:10.1016/j.waojou.2019.100021
30. Zhu L, Xu F, Wan W, et al. Gut microbial characteristics of adult patients with allergy rhinitis. Microb Cell Fact. 2020;19(1):171. doi:10.1186/s12934-020-01430-0
31. Ling Z, Li Z, Liu X, et al. Altered fecal microbiota composition associated with food allergy in infants. Appl Environ Microbiol. 2014;80(8):2546–2554. doi:10.1128/AEM.00003-14
32. Mazmanian SK, Liu CH, Tzianabos AO, et al. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122(1):107–118. doi:10.1016/j.cell.2005.05.007
33. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci USA. 2010;107(27):12204–12209. doi:10.1073/pnas.0909122107
34. Bunyavanich S, Shen N, Grishin A, et al. Early-life gut microbiome composition and milk allergy resolution. J Allergy Clin Immunol. 2016;138(4):1122–1130. doi:10.1016/j.jaci.2016.03.041
35. Abrahamsson TR, Jakobsson HE, Andersson AF, et al. Low diversity of the gut microbiota in infants with atopic eczema. J Allergy Clin Immunol. 2012;129(2):434–440, 440 e431–432. doi:10.1016/j.jaci.2011.10.025
36. Shin NR, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015;33(9):496–503. doi:10.1016/j.tibtech.2015.06.011
37. West CE, Jenmalm MC, Prescott SL. The gut microbiota and its role in the development of allergic disease: a wider perspective. Clin Exp Allergy. 2015;45(1):43–53. doi:10.1111/cea.12332
38. Ju T, Kong JY, Stothard P, et al. Defining the role of Parasutterella, a previously uncharacterized member of the core gut microbiota. ISME J. 2019;13(6):1520–1534. doi:10.1038/s41396-019-0364-5
39. Maslowski KM, Vieira AT, Ng A, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–1286. doi:10.1038/nature08530
40. Trompette A, Gollwitzer ES, Yadava K, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. 2014;20(2):159–166. doi:10.1038/nm.3444
41. Holscher HD, Caporaso JG, Hooda S, et al. Fiber supplementation influences phylogenetic structure and functional capacity of the human intestinal microbiome: follow-up of a randomized controlled trial. Am J Clin Nutr. 2015;101(1):55–64. doi:10.3945/ajcn.114.092064
42. Jang SE, Lim SM, Jeong JJ, et al. Gastrointestinal inflammation by gut microbiota disturbance induces memory impairment in mice. Mucosal Immunol. 2018;11(2):369–379. doi:10.1038/mi.2017.49
43. Garcia PM, Moore J, Kahan D, et al. Effects of vitamin D supplementation on inflammation, colonic cell kinetics, and microbiota in colitis: a review. Molecules. 2020;25(10):2300. doi:10.3390/molecules25102300
44. Mahdavinia M, Engen PA, LoSavio PS, et al. The nasal microbiome in patients with chronic rhinosinusitis: analyzing the effects of atopy and bacterial functional pathways in 111 patients. J Allergy Clin Immunol. 2018;142(1):287–290 e284. doi:10.1016/j.jaci.2018.01.033
45. Goldberg MR, Mor H, Magid Neriya D, et al. Microbial signature in IgE-mediated food allergies. Genome Med. 2020;12(1):92. doi:10.1186/s13073-020-00789-4
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.