")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Alpha-1 proteinase inhibitors for the treatment of alpha-1 antitrypsin deficiency: safety, tolerability, and patient outcomes
Authors Chotirmall S , Al-Alawi M, McEnery T , McElvaney NG
Received 17 November 2014
Accepted for publication 20 December 2014
Published 29 January 2015 Volume 2015:11 Pages 143—151
DOI https://doi.org/10.2147/TCRM.S51474
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Sanjay H Chotirmall,1 Mazen Al-Alawi,2 Thomas McEnery,2 Noel G McElvaney2
1Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore; 2Department of Respiratory Medicine, Beaumont Hospital, Dublin, Republic of Ireland
Abstract: Alpha-1 antitrypsin (AAT) deficiency remains an underrecognized genetic disease with predominantly pulmonary and hepatic manifestations. AAT is derived primarily from hepatocytes; however, macrophages and neutrophils are secondary sources. As the natural physiological inhibitor of several proteases, most importantly neutrophil elastase (NE), it plays a key role in maintaining pulmonary protease–antiprotease balance. In deficient states, unrestrained NE activity promotes damage to the lung matrix, causing structural defects and impairing host defenses. The commonest form of AAT deficiency results in a mutated Z AAT that is abnormally folded, polymerized, and aggregated in the liver. Consequently, systemic levels are lower, resulting in diminished pulmonary concentrations. Hepatic disease occurs due to liver aggregation of the protein, while lung destruction ensues from unopposed protease-mediated damage. In this review, we will discuss AAT deficiency, its clinical manifestations, and augmentation therapy. We will address the safety and tolerability profiles of AAT replacement in the context of patient outcomes and cost-effectiveness and outline future directions for work in this field.
Keywords: alpha-1, augmentation, deficiency, replacement, emphysema
Introduction
Alpha-1 antitrypsin deficiency (AATD) is an autosomal codominant condition that remains the only consistent genetic risk factor for the development of chronic obstructive pulmonary disease (COPD). Historically, the deficient state was first identified by Laurell and Eriksson1 in the 1960s, who associated disease with the occurrence of emphysema. Following this, and less than a decade later, an association with liver disease was made.2
Since then, our understanding of the condition has exponentially increased. The gene, which is located on chromosome 14q, codes for AAT and is a “serpin” family member, possessing key physiological roles in serine protease inhibition. While numerous AAT alleles have been described, nomenclature is based on gel electrophoresis migration patterns. For instance, those with higher isoelectric points are allocated letters nearer the start of the alphabet. While >100 alleles have been described, they are generally divided into four groups based on serum level and function (Table 1).3
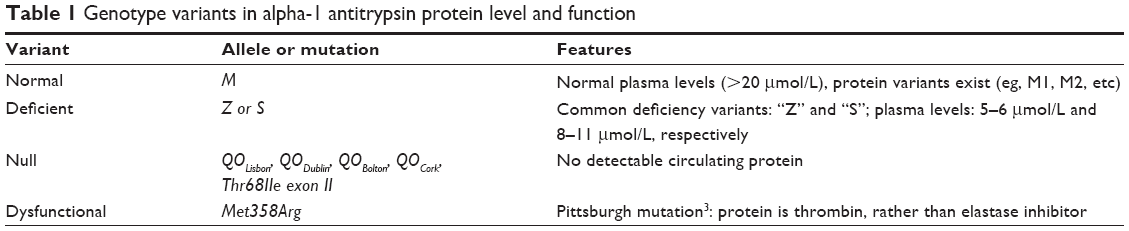 | Table 1 Genotype variants in alpha-1 antitrypsin protein level and function |
The highest incidence of AATD remains within Europe,4,5 with the first diagnostic step involving a screen for low serum concentrations of AAT. When levels are low, further analysis is performed to determine phenotype. This can involve phenotyping using isoelectric focusing or genotyping, or in some cases gene sequencing. Most mutations detected in clinical practice are deficient alleles homo- or heterozygous for the “S” or “Z” variants that correlate with the severity of the clinical manifestations observed. Null and dysfunctional states are less frequently encountered; however, increasing numbers of “null” mutations are described and named based on their first known location of detection (Table 1).
While a variety of genetic mutations can occur within the AAT gene, the most common deficient states result from single amino acid substitutions: valine for glutamate at position 264 (Glu264Val) in the case of “S” variants and lysine for glutamate at position 342 (Glu342Lys) for “Z” variants.6 Owing to a reduced propensity to polymerize, the “S” protein variant results in milder deficiency states with reduced hepatocellular accrual as compared to the “Z” allele.7–9
Hepatic polymerization of Z AAT entraps the abnormal protein within the liver, with a subsequent diminished systemic release. As a result, pulmonary protease–antiprotease imbalance occurs, and proteases such as neutrophil elastase (NE) are unopposed in their actions in the lungs, resulting in pulmonary damage and emphysema. Pulmonary immunity is further compromised by the NE-mediated effects on complement receptors,10 CXCR1 receptors,11 immunoglobulins,12 ciliary motility,13 and antiproteases such as secretory leucoprotease inhibitor, and elafin.14,15 AATD-associated liver disease is linked to the hepatic polymerization of the Z AAT protein. In the normal physiological state, cells clear abnormally folded proteins after they are identified. In AATD, however, there is a significant imbalance between the abnormal protein folding load and the cell’s ability to process this load. Hepatic endoplasmic reticulum (ER) stress results, causing a multitude of effects including the ER overload response, the unfolded protein response, and apoptosis.16
Clinical manifestations of AATD
Pulmonary disease
The major clinical features in AATD include pulmonary and hepatic diseases. The fundamental basis of AATD-associated pulmonary disease is early-onset panacinar emphysema observed in a basal distribution.17,18
Most pulmonary disease associated with AATD coupled with either a current or previous history of cigarette smoking, usually presents in the third decade of life.19–21 COPD has been demonstrated in three-quarters of Z AATD subjects with a median age of 52 years.22 While the natural history of the condition is unclear, it is predicted that a significant proportion of individuals with a current diagnosis of smoking-related emphysema remain undiagnosed with regard to AATD. The majority of individuals with this disease do have some history of smoking, which in itself has been shown to reduce survival in AATD sufferers by up to two decades.19 However, it should be noted that individuals with AATD can have significant obstructive lung disease even in the absence of a smoking history.
Symptom frequency in individuals with AATD was assessed in the population recorded within the National Heart, Lung, and Blood Institute (NHLBI) registry. Dyspnea was most common, followed by wheeze and cough. Occurrence of wheeze must be considered carefully as up to one third in the registry had documented asthma and half had significant bronchodilator-associated reversibility on spirometry.23 To further study this, Eden et al24 looked for the presence of asthma in patients with AATD. They performed an assessment of wheezing, bronchodilator responsiveness, occurrence of atopy, and the presence of an increased serum immunoglobulin E (IgE) level. The study illustrated that three or more of such markers were present in 22% of AAT-deficient patients as compared to 5% of COPD patients without AATD.24
Pulmonary function usually illustrates an obstructive pattern, while arterial blood gas sampling shows normo–hypoxia at rest, with hypercapnia as disease advances.23 Importantly, chest radiography in early disease may be normal or may show minimal change. This has led to interest in detecting disease earlier and permitting greater quantitative assessment through computed tomography (CT) scans of the thorax. Consequently, the use of CT imaging in AATD has intensified for several reasons. First, it allows the detection of early disease before any appreciable loss of lung function by spirometry. Second, it permits an evaluation of the efficacy of therapeutic interventions such as augmentation therapy, and finally, it has increased our recognition of other associated pulmonary conditions such as bronchiectasis. While initial studies did not clearly reveal this association, work that followed showed that up to 40% of AATD individuals have some degree of coexistent bronchiectasis.25–27
Hepatic disease
Up to 15% of Z AATD individuals have demonstrated significant liver disease in the first two decades of life.2,28 Hepatocyte inclusion bodies, an effect of Z AAT protein polymerization, has been described from a histological perspective.
With its incidence being greatest in children, Z AAT-associated liver disease can present as abnormal serum liver enzymes, neonatal jaundice (most common), or cirrhosis. Importantly, while infants with the SZ phenotype show no signs of early liver disease, up to 8% may show abnormal liver function in their teenage years. Adults with documented liver disease in infancy were also found to be clinically well in later life.28,29
Adult-associated Z AATD liver disease may present as chronic hepatic impairment or hepatocellular carcinoma.28,30 Up to one in ten AATD patients >50 years of age will develop cirrhosis associated with late-onset symptomatic hepatitis. The relative age of onset for liver disease is increased in heterozygotes (MZ) when compared to homozygotes (ZZ) by almost fifteen years.31
Other disease
AAT naturally inhibits proteinase 3 and therefore a link to Wegener’s granulomatosis is described.31,32 A rare necrotizing skin panniculitis is also observed in clinical practice in some individuals with AATD. The presence of intracranial or abdominal vascular aneurysms and fibromuscular dysplasia has been suggested as being associated with AATD; however, direct causality has not been established.33 Renal disease ranging from IgA nephropathy to membranoproliferative glomerulonephritis can similarly occur in AAT-deficient states, with particularly increased rates in AATD patients with liver cirrhosis.34,35
AAT augmentation therapy
The rationale for AAT augmentation originated from our understanding of disease pathophysiology. Intravenous treatment with pooled purified human AAT with the intention of correcting the deficient state augmented both serum and lung epithelial levels of AAT.36
In the late 1980s, plasma-purified AAT was safely delivered intravenously to patients with deficiency in order to achieve plasma levels exceeding a protective threshold of 11 μm.37 This target concentration was derived from levels detected in AATD SZ individuals, who if nonsmokers very rarely develop pulmonary disease.38 Increased levels of AAT coupled with increased anti-NE capacity both in serum and on the pulmonary epithelial surface were shown following intravenous AAT administration in these studies. While these early studies illustrated biochemical efficacy, there remained a need to demonstrate clinical benefit.39
Potential clinical benefit from intravenous AAT replacement was demonstrated when a Danish group of AAT-deficient ex-smokers were studied against a comparable German cohort who had received augmentation. Those with a moderately reduced FEV1 (31%–65%) illustrated a significant but modest reduction in the annual rate of FEV1 decline (21 mL/year) with augmentation.40 While comparable results were noted within the NHLBI registry, importantly, this latter data set also illustrated a mortality benefit not identified in previous work.41
Additional radiology-based work was performed by Dirksen et al42 who assessed thoracic CT changes in those receiving augmentation. While their data detected no significant difference (P=0.07), it importantly provided enough information to develop a power statistic and critically determine the number of AAT individuals needed for future clinical trials to conclusively illustrate a clinical effect using radiological end points.42 It was determined that approximately n=550 AAT-deficient individuals would be needed over a 24-month period to illustrate significant benefit from intravenous AAT augmentation on spirometry alone. Conversely, only n=130 would need to be examined to demonstrate radiological impact focused on CT indexes of emphysema. This was a significant breakthrough in the field, acknowledged by the US Food and Drugs Administration.43 Consequently, spirometry was considered a secondary efficacy end point in the study of augmentation therapy.
The EXAcerbations and Computed Tomography scan as Lung End points (EXACTLE) trial followed. This multicenter, randomized, placebo-controlled, double-blind, exploratory trial utilized CT densitometry and occurrence of exacerbations to assess the therapeutic effectiveness of weekly intravenous AAT augmentation over an approximately 2-year period. Radiological progression of emphysema was defined by a 15th-percentile change in lung density as derived from the CT voxel distribution histogram of the entire lung. Seventy-seven ZZ AATD patients were randomized to either 60 mg/kg AAT (Prolastin®; Talecris) or placebo. This study illustrated that CT was a sensitive and effective measure of emphysema progression. A number of statistical analyses were utilized in this study, with P-values ranging from 0.049 to 0.084, but all suggested at least a trend toward efficacy of augmentation therapy in reducing loss of lung density by AAT augmentation. It was felt, however, that this study suffered from being underpowered.44
Following this, a multicenter, multinational, randomized controlled trial was conducted and recently completed. This study randomized ZZ AATD patients to receive AAT augmentation therapy (Zemaira, CSL Behring) intravenously 60 mg/kg weekly or placebo over 2 years, measuring CT scan lung density at regular study intervals (RAPID). Secondary end points included spirometry, exacerbations, and quality of life measurements. This study evaluated 180 subjects over the 2-year period and was followed with an extension study in which all study participants received active drug. The results showed that intravenous augmentation slowed emphysema progression by decreasing loss of lung density as measured by CT imaging. This is the first prospective data utilizing a novel radiological end point to conclusively demonstrate clinical efficacy of augmentation therapy.39
Concerns about product purity and risks of transmissibility of infection through use of human plasma-derived AAT led to evaluation of transgenic and recombinant sources of AAT. Recombinant AAT was successfully produced in bacteria, such as Escherichia coli, and in yeast, as well as in transgenic sheep that were engineered to produce AAT in their milk.45–47
A major disadvantage to these recombinant protein forms of AAT is lack of glycosylation or abnormal glycosylation, which is associated with altered renal clearance. Consequently, when compared with plasma-purified AAT following intravenous administration, such forms are eliminated more rapidly. To overcome this particular issue, an inhaled product with an appropriate half-life on the pulmonary epithelial surface was considered and subsequently investigated. Aerosolization of plasma-purified AAT (Prolastin) and recombinant AAT were effective at delivery not solely to the alveolar surface but, critically, to the alveolar interstitium.48 Whether that is sufficient for clinical efficacy remains to be evaluated.
Currently available AAT preparations for augmentation include Aralast/Glassia (Baxter®), Prolastin-C (Grifols®), and Zemira (CSL Behring®), all of which are purified protein intravenous formulations taken from pooled human plasma.
Gene-based therapeutics has also been researched as an alternative to augment AAT levels. Thus far, the normal AAT gene has been successfully introduced into striated muscle cells in animal models with the use of an adeno-associated virus.49–51 Alternatively, plasmid and recombinant adeno-associated virus (rAAV) vectors have been assessed for effective transgene delivery and some success is observed with intrapleural delivery of AAV vectors in murine and primate model systems. Long-term pleural expression of AAT mRNA has been shown to last up to 1 year posttreatment in such data sets.52 Brantly et al53 demonstrated that intramuscular injection of recombinant AAV in a dose-escalating fashion was not associated with any vector-associated adverse events. Sustained AAT levels at 12 months have now been proven and Phase II clinical trials show promise.54,55 Additionally, evidence by Brigham et al56 illustrate that plasmid–cationic liposomal delivery of a normal AAT gene to the respiratory epithelium of deficient patients produces potentially local therapeutic AAT concentrations and that AAT gene therapy, just like AAT protein therapy, may provide anti-inflammatory benefits. While significant progress has been made in this field, the relative roles of intrapleural and targeted airway delivery, micro-RNA-expressing vectors, and genome editing are all eagerly awaited.57
Patient safety and tolerability
Distinguishing between safety and tolerability of any medicinal product is important. The safety of a medicine pertains to the medical risks conferred to the patient from its administration, which is commonly assessed in clinical trials through laboratory tests, measurement of vital signs, clinical adverse events, and other special safety tests, such as electrocardiograms, arterial blood gas levels, and radiology.58 Tolerability, however, represents the degree to which a subject can tolerate adverse effects. The investigation of safety and tolerability of any new therapy is a multidimensional challenge. It demands strict scope of evaluation coupled with an anticipated choice of variables to assess utilization of appropriate methods of data collection. Commonly used laboratory tests concerning clinical chemistry and hematology, vital signs, and clinical adverse events (diseases, signs, and symptoms) predominantly form the main body of the safety and tolerability data described for a particular drug or therapeutic intervention.
Patient outcomes from two major studies of patients following AAT augmentation therapy suggest that it appears well tolerated and is generally safe.59,60 Wencker et al60 reported the trial of n=443 augmentation therapy recipients, of whom 65 experienced a total of 124 adverse events. Most commonly encountered adverse reactions were relatively benign and self-resolving. These included chills, urticarial rashes, fatigue, nausea, and vomiting. Although specific adverse effects may be anticipated and specifically monitored for during therapy, the range of possible adverse effects is large, and new potentially unforeseeable effects remain possible. The trial data however are reassuring, given that 58,000 infusions resulted in only five severe side effects that necessitated hospital admission but importantly all had a complete recovery. Four patients suffered anaphylactic reactions and one patient developed congestive cardiac failure. Critically, no deaths or instances of hematologic viral transmission were observed.60
Stoller et al59 reported on the NHLBI experience of n=747 patients receiving weekly AAT augmentation therapy for a period of seven years. The overall rate of adverse events was low at 0.02 per patient-month, with 83% of patients reporting no events. There were a total of 720 recorded adverse events, the most common of which included headache (47%), dizziness (17%), nausea (9%), and dyspnea (9%). It is important to note however that the data highlighted a lower rate of adverse events in participants receiving augmentation therapy less frequently than weekly. The rate of total adverse events decreased from 0.03 events per patient-month at weekly infusions to 0.024 events per patient-month at 2–3 weeks (P=0.020) and 0.005 events per patient-month at monthly infusions (P<0.001). The study importantly was complicated by the recall of two α1-antiprotease batches in 1989 and 1991, which were associated with a clustering of adverse events. The events related to the flawed lots were excluded from the analysis of the NHLBI registry, and this may affect its comparability with other studies. The occurrence of serious adverse events and treatment discontinuations due to them are particularly important to register.
Meyer et al61 reported a rare IgE anaphylactic reaction following the third intravenous infusion of AAT. Specific IgE antibodies against Prolastin-HS were identified on serum analysis of the patient. Consequently, manufacturing modifications to Prolastin have subsequently led to the development of an updated α(1)-proteinase inhibitor product, designated Prolastin-C, which has demonstrated pharmacokinetic equivalence and a comparable safety profile to Prolastin without any reported life-threatening adverse events to date.62
Adverse event monitoring is particularly important with augmentation therapy as long-term treatment is envisioned and an expected proportion of treatment withdrawals or natural deaths are expected. For such situations, survival analysis methods should be considered and cumulative adverse event rates calculated in order to avoid the risk of underestimation. Alternatively any new adverse effects may be screened by later-phase controlled trials, even if such trials generally would lack power for this purpose.58
Patient outcomes and cost-effectiveness
As shown above, augmentation therapy has been clearly shown to raise anti-protease serum and epithelial lining fluid levels above putative “protective threshold” values. Evidence suggests that such an approach slows lung function declines, may reduce infection or exacerbation rates, potentially enhance survival, and is generally well tolerated (Table 2). In addition, a meta-analysis by Chapman et al63 supports the use of augmentation therapy in slowing the rate of lung function decline in patients with AATD, highlighting that patients with moderate obstructive disease as determined by spirometry are most likely to benefit. A further integrated analysis of two randomized controlled trials by Stockley et al64 demonstrated that intravenous augmentation therapy significantly reduced declines in lung density on CT imaging. Once-monthly and twice-weekly augmentation has also been studied, and most data to date support a weekly regimen.60
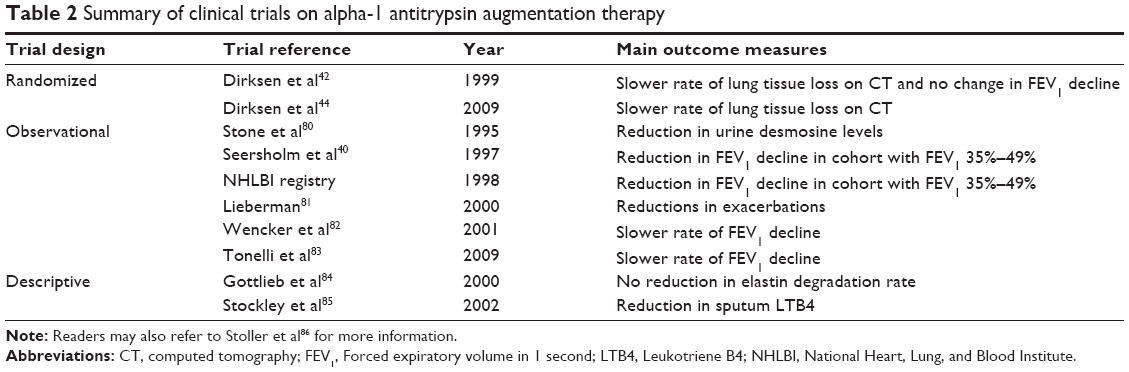 | Table 2 Summary of clinical trials on alpha-1 antitrypsin augmentation therapy |
However, while augmentation therapy has been associated with a significant potential increase in gain of life years, this benefit is adversely affected by smoking and comes at high financial cost. For instance, female ex-smokers gain an estimated mean of 7.14 years at a cost of US$248,361/year, compared to female nonsmokers who gain 9.19 years at a cost of US$160,502/year. On the other hand, male ex-smokers gain 5.93 years at US$142,250/year and nonsmokers 10.6 years at US$59,234/year.65 In one publication, Gildea et al66 suggested that the cost of augmentation therapy would need to be reduced from US$54,765 to US$4,900 to be deemed a lifelong cost-effective therapy.
In order to address concerns over such high costs of therapy, the efficacy of longer intervals between therapy has been considered. Hubbard et al67 administered A1AT at 250 mg/kg monthly and demonstrated serum A1AT levels and anti-elastase activity within epithelial lining fluid within the protective range at 28 days postinfusion. In contrast, Barker et al68 adopted a fortnightly regimen and failed to demonstrate protective serum levels. Similarly, Cammarata et al69 failed to achieve adequate serum A1AT levels with monthly replacement regimes. While the flexibility of an increased-interval dosing regimen of augmentation therapy would significantly reduce the cost burden associated with augmentation and is attractive, this is unlikely to be fruitful as several studies have demonstrated high levels of variability in the trough concentrations of serum AAT when administered at intervals exceeding once weekly, which now remains the regimen of choice.70,71 Other methods of administration such as gene therapy, subcutaneous administration, or aerosolization of AAT may yet yield benefits in this regard.
An assessment of the direct medical costs of simply having AATD was performed by surveying members within the US Alpha One Foundation Registry. The mean annual estimated costs were found to be highest for ZZ AAT patients, at approximately US$30,948/year but were even higher for the subgroup on augmentation therapy (US$40,123/year).72 This initial study only reported total costs rather than the specific components of the direct medical costs. Use of augmentation accounted for more than half of all costs for the ZZ AAT individual, with other major contributors being prescription drugs, hospitalizations, health insurance, and physician visits.73
In general, it is cited that the “estimated” annual cost for patients receiving augmentation therapy in the US is however even greater than these figures suggest and may be as high as approximately US$120,000 annually. This is thrice the yearly cost for patients with COPD alone. While use of augmentation slows disease progression, consequently positively affecting hospital admission rates and lung transplantation, it is clearly an expensive therapy, with issues of access complicated by reported shortages and reimbursement concerns.74
The benefit of AAT augmentation in ZZ-homozygote AAT patients is now becoming clearer; however, its role in Z heterozygotes (eg, MZ and SZ) remains uncertain. It is currently not recommended for MZ heterozygotes who have COPD.75 This is based on the absence of supporting evidence for efficacy in this cohort, and the fact that the target nadir serum levels for augmentation in ZZ homozygotes are below the usual AAT serum levels for MZ individuals. Recent evidence by Molloy et al76 has shown that MZ heterozygotes have significantly more airflow obstruction and COPD than MM individuals and that cigarette smoke exposure significantly augments this effect. Importantly, however, up to 10% of SZ heterozygotes have serum levels below the threshold of 11 μM and in this setting, the potential benefit of augmentation might be raised.77 The current pool of available evidence does not address this issue, and future work is required to examine potential benefits of augmentation in SZ individuals at risk of accelerated lung function decline and increased mortality.
The role for augmentation therapy in the setting of lung transplantation is a further challenge for future work.78 Limited evidence by King et al79 demonstrates an increased free elastase activity in bronchoalveolar lavage fluid of lung transplant recipients during lower respiratory tract inflammation, a phenomenon absent in the healthy state. Subsequent augmentation therapy inhibits elastase-mediated injury to the transplanted lung and hence should be considered for even the non-AATD setting. While increased oxidative stress in the AATD lung necessitates a double rather than single lung transplant in AATD, there persists a paucity of clinical evidence to inform transplant physicians about the merit of augmentation strategies for such patients during peri- or posttransplant clinical care.
Future challenges and conclusion
Determining the efficacy of currently available AAT augmentation strategies for the treatment of deficiency remains a clinical priority. This will inevitably involve assessing the present dosage strategy to see whether better clinical effects can be achieved with higher doses. Trials assessing superiority or equivalence among the various forms of commercially available pooled AAT remain to be performed. Expanding the available evidence base with focus on safety and tolerability data for the various therapeutic options is important. With expected future increases in the use of AAT augmentation, not just for AATD-related lung disease but also for its use in AAT-related panniculitis and its emerging profile as an anti-inflammatory agent, specific concerns of safety and tolerability in at-risk populations (including immune-suppressed individuals, females of child-bearing potential, the elderly, and posttransplant population) will need to be considered. Finally, cost-effectiveness of intravenous augmentation therapy needs to be improved and attention given to other strategies such as gene therapy, aerosol administration, or subcutaneous administration of AAT to see whether they have comparable or improved efficacy and cost-effectiveness to what is now currently the gold standard of once-weekly intravenous therapy with plasma-purified AAT.
Disclosure
The authors report no conflicts of interest in this work.
References
Laurell CB, Eriksson S. The electrophoretic alpha1-globulin pattern of serum in alpha 1-antitrypsin deficiency. 1963. COPD. 2013;10(suppl 1):3–8. | ||
Sharp HL, Bridges RA, Krivit W, Freier EF. Cirrhosis associated with alpha-1-antitrypsin deficiency: a previously unrecognized inherited disorder. J Lab Clin Med. 1969;73(6):934–939. | ||
Owen MC, Brennan SO, Lewis JH, Carrell RW. Mutation of antitrypsin to antithrombin. Alpha 1-antitrypsin Pittsburgh (358 Met leads to Arg), a fatal bleeding disorder. N Engl J Med. 1983;309(12):694–698. | ||
de Serres FJ. Worldwide racial and ethnic distribution of alpha 1-antitrypsin deficiency: summary of an analysis of published genetic epidemiologic surveys. Chest. 2002;122(5):1818–1829. | ||
Hutchison DC. Alpha 1-antitrypsin deficiency in Europe: geographical distribution of Pi types S and Z. Respir Med. 1998;92(3):367–377. | ||
Curiel DT, Chytil A, Courtney M, Crystal RG. Serum alpha 1-antitrypsin deficiency associated with the common S-type (Glu264----Val) mutation results from intracellular degradation of alpha 1-antitrypsin prior to secretion. J Biol Chem. 1989;264(18):10477–10486. | ||
Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature. 1992;357(6379):605–607. | ||
Janciauskiene S, Dominaitiene R, Sternby NH, Piitulainen E, Eriksson S. Detection of circulating and endothelial cell polymers of Z and wild type alpha 1-antitrypsin by a monoclonal antibody. J Biol Chem. 2002; 277(29):26540–26546. | ||
Mulgrew AT, Taggart CC, Lawless MW, et al. Z alpha 1-antitrypsin polymerizes in the lung and acts as a neutrophil chemoattractant. Chest. 2004;125(5):1952–1957. | ||
Tosi MF, Zakem H, Berger M. Neutrophil elastase cleaves C3bi on opsonized pseudomonas as well as CR1 on neutrophils to create a functionally important opsonin receptor mismatch. J Clin Invest. 1990;86(1):300–308. | ||
Hartl D, Latzin P, Hordijk P, et al. Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat Med. 2007;13(12):1423–1430. | ||
Fick RB Jr, Naegel GP, Squier SU, Wood RE, Gee JB, Reynolds HY. Proteins of the cystic fibrosis respiratory tract. Fragmented immunoglobulin G opsonic antibody causing defective opsonophagocytosis. J Clin Invest. 1984;74(1):236–248. | ||
Smallman LA, Hill SL, Stockley RA. Reduction of ciliary beat frequency in vitro by sputum from patients with bronchiectasis: a serine proteinase effect. Thorax. 1984;39(9):663–667. | ||
Guyot N, Butler MW, McNally P, et al. Elafin, an elastase-specific inhibitor, is cleaved by its cognate enzyme neutrophil elastase in sputum from individuals with cystic fibrosis. J Biol Chem. 2008;283(47):32377–32385. | ||
Taggart CC, Lowe GJ, Greene CM, et al. Cathepsin B, L, and S cleave and inactivate secretory leucoprotease inhibitor. J Biol Chem. 2001;276(36):33345–33352. | ||
Miller SD, Greene CM, McLean C, et al. Tauroursodeoxycholic acid inhibits apoptosis induced by Z alpha-1 antitrypsin via inhibition of bad. Hepatology. 2007;46(2):496–503. | ||
Eriksson S. Pulmonary emphysema and alpha 1-antitrypsin deficiency. Acta Med Scand. 1964;175:197–205. | ||
Gishen P, Saunders AJ, Tobin MJ, Hutchison DC. Alpha 1-antitrypsin deficiency: the radiological features of pulmonary emphysema in subjects of Pi type Z and Pi type SZ: a survey by the British Thoracic Association. Clin Radiol. 1982;33(4):371–377. | ||
Brantly ML, Paul LD, Miller BH, Falk RT, Wu M, Crystal RG. Clinical features and history of the destructive lung disease associated with alpha-1-antitrypsin deficiency of adults with pulmonary symptoms. Am Rev Respir Dis. 1988;138(2):327–336. | ||
Tobin MJ, Cook PJ, Hutchison DC. Alpha 1 antitrypsin deficiency: the clinical and physiological features of pulmonary emphysema in subjects homozygous for Pi type Z. A survey by the British thoracic association. Br J Dis Chest. 1983;77(1):14–27. | ||
Janus ED, Phillips NT, Carrell RW. Smoking, lung function, and alpha 1-antitrypsin deficiency. Lancet. 1985;1(8421):152–154. | ||
McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the national heart, lung and blood institute registry of alpha 1-antitrypsin deficiency. Alpha 1-antitrypsin deficiency registry study group. Chest. 1997;111(2):394–403. | ||
Seersholm N, Kok-Jensen A. Survival in relation to lung function and smoking cessation in patients with severe hereditary alpha 1-antitrypsin deficiency. Am J Respir Crit Care Med. 1995;151(2 pt 1):369–373. | ||
Eden E, Mitchell D, Mehlman B, et al. Atopy, asthma, and emphysema in patients with severe alpha-1-antitrypysin deficiency. Am J Respir Crit Care Med. 1997;156(1):68–74. | ||
Larsson C. Natural history and life expectancy in severe alpha 1-antitrypsin deficiency, Pi Z. Acta Med Scand. 1978;204(5):345–351. | ||
Guest PJ, Hansell DM. High resolution computed tomography (HRCT) in emphysema associated with alpha-1-antitrypsin deficiency. Clin Radiol. 1992;45(4):260–266. | ||
King MA, Stone JA, Diaz PT, Mueller CF, Becker WJ, Gadek JE. Alpha 1-antitrypsin deficiency: evaluation of bronchiectasis with CT. Radiology. 1996;199(1):137–141. | ||
Sveger T, Eriksson S. The liver in adolescents with alpha 1-antitrypsin deficiency. Hepatology. 1995;22(2):514–517. | ||
Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 1976;294(24):1316–1321. | ||
Eriksson S. Alpha 1-antitrypsin deficiency and liver cirrhosis in adults. An analysis of 35 Swedish autopsied cases. Acta Med Scand. 1987;221(5):461–467. | ||
Rakela J, Goldschmiedt M, Ludwig J. Late manifestation of chronic liver disease in adults with alpha-1-antitrypsin deficiency. Dig Dis Sci. 1987;32(12):1358–1362. | ||
Savige JA, Chang L, Cook L, Burdon J, Daskalakis M, Doery J. Alpha 1-antitrypsin deficiency and anti-proteinase 3 antibodies in anti-neutrophil cytoplasmic antibody (ANCA)-associated systemic vasculitis. Clin Exp Immunol. 1995;100(2):194–197. | ||
Schievink WI, Puumala MR, Meyer FB, Raffel C, Katzmann JA, Parisi JE. Giant intracranial aneurysm and fibromuscular dysplasia in an adolescent with alpha 1-antitrypsin deficiency. J Neurosurg. 1996;85(3):503–506. | ||
Davis ID, Burke B, Freese D, Sharp HL, Kim Y. The pathologic spectrum of the nephropathy associated with alpha 1-antitrypsin deficiency. Hum Pathol. 1992;23(1):57–62. | ||
Noble-Jamieson G, Thiru S, Johnston P, Friend P, Barnes ND. Glomerulonephritis with end-stage liver disease in childhood. Lancet. 1992;339(8795):706–707. | ||
Gadek JE, Klein HG, Holland PV, Crystal RG. Replacement therapy of alpha 1-antitrypsin deficiency. Reversal of protease-antiprotease imbalance within the alveolar structures of PiZ subjects. J Clin Invest. 1981;68(5):1158–1165. | ||
Wewers MD, Casolaro MA, Sellers SE, et al. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N Engl J Med. 1987;316(17):1055–1062. | ||
Crystal RG. Alpha 1-antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. J Clin Invest. 1990;85(5):1343–1352. | ||
Chapman KR, Burdon JGW, Piitulainen E, et al. IV alpha1 antitrypsin (A1AT) preserves lung density in homozygous alpha1 antitrypsin deficiency (A1ATD); a randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2013;187:A6069. | ||
Seersholm N, Wencker M, Banik N, et al. Does alpha1-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha1-antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL) alpha1-AT study group. The Eur Respir J. 1997;10(10):2260–2263. | ||
Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. The Alpha-1-Antitrypsin Deficiency Registry Study Group. Am J Respir Crit Care Med. 1998;158(1):49–59. | ||
Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med. 1999;160(5 pt 1):1468–1472. | ||
FDA. Proceedings of Meeting of Blood and Plasma Advisory Committee. Rockville, Maryland: FDA; 1998. | ||
Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J. 2009;33(6):1345–1353. | ||
Ziomek CA. Commercialization of proteins produced in the mammary gland. Theriogenology. 1998;49(1):139–144. | ||
Wright G, Carver A, Cottom D, et al. High level expression of active human alpha-1-antitrypsin in the milk of transgenic sheep. Biotechnology. 1991;9(9):830–834. | ||
Casolaro MA, Fells G, Wewers M, et al. Augmentation of lung antineutrophil elastase capacity with recombinant human alpha-1-antitrypsin. J Appl Physiol. 1987;63(5):2015–2023. | ||
Hubbard RC, McElvaney NG, Sellers SE, Healy JT, Czerski DB, Crystal RG. Recombinant DNA-produced alpha 1-antitrypsin administered by aerosol augments lower respiratory tract antineutrophil elastase defenses in individuals with alpha 1-antitrypsin deficiency.J Clin Invest. 1989;84(4):1349–1354. | ||
Kolodka TM, Finegold M, Kay MA, Woo SL. Hepatic gene therapy: efficient retroviral-mediated gene transfer into rat hepatocytes in vivo. Somat Cell Mol Genet. 1993;19(5):491–497. | ||
Song S, Morgan M, Ellis T, et al. Sustained secretion of human alpha-1-antitrypsin from murine muscle transduced with adeno-associated virus vectors. Proc Natl Acad Sci U S A. 1998;95(24):14384–14388. | ||
Chulay JD, Ye GJ, Thomas DL, et al. Preclinical evaluation of a recombinant adeno-associated virus vector expressing human alpha-1 antitrypsin made using a recombinant herpes simplex virus production method. Hum Gene Ther. 2011;22(2):155–165. | ||
Chiuchiolo MJ, Kaminsky SM, Sondhi D, et al. Intrapleural administration of an AAVrh.10 vector coding for human alpha1-antitrypsin for the treatment of alpha1-antitrypsin deficiency. Human gene therapy. Clin Dev. 2013;24(4):161–173. | ||
Brantly ML, Spencer LT, Humphries M, et al. Phase I trial of intramuscular injection of a recombinant adeno-associated virus serotype 2 alphal-antitrypsin (AAT) vector in AAT-deficient adults. Hum Gene Ther. 2006;17(12):1177–1186. | ||
Brantly ML, Chulay JD, Wang L, et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc Natl Acad Sci U S A. 2009;106(38):16363–16368. | ||
Flotte TR, Trapnell BC, Humphries M, et al. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing alpha1-antitrypsin: interim results. Hum Gene Ther. 2011;22(10):1239–1247. | ||
Brigham KL, Lane KB, Meyrick B, et al. Transfection of nasal mucosa with a normal alpha1-antitrypsin gene in alpha1-antitrypsin-deficient subjects: comparison with protein therapy. Hum Gene Ther. 2000;11(7):1023–1032. | ||
Loring HS, Flotte TR. Current status of gene therapy for alpha-1 antitrypsin deficiency. Expert Opin Biol Ther. 2014:1–8. | ||
E9 Statistical Principles for Clinical Trials. Guidance for Industry; 1998. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073137.pdf | ||
Stoller JK, Fallat R, Schluchter MD, et al. Augmentation therapy with alpha1-antitrypsin: patterns of use and adverse events. Chest. 2003;123(5):1425–1434. | ||
Wencker M, Banik N, Buhl R, Seidel R, Konietzko N. Long-term treatment of alpha1-antitrypsin deficiency-related pulmonary emphysema with human alpha1-antitrypsin. Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL)-alpha1-AT-study group. Eur Respir J. 1998;11(2):428–433. | ||
Meyer FJ, Wencker M, Teschler H, et al. Acute allergic reaction and demonstration of specific IgE antibodies against alpha-1-protease inhibitor. Eur Respir J. 1998;12(4):996–997. | ||
Stocks JM, Brantly ML, Wang-Smith L, et al. Pharmacokinetic comparability of Prolastin(R)-C to Prolastin(R) in alpha(1)-antitrypsin deficiency: a randomized study. BMC Clin Pharmacol. 2010;10:13. | ||
Chapman KR, Stockley RA, Dawkins C, Wilkes MM, Navickis RJ. Augmentation therapy for alpha1 antitrypsin deficiency: a meta-analysis. COPD. 2009;6(3):177–184. | ||
Stockley RA, Parr DG, Piitulainen E, Stolk J, Stoel BC, Dirksen A. Therapeutic efficacy of alpha-1 antitrypsin augmentation therapy on the loss of lung tissue: an integrated analysis of 2 randomised clinical trials using computed tomography densitometry. Respir Res. 2010;11:136. | ||
Sclar DA, Evans MA, Robison LM, Skaer TL. Alpha1-proteinase inhibitor (human) in the treatment of hereditary emphysema secondary to alpha1-antitrypsin deficiency: number and costs of years of life gained. Clin Drug Investig. 2012;32(5):353–360. | ||
Gildea TR, Shermock KM, Singer ME, Stoller JK. Cost-effectiveness analysis of augmentation therapy for severe alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2003;167(10):1387–1392. | ||
Hubbard RC, Sellers S, Czerski D, Stephens L, Crystal RG. Biochemical efficacy and safety of monthly augmentation therapy for alpha 1-antitrypsin deficiency. JAMA. 1988;260(9):1259–1264. | ||
Barker AF, Iwata-Morgan I, Oveson L, Roussel R. Pharmacokinetic study of alpha1-antitrypsin infusion in alpha1-antitrypsin deficiency. Chest. 1997;112(3):607–613. | ||
Cammarata SK, Stone CL, Carey JL, Eichenhorn MS. Failure to achieve adequate serum levels with monthly replacement therapy in alpha 1-antitrypsin deficiency. Chest. 1994;106(2):651–652. | ||
Zamora NP, Pla RV, Del Rio PG, Margaleff RJ, Frias FR, Ronsano JB. Intravenous human plasma-derived augmentation therapy in alpha 1-antitrypsin deficiency: from pharmacokinetic analysis to individualizing therapy. Ann Pharmacother. 2008;42(5):640–646. | ||
Soy D, de la Roza C, Lara B, Esquinas C, Torres A, Miravitlles M. Alpha-1-antitrypsin deficiency: optimal therapeutic regimen based on population pharmacokinetics. Thorax. 2006;61(12):1059–1064. | ||
Mullins CD, Huang X, Merchant S, Stoller JK; Alpha One Foundation Research Network Registry I. The direct medical costs of alpha(1)-antitrypsin deficiency. Chest. 2001;119(3):745–752. | ||
Mullins CD, Wang J, Stoller JK. Major components of the direct medical costs of alpha1-antitrypsin deficiency. Chest. 2003;124(3):826–831. | ||
Mullins CD, Blatt L, Wang J. Societal implications of the pharmacoeconomics of alpha1-antitrypsin deficiency. Expert Rev Pharmacoecon Outcomes Res. 2002;2(3):243–249. | ||
Sandhaus RA, Turino G, Stocks J, et al; Medical and Scientific Advisory Committee of the Alpha-1 Foundation. Alpha1-antitrypsin augmentation therapy for PI*MZ heterozygotes: a cautionary note. Chest. 2008;134(4):831–834. | ||
Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in alpha1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med. 2014;189(4):419–427. | ||
Turino GM, Barker AF, Brantly ML, et al. Clinical features of individuals with PI*SZ phenotype of alpha 1-antitrypsin deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Am J Respir Crit Care Med. 1996;154(6 pt 1):1718–1725. | ||
Trulock EP. Lung transplantation for alpha 1-antitrypsin deficiency emphysema. Chest. 1996;110(6 suppl):284S–294S. | ||
King MB, Campbell EJ, Gray BH, Hertz MI. The proteinase-antiproteinase balance in alpha-1-proteinase inhibitor-deficient lung transplant recipients. Am J Respir Crit Care Med. 1994;149(4 pt 1):966–971. | ||
Stone PJ, Morris TA 3rd, Franzblau C, Snider GL. Preliminary evidence that augmentation therapy diminishes degradation of cross-linked elastin in alpha-1-antitrypsin-deficient humans. Respiration. 1995;62(2):76–79. | ||
Lieberman J. Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting data. Chest. 2000;118(5):1480–1485. | ||
Wencker M, Fuhrmann B, Banik N, Konietzko N; Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen. Longitudinal follow-up of patients with alpha(1)-protease inhibitor deficiency before and during therapy with IV alpha(1)-protease inhibitor. Chest. 2001;119(3):737–744. | ||
Tonelli AR, Rouhani F, Li N, Schreck P, Brantly ML. Alpha-1-antitrypsin augmentation therapy in deficient individuals enrolled in the Alpha-1 Foundation DNA and Tissue Bank. Int J Chron Obstruct Pulmon Dis. 2009;4:443–452. | ||
Gottlieb DJ, Luisetti M, Stone PJ, et al. Short-term supplementation therapy does not affect elastin degradation in severe alpha(1)-antitrypsin deficiency. The American–Italian AATD Study Group. Am J Respir Crit Care Med. 2000;162(6):2069–2072. | ||
Stockley RA, Bayley DL, Unsal I, Dowson LJ. The effect of augmentation therapy on bronchial inflammation in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2002;165(11):1494–1498. | ||
Stoller JK, Aboussouan LS. Alpha1-antitrypsin deficiency. Lancet. 2005;365(9478):2225–2236. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.