Back to Journals » Drug Design, Development and Therapy » Volume 12
α-hemihydrate calcium sulfate/octacalcium phosphate combined with sodium hyaluronate promotes bone marrow-derived mesenchymal stem cell osteogenesis in vitro and in vivo
Authors Chen C, Zhu C, Hu X, Yu Q, Zheng Q, Tao S, Fan L
Received 7 May 2018
Accepted for publication 27 July 2018
Published 2 October 2018 Volume 2018:12 Pages 3269—3287
DOI https://doi.org/10.2147/DDDT.S173289
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Changshun Chen,1 Chen Zhu,2 Xiang Hu,1 Qiuli Yu,3 Qianjin Zheng,1 Shengxiang Tao,1 Lihong Fan2
1Department of Orthopedics, Zhongnan Hospital of Wuhan University, Wuhan, Hubei, China; 2School of Chemistry, Chemical Engineering and Life Sciences, Wuhan University of Technology, Wuhan, Hubei, China; 3School of Health Sciences, Wuhan University, Wuhan, Hubei, China
Purpose: The aims of this research were to combine a-hemihydrate calcium sulfate/octacalcium phosphate (α-CSH/OCP) with sodium hyaluronate (SH) or SH sulfate (SHS) to determine whether these composites can be used as a new type of bone repair material. This study may provide a theoretical basis and new ideas for the construction of active bone repair materials and their clinical application.
Methods: In this study, we combined α-CSH/OCP with SH or SHS. Scanning electron microscopy (SEM), Fourier-transform infrared (FTIR) spectroscopy, X-ray diffraction (XRD), thermogravimetric analysis (TGA), and the wettability test were performed, and porosity, setting time, in vitro degradation, and the mechanical properties of these composite materials were analyzed to evaluate the ultrastructural and physicochemical properties. We evaluated the histocompatibility of these composites by MTT assay, hemolysis, acute toxicity, and pyrogenic and intracutaneous stimulation tests. In addition, the osteogenic differentiation ability of these materials was detected in vitro using Western blot analysis and in vivo using an animal model of bone defect.
Results: The α-CSH/OCP/SH composite had a compressive strength of 13.72 MPa, a porous rate of 27.45%, and the 28-day degradation rate of 64%. The MTT assay results showed that the relative proliferation rates of the α-CSH/OCP/SH group were greater than 90%. The results of the α-CSH/OCP/SH composite in the hemolysis, acute toxicity, pyrogenic, and intracutaneous stimulation tests were within the normal range. Western blot analysis indicated that the expression of bone extracellular matrix (ECM) proteins was notably upregulated and always higher in the α-CSH/OCP/SH group than in the other groups. XRD of the rabbit radius-defect model indicated that bone healing in the area implanted with α-CSH/OCP/SH was excellent approximately 9 weeks after repair.
Conclusion: α-CSH/OCP/SH has very good biocompatibility and exhibits clear advantages in the induction of bone regeneration and self-repair, and this compound shows promise in the field of bone tissue engineering.
Keywords: sodium hyaluronate, characterization, BMSCs, biocompatibility, osteogenic differentiation
Introduction
Bone defects have become a serious clinical problem1 due to their prevalence in clinical orthopedics cases and the common need to resect the affected parts of the bone.2 Numerous methods are used to treat bone defects, such as autografts, alloplastic materials, and allografts.3 At present, the gold-standard treatment for bone defects is autogenous bone grafting. However, autogenous bone graft material is limited and donor site complications include pain and infection.4,5 Bone tissue engineering provides an alternative method for the treatment of bone defects that aims to regenerate damaged bone tissues by combining cells with bone repair materials, which act as templates for tissue regeneration and guide new tissue growth.6 Because of its unlimited supply and lack of risk for spreading disease, engineered bone is considered as a potential alternative to traditional bone grafts.7 In the field of bone tissue engineering, synthetic materials show promise due to their potential bone engineering characteristics; these materials include composites, stimuli-responsive materials, gels, and scaffolding. Stimuli-responsive materials can change their shapes from a 2D flat-planar structure to 3D structure with various morphologies with small or modest variations in their physical environment, such as temperature, pH, and magnetic fields. However, there are some concerns regarding the use of programmable adaptive materials in clinical applications.8 Gels and scaffold materials show good biocompatibility, biodegradability, mechanical properties, vascularization, and innervation. However, these materials are imperfect and require additional studies.9
Bone tissue engineering has become an attractive therapeutic approach with great potential for repairing bone defects.10 In bone tissue engineering, bone repair materials serve as matrices for tissue formation, thus playing a pivotal role in bone repair. An excellent bone repair material should be three-dimensional porous structure with good biological conductivity, biocompatibility, biodegradation, and non-cytotoxic, and have desirable mechanical properties and surface properties that promote cell adhesion, proliferation and differentiation.11,12 The new generation of bone repair materials should also be able to participate in the physiological activities of the body, activate genes on the molecular level, stimulate cells, and induce bone tissue formation, being actively involved in cell processes and gene expression. However, fabricating such an ideal material remains a major challenge in the field of bone tissue engineering.13 There are many types of bone tissue repair materials, and the research is extensive. With further study of bone tissue engineering materials, researchers have discovered deficiencies in all types of single materials, suggesting that single materials are not the ideal solution to repair bone defects.14 Recently, studies of biomimetic composite material have become a crucial element of bone tissue engineering research. Composite materials are designed to mimic one or more bone-forming components to elicit specific cellular responses and provide an ideal environment for bone formation.15,16 However, many problems remain to be solved in the clinical application of composite materials. The main issues are the optimization of the ratio of the composite materials to adapt their degradation rate to the growth rate of tissue cells and the maintenance of the porosity and high mechanical strength of the material.17
Inorganic crystals, mainly hydroxyapatite (HA), account for 65%–70% of bone tissue weight.18 HA has the same mineral composition as bone and excellent osteoconductive properties; however, HA does not remodel or readily degrade.19,20 α-Hemihydrate calcium sulfate (α-CSH) can self-solidify and degrade in a suitable manner. The pore structure can be changed through different production processes to enable satisfactory vessel ingrowth with an appropriate degradation rate.21 Octacalcium phosphate (OCP) has excellent biological activity and biocompatibility, but its mechanical properties are insufficient. Among the bioceramics employed as precursors in bone repair materials, the calcium phosphate family, and especially OCP with its higher physical and chemical absorbance,22 has a considerable effect on the mineralization of tooth bone and skeletal bone because the apatite layers of OCP and HA in the extracellular matrix (ECM) share many structural similarities.23 OCP is an unstable composite that can be converted rapidly to biological apatite. Notably, nanocrystalline HA is the major constituent (nearly 70%) of bone ECM. Moreover, calcium in this structure induces cell homing and recalls bone marrow cells that induce more bone cell resorption.24,25
Sodium hyaluronate (SH) is a glycosaminoglycan that is found in the ECM of mammalian tissues.26 SH stimulates osteoprogenitor cells to migrate, proliferate, and differentiate into osteoblasts by binding to cell surface receptors, such as CD44 and the receptor for hyaluronic acid-mediated motility.27,28 Thus, SH appears to be able to actively induce bone formation by activating osteoblasts. Therefore, we speculate that α-CSH/OCP combined with SH should be highly porous and noncytotoxic and have desirable mechanical properties and surface properties that promote cell adhesion, proliferation, and differentiation.
The aims of this research were to combine α-CSH and OCP with SH or SH sulfate (SHS) and evaluate the structure, biocompatibility, and performance of these materials in promoting bone marrow-derived mesenchymal stem cell (BMSC) osteogenesis in vitro. In addition, a New Zealand White rabbit bone defect model was generated. X-ray diffraction (XRD), H&E staining, and immunohistochemistry were performed. This composite has potential as a new type of bone repair material, and this study provides a theoretical basis and new ideas for the construction and clinical application of active bone repair materials.
Methods
Preparation of materials
Preparation of SHS
Sodium nitrite (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) aqueous solution was added to a constant-pressure funnel at a molar ratio of 4.25:1, and the sodium nitrite solution was added to a three-mouth flask dropwise to react with sodium bisulfite (Sinopharm Chemical Reagent Co., Ltd.) at 90°C. After a 90-minute isothermal reaction, the esterification agent sodium trisulfonate (N(SO3Na)3) was prepared.29 The pH was adjusted to 7 with 1 mol/L of sodium hydroxide solution at 40°C; then, SH (Liyang Biomaterials Co., China) was added (nNaNO2/mSH = 6/403 mol/g), and the mixture was magnetically agitated for 6 hours. The solution was washed with anhydrous ethanol and then dialyzed for 72 hours. Finally, SHS was obtained by isothermal drying. The reaction steps are shown in Figure 1.
 | Figure 1 Reaction principle of SHS. |
Preparation of α-CSH and OCP
α-CSH was prepared using the hydrothermal method. In brief, 20 mL of crystal modifier containing 2 wt% magnesium sulfate heptahydrate (Sinopharm Chemical Reagent Co., Ltd.) and 0.15 wt% sodium citrate (Sinopharm Chemical Reagent Co., Ltd.) was added to 6.7 g of calcium sulfate dihydrate (Sinopharm Chemical Reagent Co., Ltd.) at 0.15 MPa and 130°C for 5 hours.46 The material was dried in the oven for 4 hours at 110°C, and a powder was generated by grinding and sifting.
OCP was prepared by the homogeneous precipitation method.47 The 0.04 M calcium acetate (Sinopharm Chemical Reagent Co., Ltd.) solution was trickled slowly into the 0.04 M sodium dihydrogen phosphate (Sinopharm Chemical Reagent Co., Ltd.) solution with a drop funnel, and the mixture was stirred at 68°C for 1 hour and reacted for 2 hours. The sediment was filtered, and the powder was washed with distilled water and dried.
α-CSH/OCP, α-CSH/OCP/SH, and α-CSH/OCP/SHS composite preparation
The composite bone repair materials α-CSH/OCP/SH and α-CSH/OCP/SHS were prepared by freeze-drying. α-CSH/OCP composite was prepared by adding 80 wt% α-CSH to 20 wt% OCP powder, and the resulting powder was added to absolute ethyl alcohol for ultrasonic dispersion. α-CSH/OCP/SH and α-CSH/OCP/SHS composites were prepared by mixing with liquid containing 1 wt% SH or 1 wt% SHS at a final ratio of 0.5 mL/g. The composites were mixed well and allowed to dry at room temperature.
Molecular weight (MW) of SHS
The degree of SHS substitution was measured by BaSO4-gelatin spectrophotometry, and the viscosity average MW of SHS was determined using the Wurtzmann viscosity method and the gradual dilution method.30 SHS with different degrees of substitution was diluted with sodium chloride solution of 0.2 mol/L to a sample solution of 0.02, 0.03, 0.04, and 0.05 g/100 mL. According to effluent time Ti at different concentrations of the sample solution, relative viscosity (ηr=T/T0) and specific viscosity (ηsp=ηr−1) were calculated. Using the ηsp/C to map, another line (C) was obtained and extrapolated to C=0 and, then, another characteristic viscosity number [η] was obtained. Finally, the viscosity average MW was obtained by calculation. The control group followed the same steps using SH.
Material characterization
NMR of SHS
SHS was dissolved in D2O and placed in a 5 mm tube. The 13C NMR spectra (100.5924 MHZ) were measured at 26°C with Bruker AMX-500 Superconducting Magnetic Resonance Spectrometer (BrukerOptik GmbH, Ettlingen, Germany). The sampling time was 0.9 seconds, the delay time was 1 second, and the pulse width was 90° (9.3 seconds); DMSO was used as the internal standard (39.4 ppm).
Fourier-transform infrared (FTIR) spectroscopy of SH, SHS, α-CSH, OCP, and α-CSH/OCP
Samples of SH, SHS, α-CSH, OCP, and α-CSH/OCP were milled and dried in a constant temperature drying box at 40°C. A Nicolet-170sx infrared spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) was used to collect FTIR spectra. The spectrometer is equipped with a KBR beam splitter, and the spectral resolution is 400–4,000 cm−1 in transmission mode.
Scanning electron microscopy (SEM)
The materials were cut into powder and then coated with gold for ultrastructure analysis. The morphology of the powdered α-CSH/OCP/SH and α-CSH/OCP/SHS was observed by a high-resolution SEM (SEMMO-JSM-IT 300; Hitachi Ltd., Tokyo, Japan) at 20 kV acceleration voltage.
XRD
Wide-angle XRD measurements of α-CSH/OCP/SH and α-CSH/OCP/SHS were obtained at room temperature on a Bruker D8-Advance X’Pert PRO AXS diffractometer (BrukerOptik GmbH) (λ=1.5406 nm, operating at 50 kV and 180 mA).
Thermogravimetric analysis (TGA)
TGA was conducted with an STA 449 F3 instrument (TA Instruments, Hüllhorst, Germany), and experiments were conducted on powdered α-CSH/OCP/SH and α-CSH/OCP/SHS bone repair materials at a heating rate of 10°C·min−1 from room temperature to 1,000°C.
Porosity
The porosity of the bone repair materials was measured based on Archimedes’ principle, and distilled water was used as the liquid medium. The porosity was calculated via the following formula:
|
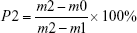
where m0 is the dry weight of bone repair materials, m1 is the weight of bone repair materials suspended in ethanol, and m2 is the weight of bone repair materials saturated with distilled water. Five samples were tested to calculate the average porosity.
Setting time
Polytetrafluoroethylene Teflon molds (20 mm high and 10 mm in diameter) were filled with α-CSH/OCP/SH and α-CSH/OCP/SHS powders. The initial and final setting times of the composites were measured by Vicat needle. When the permeability of the hydrated cement paste reached a certain value, the initial and the final setting times of the cement stone were determined. The test was repeated at least five times, and the average value is reported.
In vitro degradation
α-CSH, α-CSH/OCP/SH, and α-CSH/OCP/SHS were weighed (W0) and incubated in 50 mL of PBS (0.01 M, pH 7.4) at 37°C with shaking (60 rpm). The PBS solution was changed every 2 days. On days 0, 1, 3, 7, 14, 21, and 28, three samples (N=3) were washed thoroughly with distilled water and vacuum-dried to a constant weight (Wt) at room temperature. Degradation was defined as WLOSS =(W0−Wt)/W0×100%.
Mechanical properties
As in the in vitro degradation analysis, samples of α-CSH, α-CSH/OCP/SH, and α-CSH/OCP/SHS were immersed in PBS (pH 7.4) and the environment was maintained at 37°C in a water bath with 100% relative humidity for 28 days. The composites immersed in PBS were removed at different time intervals (0, 1, 3, 7, 14, 21, and 28 days) and dried at 60°C for 24 hours. For compressive strength analysis, the specimens were tested in a universal testing machine (WDW-E; Baihe Instrument Technology Co., Ltd., Shanghai, China) equipped with a 500 N load cell at a crosshead speed of 2 mm/min. The compressive strength of the composite was observed.
Wettability
The wettability test was conducted based on the change in the contact angle with distilled water. In brief, 20 μL of distilled water was added to the surface of different fiber samples.
Biocompatibility study
BMSC harvesting and culture and MTT assay
- The composite bone repair materials of OCP, α-CSH/OCP, α-CSH/OCP/SH, and α-CSH/OCP/SHS were sterilized by high-pressure steam sterilization. Normal saline was added at a final ratio of 0.1 kg/L. After the solution was incubated at 37°C and 5% CO2 for 24 hours, it was passed through a 0.22 μm filter membrane. The concentration of this extract was considered to be 100%. Therefore, when the material extract and DMEM were mixed at 1:1, the concentration of the materials extract was 50%. Similarly, when the material extract and DMEM were mixed at 1:3, the concentration of the material extract was 25%.
The experimental groups are extracts of different concentrations of materials, the positive control group is carbolic acid solution, and the negative control group is PBS.
The relative growth rate (RGR) was calculated according to the following equation:
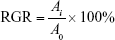
where Ai is the average absorbance of the experimental group and A0 is the average absorbance of negative control group.
- Three adult male New Zealand White rabbits were selected, and 5 mL of bone marrow was extracted from the ilium under sterile conditions using a 1 mL syringe prefilled with heparin. Primary cell culture was performed using the whole bone marrow adherence method. The harvested bone marrow was passed through a 100-mesh stainless steel screen and resuspended in DMEM. The supernatant was discarded after centrifugation at 1,000 rpm for 5 minutes. The remaining suspension was mixed with DMEM (containing 10% FBS) at a 1:3 ratio and then placed in a 75 mL culture flask. Primary culture cell was conducted at 37°C in a humidified 5% CO2 incubator. The medium was replaced for the first time after 24 hours. The cells were washed with PBS twice and then cultured in 5 mL of DMEM (containing 10% FBS). The medium was replaced every other day. When the cells reached 80% confluence on the bottom of the culture flask, they were digested with 0.25% trypsin, passaged at a 1:2 ratio, and further cultured.
- Third-generation cells with a good growth status were harvested, and CD14-FITC and CD44-CY3 were detected by flow cytometry (FACSCalibur; BD, Franklin Lakes, NJ, USA). CD44-positive purified rabbit BMSCs were suspended at a concentration of 5×104 cells/mL. Five 96-well plates were used for the experimental and control groups, with 40 wells for each group, for a total of 320 wells. The cells were plated into 96-well plates at 2×103 cells/well and then cultured at 37°C in a 5% CO2 incubator for 24 hours. When inverted phase contrast microscopy (IX51; Olympus Corporation, Tokyo, Japan) revealed that the cells adhered to the plates, the medium was discarded. Then, 100 μL of materials in solution, 50 μL of materials in solution and 50 μL of DMEM or 25 μL of materials in solution, and 75 μL of DMEM were added to the experimental wells. The positive control wells contained 100 μL of 0.64% carbolic acid solution, and the negative control wells contained 100 μL of 10% PBS. The medium was replaced once every 2 days, and the cells were cultured for 8 days. On days 1, 3, 5, and 7 of cell culture, 20 μL of MTT solution (5 mg/mL) was added to five wells. Cells were further cultured for 4 hours under standard conditions (37°C, 5% CO2, saturating humidity). Then, the medium was removed and 200 μL of DMSO was added to each well. The plates were loaded into a multimode microplate reader and oscillated at 100 times/min for 10 minutes to fully dissolve the crystals on the surface of the bone repair materials. The absorbance (A) at 490 nm was measured in each well. The data were subjected to statistical analysis.
Hemolysis test
A hemolysis test was performed. Briefly, solutions of OCP, α-CSH/OCP, α-CSH/OCP/SH, and α-CSH/OCP/SHS (n=3) were incubated at 37°C for 24 hours. Distilled water and saline were used as the positive and negative controls, respectively. Ten milliliters of saline containing 0.5 mL of potassium oxalate anticoagulant (20 g/L) was used to dilute 8 mL of fresh venous blood from New Zealand White rabbits. After incubation at 37°C for 60 minutes, the mixture was centrifuged and the OD of the supernatant was determined at 545 nm. The hemolysis ratio (HR) was calculated according to the following equation:
|
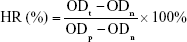
where ODt, ODn, and ODp are the optical densities of the samples, negative control, and positive control, respectively. Test materials with an HR of more than 5% were considered hemolytic.
Acute toxicity test
An acute toxicity test was performed. Twenty Kunming mice were randomly divided into five groups of four mice each.48 The body weight of each mouse was measured. Solutions of OCP, α-CSH/OCP, α-CSH/OCP/SH, and α-CSH/OCP/SHS (10 mg/kg; experimental groups) and of normal saline (control group) were preheated to 38.5°C and administered via intraperitoneal injection. At 12, 24, 48, and 72 hours after injection, the mice were evaluated for overall condition, toxicity, and death.
Pyrogenic test
A pyrogenic test was performed. The body temperature of 27 New Zealand White rabbits was measured over 3 days. The rabbits were fasted from food and water for 2 hours before measuring the rectal temperature. After immobilization, a thermometer was inserted into the anus at an insertion depth of 5–6 cm. The temperature was read after 5 minutes. Readings were made once every hour for a total of three readings. The normal temperature range was set as 38.7°C–39.5°C, and the difference between the highest and lowest temperature readings should not exceed 0.4°C. Fifteen rabbits that satisfied the body temperature requirement were selected and numbered. The rabbits were immobilized, and the rectal temperature was measured once daily for 3 days. On the fourth day, the pyrogenic test was performed under the same room temperature and humidity conditions. The materials solutions were preheated to 38.5°C and then injected via the marginal ear vein at a dose of 10 mL/kg. Rectal temperature was measured at 1, 2, and 3 hours. The rise in body temperature was calculated as the highest reading minus the normal body temperature. Bone repair materials were considered nonpyrogenic if the rise in body temperature was less than 0.6°C for each rabbit.
Intracutaneous stimulation test
An intracutaneous stimulation test was performed. Two New Zealand White rabbits were selected to receive injections 1.5 cm from the spine. The spacing between two adjacent injection sites was 2 cm. Five injection sites that received 0.2 mL of normal saline on the left side of the head were chosen as negative controls, and 0.2 mL of materials solution was injected into five sites on the right side of the head. Local skin and tissues were observed for signs of irritation at 24, 48, and 72 hours. Reactions such as red spots and edema at each injection site were observed.
BMSC osteogenesis in vitro
The bone materials promoting BMSC osteogenesis in vitro was tested by Western blot analysis and immunofluorescence staining. After culturing with or without the bone materials (25% OCP, α-CSH/OCP, α-CSH/OCP/SH, or α-CSH/OCP/SHS) for 7 days, the cells were lysed using the protein extraction reagent RIPA (AS1004; Aspen, Inc., San Mateo, CA, USA). Samples were separated by SDS polyacrylamide gel electrophoresis and electrotransferred onto hybond-polyvinylidene fluoride (PVDF) membranes. The membranes were incubated overnight at 4°C with rabbit anti-GAPDH antibody (1:10,000; Abcam, Cambridge, UK), rabbit anti-ALP antibody (1:10,000; Abcam, Inc.), mouse anti-collagen type 1 (COL1) antibody (1:500; Abcam, Inc.), rabbit anti-osteopontin (OPN) antibody (1:1,000; Abcam, Inc.), rabbit anti-osteocalcin (OCN) antibody (1:500; Abcam, Inc.), or rabbit anti-runt-related transcription factor 2 (RUNX2) antibody (1:500 Bioss Antibodies Inc., Woburn, MA, USA) in Tris buffered saline (TBST). Horseradish peroxidase-labeled goat anti-rabbit (1:10,000; Aspen, Inc.) and goat anti-mouse (1:10,000; Aspen, Inc.) secondary antibodies were used. The protein bands were detected by ECL. The results were quantified by ImageJ software (MicroPublisher; Q-IMAGING, Inc.).
After being cultured with or without materials (25% OCP, α-CSH/OCP, α-CSH/OCP/SH, or α-CSH/OCP/SHS) for 7 days, the cell clots were collected and fixed in 4% paraformaldehyde at room temperature. Then, the samples were blocked with BSA and incubated overnight at 4°C with rabbit anti-ALP antibody (1:200; Abcam, Inc.), rabbit anti-COL1 antibody (1:200; Abcam, Inc.), rabbit anti-OPN antibody (1:200; Abcam, Inc.), rabbit anti-OCN antibody (1:150; Abcam, Inc.), or rabbit anti-RUNX2 antibody (1:100; Bioss, Inc.) in TBST. Next, the cells were incubated with the following secondary antibodies for 50 minutes at 37°C: FITC-labeled goat anti-rabbit, FITC-labeled goat anti-mouse, CY3-labeled goat anti-rabbit, CY3-labeled goat anti-mouse, and CY3-labeled goat anti-donkey (Aspen, Inc.). The nuclei were stained with DAPI. After being washed three times with PBS, the clots were viewed under a fluorescence microscope (IX51).
In vivo osteogenesis in the animal model, X-ray detection, and histological examination
For in vivo studies, 24 New Zealand White rabbits were divided into six groups (OCP, α-CSH/OCP, α-CSH/OCP/SH, α-CSH/OCP/SHS, negative control, and positive control) of four rabbits each. For the operation, sodium pentobarbital (100 mg/kg) anesthesia was administered through the auricular vein. After exposing the middle segment of the right radius, a 1.5 cm osteotomy was performed at the same position on the right radius. An artificial bone repair material that is commonly used in clinical practice (BonGold™; Allgens Medical, Inc.™, Basking Ridge, NJ, USA) was used as a positive control. Then, OCP, α-CSH/OCP, α-CSH/OCP/SH, and α-CSH/OCP/SHS were used to fix the defects in the experimental group; the negative control did not receive treatment for the defect. The cut was sutured and properly cleaned with povidone iodine antiseptic. Penicillin was administered daily during the postoperative period (3 days). XRD was performed on the day of the operation and every 3 weeks thereafter. Rabbits were sacrificed at 6 and 9 weeks after the operation, and the liver and kidney were removed. The harvested specimens were fixed in 10% formalin and stored at room temperature before histological analysis.
The radius of the animal model of tissue-engineered bone formation was evaluated by X-ray scanning. A digital medical X-ray photography system (DigitalDiagnost Pro; Philips, Shanghai, China) was used for scanning at 0, 3, 6, and 9 weeks after surgery.
At 6 and 9 weeks after surgery, the rabbits were anesthetized with sodium pentobarbital (100 mg/kg). The liver and kidneys were excised and fixed in 4% paraformaldehyde. The samples were dehydrated in ethanol and washed and embedded in paraffin wax. A microtome was used to prepare 4 μm sections, which were stained with H&E and examined by digital microscopy.
Statistical analyses
SAS 9.4 was used for all statistical analyses, and P<0.05 was considered statistically significant.
Results
Determination of the MW of SHS
The viscosity average MW was calculated according to the following equation:
|
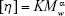
where [η] is the characteristic viscosity number, K=3.6×10−4, α=0.78.
The results show that the higher the degree of substitution, the smaller the MW (Table 1), because the esterification reaction leads to the degradation of the SH chain.
 | Table 1 The viscosity average MW of SHS |
Characterization of the bone repair materials
SEM results of materials
The blend system exhibited a 3D lamellar porous structure (Figure 2). α-CSH forms fine needle-like crystals with larger pores, and OCP forms typical thin crystal slices. As observed in 2, 4, and 8 kx, the α-CSH/OCP/SH and α-CSH/OCP/SHS composite materials exhibited typical α-CSH and OCP crystal morphology.
 | Figure 2 SEM analysis of bone repair materials: (A) α-CSH/OCP/SH and (B) α-CSH/OCP/SHS. |
The 13C nuclear magnetic resonance (NMR) spectrum of SHS
The 13C NMR spectrum of SHS is shown in Figure 3A. The chemical shift of the C atom of SH shifted to the lower field after the sulfate reaction. The C-6 position of G4S moved to the low field (δ=68.6 ppm), but the chemical shift of other positions was not obvious. This result shows that the main position of the sulfate reaction of the hydroxyl group of SH is the C-6 position of the 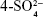 -galactose unit (G4S).31 The results further show that the sulfate group was successfully introduced into the SH structure without introducing other hydroxyl sites or destroying the SH structure.
-galactose unit (G4S).31 The results further show that the sulfate group was successfully introduced into the SH structure without introducing other hydroxyl sites or destroying the SH structure.
 | Figure 3 (A) The 13C NMR spectrum of SHS; (B) X-ray diffraction pattern of composite materials: (a) α-CSH, (b) α-CSH/OCP/SH, and (c) α-CSH/OCP/SHS; and (C1 and C2) Fourier-transform infrared spectra of materials: (C1a) SH, (C1b) SHS, (C2a) α-CSH, (C2b) α-CSH/OCP, and (C2c) OCP. |
Fourier transform infrared spectroscopy results
Figure 3C2 shows the FTIR spectra of (a) α-CSH, (b) α-CSH/OCP, and (c) OCP. The characteristic peaks of HA at 3,594 and 3,552/cm are caused by the absorption of H2O, while peaks at 1,093, 659, and 603/cm are caused by the absorption of 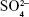 .32 For OCP, the P−O characteristic peaks appear at 1,135, 1,068, and 983/cm; HPO42− absorption appears at 871/cm, and PO43− absorption is shown at 576 and 661/cm.33 For α-CSH/OCP, the FTIR spectra (b) appear to be superpositions of the α-CSH and OCP spectra, which may be evidence that α-CSH/OCP composite powder has been successfully prepared by the chemical coprecipitation method.
.32 For OCP, the P−O characteristic peaks appear at 1,135, 1,068, and 983/cm; HPO42− absorption appears at 871/cm, and PO43− absorption is shown at 576 and 661/cm.33 For α-CSH/OCP, the FTIR spectra (b) appear to be superpositions of the α-CSH and OCP spectra, which may be evidence that α-CSH/OCP composite powder has been successfully prepared by the chemical coprecipitation method.
Figure 3C1 shows the FTIR spectra of SH (A) and SHS (B). There are multiple absorption peaks of N−H and O−H stretching vibration that overlap at 3,438/cm, indicating that SH has a multi-hydroxyl structure. The absorption peaks produced by the coupling of the O−H flexural vibration and the stretching vibration of C−O and C=O at 1,627, 1,407, and 1,328 cm−1 indicate that there are hydroxyl groups and carboxyl groups in the glycosylated polysaccharides. There are characteristic absorption peaks of C−N and C=O stretching vibration at 1,577 cm−1, combined with an N−H vibration absorption peak at 3,438 cm−1, indicating the presence of an acetyl amino structure. There is a C−O stretching vibration absorption peak at 1,047 cm−1and 1,153 cm−1, which is in accordance with the structure of SH.34 The intensity of the absorption peak at 3,438 cm−1 was obviously weakened in the infrared spectrum of SHS, and the asymmetrical stretching vibration peak of the sulfate group O=S=O at 1,267 cm−1 was obviously enhanced. Furthermore, the characteristic peaks of C−O−S stretching vibration of the C-6 position were observed at 802 cm−1. These results fully show that the sulfate group was successfully introduced into the SH structure. No characteristic peaks of other functional groups were found in the spectra, indicating that the sulfate groups were not introduced into other hydroxyl sites and that the structure of SH was not destroyed.
XRD
The XRD spectra of (a) α-CSH, (b) α-CSH/OCP/SH and (c) α-CSH/OCP/SHS samples are shown in Figure 3B. The peaks at 2θ=14.8, 32.0, 25.7, 29.8, and 42.3° are the characteristic peaks of α-CSH. For α-CSH/OCP/SH (b) and α-CSH/OCP/SHS (c) samples, the observed spectra can be considered combinations of OCP and SH/SHS. These findings indicate that SH/SHS filled the gaps in the α-CSH/OCP network, and the lack of apparent interruptions indicates crystalline SH/SHS and amorphous α-CSH/OCP.
TGA
Figure 4A presents TGA curves of α-CSH/OCP/SH and α-CSH/OCP/SHS samples. As shown in Figure 4A1, there are three sections of the TG curves for α-CSH/OCP/SH samples. The first section is at approximately 100°C–146°C, which can be explained by the loss of free water and crystal water. Weight loss occurs at approximately 200°C, caused by the thermal decomposition of SH. Similarly, TG curves for α-CSH/OCP/SHS samples have three sections (Figure 4A2). The section at approximately 100°C–150°C can be attributed to the loss of free water and crystal water, and the section at 200°C is caused by the thermal decomposition of SHS.
 | Figure 4 (A) Thermogravimetric analysis of composite materials: (A1) α-CSH/OCP/SH and (A2) α-CSH/OCP/SHS. (B) In vitro degradation analysis of composite materials. (C) Mechanical property analysis of α-CSH/OCP/SH and α-CSH/OCP/SHS composites after different aging periods. |
Porosity and pore size
The porosity was 27.45% for α-CSH/OCP/SH and 30.63% for α-CSH/OCP/SHS, indicating that α-CSH/OCP/SH is denser than α-CSH/OCP/SHS.
Setting time
The initial and final setting times of the α-CSH/OCP/SH composite were 4.456 and 43.544 minutes, respectively. The initial and final setting times of the α-CSH/OCP/SHS composite were 5.258 and 51.357 minutes, respectively. In contrast, α-CSH had an initial setting time of 2.492 minutes and a final setting time of 4.211 minutes (Table 2).
 | Table 2 Setting time analysis |
In vitro degradation
As shown in Figure 4B, the three materials were degradable. In the first 5 days, the three materials degraded quickly. At 28 days, the degradation rates of the α-CSH/OCP/SHS composite, the α-CSH/OCP/SH composite, and CSH were 40.0, 64.0, and 77.0%, respectively. The analysis shows that α-CSH degradation was too rapid, α-CSH/OCP/SHS degradation was too slow, and the α-CSH/OCP/SH degradation rate was ideal.
Mechanical property test
Figure 4C depicts the compressive strength analysis of α-CSH/OCP/SH and α-CSH/OCP/SHS composites at different time points. Overall, the mechanical strength of the two composite artificial bone materials gradually decreased as they degraded. The compressive strength value of the α-CSH/OCP/SH composite was 13.72 MPa on day 1 but decreased to 29% of this value after 28 days. The compressive strength value of the α-CSH/OCP/SHS composite was 14.21 MPa on day 1 but decreased to 32% of this value after 28 days. The mechanical strength of α-CSH/OCP/SHS composite artificial bone material was greater than that of α-CSH/OCP/SH. In general, the mechanical strength of the α-CSH/OCP/SH and α-CSH/OCP/SHS composites gradually stabilized, which fits the requirements for the degradation rate of ideal bone repair materials.
Wettability
The wettability of α-CSH, α-CSH/OCP/SH, and α-CSH/OCP/SHS was evaluated using the contact angle method. The contact angles of α-CSH, α-CSH/OCP/SH and α-CSH/OCP/SHS were 18.9, 16.9, and 13.0°, respectively. These results indicated that the addition of SH/SHS to the hydrophobic α-CSH/OCP composite material improved the hydrophilic properties of α-CSH/OCP/SH and α-CSH/OCP/SHS bone repair materials.
Biocompatibility
MTT assay results
ANOVA was used to compare differences among groups. The effects of the materials on rabbit BMSC proliferation are shown in Figure 5. The relative proliferation rate in the 25% α-CSH/OCP/SH group was higher than that in the control, 25% OCP, 25% α-CSH/OCP, and 25% α-CSH/OCP/SHS groups from days 1 to 7. The relative proliferation rates of the α-CSH/OCP/SH groups were greater than 90%. Thus, the cytotoxicity of α-CSH/OCP/SH on rabbit BMSCs was characterized as class 0–1, indicating no obvious cytotoxicity. The relative proliferation rates of the α-CSH/OCP/SHS groups were less than 50%.
 | Figure 5 The RGR of rabbit BMSCs. |
Acute toxicity test results
At 72 hours after the injection of materials solutions, all rabbits showed normal eating and movement behavior and appeared to be in a good mental state. There were no signs of toxicity, such as paralysis, respiratory inhibition, and reduced activity.
Pyrogenic test results
At 1, 2, and 3 hours after the injection of the material solutions, the body temperature increase in all experimental rabbits was less than 0.6°C. There was no abnormal increase in the body temperature of rabbits that received chemical treatment, indicating that the materials in the physical and chemical treatments did not induce a pyrogenic reaction. ANOVA of the completely randomized experiment showed no significant changes in body temperature before or after the pyrogenic test (P>0.05, Table 3).
 | Table 3 Changes in body temperature (X±S, n=3, °C) |
Intracutaneous stimulation test results
The local skin of rabbits in the experimental and control groups was observed for red spots, edema, and necrosis immediately after injection and at 24, 48, and 72 hours postinjection. No obvious red spots were observed on experimental animals, indicating mild irritation at most.
Hemolysis test results
In the hemolysis test, the HR values of α-CSH/OCP/SH, α-CSH/OCP, and OCP were below 5% and the HR value of α-CSH/OCP/SHS was greater than 5% (Table 4). Thus, α-CSH/OCP/SH did not lead to erythrocyte rupture according to the ISO standard.
 | Table 4 Hemolysis ratio |
BMSC osteogenesis in vitro
Western blot analysis results
ANOVA was used to compare differences among groups. Figure 6A shows the protein expression levels of ALP, COL1, OPN, OCN, and RUNX2, which are quantified in Figure 6B–F. The results showed that the expression of these proteins was always higher in the 25% α-CSH/OCP/SH group than in the control, 25% OCP, 25% α-CSH/OCP, and 25% α-CSH/OCP/SHS groups.
 | Figure 6 Western blot assays. |
Immunofluorescence staining results
After incubation, confocal microscopy was performed to detect immunofluorescence for COL1, ALP, OPN, OCN, and RUNX2. The expression of COL1, ALP, OPN, OCN, and RUNX2 was absent or weak in the negative control group but strong in the OCP, α-CSH/OCP, and α-CSH/OCP/SHS groups. COL1, ALP, OPN, OCN, and RUNX2 expressions were highest in the α-CSH/OCP/SH group (Figure 7).
 | Figure 7 Immunofluorescence staining for COL1, ALP, OPN, OCN, and RUNX2. |
In vivo osteogenesis
As shown in Figure 8, the α-CSH/OCP, α-CSH/OCP/SH, and positive control groups had significantly more new calluses than the other groups at 3 weeks after the operation. At 6 weeks after the operation, there was significant callus formation in all but the negative control and OCP groups; furthermore, the α-CSH/OCP/SH group and the positive control group had formed a medullary cavity in the bone defect. Nine weeks after the operation, the bone defects in the α-CSH/OCP/SH group and the positive control group were completely healed, including callus and medullary cavity formation. Significant bone defects remained in the negative control group. Although there was no obvious bone defect in the other groups, there was no medullary cavity. The histological examination results showed no presence of inflammatory cells, dead cells, differentiated cells, or abnormal neonatal tissue in the liver or kidney (Figure 9).
 | Figure 8 X-ray detection of bone repair in the animal model. |
 | Figure 9 Histology of the liver (A) and kidney (B) 6 and 9 weeks after implantation. |
Discussion
In this study, a novel and functional bone tissue engineering material was prepared. The porous composite bone tissue engineering material α-CSH/OCP/SH has a specific degradation rate and favorable mechanical strength. MTT, hemolysis, acute toxicity, pyrogenic, and intracutaneous stimulation assays were performed. The results show that the composite materials have excellent biocompatibility. The capability of the materials to induce the osteoblast differentiation of BMSCs and to repair bone defects in an animal model was also evaluated. The composite materials have demonstrated abilities to induce bone regeneration and self-repair. Therefore, these materials can be used in bone tissue engineering.
When the surface morphology of the bone repair materials was observed by SEM, the α-CSH/OCP/SH and α-CSH/OCP/SHS composite materials exhibited typical α-CSH and OCP crystal morphology with larger pores. Open pores are beneficial for osseointegration as they facilitate the transport of nutrients and oxygen required for vascularization during bone tissue development.35 The setting time is known to be important for bone repair materials in the bone tissue engineering field. Setting time analysis revealed that the addition of SH/SHS improved the initial setting time (t1), final setting time (t2), and t2−t1 value. The setting time of α-CSH/OCP/SHS composite artificial bone was longer, which is beneficial for the clinical application of artificial bone materials. Compressive strength analysis provides information on the mechanical properties of materials and is an important tool for modern orthopedic applications. A higher compressive strength indicates increased material strength.7 The materials had initial compressive strength values 13.72 and 14.21, but after 28 days, these values decreased to 29 and 32% of the initial values, respectively, which meets the requirements for bone regeneration. In body fluids, which contain bonding agents and hydrolyzers, the material is decomposed by physical dissolution into particles, molecules, and ions; this process dissolves the material.36 The degradation of α-CSH was too fast in vitro. As a calcium phosphate bone graft material, OCP has a low solubility, resulting in a degradation rate that is too slow for ready absorption by the body and not conducive to new bone growth. In addition, an acidic environment is generated in the process of α-CSH degradation, which may cause local inflammation when applied clinically. Conversely, OCP is alkaline, and its aqueous solution is weakly alkaline; therefore, the complex can help body fluids remain at an appropriate pH for bone tissue repair37 and exhibits an appropriate degradation rate. In our study, the in vitro degradation rate of the composites was decreased by the addition of OCP; this result was in line with the above results. Both α-CSH/OCP/SH and α-CSH/OCP/SHS had an ideal degradation rate. The surface of materials will contact and interact with cells first, so the superficial physicochemical properties of materials, such as hydrophilicity and hydrophobicity, can influence a series of cellular responses, including adhesion, proliferation, and differentiation.38 The surface hydrophilicity of materials can be represented by the contact angle with water, with better surface hydrophilicity indicated by a smaller contact angle with water. The contact angles of α-CSH, α-CSH/OCP/SH, and α-CSH/OCP/SHS were 18.9, 16.9, and 13.0°, respectively, indicating that the addition of SH/SHS to hydrophobic α-CSH/OCP materials improved the hydrophilicity of α-CSH/OCP/SH and α-CSH/OCP/SHS bone repair materials. α-CSH/OCP/SH and α-CSH/OCP/SHS had good bioactivity. In our study, the porosity was 30.63% for α-CSH/OCP/SH and 27.45% for α-CSH/OCP/SHS. These results indicate that α-CSH/OCP/SH is promising for biomaterial applications, because the ideal porosity of implant materials is in the range of 30–90 vol%.39
The biocompatibility of bone repair materials is defined as the ability of the material to support cellular activity. Thus, biomaterials should be nontoxic.35 The biocompatibility of the materials was assessed by acute toxicity, pyrogenic, and intracutaneous stimulation tests. The results showed that the bone repair materials caused no acute toxicity, pyrogenic reaction, or stimulation.
BMSCs are an attractive choice for bone tissue engineering because they can differentiate into cells in the mesoderm lineage, including osteoblasts, chondrocytes, and adipocytes,40,41 as they are multipotent cells characterized by high proliferation and differentiation abilities and limited immunosuppression and immunogenicity. Under different induction conditions, BMSCs can be differentiated into osteoblasts, fibroblasts, and chondrocytes.42 MTT cytotoxicity assays were performed on the leachate of the bone repair materials; the results showed that the relative proliferation rates in the α-CSH/OCP/SH groups were greater than 90%. The safety of the bone repair materials was evaluated according to the International Standard ISO-10993 “Biological Evaluation of Medical Devices Part 1”. The cytotoxicity of the leachate of α-CSH/OCP/SH bone repair materials on rabbit BMSCs was class 0–1, indicating no obvious cytotoxicity. The relative proliferation rates in the α-CSH/OCP/SHS groups were less than 50%, indicating obvious cytotoxicity. It is important to develop a biomaterial that mimics natural bone ECM both chemically and structurally.43 Bone ECM proteins include OPN and OCN. RUNX2 regulates cell growth, angiogenesis and the production of signaling proteins, which are involved in the formation of osteoblast phenotypes.44 Bone is rich in collagen I. The cell surface glycoprotein ALP is involved in the mineralization of bone tissue and is the most widely recognized marker of osteoblast differentiation.45 Therefore, these proteins are commonly used as specific markers of osteoblast activity in vitro and in vivo. After inducing osteoblast differentiation for 7 days, Western blot analysis and immunofluorescence staining revealed that the expression levels of these proteins were higher in the α-CSH/OCP/SH and α-CSH/OCP/SHS groups than in the control group. These results suggest that the α-CSH/OCP/SH and α-CSH/OCP/SHS materials can induce bone-forming protein expression and promote cell adhesion and spreading.
The in vitro experiment results prompted us to investigate the properties of the bone repair materials in vivo. A rabbit radius-defect model was used to investigate the ability of the α-CSH/OCP/SH and α-CSH/OCP/SHS materials to promote osteogenesis. The rabbit defect model was adopted to allow defect repair in a short period of time. Active bone regeneration in the defects occurred in both the experimental and positive control groups. The results further showed that the α-CSH/OCP/SH material promoted the differentiation of BMSCs into osteoblasts in vivo. The histological analyses suggested that the α-CSH/OCP/SH materials are biocompatible. Taken together, the results indicate that the implanted materials and cells are able to appropriately promote osteogenesis.
Conclusion
In this study, we developed the porous composite α-CSH/OCP/SH for use as bone tissue engineering material. The composite material is a noncytotoxic 3D porous structure with good biological conductivity and excellent biocompatibility and biodegradation that exhibited clear advantages regarding the induction of bone regeneration and self-repair. Moreover, α-CSH/OCP/SH closely mimics natural bone matrix and is a promising biomimetic bone repair material.
Ethics approval
All experiments were approved by the Commission for Animal Protection and Utilization of Wuhan University, and animal procedures were conducted in strict accordance with national and institutional guidelines.
Acknowledgments
First and foremost, I, Changshun Chen, would like to show my deepest gratitude to my supervisor, Professor Shengxiang Tao, who has provided me with valuable guidance in every stage of the writing of this thesis. I also extend my thanks to Professor Lihong Fan and Dr Xiang Hu for guidance and help. My sincere appreciation goes to the teachers and students from School of Chemistry, Chemical Engineering and Life Sciences, Wuhan University of Technology, who participated this study with great cooperation.
This work was supported by a grant from the Project of Science and Technology Innovation Training Fund of Central South Hospital of Wuhan University (znpy2017030), the Autonomous Scientific Research Project of Wuhan University (2042018kf0216), the Training Project on the Surface of Zhongnan Hospital of Wuhan University (znpy20160045), the Training Program for Middle-age and Young Medical Personnel in Wuhan, and Training Plan for Young and Middle-aged Technical Talents in Zhongnan Hospital of Wuhan University.
Disclosure
The authors report no conflicts of interest in this work.
References
Holzwarth JM, Ma PX. Biomimetic nanofibrous scaffolds for bone tissue engineering. Biomaterials. 2011;32(36):9622–9629. | ||
Amstein CF, Hartman PA. Adaptation of plastic surfaces for tissue culture by glow discharge. J Clin Microbiol. 1975;2(1):46–54. | ||
Frohbergh ME, Katsman A, Botta GP, et al. Electrospun hydroxyapatite-containing chitosan nanofibers crosslinked with genipin for bone tissue engineering. Biomaterials. 2012;33(36):9167–9178. | ||
Al-Sayyad MJ, Abdulmajeed TM. Fracture of the anterior iliac crest following autogenous bone grafting. Saudi Med J. 2006;27(2):254–258. | ||
Seiler JG 3rd, Johnson J. Iliac crest autogenous bone grafting: donor site complications. J South Orthop Assoc. 2000;9(2):91. | ||
O’Brien FJ. Biomaterials & scaffolds for tissue engineering. Materials Today. 2011;14(3):88–95. | ||
Amini AR, Laurencin CT, Nukavarapu SP. Bone tissue engineering: recent advances and challenges. Crit Rev Biomed Eng. 2012;40(5):363–408. | ||
Fan X, Chung JY, Lim YX, Li Z, Loh XJ. Review of Adaptive Programmable Materials and Their Bioapplications. ACS Appl Mater Interfaces. 2016;8(49):33351–33370. | ||
Lim J, You M, Li J, Li Z. Emerging bone tissue engineering via Polyhydroxyalkanoate (PHA)-based scaffolds. Mater Sci Eng C Mater Biol Appl. 2017;79:917–929. | ||
Xu C, Su P, Chen X, et al. Biocompatibility and osteogenesis of biomimetic Bioglass-Collagen-Phosphatidylserine composite scaffolds for bone tissue engineering. Biomaterials. 2011;32(4):1051–1058. | ||
Salgado AJ, Coutinho OP, Reis RL. Bone tissue engineering: state of the art and future trends. Macromol Biosci. 2004;4(8):743–765. | ||
Porter JR, Ruckh TT, Popat KC. Bone tissue engineering: a review in bone biomimetics and drug delivery strategies. Biotechnol Prog. 2009;25(6):1539–1560. | ||
Mastrogiacomo M, Muraglia A, Komlev V, et al. Tissue engineering of bone: search for a better scaffold. Orthod Craniofac Res. 2005;8(4):277–284. | ||
Li X, Xie J, Yuan X, Xia Y. Coating electrospun poly(epsilon-caprolactone) fibers with gelatin and calcium phosphate and their use as biomimetic scaffolds for bone tissue engineering. Langmuir. 2008;24(24):14145–14150. | ||
Rezwan K, Chen QZ, Blaker JJ, Boccaccini AR. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials. 2006;27(18):3413–3431. | ||
Liao SS, Cui FZ, Zhang W, Feng QL. Hierarchically biomimetic bone scaffold materials: nano-HA/collagen/PLA composite. J Biomed Mater Res B Appl Biomater. 2004;69(2):158–165. | ||
Ma XF, Zhang JY. Development of bone tissue engineering scaffold materials. Chinese Journal of Tissue Engineering Research. 2014;18(30):4895–4899. | ||
Glimcher MJ. Molecular Biology of Mineralized Tissues with Particular Reference to Bone. Rev Modern Phy. 1959;31(2):359–393. | ||
Whang PG, Wang JC. Bone graft substitutes for spinal fusion. Spine J. 2003;3(2):155–165. | ||
Thomas MV, Puleo DA, Al-Sabbagh M. Calcium sulfate: a review. J Long Term Eff Med Implants. 2005;15(6):599–607. | ||
Christensen AN, Jensen TR, Nonat A. A new calcium sulfate hemi-hydrate. Dalton Trans. 2010;39(8):2044–2048. | ||
Kamakura S, Sasano Y, Suzuki O. Synthetic octacalcium phosphate (OCP) is an effective scaffold to regenerate bone. International Congress. 2005;1284(1284):290–295. | ||
Mondrinos MJ, Dembzynski R, Lu L, et al. Porogen-based solid freeform fabrication of polycaprolactone-calcium phosphate scaffolds for tissue engineering. Biomaterials. 2006;27(25):4399–4408. | ||
Rajzer I, Menaszek E, Kwiatkowski R, Planell JA, Castano O. Electrospun gelatin/poly(ε-caprolactone) fibrous scaffold modified with calcium phosphate for bone tissue engineering. Mater Sci Eng C Mater Biol Appl. 2014;44:183–190. | ||
Ouyang HW, Cao T, Zou XH, et al. Mesenchymal stem cell sheets revitalize nonviable dense grafts: implications for repair of large-bone and tendon defects. Transplantation. 2006;82(2):170–174. | ||
Collins MN, Birkinshaw C. Hyaluronic acid based scaffolds for tissue engineering – a review. Carbohydr Polym. 2013;92(2):1262–1279. | ||
Pasquinelli G, Orrico C, Foroni L, et al. Mesenchymal stem cell interaction with a non-woven hyaluronan-based scaffold suitable for tissue repair. J Anat. 2008;213(5):520–530. | ||
Turley EA, Noble PW, Bourguignon LY. Signaling properties of hyaluronan receptors. J Biol Chem. 2002;277(7):4589–4592. | ||
Cui D, Liu M, Liang R, Bi Y. Synthesis and Optimization of the Reaction Conditions of Starch Sulfates in Aqueous Solution. Starch/Stärke. 2007;59(2):91–98. | ||
Pal S, Sen G, Mishra S, Dey RK, Jha U. Carboxymethyl tamarind: Synthesis, characterization and its application as novel drug-delivery agent. J Appl Polym Sci. 2010;110(1):392–400. | ||
Melo MRS, Feitosa JPA, Freitas ALP, Paula R. Isolation and characterization of soluble sulfated polysaccharide from the red seaweed Gracilaria cornea. Carbohydrate Polymers. 2002;49(4):491–498. | ||
Wang Pet al. Physicochemical properties of chitosan microspheres/silk fibroin/calcium sulfate bone cement. Chinese Journal of Tissue Engineering Research. 2014;18(12):1831p–1838p. | ||
Yb H. Study on the Osteogenic Properties of Silk Fibroin Octacalcium Phosphate Compound Porous Scaffolds. Suzhou University: Suzhou; 2016. | ||
Shahong W, Jiye C, Yi H. Research on Hyaluronic Acid and Pectin Complex. Journal of Biomedical Engineering. 2009;33:561561–565565. | ||
Bose S, Roy M, Bandyopadhyay A. Recent advances in bone tissue engineering scaffolds. Trends Biotechnol. 2012;30(10):546–554. | ||
Siegel HJ, Baird RC 3rd, Hall J, Lopez-Ben R, Lander PH. The outcome of composite bone graft substitute used to treat cavitary bone defects. Orthopedics. 2008;31(8):754. | ||
Mao KY, Hao LB, Tang PF, et al. Osteoblast MC3T3-E1 Culture on a Fast-setting Carbonated Hydroxyapatite Bone-like Material. Journal of Bioactive & Compatible Polymers. 2005;20(6):541–555. | ||
Wang C, Meng G, Zhang L, Xiong Z, Liu J. Physical properties and biocompatibility of a core-sheath structure composite scaffold for bone tissue engineering in vitro. J Biomed Biotechnol. 2012;2012(1):579141. | ||
Sobieszczyk S. Optimal Features of Porosity of Ti Alloys Considering their Bioactivity and Mechanical Properties. Advances in Materials Sciences. 2010;10(2):20–30. | ||
Song S, Liu S, Qu X, et al. BMP2 and VEGF promote angiogenesis but retard terminal differentiation of osteoblasts in bone regeneration by up-regulating Idl. Acta Biochim Biophys Sin (Shanghai). 2011;43(10):796–804. | ||
Connelly JT, Petrie TA, García AJ, Levenston ME. Fibronectin- and collagen-mimetic ligands regulate bone marrow stromal cell chondrogenesis in three-dimensional hydrogels. Eur Cell Mater. 2011;22(7):168–76; discussion 176–177. | ||
Oyane A, Uchida M, Choong C, Triffitt J, Jones J, Ito A. Simple surface modification of poly(epsilon-caprolactone) for apatite deposition from simulated body fluid. Biomaterials. 2005;26(15):2407–2413. | ||
Liu H, Peng H, Wu Y, et al. The promotion of bone regeneration by nanofibrous hydroxyapatite/chitosan scaffolds by effects on integrin-BMP/Smad signaling pathway in BMSCs. Biomaterials. 2013;34(18):4404–4417. | ||
Lian JB, Stein GS, Javed A, et al. Networks and hubs for the transcriptional control of osteoblastogenesis. Rev Endocr Metab Disord. 2006;7(1–2):1–16. | ||
Xie XH, Wang XL, Zhang G, et al. Structural and degradation characteristics of an innovative porous PLGA/TCP scaffold incorporated with bioactive molecular icaritin. Biomed Mater. 2010;5(5):054109. | ||
Suzuki O, Nakamura M, Miyasaka Y, Kagayama M, Sakurai M. Bone formation on synthetic precursors of hydroxyapatite. Tohoku J Exp Med. 1991;164(1):37–50. | ||
Chen YJ. Research on an Injectable Calcium Sulfate Bone Cement Product. Zhejiang: Zhejiang University; 2012. | ||
He HY, Zhang JY, Mi X, Hu Y, Gu XY. Rapid prototyping for tissue-engineered bone scaffold by 3D printing and biocompatibility study. Int J Clin Exp Med. 2015;8(7):11777–11785. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.