Back to Journals » OncoTargets and Therapy » Volume 13
Alpha-1 Antitrypsin Induces Epithelial-to-Mesenchymal Transition, Endothelial-to-Mesenchymal Transition, and Drug Resistance in Lung Cancer Cells
Authors Wu D, Liu T ![]() , Deng S
, Deng S ![]() , Han R, Zhang T, Li J, Xu Y
, Han R, Zhang T, Li J, Xu Y ![]()
Received 16 December 2019
Accepted for publication 7 April 2020
Published 4 May 2020 Volume 2020:13 Pages 3751—3763
DOI https://doi.org/10.2147/OTT.S242579
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Dong-ming Wu,* Teng Liu,* Shi-hua Deng, Rong Han, Ting Zhang, Jing Li, Ying Xu
Clinical Laboratory, The First Affiliated Hospital of Chengdu Medical College, Chengdu, Sichuan 610041, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ying Xu Tel +86 02883016723
Email [email protected]
Purpose: Alpha-1 antitrypsin (A1AT) is a secreted protein that plays an important role in various diseases. However, the role of A1AT in non-small cell lung cancer is obscure.
Materials and Methods: A1AT expression in non-small cell lung cancer was analyzed using quantitative reverse transcription PCR, Western blotting (WB), immunohistochemistry (IHC), and ELISA. WB and IF were used to analyze markers of epithelial-to-mesenchymal transition (EMT), EndoMT, and cancer stem cell (CSC). Transwell and cell wound healing assays were used to analyze migration and invasion abilities. Colony formation and CCK-8 assays were used to analyze cell proliferation following cisplatin treatment.
Results: A1AT expression was higher in lung cancer samples than in normal tissues and the increased expression was correlated with poor overall survival of patients. In vitro experiments showed that A1AT overexpressed by plasmid transfection significantly promoted migration, invasion, EMT, EndoMT, stemness, and colony formation in lung cancer cell lines, as opposed to A1AT downregulation by siRNA transfection, which significantly inhibited all these variables.
Conclusion: A1AT is a novel therapeutic target and might be associated with tumor metastasis in lung carcinoma.
Keywords: epithelial-to-mesenchymal transition, endothelial-to-mesenchymal transition, alpha-1 antitrypsin, non-small cell lung cancer, cisplatin resistance
Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide and is associated with low patient survival rates.1,2 The vast majority of lung cancers constitute malignant epithelial tumors. Based on the size and appearance of cells, lung cancer is mainly divided into small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). NSCLC accounts for approximately 85% of lung cancer cases. Surgery, radiotherapy, and chemotherapy are common methods for treating lung cancer apart from targeted and immunotherapy, branching, and palliative care.3–5 The poor prognosis of patients with NSCLC is mainly due to the directed spreading, metastasis, recurrence, chemoresistance and other traits of the tumor cells. Multidrug resistance is a major concern that limits the success of cancer chemotherapy. Kesharwani et al made great progress in the treatment of cancer using multi-functional polymer micelles.6,7 Metastatic lung cancer cells are usually resistant to radiation and chemotherapy, which greatly affects patient prognosis.8 Therefore, identification of markers of metastasis in lung cancer is particularly important for development of effective treatments.9
Alpha-1-antitrypsin (A1AT) belongs to the serpin superfamily of proteins and is produced and secreted by cells of endodermal epithelial origin, primarily hepatocytes, and immune cells. It plays important roles in many diseases such as liver disease, emphysema, polyangiitis, and lung diseases10–15 as an anti-protease, anti-inflammatory, and anti-apoptotic agent.16 Recent studies indicate that A1AT is a key factor in epithelial-to-mesenchymal transition (EMT) in lung cancer.17 However, the role of A1AT in chemotherapy resistance is thus far unknown. Therefore, this study aimed to investigate the functional role and clinical relevance of A1AT in human lung cancer.
EMT is one of the main processes of cancer cell metastasis. In normal tissue physiology, EMT is associated with normal tissue development and organogenesis as well as tissue remodeling and wound healing.18 EMT causes polarized, immotile epithelial cells to acquire apolar, highly migratory fibroblast-like features. The role of EMT in tumor metastasis is based on the observation that acquisition of mesenchymal markers, such as N-cadherin or vimentin19 by epithelial carcinoma cells is associated with increased metastatic potential and loss of epithelial cell adhesion molecules, such as E-cadherin.20 Some evidence suggests that EMT is associated with the acquisition of stemness in cancers.21,22 Stem-cell-like cancer cells or cancer stem cells (CSCs) possess the defining characteristics of normal stem cells and have an enhanced ability to initiate tumors upon transplantation.23 CSCs are cells within a tumor that possess the capability to self-renew and differentiate into heterogeneous lineages of cancer cells that comprise the whole tumor.24
Like EMT, endothelial cells can acquire stem-cell-like properties and differentiate into many other cells in EndoMT.25 The endothelial cells lose their endothelial phenotype due to reduced expression of specific endothelial markers like VE-cadherin and gain of expression of mesenchymal markers like FSP-1 and alpha smooth muscle actin (α-SMA) during EndoMT.26 The loosening of the link between endothelial cells leads to easier passage of tumor cells through the blood vessels to the distal end in EndoMT.
Here, we show that A1AT deregulation is an independent prognostic indicator in lung carcinoma. Our findings also indicate that A1AT plays an important role in EMT and EndoMT in lung cancer. Moreover, A1AT silencing significantly enhances chemotherapy resistance. Therefore, we propose A1AT as a novel therapeutic target in lung cancer and that it might be associated with tumor metastasis in lung carcinoma.
Materials and Methods
Materials
The human microvascular endothelial cells HMVEC, NSCLC cell lines A549 (adenocarcinoma), H1650 (adenocarcinoma), SPCA-1 (adenocarcinoma), and the normal lung bronchial epithelial cell line BEAS-2B cell lines were purchased from Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and were cultured according to the manufacturer’s instructions. The antibodies used in this study were against β-actin (Cat. No. 60008-1-Ig, Proteintech) for WB; A1AT (Cat. No. 16382-1-AP, Proteintech) for WB and tissue immunohistochemistry (IHC); E-cadherin (Cat. No. 5296, Cell Signaling), N-cadherin (Cat. No. 22018-1-AP, Proteintech), FSP-1 (Cat. No. 201227, Zen BioScience), occludin (Cat. No. 13409-1-AP, Proteintech), ZO1 (Cat. No. 8193,Cell Signaling), VE-cadherin (Cat. No. 2500, Cell Signaling),CD133 (Cat. No. 18470-1-AP, Proteintech), CD44 (Cat. No. 15675-1-AP, Proteintech), and ALCAM (Cat. No. 21972-1-AP, Proteintech) for WB and immunofluorescence. Peroxidase AffiniPure goat anti-mouse IgG (Cat. No. 511103), and peroxidase AffiniPure goat anti-rabbit IgG (Cat. No. 511203) was purchased from Zen BioScience. Cy3-conjugated AffiniPure goat anti-mouse IgG (Cat. No. SA00009-1), and cy3-conjugated AffiniPure goat anti-rabbit IgG (Cat. No. SA00009-2) was purchased from Proteintech. Recombinant human alpha-1-antitrypsin (rhA1AT) (Cat. No. Ag9516) was purchased from Proteintech. The NSCLC tissue microarray (Cat. No. HLugS180Su01) was purchased from the Shanghai Outdo Biotech Company. Lung cancer tissue data were recorded in Excel named HLugS180Su01.
Cell Culture
All cells were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 10 mM L-glutamine, and 5 mgmL penicillin/streptomycin at 37 °C and 5% CO2. All media and supplements were purchased from Invitrogen.
A1AT Silencing and Overexpression
A549, H1650, and BEAS-2B cells were transfected with A1AT small interfering RNA (siRNA) (Si#01: CCCACGATATCATCACCAA, Si#02: GCCTGAAGCTAGTGGATAA, Si#03: CCAAGAAACAGATCAACGA, RIBOBIO, Guangzhou, China) using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). Opti-MEM (Gibco, Grand Island, NY, USA) transfection medium was replaced with complete culture medium 5 h after transfection. All experiments were performed 48 h after transfection. The A1AT sequence was constructed into the pcDNA3.1 vector. Ectopic expression of A1AT was achieved through pcDNA3.1-A1AT transfection using lipofectamine-3000, with an empty pCDNA3.1 vector served as a negative control. The expression levels of A1AT were measured by real-time quantitative PCR.
Total RNA Extraction and Quantitative Reverse Transcription PCR
Total RNA was extracted from small intestinal tissues using a total RNA extraction kit (Solarbio, Beijing, China), according to the manufacturer’s instructions. RNA concentrations were determined using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). One microgram of total RNA was then reverse-transcribed using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA) to synthesize cDNA. Quantitative reverse transcription PCR (qRT-PCR) was performed using a CFX96 Real-time System (Bio-Rad) with SYBR Green Supermix (Bio-Rad). Primer sense and antisense sequences of A1AT: FORWARD: AGAGCGTCCTGGGTCAACT; REVERSE: GCTTCAGTCCCTTTCTCGTC. β-actin was used as an internal control for quantification. The 2-ΔΔCT method was used to calculate relative expression levels.
Immunofluorescence
Cultured cells were fixed with 4% paraformaldehyde, washed twice with phosphate-buffered saline (PBS), and then blocked with PBS containing 10% normal goat serum. Cells were then stained with an anti-E-cadherin, anti-vimentin, or anti-FSP-1 polyclonal antibody solution for 30 min at 37 °C, washed twice with PBS, stained with a Cy3-conjugated secondary antibody for 30 min at 37 °C, and washed twice with PBS. All immunofluorescence images were captured using a DM4000 microscope (LEICA, Wetzlar, Germany) equipped with either a 20× or a 40× objective lens (LEICA) and a DFC450 C camera (LEICA). Images were processed using the LAS V4.5 controller software (LEICA).
Transwell Assays
Cells were cultured in 10-cm plates, and fresh medium was added 18 h prior to performing each assay. Cells were trypsinized, washed twice, and then resuspended in serum-free medium. The final cell density was determined using a hemocytometer. The lower wells of the transwell chamber, which had an 8 µL membrane, were loaded with RPMI-1640 containing 10% serum. A 200-μL volume of the cell suspension containing 10,000 cells was added to each upper well. The loaded chambers were incubated for 24 h at 37 °C, at which time the chambers were removed from the incubator and disassembled. Cells on the upper surface of the membrane were removed by scraping so that only cells that had migrated through the membrane remained. The membrane was then fixed with methanol, stained with 0.1% crystal violet, and air dried. Cell counts were obtained by observing the membranes under a DM4000 microscope (LEICA) and counting the cells in each field. The data are presented as an average of counts from five fields of triplicate wells for each test condition.
Co-Cultures
Cells were trypsinized, washed twice, and then resuspended in RPMI-1640 containing 10% serum. The final cell density was determined using a hemocytometer. The whole wells of the transwell chamber, which had a 3 ul membrane so that the cells in the upper wells could not migrate into the lower wells, and the apparatus was loaded with RPMI-1640 containing 10% serum. A 200-μL volume of the cell suspension containing 10,000 A549 cells that were transfected with pcDNA3.0 vector or pcDNA3.0-A1AT were added to each upper well. HMVEC cultured in the lower wells. In this way, cells were co-cultured for 24 h.
Western Blotting
For WB analysis, all protein samples were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 12% gels and then transferred to nitrocellulose membranes, which were then blocked for 1 h at 25 °C in Tris-buffered saline (TBS) containing 0.1% Tween 20 and 5% fat-free milk. Membranes were incubated with the primary antibody solutions for 18 h at 4 °C and with secondary antibody solutions at room temperature for 1 h. The secondary antibodies were conjugated with horseradish peroxidase (HRP)-conjugates, and immunoreactive signals were detected by enhanced chemiluminescence (ECL, SuperSignal; Pierce, Rockford, IL, USA) or ECL Plus (Amersham Pharmacia Biotech, Buckinghamshire, United Kingdom) according to the manufacturer’ s instructions.
Cell Viability Assays
For colony formation assay, cells (500 cells/well) were plated in 6-well plates (Corning, Corning, NY, USA) after being treated with different concentrations of cisplatin (0, 1.25, 2.5, and 5 μgmL). Cells were allowed to proliferate in the culture medium for ten days (medium was replaced every three days). Colonies were washed with PBS, fixed with methanol, and stained with crystal violet. The cell counting kit 8 (CCK-8, Beyotime, Shanghai, China) assay was performed according to the manufacturer’ s instructions.
ELISA
Firstly, serum was collected according to the CT results of a patient before treatment, then screened according to the diagnosis made by the pathologist. Sixty-three serum samples were collected from lung cancer patients from March 2017 to September 2018. All patients signed informed consent forms. All patients were newly diagnosed and untreated. There were 42 cases of carcinoma in situ and 21 cases of metastatic cancer. Twenty serum samples were also collected from healthy people. Finally, the A1AT ELISA kit (BSWS, China) was used to analyze the A1AT content in serum samples according to the manufacturer’s instructions. All patient data were recorded.
Immunohistochemistry
Tissue arrays were dewaxed, and antigens were retrieved using high pressure. Endogenous peroxidases were blocked with 3% hydrogen peroxide for 10 min. After adding normal goat serum for 30 min, tissues were incubated with the primary antibody at 4 °C overnight, washed with phosphate-buffered saline (PBS), and then incubated with a biotin-conjugated secondary antibody (ZSGB-BIO, Beijing, China) for 30 min at 37 °C. After washing, the sections were incubated with horseradish peroxidase (HRP) complex for 30 min at 37 °C and visualized using diaminobenzidine (DAB). All immunohistochemical images were obtained using an Olympus BX51 microscope equipped with a 20×, 40×, or 100× objective lens (Olympus) and a DP 50 camera (Olympus). Images were processed using the DPC controller software (Olympus).
A1AT expression was assessed by multiplying scores representing the percentage and intensity of staining. Staining intensity was graded as 0 (no staining), 1 (weak staining = light yellow), 2 (moderate staining = yellow brown), and 3 (strong staining = brown). The extent (0% –100%) of reactivity was scored as follows: 0 (<5% positive cells), 1 (5%–25% positive cells), 2 (25%–50% positive cells), 3 (51% –75% positive cells), and 4 (>75% positive cells). Scores of 0–2 were classified as low expression, whereas all other scores were classified as high expression.
Two pathologists without knowledge of the clinicopathological variables independently scored the staining on each slide. Staining assessment and the allocation of tumors by the two pathologists were similar. Cases with discrepancies were simultaneously reviewed by the original two pathologists and a senior pathologist until a consensus was reached.
Statistical Analysis
Each experiment was independently performed at least three times. All experiments were generated using a paired t-test or one-way ANOVA analyses with GraphPad Prism 5 software (GraphPad, San Diego, CA, USA). Statistical significance was defined as P < 0.05.
Results
A1AT Is Overexpressed and Correlated with Poor Prognosis in Lung Cancer
To investigate the expression pattern of A1AT in NSCLC, we first compared the level of the A1AT protein in the normal human lung cell line, BEAS-2B, with that in NSCLC cell lines (A549,SPCA-1,H1650). The A1AT protein level was significantly higher in NSCLC cell lines (Figure 1A). We further examined A1AT levels in a human NSCLC tissue microarray,which contains tumors and their overall survival in 90 patients, by immunohistochemistry (IHC). We found that the expression of A1AT was increased in tumor tissue compared to that in the adjacent tissue (Figure 1B). More importantly, patients with low A1AT expression had longer overall survival than patients with high A1AT expression (Figure 1C). Considering that A1AT is a secretory protein, we collected serum samples from 20 healthy people and serum samples from 40 patients with lung cancer. We also detected A1AT levels by ELISA. We found that serum A1AT levels were significantly higher in patients with lung cancer (Figure 1D, P<0.001). Taken together, these results demonstrated that A1AT was upregulated in NSCLC and was related to overall survival.
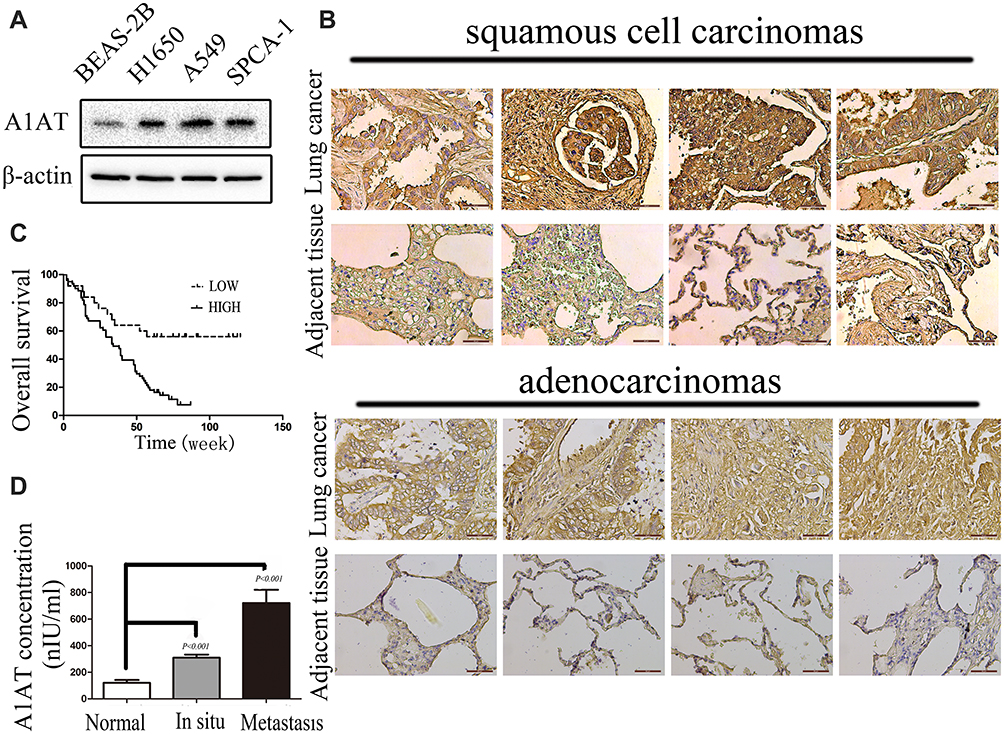 |
Figure 1 A1AT is overexpressed and correlated with poor prognosis in lung cancer. (A) A1AT is overexpressed in NSCLCs by Western blot. (B) A1AT levels in a human NSCLC tissue microarray by IHC. (C) A1AT is correlated with poor prognosis in lung cancer. Kaplan–Meier analysis of overall survival of 90 non-small cell lung cancer patients. Each subgroup was divided into low- (below or equal to the median value) and high-A1AT expression groups (above the median value); Log rank test, P < 0.001. (D) A1AT is detected by ELISA. A1AT in 63 serum samples of lung cancer patients (42 cases of carcinoma in situ and 21 cases of metastatic cancer) and 20 serum samples of healthy person are detected by ELISA kit (P < 0.001). |
A1AT Silencing Inhibits NSCLC Metastasis in vitro
EMT is a key step in the metastatic initiation of cancer cells. Because tumor metastasis is detrimental to the survival of patients with lung cancer, we investigated the effects of A1AT expression on lung cancer cell migration. A1AT was silenced by RNA interference, and the knockdown was confirmed by Western blot (WB) and quantitative PCR (qPCR) analysis for mRNA and protein expression, respectively, (Figure 2A, B, P< 0.001). Wound-healing, transwell, and Matrigel invasion assays revealed that A1AT knockdown significantly inhibited the migration of A549 adenocarcinoma cells (Figure 2C–G, P<0.01). Consistently, immunofluorescence and Western blot analysis showed a marked upregulation of the epithelial marker E-cadherin and a concomitant downregulation of the mesenchymal markers vimentin and N-cadherin in response to A1AT silencing (Figure 2H and I). Similar results were obtained for H1650 adenocarcinoma cells (Supplementary Figure 1). In addition, the function of A1AT in EMT was investigated in the normal lung epithelial cell line BEAS-2B. We found that A1AT knockdown inhibited the expression of EMT markers (Supplementary Figure 2).
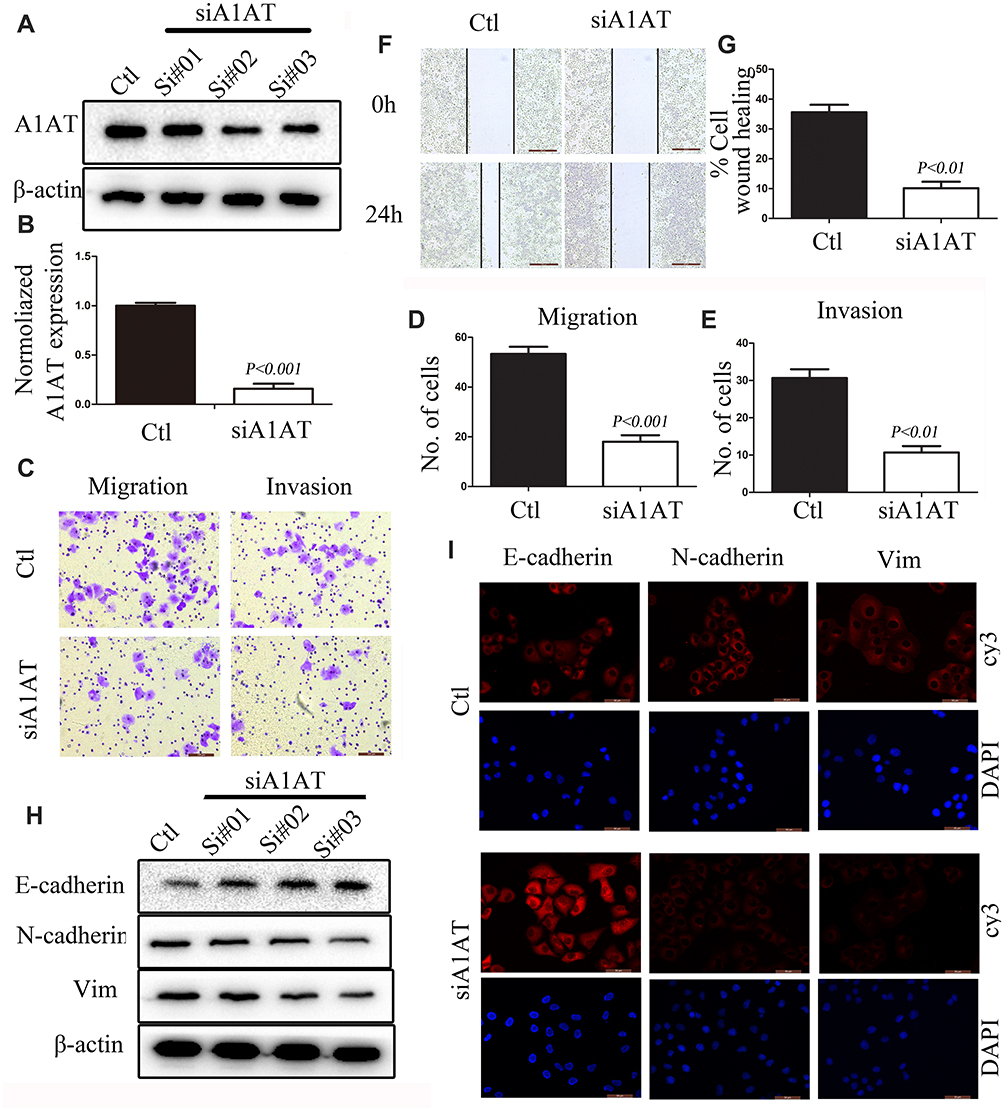 |
Figure 2 A1AT silencing inhibits NSCLC metastasis in vitro. (A, B) A1AT expression in A549 cells transfected with A1AT short interfering RNA (siRNA) or control siRNA (Ctl) by Western blot (A) and quantitative PCR (B). Si#01, Si#02, Si#03 indicate the three siRNAs used for the RNA interference. (C–E) Cell migration was monitored in transwell assays and matrigel transwell assays in A1AT-silenced and control A549 cells (scale bar, 100 μm). Representative images (C) and quantitation (D, E) are shown. (F, G) Analysis of the effects of A1AT knockdown on cell migration in wound-healing assays (scale bar, 500 μm). Representative images (F) and quantitation (G) are shown. (H, I) Analysis of the expression of E-cadherin (epithelial marker) and vimentin and N-cadherin (mesenchymal markers) in A1AT-silenced and control cells by Western blotting (H) and immunofluorescence staining (I); scale bar, 50 μm. |
A1AT Overexpression Enhances Lung Cancer Cell Metastasis in vitro
To further investigate the role of A1AT in lung cancer cells, A1AT was overexpressed in the aforementioned cell lines. First, we confirmed the overexpression of A1AT by WB and qPCR analysis (Figure 3A and B, P<0.001). In these conditions, our results were contradictory to those observed upon A1AT knockdown: overexpression of A1AT was associated with increased cell migration (Figure 3C–G, P<0.01), downregulation of the epithelial marker E-cadherin, and upregulation of the mesenchymal markers vimentin and N-cadherin (Figure 3H and I). Similar results were obtained in H1650 and BEAS-2B cells (Supplementary Figures 3 and 4, respectively,).
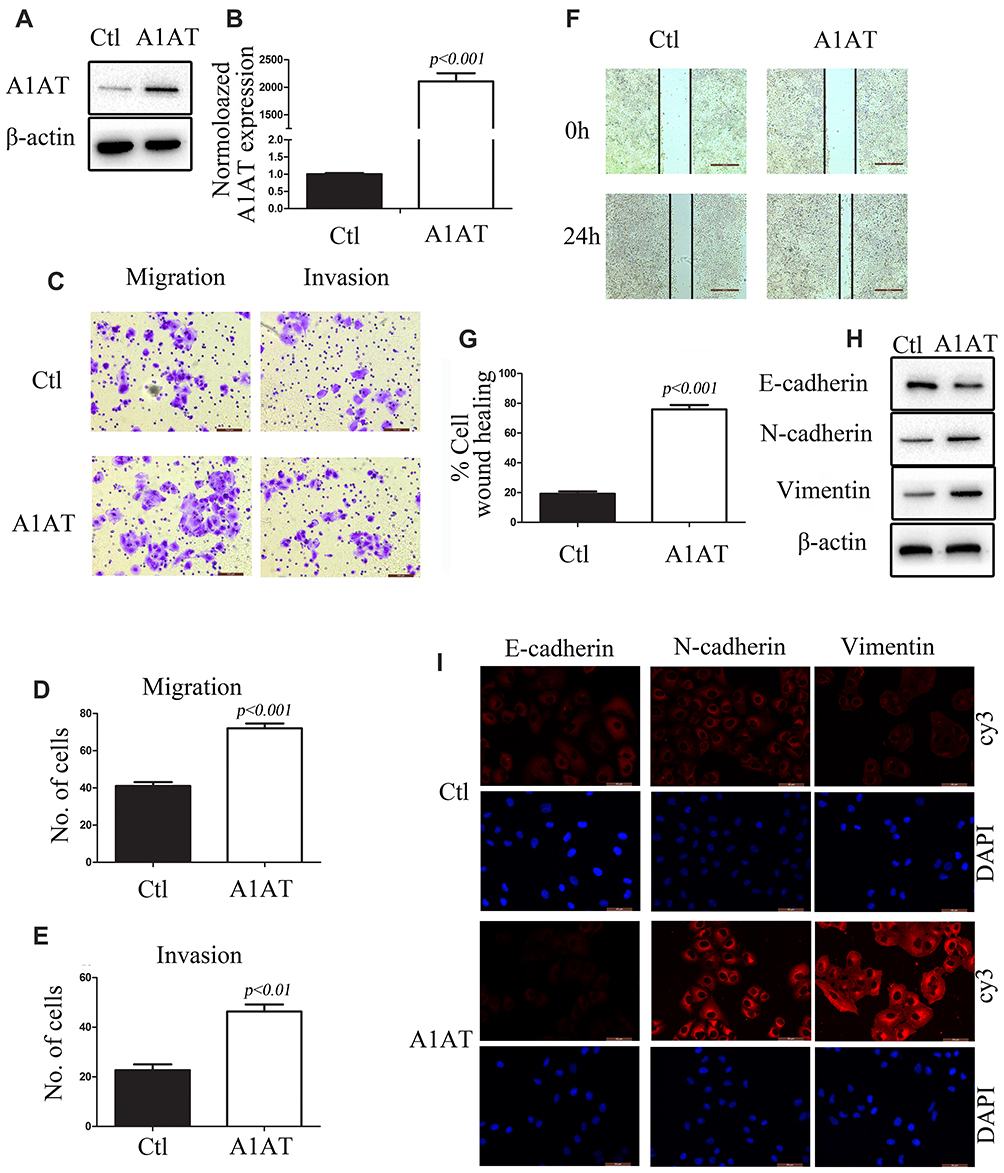 |
Figure 3 A1AT overexpression enhances lung cancer cell metastasis in vitro. (A, B) A1AT expression in A549 cells transfected with a vector overexpressing A1AT or a control vector (Ctl) by Western blot analysis (A) and quantitative PCR (B). (C–E) Cell migration was monitored in transwell assays and matrigel transwell assays in A1AT-overexpressing and control A549 cells (scale bar, 100 μm). Representative images (C) and quantitation (D, E) are shown. (F, G) Analysis of the effects of A1AT overexpression on cell migration in wound-healing assays (scale bar, 500 μm). Representative images (F) and quantitation (G) are shown. (H, I) Analysis of the expression of E-cadherin (epithelial marker) and vimentin and N-cadherin (mesenchymal markers) in A1AT-overexpressing and control cells by Western blotting (H) and immunofluorescence staining (I); scale bar, 50 μm. |
A1AT Secreted by A549 Promotes Endothelial Cell EndoMT
In our experiments, we found that metastatic carcinoma had higher A1AT expression than carcinoma in situ (Figure 1D, P<0.001), which suggests that there may be other reasons for the distant metastasis of the tumor. Considering that A1AT is a secreted protein that can affect EMT in tumor cells, will it also affect EndoMT in endothelial cells? Therefore, we set up a co-culture model of A549 and HMVEC (Figure 4A). After 24 h of cultivation, we first detected the level of A1AT in the culture medium by ELISA. The level of A1AT in the A1AT overexpression group was significantly higher than that in the control group (Figure 4B, P<0.001). We then detected VE-cadherin and FSP-1 of HMVEC by WB and IF. We found that the endothelial marker,VE-cadherin, decreased and the mesenchymal marker, FSP-1,increased (Figure 4C and D). This indicates that endothelial cells have EndoMT under the action of A1AT. Finally, we detected the expression of tight junction proteins ZO1 and occludin in HMVEC. The two proteins were all reduced under the role of A1AT. (Figure 4E and F). To ensure that the EndoMT of HMVEC is directly driven by A1AT, recombinant human alpha-1-antitrypsin (rhA1AT) was used. Cells were treated with 1000 nIUmL rhA1AT or PBS with the same volume (Ctl) for 24 h. We then detected endothelial marker VE-cadherin and the mesenchymal marker FSP-1 of HMVEC by WB and IF. We found that VE-cadherin decreased and FSP-1 increased (Supplementary Figure 5A and B). This indicates that endothelial cells have EndoMT under the action of rhA1AT. Finally, we detected the expression of tight junction proteins ZO1 and occludin in HMVEC. The two proteins were all reduced under the role of rhA1AT (Supplementary Figure 5C and D). These results are consistent with the results of the co-culture. These results demonstrated that A1AT promoted endothelial cell EndoMT and reduced tight junctions between endothelial cells.
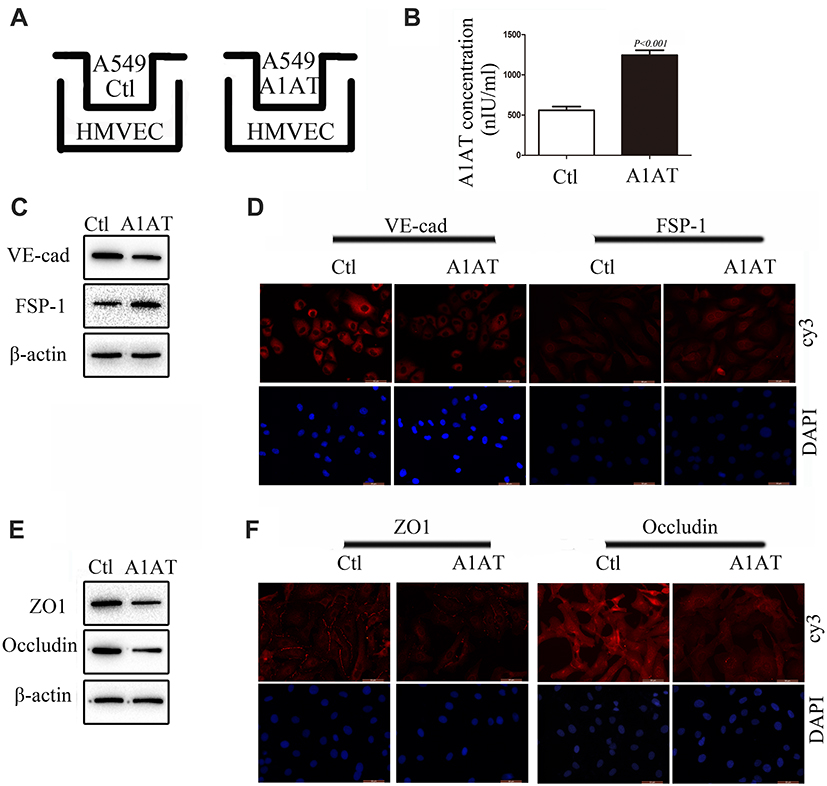 |
Figure 4 A1AT secreted by A549 promotes endothelial cells EndoMT. (A) A549 and HMVEC co-culture model. (B) A1AT levels in culture supernatant detected by (ELISA (P<0.01). (C, D) Analysis of the expression of VE-cadherin (endothelial marker) and FSP-1 (mesenchymal markers) in HMVECs by Western blotting (C) and immunofluorescence staining (D); scale bar, 50 μm. (E, F) Analysis of the expression of ZO1 and Occludin in HMVECs by Western blotting (E) and immunofluorescence staining (F); scale bar, 50 μm. |
A1AT Silencing Promotes Sensitivity to Cisplatin in Human NSCLC Cells
Our previous results suggested that A1AT knockdown inhibited the mesenchymal phenotype of the cells. Because many studies have shown that EMT imparts resistance to radiotherapy or chemotherapy,28 we speculated that A1AT silencing would affect the chemotherapeutic response by regulating stemness. First, we confirmed that A1AT knockdown is associated with increased stemness by immunofluorescence and Western blot analysis of the CSC marker cluster of differentiation-44 (CD44), CD133, and CD166 (ALCAM) expression in A1AT-knocked down and control (Ctl) A549 and H1650 cells (Figure 5A and B). Cancer stem cells are associated with chemoresistance.29 Cisplatin is the first-line drug for lung cancer therapy; however, resistance is highly prevalent in patients and significantly limits survival. Thus, we further explored the functional significance of A1AT expression in cisplatin resistance. Notably, the cell counting kit 8 (CCK-8) assay and colony formation assays revealed that A1AT knockdown sensitized A549 cells to cisplatin-induced cell death (Figure 6A, C and D, P<0.001), suggesting that A1AT enhances the resistance of the cells to cisplatin therapy.
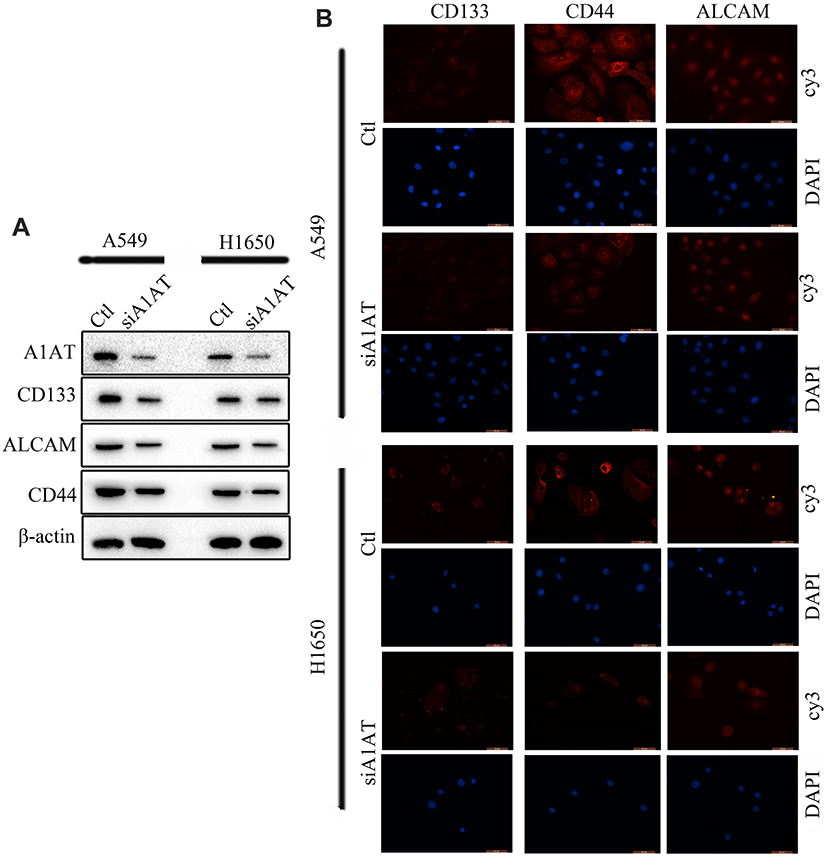 |
Figure 5 A1AT silencing inhibits stemness in NSCLC. (A, B) Analysis of CD133, ALCAM and CD44 expression by Western blotting (A) and immunofluorescence (B) in A1AT-silenced or control (Ctl) A549 and H1650 cells (scale bar, 50 μm). Si#03 indicates the siRNA used for the RNA interference. |
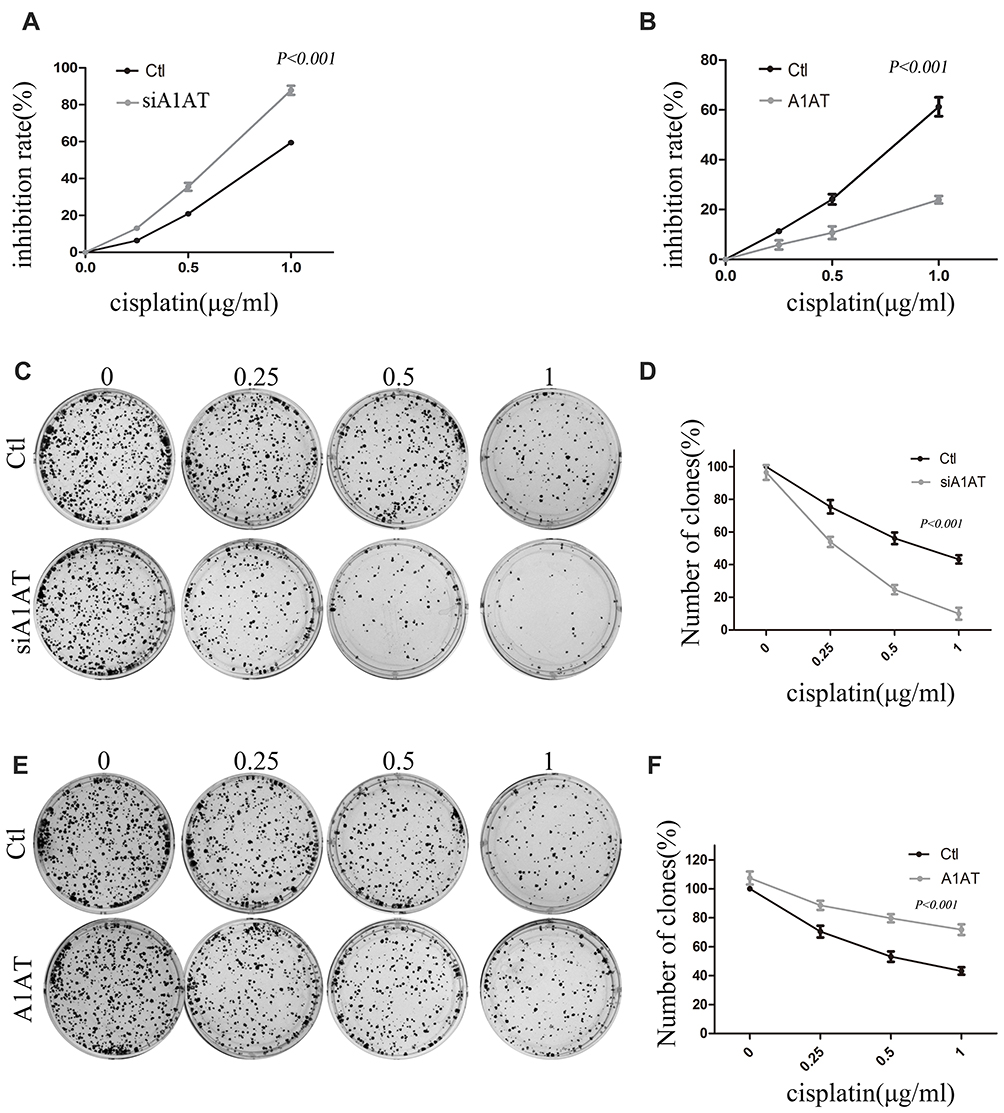 |
Figure 6 Effect of A1AT on cisplatin sensitivity in NSCLC. (A, B) Cisplatin-induced cell death was monitored by CCK-8 assays. A1AT-silenced and control (Ctl) cells treated with cisplatin. Si#03 indicates the siRNA used for the RNA interference (A). A1AT overexpressing and control cells treated with cisplatin (B). (C, D) Cisplatin-induced cell death was monitored in colony formation assays in A1AT-silenced and control A549 cells. Representative images (C) and quantitation (D) are shown. (E, F) Cisplatin-induced cell death was monitored in colony formation assays in A1AT-overexpressing and control A549 cells. Representative images (E) and quantitation (F) are shown. |
A1AT Overexpression Reduces Sensitivity to Cisplatin in Human NSCLC Cells
We confirmed the effect of A1AT on cisplatin resistance by overexpressing A1AT in A549 and H1650 cells. We found that overexpression of A1AT was associated with upregulation of CD44, CD133, and ALCAM expression (Figure 7A and B). Cell proliferation and colony formation assays revealed that A1AT overexpression decreased the sensitivity of A549 cells to cisplatin-induced cell death (Figure 6B, E and F, P<0.001).
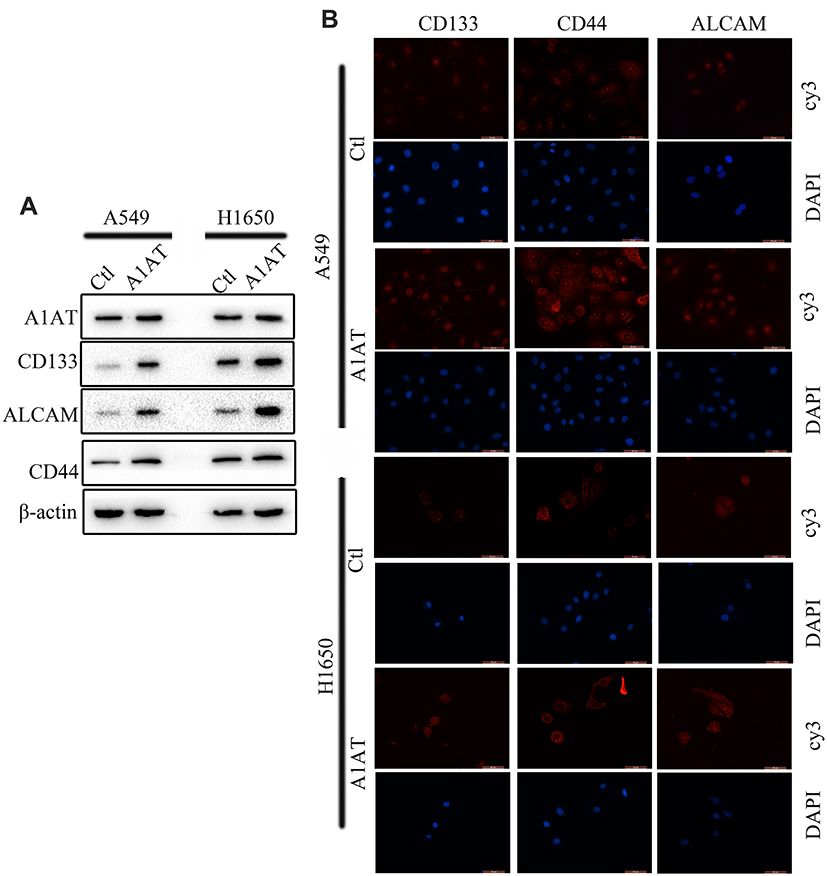 |
Figure 7 A1AT overexpression promotes stemness in NSCLC. (A, B) Analysis of CD133, ALCAM and CD44 expression by Western blotting (A) and immunofluorescence (B) in A1AT-overexpressing or control (Ctl) A549 and H1650 cells (scale bar, 50 μm). |
Discussion
Lung cancer is the primary cause of cancer-related death,30 with millions of new cases diagnosed each year. Although advances have been made in the treatment of this disease through surgery, chemotherapy, and radiotherapy, cancer metastasis remains the main cause of poor prognosis, with a low overall 5-year survival rate.1,2 Thus, it is important to identify new cancer metastasis-related genes and to study the mechanism through which they promote the development of cancers.
A1AT is highly expressed in cells of endodermal epithelial origin. A1AT protein is secreted into the blood, where it inhibits neutrophil proteases, thereby protecting host tissues from non-specific injury associated with inflammation.8 In our study, we found that A1AT influences EMT and drug resistance in human NSCLC.
It is widely accepted that EMT plays a significant role during tumor invasion and metastasis.31–33 EMT is associated with the expression of mesenchymal markers such as N-cadherin or vimentin and loss of epithelial cell adhesion molecules such as E-cadherin.27,34,35 In our results, silencing of A1AT leads to the downregulation of epithelial markers (E-cadherin) and the upregulation of mesenchymal markers (vimentin), implying that A1AT silencing inhibits EMT in lung cancer cells. Similar results were obtained in immunofluorescence assays. Therefore, the downregulation of A1AT reverses EMT in vitro. Tumor metastasis includes several steps: loss of cellular adhesion, increased motility and invasiveness, intravasation, extravasation, and colonization at a distant site.36 In our transwell and wound-healing assays, A1AT downregulation significantly inhibited the migration of lung cancer cells, and A1AT overexpression had the opposite effect, indicating that A1AT silencing inhibits lung cell metastasis in vitro.
Similar to EMT, endothelial cells can also dedifferentiate into mesenchymal cells. In our results, secreted A1AT led to the downregulation of endothelial markers (VE-cadherin) and the upregulation of mesenchymal markers (FSP-1), implying that secreted A1AT promotes EndoMT in endothelial cells. There is evidence of the existence of EndoMT in many different pathological conditions such as cardiac fibrosis, cancer, vein stenosis, and retina diabetes.37–39 In EndoMT, endothelial cells dedifferentiate into mesenchymal cells, and at the same time loss of cell-cell contact.40 That makes it easier for tumor cells to pass through the blood vessel.
Many studies have shown that EMT facilitates resistance to radiotherapy and chemotherapy.41,42 Cisplatin (cis-diamminedichloro-platinum II) is a major drug used in the treatment of lung cancer, especially in NSCLC.43 Because acquired drug resistance is a common phenomenon in patients with NSCLC,44 a detailed understanding of the molecular mechanism of cisplatin resistance is necessary to develop new therapeutic strategies that circumvent it. Consistently, A1AT-knockdown cells displayed increased sensitivity to cisplatin-induced cell death when compared to control cells.
In summary, A1AT induces EMT, EndoMT, and drug resistance in human NSCLC and is a promising therapeutic target for lung cancer.
Ethical Approval
In accordance with the Declaration of Helsinki, written informed consent was obtained from each patient. The Ethics Committee of the First Affiliated Hospital of Chengdu Medical College approved this study. All experiments were performed following the guidelines and regulations of the Ethics Committee of the First Affiliated Hospital of Chengdu Medical College.
Acknowledgments
Dong-ming Wu and Teng Liu are co-first authors. We would like to thank the Clinical Laboratory of the First Affiliated Hospital of Chengdu Medical College, China, for their help in the execution of the experiments.
Funding
This study was funded by the National Natural Science Foundation of China (81802955, 81972977), the Foundation of Sichuan Science and Technology Agency (2018JY0648, 2019YJ0589), the Foundation of The First Affiliated Hospital of Chengdu Medical College (CYFY2017ZD03, CYFY2018ZD02, CYFY2017YB08), the Foundation of Chengdu Medical College (CYCG16-04), and the Foundation of Collaborative Innovation Center of Sichuan for Elderly Care and Health, Chengdu Medical College (19Z01).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi:10.1002/ijc.29210
2. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
3. Antonio CD, Antonio P, Gori B, et al. Bone and brain metastasis in lung cancer: recent advances in therapeutic strategies. Ther Adv Med Oncol. 2014;6(3):101–114. doi:10.1177/1758834014521110
4. Khemasuwan D, Atul CM, Ko-Pen W. Past, present, and future of endobronchial laser photoresection. J Thorac Dis. 2015;7(Suppl 4):S380–S388. doi:10.3978/j.issn.2072-1439.2015.12.55
5. Parikh RB, Kirch RA, Smith TJ, Temel JS. Early specialty palliative care–translating data in oncology into practice. N Engl J Med. 2013;369(24):2347–2351. doi:10.1056/NEJMsb1305469
6. Kesharwani SS, Kaur S, Tummala H, Sangamwar AT. Overcoming multiple drug resistance in cancer using polymeric micelles. Exp Opinion Drug Delivery. 2018;15(11):1127–1142. doi:10.1080/17425247.2018.1537261
7. Kesharwani SS, Kaur S, Tummala H, Sangamwar AT. Multifunctional approaches utilizing polymeric micelles to circumvent multidrug resistant tumors. Colloids Surf B Biointerfaces. 2018;173(1):581–590. doi:10.1016/j.colsurfb.2018.10.022
8. Custodio A, de Castro J. Strategies for maintenance therapy in advanced non-small cell lung cancer: current status, unanswered questions and future directions. Crit Rev Oncol Hematol. 2012;82(3):338–360. doi:10.1016/j.critrevonc.2011.08.003
9. Liu L, Zhao E, Li C, et al. TRIM28, a new molecular marker predicting metastasis and survival in early-stage non-small cell lung cancer. Cancer Epidemiol. 2013;37(1):71–78. doi:10.1016/j.canep.2012.08.005
10. Teckman JH, Blomenkamp KS. Pathophysiology of Alpha-1 antitrypsin deficiency liver disease. Methods Mol Biol. 2017;1639:1–8.
11. Jaberie H, Naghibalhossaini F. Recombinant production of native human α-1-antitrypsin protein in the liver HepG2 cells. Biotechnol Lett. 2016;38(10):1683–1690. doi:10.1007/s10529-016-2150-z
12. Motawi T, Shaker OG, Hussein RM, Houssen M. Polymorphisms of α1-antitrypsin and Interleukin-6 genes and the progression of hepatic cirrhosis in patients with a hepatitis C virus infection. Balkan J Med Genet. 2017;19(2):35–44. doi:10.1515/bjmg-2016-0034
13. Pervakova MY, Emanuel VL, Titova ON, et al. The diagnostic value of alpha-1-antitrypsin phenotype in patients with granulomatosis with polyangiitis. Int J Rheumatol. 2016;2016:7831410. doi:10.1155/2016/7831410
14. Ni K, Umair Mukhtar Mian M, Meador C, et al. Oncostatin M and TNF-α induce alpha-1 antitrypsin production in undifferentiated adipose stromal cells. Stem Cells Dev. 2017;26(20):1468–1476. doi:10.1089/scd.2017.0099
15. Buggio M, Towe C, Annan A, et al. Pulmonary vasculature directed adenovirus increases epithelial lining fluid alpha-1 antitrypsin levels. J Gene Med. 2016;18(1–3):38–44. doi:10.1002/jgm.2874
16. Ebrahimi T, Rust M, Kaiser SN, et al. α1-antitrypsin mitigates NLRP3-inflammasome activation in amyloid β1-42-stimulated murine astrocytes. J Neuroinflammation. 2018;15(1):282. doi:10.1186/s12974-018-1319-x
17. Shakya R, Tarulli GA, Sheng L, et al. Mutant p53 upregulates alpha-1 antitrypsin expression and promotes invasion in lung cancer. Oncogene. 2017;36(31):4469–4480. doi:10.1038/onc.2017.66
18. Wu DM, Zhang P, Liu RY, et al. Phosphorylation and changes in the distribution of nucleolin promote tumor metastasis via the PI3K/Akt pathway in colorectal carcinoma. FEBS Lett. 2014;588(10):1921–1929. doi:10.1016/j.febslet.2014.03.047
19. Österreicher CH1, Penz-Österreicher M, Grivennikov SI, et al. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc Natl Acad Sci U S A. 2011;108(1):308–313. doi:10.1073/pnas.1017547108
20. Wang H, Shi J, Luo Y, et al. LIM and SH3 protein 1 induces TGFbeta-mediated epithelial-mesenchymal transition in human colorectal cancer by regulating S100A4 expression. Clin Cancer Res. 2014;20(22):5835–5847. doi:10.1158/1078-0432.CCR-14-0485
21. Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi:10.1016/j.cell.2008.03.027
22. Ye X, Tam WL, Shibue T, et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 2015;525(7568):256–260. doi:10.1038/nature14897
23. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10(6):717–728. doi:10.1016/j.stem.2012.05.007
24. Kong D, Li Y, Wang Z, Sarkar FH. Cancer stem cells and epithelial-to-mesenchymal transition (EMT)-phenotypic cells: are they cousins or twins? Cancers (Basel). 2011;3(1):716–729. doi:10.3390/cancers30100716
25. Zeisberg EM, Tarnavski,M O, Zeisberg AL, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13(8):952–961. doi:10.1038/nm1613
26. Medici D, Kalluri R. Endothelial.Mesenchymal Transition and Its Contribution to the Emergence of Stem Cell Phenotype. Elsevier. 2012:379–384.
27. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110. doi:10.1038/nrc3447
28. Tièche CC, Peng RW, Dorn P, Froment L, Schmid RA, Marti TM. Prolonged pemetrexed pretreatment augments persistence of cisplatin-induced DNA damage and eliminates resistant lung cancer stem-like cells associated with EMT. BMC Cancer. 2016;16(1):125. doi:10.1186/s12885-016-2117-4
29. Leung EL, Fiscus RR, Tung JW, et al. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS One. 2010;5(11):e14062. doi:10.1371/journal.pone.0014062
30. Torre LA1, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21262
31. Nam S, Park T. Pathway-based evaluation in early onset colorectal cancer suggests focal adhesion and immunosuppression along with epithelial-mesenchymal transition. PLoS One. 2012;7(4):e31685. doi:10.1371/journal.pone.0031685
32. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi:10.1172/JCI39104
33. Zhang Q, Wang SY, Nottke AC, Rocheleau JV, Piston DW, Goodman RH. Redox sensor CtBP mediates hypoxia-induced tumor cell migration. Proc Natl Acad Sci U S A. 2006;103(24):9029–9033. doi:10.1073/pnas.0603269103
34. Scheel C, Weinberg RA. Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int J Cancer. 2011;129(10):2310–2314. doi:10.1002/ijc.26311
35. Zhang XL, Huang CX, Zhang J, Inoue A, Zeng SE, Xiao SJ. CtBP1 is involved in epithelial-mesenchymal transition and is a potential therapeutic target for hepatocellular carcinoma. Oncol Rep. 2013;30(2):809–814. doi:10.3892/or.2013.2537
36. Holz C, Niehr F, Boyko M, et al. Epithelial-mesenchymal-transition induced by EGFR activation interferes with cell migration and response to irradiation and cetuximab in head and neck cancer cells. Radiother Oncol. 2011;101(1):158–164. doi:10.1016/j.radonc.2011.05.042
37. Cao Y, Feng B, Chen S, Chu Y, Chakrabarti S. Mechanisms of endothelial to mesenchymal transition in the retina in diabetes. Invest Ophthalmol Vis Sci. 2014;55(11):7321–7331. doi:10.1167/iovs.14-15167
38. Cooley BC, Nevado J, Mellad J, et al. TGF-beta signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci Transl Med. 2014;6(227):227ra34. doi:10.1126/scitranslmed.3006927
39. Maddaluno L, Rudini N, Cuttano R, et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature. 2013;498(7455):492–496. doi:10.1038/nature12207
40. Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. 2010;16(12):1400–1406. doi:10.1038/nm.2252
41. Theys J, Jutten B, Habets R, et al. E-Cadherin loss associated with EMT promotes radioresistance in human tumor cells. Radiother Oncol. 2011;99(3):392–397. doi:10.1016/j.radonc.2011.05.044
42. Grossi F, Bennouna J, Havel L, Hochmair M, Almodovar T. Oral vinorelbine plus cisplatin versus pemetrexed plus cisplatin as first-line treatment of advanced NS-NSCLC: cost minimization analysis in 12 European countries. Curr Med Res Opin. 2016;32(9):1577–1584. doi:10.1080/03007995.2016.1190700
43. Chang A. Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer. 2011;71(1):3–10. doi:10.1016/j.lungcan.2010.08.022
44. Chen X, Jiang Y, Huang Z, et al. miRNA-378 reverses chemoresistance to cisplatin in lung adenocarcinoma cells by targeting secreted clusterin. Sci Rep. 2016;6(1):19455. doi:10.1038/srep19455
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.