Back to Journals » Drug Design, Development and Therapy » Volume 10
α-Aminoadipic acid protects against retinal disruption through attenuating Müller cell gliosis in a rat model of acute ocular hypertension
Authors Wang X, Su J, Ding J, Han S, Ma W, Luo H, Hughes G, Meng Z, Yin Y, Wang Y, Li J
Received 29 January 2016
Accepted for publication 10 March 2016
Published 20 October 2016 Volume 2016:10 Pages 3449—3457
DOI https://doi.org/10.2147/DDDT.S105362
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Xiaolei Wang,1,2,* Jier Su,2,3,* Jingwen Ding,4 Song Han,2 Wei Ma,2,5 Hong Luo,2 Guy Hughes,6 Zhaoyang Meng,1 Yi Yin,1 Yanling Wang,1 Junfa Li2
1Department of Ophthalmology, Beijing Friendship Hospital, 2Department of Neurobiology, Beijing Institute for Brain Disorders, Capital Medical University, Beijing, 3Ningbo College of Health Sciences, Ningbo, 4Department of Ophthalmology, Beijing Tongren Hospital, Capital Medical University, Beijing, 5Beijing Stomatological Hospital, Capital Medical University, Beijing, People’s Republic of China; 6University of California, Irvine School of Medicine, Irvine, CA, USA
*These authors contributed equally to this work
Objective: Ocular hypertension is an important risk factor for glaucoma. The purpose of this study was to investigate the gliotoxic effects of α-aminoadipic acid (AAA) in a rat model of AOH and its underlying mechanisms.
Materials and methods: In the rat model of acute ocular hypertension (AOH), intraocular pressure was increased to 110 mmHg for 60 minutes. Animals were divided into four groups: sham operation (Ctrl), AOH, AOH + phosphate-buffered saline (PBS), and AOH + AAA. Cell apoptosis in the ganglion cell layer was detected with the terminal deoxynucleotidyl transferase-mediated uridine 5'-triphosphate-biotin nick end labeling (TUNEL) assay, and retinal ganglion cells (RGCs) immunostained with Thy-1 were counted. Müller cell activation was detected using immunostaining with glutamine synthetase and glial fibrillary acidic protein. Tumor necrosis factor-α (TNF-α) was examined using Western blot.
Results: In the rat model of AOH, cell apoptosis was induced in the ganglion cell layer and the number of RGCs was decreased. Müller cell gliosis in the retinas of rats was induced, and retinal protein levels of TNF-α were increased. Intravitreal treatment of AAA versus PBS control attenuated these retinal abnormalities to show protective effects in the rat model of AOH.
Conclusion: In the retinas of the rat model of AOH, AAA treatment attenuated retinal apoptosis in the ganglion cell layer and preserved the number of RGCs, likely through the attenuation of Müller cell gliosis and suppression of TNF-α induction. Our observations suggest that AAA might be a potential therapeutic target in glaucoma.
Keywords: glaucoma, acute ocular hypertension, α-aminoadipic acid, retina, Müller cells, retinal ganglion cells, TNF-α
Introduction
Glaucoma, a neurodegenerative disease, is characterized by gradual loss of retinal ganglion cells (RGCs) and optic nerve atrophy.1 Multiple factors are related to glaucoma, such as high intraocular pressure (IOP)2 and low cerebral spinal fluid pressure.3 Müller cells are the major type of glial cells in the mammalian retina that can support and nourish retinal neurons, maintain extracellular ion homeostasis, glutamate recycling, and interaction in synaptic transmission.4,5 Therefore, Müller cells are involved in retinal function. Investigations indicate that Müller cells not only play an important physiological role, but they are also involved in multiple pathological retinal diseases such as glaucoma.6–8 It has been reported that reactive Müller cells can aggravate retinal damage by releasing cytokines such as tumor necrosis factor-α (TNF-α).9 Growing evidence supports that increased glial production of TNF-α contributes to the neurodegeneration in glaucoma.10 TNF-α is a secreted inflammatory cytokine that is responsible for apoptosis, necrosis, and inflammation.11,12 TNF-α is increased in the retina following ischemia or damage and other neurodegenerative disorders.13–17
α-Aminoadipic acid (AAA), a six-carbon homolog of glutamate, is a well-known compound that induces specific glial toxicity through blocking the glutamate uptake.18–20 The use of AAA to interfere with glial influence on neuronal tissues is reported.21,22 However, the effects of AAA inhibition on Müller cell gliosis in glaucoma are unknown. The current study aimed to determine if AAA treatment inhibits Müller cell gliosis and protects against retinal abnormalities induced in a rat model of acute ocular hypertension (AOH) mimicking glaucoma. Müller cell activation of RGCs and TNF-α induction were examined.
Materials and methods
Animals
Adult male Sprague Dawley rats (weight range, 200–250 g, 8–10 weeks) were used in this study in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and followed the People’s Republic of China animal care guidelines. This study was monitored and approved by the Animal Care Committee of the Capital Medical University, SCXK (Jing) 2012-0001. The animals were housed with a 12-hour light/12-hour dark cycle with standard chow and water. All surgical procedures were performed under anesthesia with intraperitoneal chloral hydrate (450 mg/kg) and topical 0.5% proparacaine hydrochloride eye drops (Alcon, Inc., Hünenberg, Switzerland).
Rat model of AOH
The IOP was increased to 110 mmHg (14.63 kPa) for 60 minutes by using an elevated 500 mL plastic container of sterile physiological saline connected to a 27-gauge needle placed in the anterior chamber of the eye. Sham procedure eyes were treated similarly but without the elevation of the bottle; hence, the normal IOP was maintained. Time-course examination was performed at 1 day, 3 days, and 5 days after acute IOP elevation. Animals were divided into sham operation (Ctrl) group, AOH group, AOH + phosphate-buffered saline (PBS) control group, and AOH + AAA-treated group. AAA (250 μg) was injected into vitreous humor in 5 μL at 50 g/L concentration after 3 hours of acute IOP elevation according to the literature.23,24
Immunohistochemistry
Tissues were fixed with 4% paraformaldehyde. Eyeballs were cryoprotected in 30% sucrose overnight at 4°C and then embedded in optimal cutting temperature compound (Sakura Finetechinical Co. Ltd., Tokyo, Japan). Sections (14 μm) were incubated in 3% bovine serum albumin and 0.3% Triton X-100 (Sigma-Aldrich Co., St Louis, MO, USA) to block nonspecific binding and then incubated with primary mouse monoclonal antibodies against Thy-1 (1:200, RGC marker; Abcam, Cambridge, UK), primary mouse monoclonal antibodies against glutamine synthetase (GS; 1:100, Müller glial cell maker; Abcam), rabbit polyclonal antibodies against glial fibrillary acidic protein (GFAP; 1:200, gliosis maker; Cell Signaling Technology Inc., Danvers, MA, USA) overnight at 4°C, followed by incubation with secondary antibodies for 1 hour at room temperature. Nuclei were stained by Hoechst 33342. Sections were mounted with Fluoromount-G (SouthernBiotech, Birmingham, AL, USA). Negative controls were incubated without primary antibodies. Slides were visualized using a confocal microscope (Leica Microsystems, Wetzlar, Germany). The intensity of immunoreactivity from photographs was analyzed using Image-Pro®Plus 6.0 (Media Cybernetics Inc., Silver Spring, MD, USA). Optical densities obtained from immunohistochemistry images were corrected by subtracting the average value of background noise from five image inputs.
Terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling assay
The terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL) assay was conducted according to the manufacturer’s protocol. Briefly, tissue sections were fixed in paraformaldehyde and were subsequently incubated in a TUNEL reaction medium. The immune-labeled RGCs were costained with Hoechst 33342 for 5 minutes. The sections were then mounted on microscope slides and examined under ultraviolet light using an epifluorescence microscope. Ten visual fields were analyzed per retina; the experiment was replicated three times.
Western blot
Protein extracts were generated from retinas. Retinas from sham operation group (Ctrl), AOH group, AOH + PBS control group, and AOH + AAA group were homogenized in lysis buffer and sonicated to dissolve the tissue completely. In all, 40 μg of total proteins from each sample were loaded per lane for sodium dodecyl sulfate polyacrylamide gel electrophoresis. The proteins were transferred onto the nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany) and the blocked nitrocellulose membrane was incubated with a primary rabbit polyclonal antibody against TNF-α (1:1,000; Abcam) for 3 hours (room temperature). After washing in Tris Buffered Saline, with Tween-20 (TBST), the membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit or mouse IgG prior to detection of the labeled proteins by using ECL-plus kit (PerkinElmer Inc., Waltham, MA, USA) followed by exposure of blots to X-Omat (Kodak, Rochester, NY, USA) imaging film. The images were quantified using ImageJ software (National Institutes of Health, Maryland, USA). The Gel Doc-2000 imaging system (Bio-Rad Laboratories Inc., Hercules, CA, USA) was used to perform the quantitative analysis of Western blot results. The ratio of TNF-α was determined by normalizing with the levels of endogenous β-actin. The β-actin band density was expressed as 100%, and the other group was expressed as percentage of that from the control group.
Statistical analysis
The normality of the data was tested using the Shapiro–Wilk method. Analysis of variance was used to determine the difference between TNF-α and fluorescence intensity among three or more independent (unrelated) groups, followed by multiple comparisons using the Student–Newman–Keuls test. The Kruskal–Wallis test was used to determine the difference between TUNEL-positive cells and Thy-1-positive RGCs among three or more independent (unrelated) groups, followed by multiple comparisons using the Nemenyi test. Statistical analysis was performed using SAS 9.2 (SAS Institute Inc., Cary, NC, USA). Values were reported as mean ± SD. A value of P<0.05 was considered as statistically significant.
Results
AAA attenuates AOH-induced cell apoptosis in RGCs
To determine the effects of AAA on RGCs in the AOH model, AOH was induced in SD rats and rats were intravitreally treated with PBS, AAA, AOH + PBS, and AOH + AAA. Significantly reduced retinal thickness in the inner plexiform layer (IPL) and inner nuclear layer was observed after AOH induction versus control (Figure S1). TUNEL assay was used to examine the cell apoptosis in the retinal sections. The mean values of TUNEL-positive cells per visual field were 0.1±0.3 in Control (Figure 1A), 0.2±0.4 in AAA(Figure 1B), 4.6±0.8 in AOH + PBS, and 1.7±0.7 in AOH + AAA. AOH induced the apoptotic death of cells in the ganglion cell layer(GCL; Figure 1C). In the AOH model, intravitreal treatment with AAA attenuated the TUNEL-positive RGCs more significantly than the intravitreal treatment with PBS (P<0.05; Figure 1D), indicating that AAA treatment rescued RGCs from the cell apoptosis induced in the AOH model.
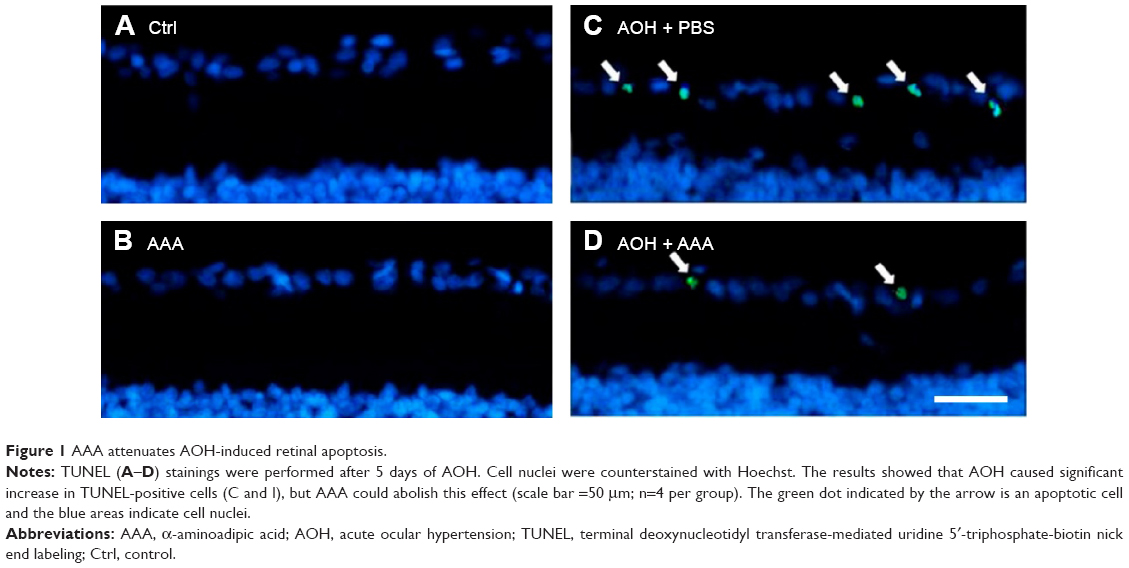 | Figure 1 AAA attenuates AOH-induced retinal apoptosis. |
AAA preserves the AOH-reduced number of RGCs
In addition, retinal sections were immunostained with antibody against Thy-1, a marker for RGCs.25 The mean values of Thy-1-positive RGCs per visual field in Ctrl, AAA, AOH + PBS, and AOH + AAA groups were 8.2±0.9, 8.1±0.8, 3.9±0.7, and 6±0.6, respectively. AAA treatment did not change the number of RGCs in normal conditions (Figure 2A and B). When compared with the Ctrl group (Figure 2A), the number of Thy-1-positive RGCs was decreased in the AOH + PBS group (Figure 2C), and AAA treatment significantly rescued Thy-1-positive RGCs in AOH (P<0.05; Figure 2D), corresponding to AAA-attenuated apoptosis of RGCs in AOH (AOH + PBS versus AOH + AAA, P<0.05; Figure 1).
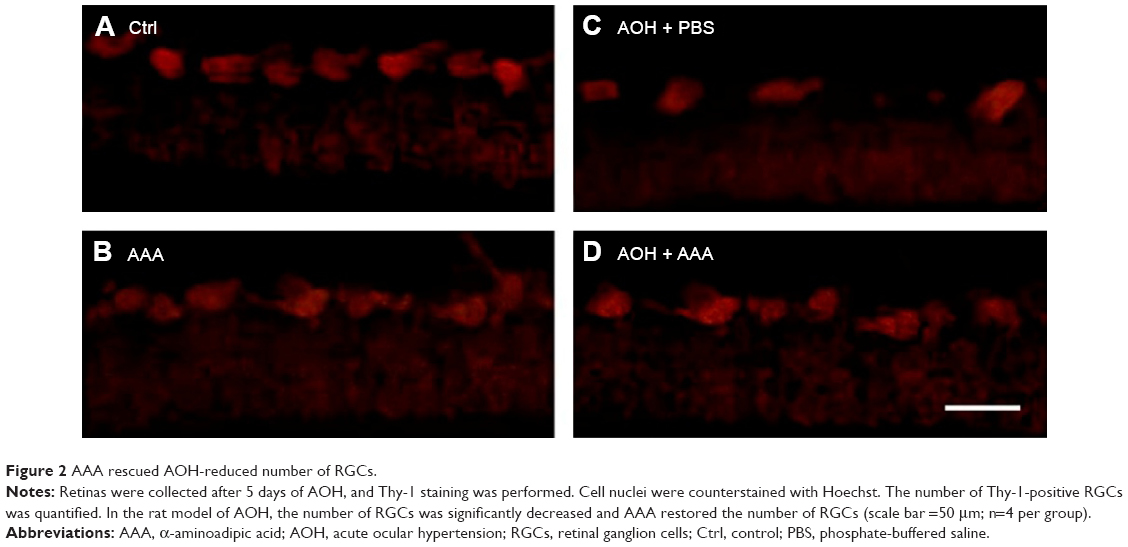 | Figure 2 AAA rescued AOH-reduced number of RGCs. |
AAA attenuates AOH-induced Müller cell gliosis
In the rat model of AOH, AAA rescued RGCs. To determine if AAA protects against AOH-induced RGC death through modulating Müller cell gliosis, immonohistochemistry for GFAP (a marker for activated glial cells) and GS (Müller cell-specific marker) was conducted.26 Müller cells cover the whole retina from the nerve fiber layer to the photoreceptor layer,27 and astrocytes mainly locate along GCL. GFAP was mainly expressed along nerve fiber layer and GCL and was less or absent at other layers in the control group (Figure S2). In the rat model of AOH, colocalization of GFAP and GS was observed across IPL (Figure S2), indicating Müller cell activation. After 1 day, 3 days, and 5 days of AOH induction in rat, GFAP expression was also observed across IPL (Figure S3), indicating Müller cell gliosis. AAA treatment versus PBS control markedly reduced GFAP immunoreactivity across IPL (Figure 3), indicating attenuation in Müller cell gliosis.
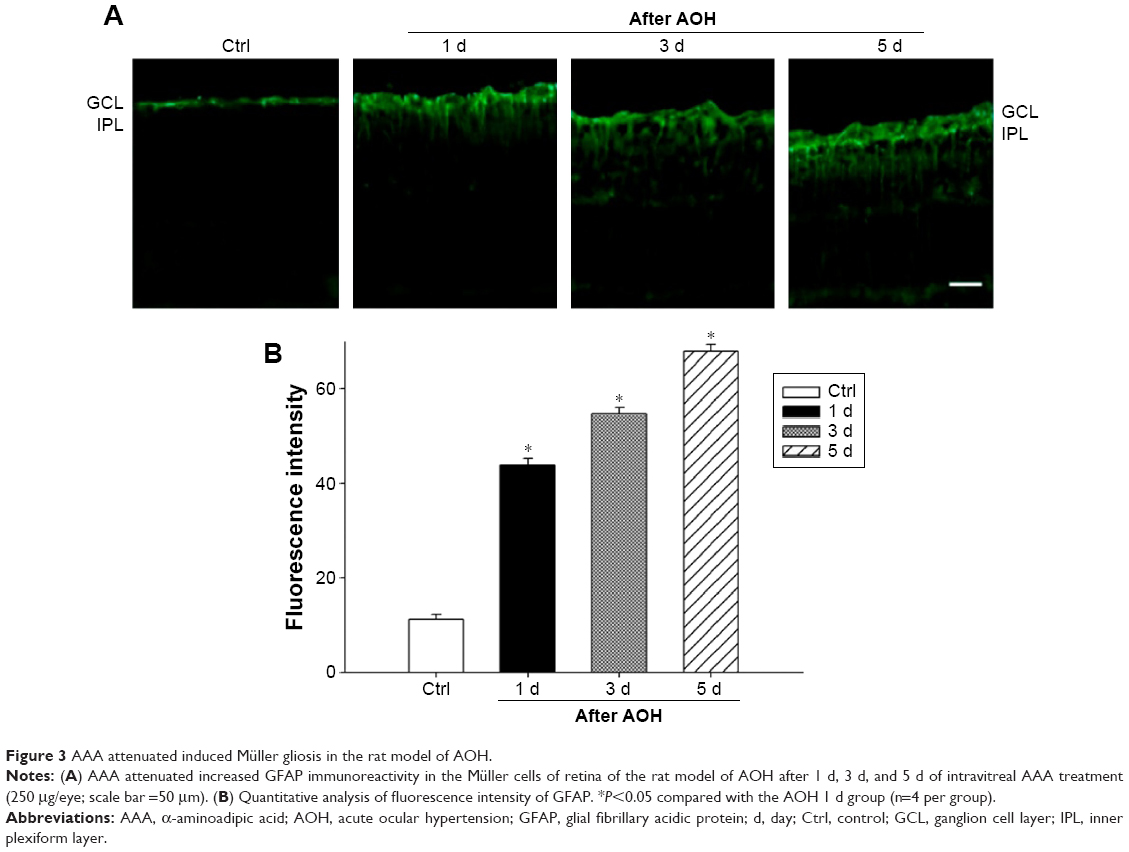 | Figure 3 AAA attenuated induced Müller gliosis in the rat model of AOH. |
AAA decreases AOH-induced TNF-α production
As mentioned earlier, AAA rescued RGCs likely through the inhibition of Müller cells gliosis in the rat model with AOH. Activation of Müller cells may induce TNF-α production, leading to neurodegeneration in glaucoma.10 To determine if AAA modulates TNF-α production, Western blot was performed for the investigation of retinal protein levels of TNF-α. Significantly upregulated protein levels of retinal TNF-α were observed in AOH versus sham operation (Ctrl) group (P<0.05; Figure 4). Importantly, AAA treatment versus PBS control significantly attenuated TNF-α levels in the retinas of the rat model of AOH (P<0.05; Figure 4).
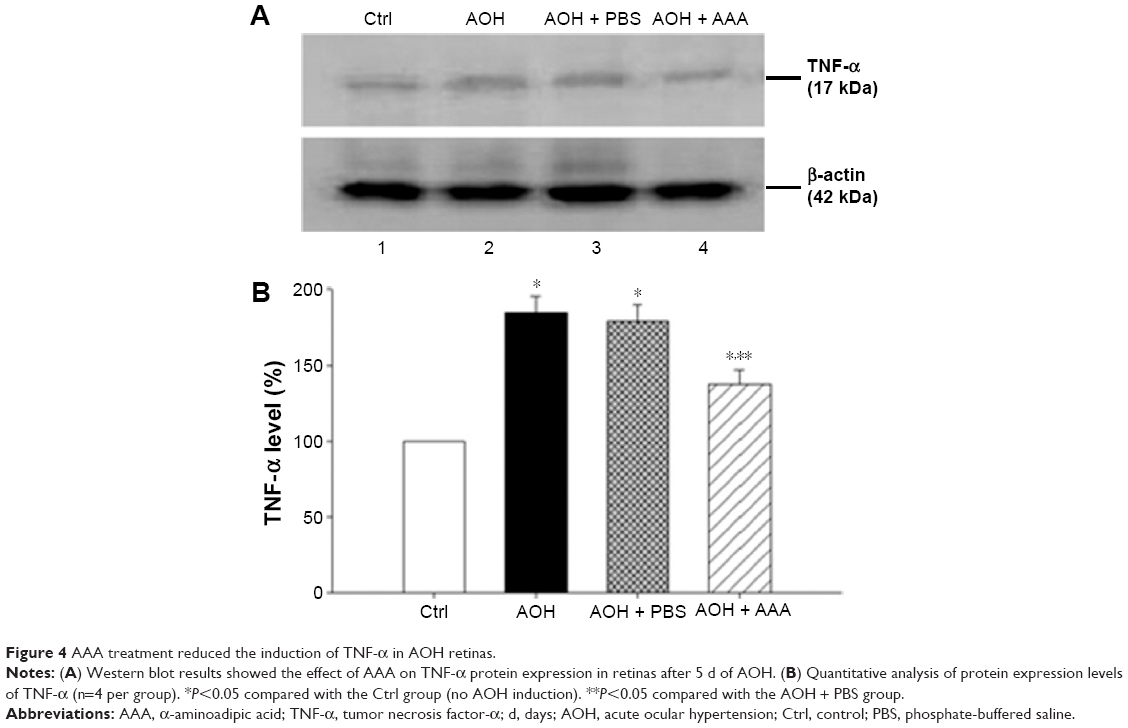 | Figure 4 AAA treatment reduced the induction of TNF-α in AOH retinas. |
Discussion
Glaucoma is a leading cause of irreversible vision loss characterized by progressive death of RGCs, and elevated IOP is a major risk factor.28 A rodent model of AOH is well established for acute angle closure glaucoma, and it has been widely used to investigate the pathogenesis of death of RGCs.29 In the current rat model of AOH, we found induced cell apoptosis and decreased number of RGCs with IOP induction versus sham operation control. Importantly, treatment of AAA versus PBS control significantly attenuated RGC apoptosis and rescued the reduced number of RGCs, demonstrating protective effects on AOH retinas (Figure 5).
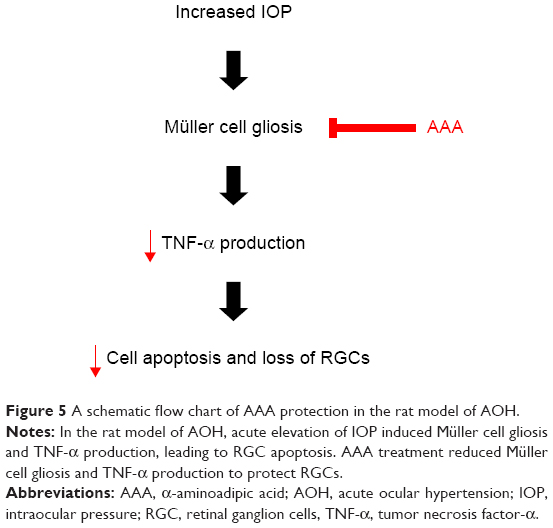 | Figure 5 A schematic flow chart of AAA protection in the rat model of AOH. |
Activated Müller cells contribute to the progression of glaucoma.30 Müller cells perform a multitude of important regulatory and supportive roles, including secretion of trophic factors, removal of metabolic waste, and neurotransmitter recycling.31,32 We found that Müller cells were activated with induced GFAP immunoreactivity in the rat model of AOH versus control retinas. GFAP, an intermediate filament protein, is considered as a marker of reactive Müller cell gliosis,27 which is not or less expressed in Müller cells in normal retinas and expressed highly at ischemic,33 light-induced retinal degeneration,34 and retinal detachment.35 Activation of Müller cells so far was demonstrated to have both protective and detrimental effects.36–38 Especially early after injury, Müller cell gliosis is believed to be neuroprotective and promotes the repair of neurons in response to injury,36 with GFAP upregulation to provide additional structural integrity to the retina at the site of injury.39 However, activated Müller cells also express TNF-α,40 monocyte chemoattractant protein,41 and nitric oxide42 to induce death of RGCs. TNF-α induction contributes to inflammation, apoptosis, and necrosis, leading to cell death. We observed that AAA treatment attenuated GFAP immunoreactivity in Müller cell processes across IPL and reduced retinal TNF-α levels in the rat model of AOH. These findings suggested that AAA promoted survival of RGCs in the rat model of AOH, likely through the inhibition of AOH-induced Müller cell gliosis and in turn downregulation of TNF-α protein production.
Conclusion
We found that AAA could effectively protect retina against loss of RGCs and apoptosis in AOH retinas through attenuating Müller cell gliosis and downregulating TNF-α production. These observations suggested that AAA might be a potential therapeutic target in the treatment of neurodegeneration in glaucoma.
Acknowledgments
This study was funded by Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (No XM201305) and Clinical-Basic Cooperation Program from Capital Medical University (Nos 14JL32 and 2015YKSJ02).
Disclosure
The authors report no conflicts of interest in this work.
References
Gugleta K, Turksever C, Polunina A, Orgul S. Effect of ageing on the retinal vascular responsiveness to flicker light in glaucoma patients and in ocular hypertension. Br J Ophthalmol. 2013;97(7):848–851. | ||
Husain S, Abdul Y, Crosson CE. Preservation of retina ganglion cell function by morphine in a chronic ocular-hypertensive rat model. Invest Ophthalmol Vis Sci. 2012;53(7):4289–4298. | ||
Ren R, Jonas JB, Tian G, et al. Cerebrospinal fluid pressure in glaucoma: a prospective study. Ophthalmology. 2010;117(2):259–266. | ||
Chung SH, Shen W, Jayawardana K, et al. Differential gene expression profiling after conditional Muller-cell ablation in a novel transgenic model. Invest Ophthalmol Vis Sci. 2013;54(3):2142–2152. | ||
Coorey NJ, Shen W, Chung SH, Zhu L, Gillies MC. The role of glia in retinal vascular disease. Clin Exp Optom. 2012;95(3):266–281. | ||
Tezel G; Fourth ARVO/Pfizer Ophthalmics Research Institute Conference Working Group. The role of glia, mitochondria, and the immune system in glaucoma. Invest Ophthalmol Vis Sci. 2009;50(3):1001–1012. | ||
Johnson EC, Morrison JC. Friend or foe? Resolving the impact of glial responses in glaucoma. J Glaucoma. 2009;18(5):341–353. | ||
Bringmann A, Wiedemann P. Muller glial cells in retinal disease. Ophthalmologica. 2012;227(1):1–19. | ||
Wang X, Ng YK, Tay SS. Factors contributing to neuronal degeneration in retinas of experimental glaucomatous rats. J Neurosci Res. 2005;82(5):674–689. | ||
Tezel G. TNF-alpha signaling in glaucomatous neurodegeneration. Prog Brain Res. 2008;173:409–421. | ||
Gupta S. Molecular steps of tumor necrosis factor receptor-mediated apoptosis. Curr Mol Med. 2001;1(3):317–324. | ||
Gesslein B, Hakansson G, Gustafsson L, Ekstrom P, Malmsjo M. Tumor necrosis factor and its receptors in the neuroretina and retinal vasculature after ischemia-reperfusion injury in the pig retina. Mol Vis. 2010;16:2317–2327. | ||
Tezel G, Li LY, Patil RV, Wax MB. TNF-alpha and TNF-alpha receptor-1 in the retina of normal and glaucomatous eyes. Invest Ophthalmol Vis Sci. 2001;42(8):1787–1794. | ||
Gustavsson C, Agardh CD, Hagert P, Agardh E. Inflammatory markers in nondiabetic and diabetic rat retinas exposed to ischemia followed by reperfusion. Retina. 2008;28(4):645–652. | ||
Nakazawa T, Nakazawa C, Matsubara A, et al. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J Neurosci. 2006;26(49):12633–12641. | ||
Cheong CU, Chang CP, Chao CM, Cheng BC, Yang CZ, Chio CC. Etanercept attenuates traumatic brain injury in rats by reducing brain TNF-alpha contents and by stimulating newly formed neurogenesis. Mediators Inflamm. 2013;2013:620837. | ||
Maddahi A, Kruse LS, Chen QW, Edvinsson L. The role of tumor necrosis factor-alpha and TNF-alpha receptors in cerebral arteries following cerebral ischemia in rat. J Neuroinflammation. 2011;8:107. | ||
Pannicke T, Stabel J, Heinemann U, Reichelt W. alpha-Aminoadipic acid blocks the Na(+)-dependent glutamate transport into acutely isolated Muller glial cells from guinea pig retina. Pflugers Arch. 1994;429(1):140–142. | ||
Ishikawa Y, Mine S. Aminoadipic acid toxic effects on retinal glial cells. Jpn J Ophthalmol. 1983;27(1):107–118. | ||
Khurgel M, Koo AC, Ivy GO. Selective ablation of astrocytes by intracerebral injections of alpha-aminoadipate. Glia. 1996;16(4):351–358. | ||
Linser PJ, Moscona AA. Induction of glutamine synthetase in embryonic neural retina: its suppression by the gliatoxic agent alpha-aminoadipic acid. Brain Res. 1981;227(1):103–119. | ||
Huck S, Grass F, Hatten ME. Gliotoxic effects of alpha-aminoadipic acid on monolayer cultures of dissociated postnatal mouse cerebellum. Neuroscience. 1984;12(3):783–791. | ||
Rich KA, Figueroa SL, Zhan Y, Blanks JC. Effects of Muller cell disruption on mouse photoreceptor cell development. Exp Eye Res. 1995;61(2):235–248. | ||
Ganesh BS, Chintala SK. Inhibition of reactive gliosis attenuates excitotoxicity-mediated death of retinal ganglion cells. PLoS One. 2011;6(3):e18305. | ||
Barnstable CJ, Drager UC. Thy-1 antigen: a ganglion cell specific marker in rodent retina. Neuroscience. 1984;11(4):847–855. | ||
Chen H, Weber AJ. Expression of glial fibrillary acidic protein and glutamine synthetase by Muller cells after optic nerve damage and intravitreal application of brain-derived neurotrophic factor. Glia. 2002; 38(2):115–125. | ||
Bringmann A, Iandiev I, Pannicke T, et al. Cellular signaling and factors involved in Muller cell gliosis: neuroprotective and detrimental effects. Prog Retin Eye Res. 2009;28(6):423–451. | ||
Gupta N, Yucel YH. Glaucoma as a neurodegenerative disease. Curr Opin Ophthalmol. 2007;18(2):110–114. | ||
Mi XS, Feng Q, Lo AC, et al. Protection of retinal ganglion cells and retinal vasculature by Lycium barbarum polysaccharides in a mouse model of acute ocular hypertension. PLoS One. 2012;7(10):e45469. | ||
Chong RS, Martin KR. Glial cell interactions and glaucoma. Curr Opin Ophthalmol. 2015;26(2):73–77. | ||
Bringmann A, Pannicke T, Grosche J, et al. Muller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25(4):397–424. | ||
Reichenbach A, Bringmann A. New functions of Muller cells. Glia. 2013;61(5):651–678. | ||
Wurm A, Iandiev I, Uhlmann S, et al. Effects of ischemia-reperfusion on physiological properties of Muller glial cells in the porcine retina. Invest Ophthalmol Vis Sci. 2011;52(6):3360–3367. | ||
Torbidoni V, Iribarne M, Suburo AM. Endothelin receptors in light-induced retinal degeneration. Exp Biol Med (Maywood). 2006;231(6):1095–1100. | ||
Ghosh F, Johansson K. Neuronal and glial alterations in complex long-term rhegmatogenous retinal detachment. Curr Eye Res. 2012;37(8):704–711. | ||
Kim JH, Kim JH, Park JA, et al. Blood-neural barrier: intercellular communication at glio-vascular interface. J Biochem Mol Biol. 2006;39(4):339–345. | ||
Bringmann A, Pannicke T, Biedermann B, et al. Role of retinal glial cells in neurotransmitter uptake and metabolism. Neurochem Int. 2009;54(3–4):143–160. | ||
Martin KR, Levkovitch-Verbin H, Valenta D, Baumrind L, Pease ME, Quigley HA. Retinal glutamate transporter changes in experimental glaucoma and after optic nerve transection in the rat. Invest Ophthalmol Vis Sci. 2002;43(7):2236–2243. | ||
Dyer MA, Cepko CL. Control of Muller glial cell proliferation and activation following retinal injury. Nat Neurosci. 2000;3(9):873–880. | ||
Lei X, Zhang J, Shen J, et al. EPO attenuates inflammatory cytokines by Muller cells in diabetic retinopathy. Front Biosci (Elite Ed). 2011;3:201–211. | ||
Nakazawa T, Hisatomi T, Nakazawa C, et al. Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc Natl Acad Sci U S A. 2007;104(7):2425–2430. | ||
Zeng K, Xu H, Chen K, et al. Effects of taurine on glutamate uptake and degradation in Muller cells under diabetic conditions via antioxidant mechanism. Mol Cell Neurosci. 2010;45(2):192–199. |
Supplementary materials
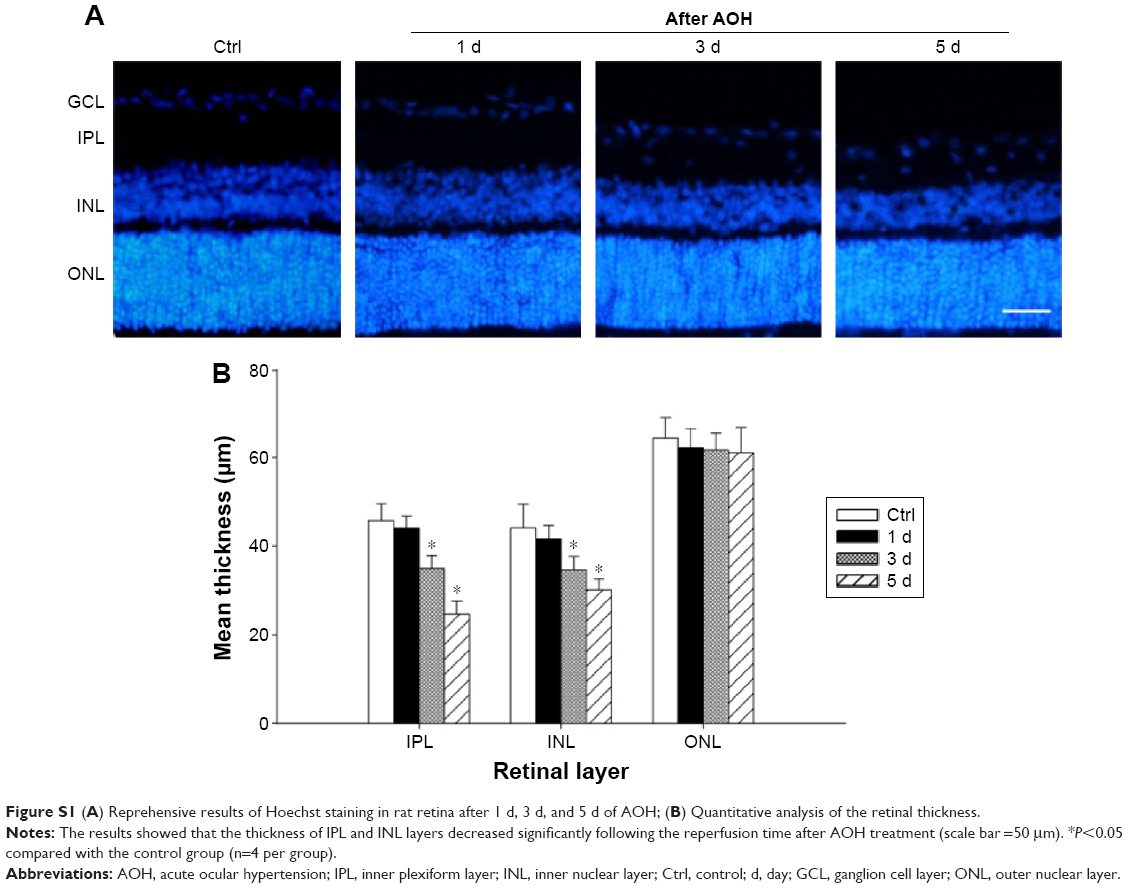 | Figure S1 (A) Reprehensive results of Hoechst staining in rat retina after 1 d, 3 d, and 5 d of AOH; (B) Quantitative analysis of the retinal thickness. |
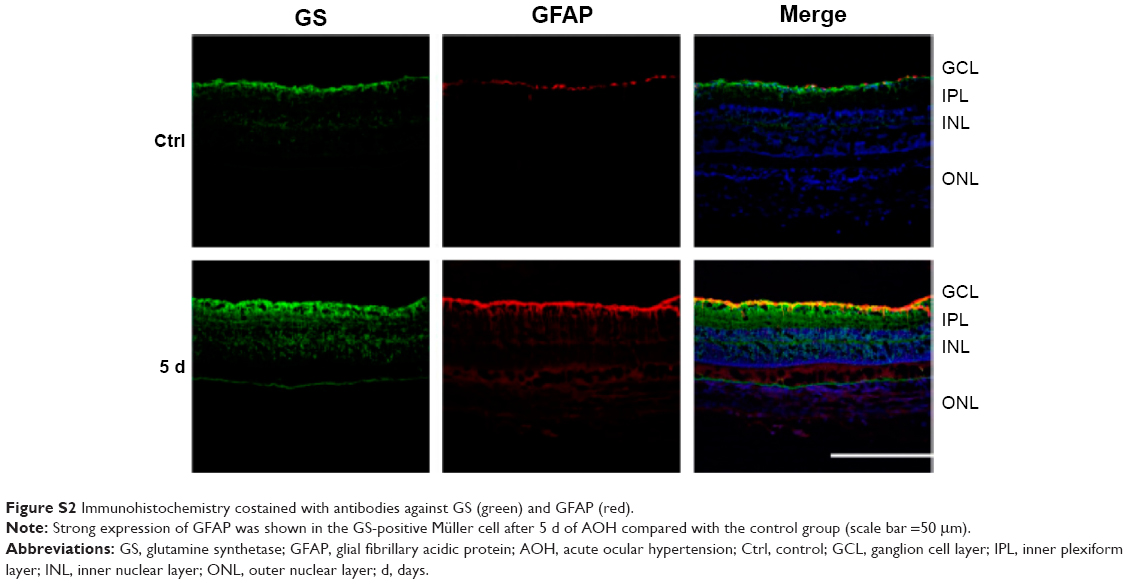 | Figure S2 Immunohistochemistry costained with antibodies against GS (green) and GFAP (red). |
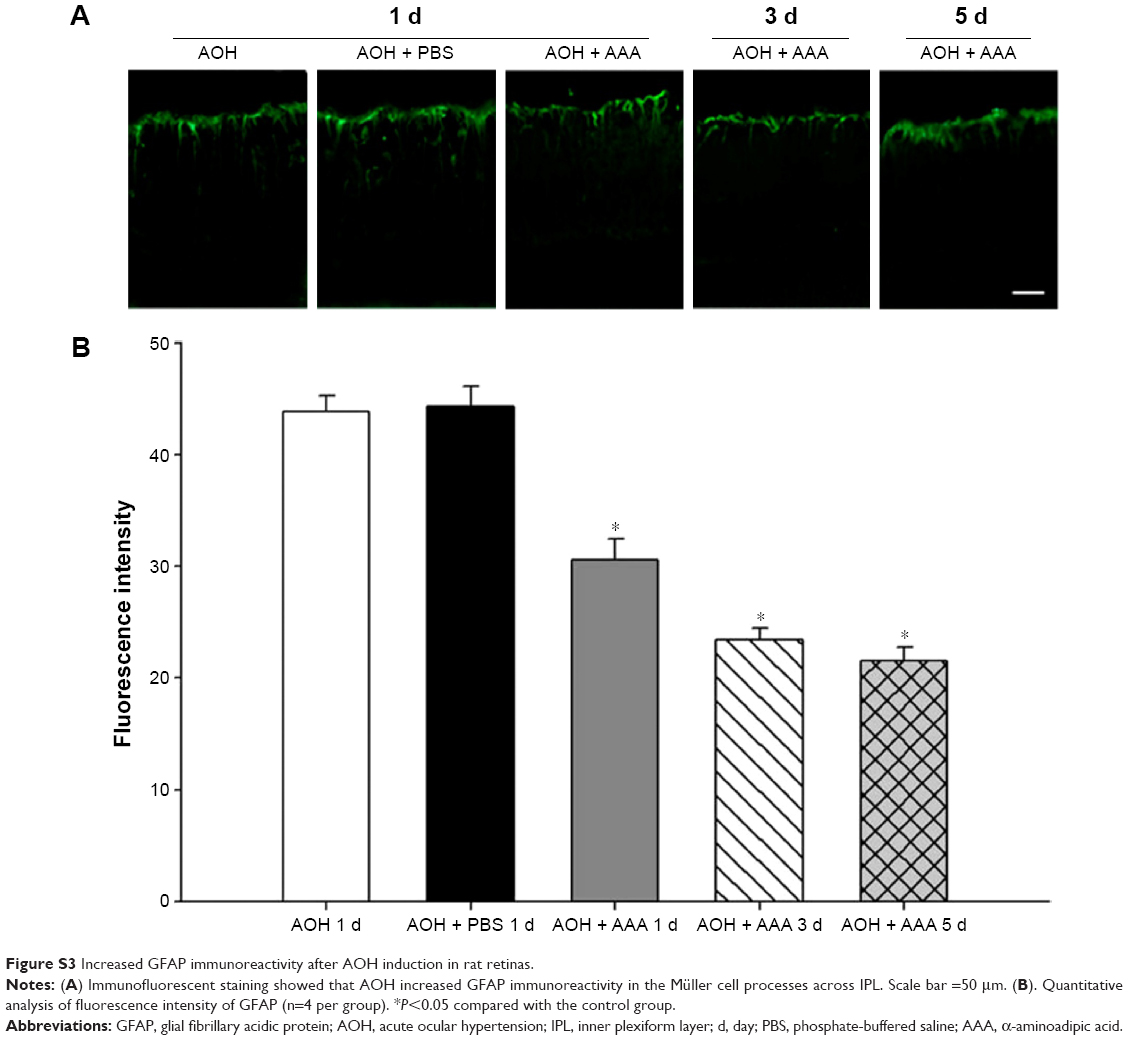 | Figure S3 Increased GFAP immunoreactivity after AOH induction in rat retinas. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.