Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
Alpha-1 Antitrypsin Augmentation Therapy Improves Survival in Severely Deficient Patients with Predicted FEV1 Between 10% and 60%: A Retrospective Analysis of the NHLBI Alpha-1 Antitrypsin Deficiency Registry
Authors Rahaghi FF, Monk R, Ramakrishnan V, Beiko T, Strange C ![]()
Received 30 July 2020
Accepted for publication 26 October 2020
Published 3 December 2020 Volume 2020:15 Pages 3193—3199
DOI https://doi.org/10.2147/COPD.S263725
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Richard Russell
Franck F Rahaghi,1 Richard Monk,2 Viswanathan Ramakrishnan,3 Tatsiana Beiko,2 Charlie Strange2
1Department of Pulmonary and Critical Care, Cleveland Clinic Florida, Weston, FL, USA; 2Division of Pulmonary, Critical Care, Allergy and Sleep Medicine, Medical University of South Carolina, Charleston, SC, USA; 3Department of Public Health Sciences, Medical University of South Carolina, Charleston, SC, USA
Correspondence: Franck F Rahaghi
Department of Pulmonary and Critical Care, Cleveland Clinic Florida, Weston, FL, USA
Tel +1 954 659 5450
Fax +1 954 6595451
Email [email protected]
Purpose: The extent of the survival benefit of augmentation therapy for alpha-1 antitrypsin deficiency (AATD) in individuals with advanced COPD is difficult to define. We performed a retrospective analysis using all available data from the observational registry of individuals with severe deficiency of alpha-1 antitrypsin (AAT) conducted by the NHLBI investigators.
Patients and Methods: Individuals (N=1129) with severe deficiency of AAT were evaluated for mortality using all data sources and stratified by 10% increments of baseline forced expiratory volume in 1 second (FEV1) percent predicted and by augmentation therapy status (ever receiving versus never receiving). Kaplan–Meier survival curves were constructed for each of the deciles comparing survival in treated vs non-treated groups. A multivariable model was performed to define the correlates of survival in individuals with FEV1 < 30% predicted.
Results: Amongst all subjects, augmentation was associated with improved survival (p< 0.0001). Among the individuals ever receiving augmentation therapy, survival was better than for those not receiving augmentation at all 10% increments of FEV1% predicted from 10% to 60% (P values < 0.05 in all deciles). In subgroups of participants with hyperinflation defined as residual volume (RV)> 120% predicted and in subgroups of participants with reduced diffusing capacity for carbon monoxide (DLCO) < 70% predicted, there was significantly better survival for those ever receiving augmentation therapy than for those who never received augmentation (p< 0.001). A multivariable analysis showed that mortality benefit is influenced by age, DLCO % predicted, and augmentation therapy.
Conclusion: There is a survival benefit from augmentation therapy in AATD between FEV1 values in the 10– 60% predicted range. Screening and treatment of AATD patients should therefore not be limited by the severity of illness as defined by FEV1.
Keywords: alpha-1 antitrypsin deficiency, COPD, mortality, augmentation therapy, FEV1, survival
Introduction
Since the discovery of alpha1 antitrypsin deficiency (AATD) in 1963, our understanding of the disease has advanced.1 However, controversy remains about the impact of augmentation therapy first incorporated into clinical practice at the National Heart Lung and Blood Institute (NHLBI) of the National Institute of Health (NIH).2–4 Historically, the lack of sufficient numbers of subjects to perform a randomized clinical trial of augmentation versus placebo prompted the largest therapeutics study in AATD to date, the 1988–1996 NHLBI registry of individuals with severe deficiency of alpha-1 antitrypsin. For study eligibility, serum AAT levels were set at <11 µM. To this date, meta-analyses of augmentation therapy are dominated by this 37 clinical center study that contained 1129 individuals on or off augmentation therapy.5–7
This registry data showed that a subgroup of individuals with a baseline FEV1 30–64% predicted had a slower decline of FEV1 if they had received AAT augmentation.8 There was evidence of improved survival in all participants ever on augmentation compared to those never on augmentation (p=0.02). However, further multivariate subgroup analysis showed subjects with an initial FEV1 35–49% predicted had the majority of this benefit.8 Stratified subjects were based on the then-current ATS COPD staging and excluded subjects with no follow-up visits ≥6 months after enrollment.
Since this observational study was released, controversy has persisted about the benefit of augmentation in individuals with FEV1 <30% predicted. Because of the relative lack of change in FEV1 possibly due to a floor effect on this test and the reticence to perform mortality end-point studies, individuals with FEV1 <30% predicted have been excluded from most AATD clinical trials and sometimes from therapeutic interventions.
We hypothesized that subgroup analysis of the publicly available NHLBI database might give more insight to the mortality signal and its correlates. We sought to evaluate data from all individuals in an intention to treat analysis to determine if there were other groups that objectively benefited from augmentation therapy with pooled plasma derived alpha-1 antitrypsin (alpha-1 protease inhibitor) therapy. We also evaluated whether other baseline lung function parameters have associations with a survival benefit from augmentation.
Patients and Methods
The publically available de-identified data from the Alpha-1 Antitrypsin Deficiency Registry Study Group was used with NHLBI permission. Patients were consented for de-identified collection and analysis of their data at the enrolment and the project was reviewed and approved by MUSC IRB (Pro000025833). We found baseline spirometry data for 1126 of the 1129 enrolled subjects (5 had initial spirometry labeled as follow-up spirometry, 5 had follow-up spirometry labeled as initial spirometry, and 3 had no pulmonary function testing). All 1126 had FEV1, 885 had DLCO, 879 had RV and 850 had TLC measurements with their initial lung function testing (Figure 1).
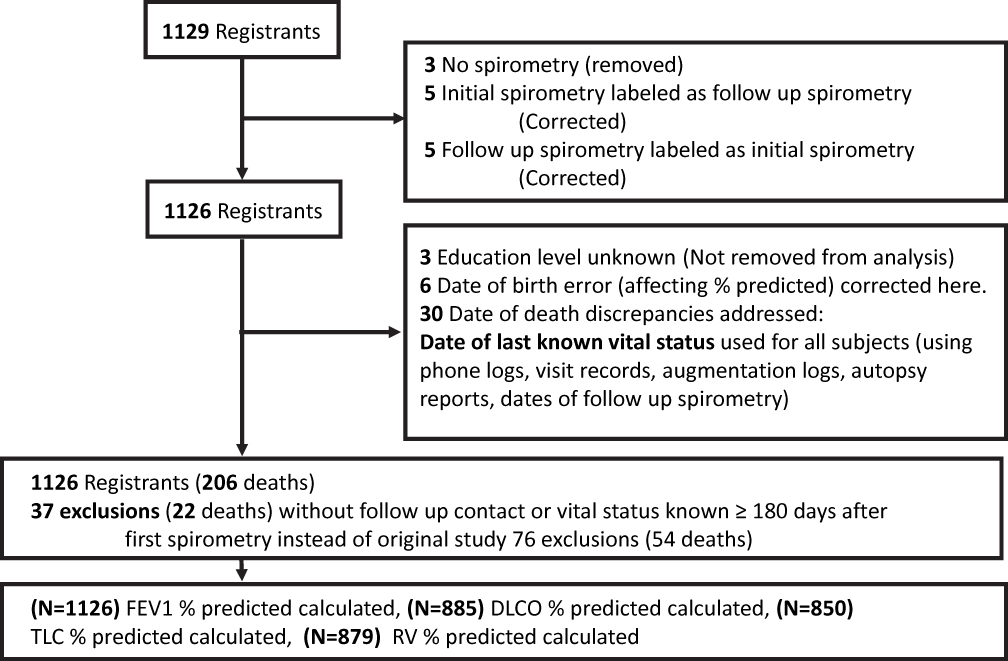 |
Figure 1 Study enrolment and changes form original analysis. |
For calculating FEV1% predicted, we used the highest FEV1 post bronchodilator divided by the FEV1 predicted by Hankinson’s model.9 To calculate DLCO% predicted, we divided the best of two values of DLCO divided by the DLCO predicted by Crapo’s model.10,11 To calculate RV% predicted, we used the post bronchodilator RV (if no post bronchodilator RV was available, we used pre bronchodilator RV) divided by the RV predicted by Crapo’s model.10 To calculate Total Lung Capacity (TLC)% predicted, we used the post bronchodilator TLC by gas diffusion (or pre bronchodilator TLC by gas diffusion, post bronchodilator TLC by plethysmography or pre bronchodilator TLC by plethysmography, in that order of priority depending on which was available) divided by the TLC predicted by Crapo’s model.10 For FEV1/Forced Vital Capacity (FVC) we divided the highest post-bronchodilator FEV1 by the highest post-bronchodilator FVC.
Subjects were stratified by highest level of education with college graduate with advanced training (n=167), college graduate (n=160), at least one year of college (n=301), high school graduate (n=389), completed 10–11 years of school (n=64), completed <10 years of school or unknown (n=45).
To classify subjects as being on augmentation during the study or not, we used data from personal augmentation administration logs, visit questionnaires and telephone questionnaires to determine whether a subject had always received augmentation for the time during the follow-up period of the study; those subjects (N=406) were considered to be on augmentation. Subjects with no reports of any augmentation therapy during follow-up (N=452) were considered to be not on augmentation. Some individuals enrolled not on augmentation therapy and began therapy during the study (N=268).
Date of death was obtained from autopsy reports, death certificates and telephone contacts (with family, friends or treating physicians). Date of last contact/censoring was obtained by using the latest date amongst lung function testing, follow-up visits, telephone contacts with the subjects or other individuals (family, friends or treating physician) who could testify to the last date they knew the subject’s vital status.
Survival analyses used Cox proportional hazards regression and were performed in JMP 5.1.2 (SAS Institute Inc., Cary, NC) with time to event censored after 2500 days (6.8 years per original protocol). Survival was compared for each decile of baseline FEV1% predicted. Because fewer subjects had post-bronchodilator lung volumes and DLCO performed, survival analyses were limited to clinically significant groupings of residual volume (RV) >120% predicted and ≤120% predicted to define groups with and without air trapping, respectively. Participants with baseline DLCO %predicted >70%, between 40–70%, and <40% were grouped for survival analyses. P-values <0.05 were considered statistically significant. Hazard ratios for mortality were calculated with 95% confidence intervals (CI).
Multivariable regressions were performed to examine the impact of baseline differences in demographics using age, sex, smoking pack-years, FEV1% predicted, DLCO % predicted, TLC % predicted, education, and BMI between augmented and non-augmented populations.
Results
Baseline FEV1 was available in 1126 individuals. Two hundred and six subjects died during the follow-up period, and mean time to death or censoring was 1597 days. Amongst all subjects, augmentation was associated with improved survival (p<0.0001). Augmentation in individuals with baseline FEV1% predicted at all increments <60% was associated with increased survival (p<0.01 in all), but not in individuals with baseline FEV1% predicted >60%. Augmentation in individuals with baseline diffusing capacity for carbon monoxide (DLCO)% predicted <60% (moderately or severely reduced) was associated with improved survival (p<0.0001). Augmentation in individuals with air trapping (baseline residual volume (RV) >120% predicted) was associated with improved survival (p=0.0005). Baseline total lung capacity had no effect on mortality.
Table 1 shows the age, sex, BMI, DLCO %predicted and TLC % predicted by baseline FEV1% predicted in 10% increments. We found a significantly better survival on augmentation in subjects with baseline FEV1% predicted 10–60% (all p<0.05) (Figure 2). None of the 6 subjects with initial FEV1% predicted <10% were on augmentation and this subgroup could not be compared. There were few deaths in the strata of FEV1% predicted >60% and we did not see significant differences in outcome based on augmentation.
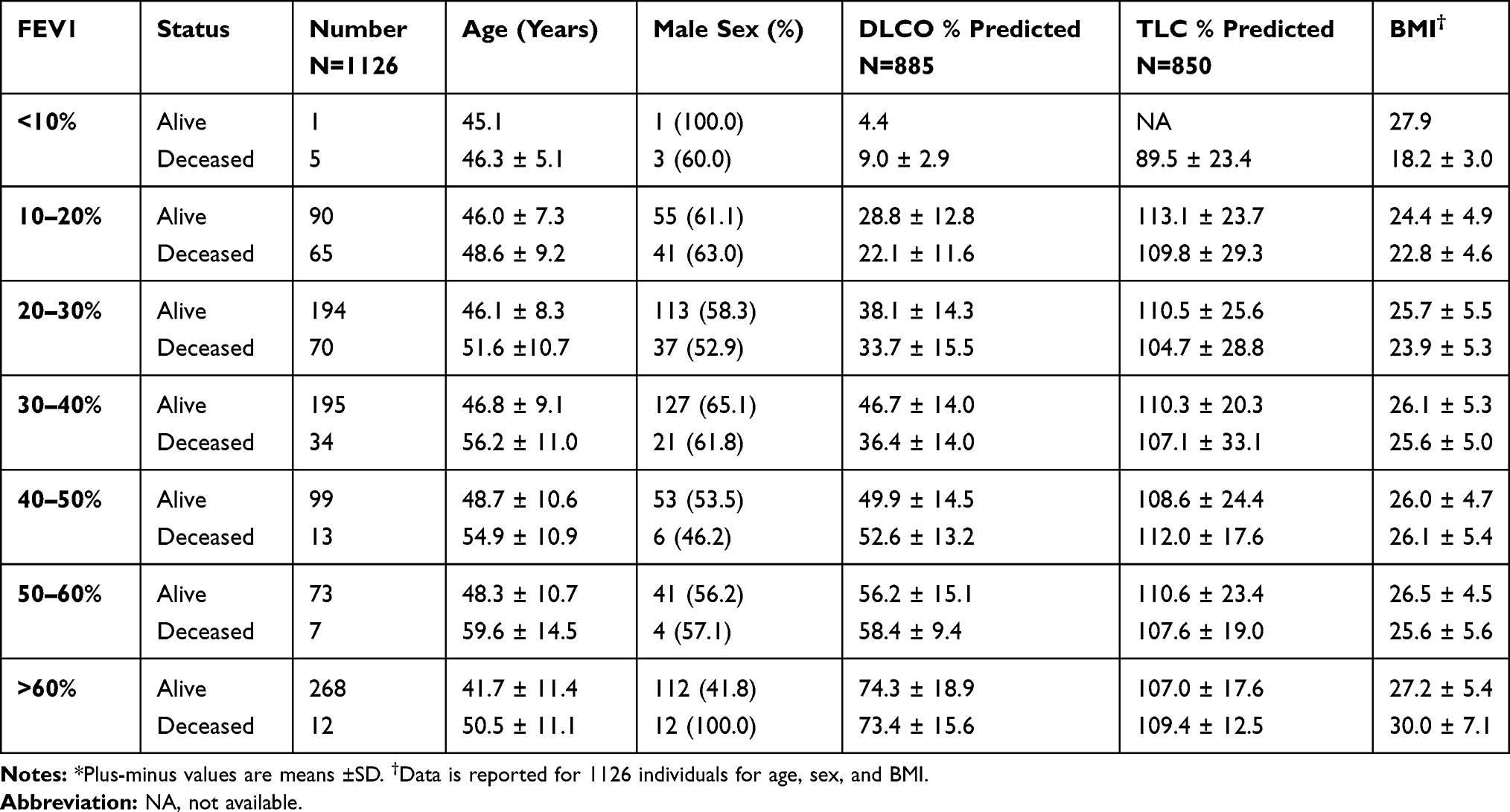 |
Table 1 Characteristics of the Study Cohort by Decile of FEV1 and Morbidity Status* |
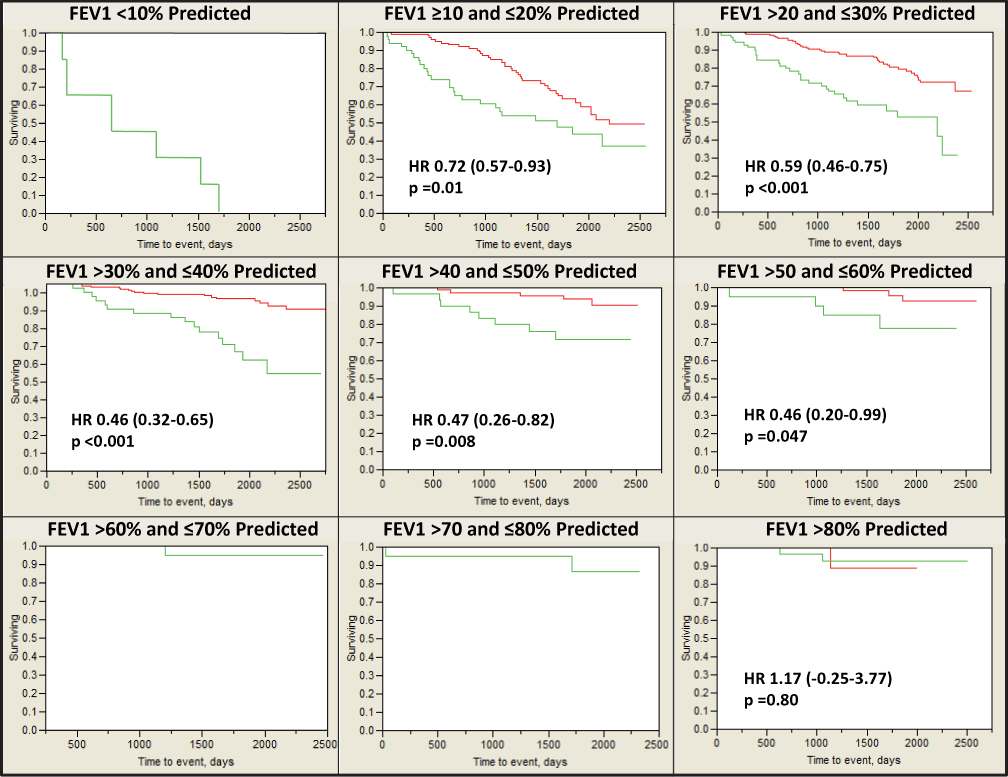 |
Figure 2 Kaplan–Meier survival curves for all study individuals with baseline FEV1 data (N=1126). Notes: The cohort treated with sometimes or always augmentation therapy (―) in red had improved survival compared to those without augmentation therapy (―) in green stratified by FEV1% predicted deciles between 10% and 60%. Graphs censored at 2500 days. |
There were significant differences in the analysis cohort from the original study report. The authors of the original paper chose not to include subjects unless a study visit was recorded ≥6 months after enrollment, arguing that for augmentation to affect mortality, this medication should be given for a sufficient time to cause biological effect. They did this to, in their words, reduce the possibility of bias toward a positive effect of augmentation therapy caused by including subjects who were not on therapy at enrollment and later died before returning for a follow-up visit and presumably before they could begin augmentation therapy. They found that 76 subjects (54 deaths) were excluded on that basis. We found only 37 subjects (15 deaths) without follow-up contact (neither office visit, spirometry visit nor telephone contact) ≥180 days after first spirometry.
The authors of the original paper also excluded 5 subjects because initial FEV1% predicted or education was missing. We found that several initial spirometry reports were erroneously labeled as follow-up. We found that only 3 subjects lacked initial FEV1 for percent predicted. Where we found subjects to have missing education data, we changed them to “unknown” as allowed by the original study. We excluded no one on the basis of missing education data.
Of the 879 subjects for which there was baseline RV, 630 had air trapping as defined by RV >120% predicted. Those subjects with air trapping had significantly better survival if they were on augmentation (p<0.001, HR 0.31 (CI 0.13–0.49)). Those without air trapping had a trend toward better survival if they were on augmentation (p=0.07), reflecting the fact that there was an overall improvement in survival on augmentation compared to off amongst subjects who happened to get RV measured with initial spirometry (p≤0.004, HR 0.24 (CI 0.08–0.40)).
DLCO was obtained from 885 subjects with initial spirometry. Amongst all 885 of these subjects, there was a significant association between augmentation and survival (p<0.01, HR 0.45 (CI 0.24–0.66)). Of these, 171 had normal diffusing capacity as defined by DLCO >70% predicted, 405 had moderately reduced diffusing capacity with DLCO 40–70% predicted, and 309 had substantially reduced diffusing capacity based on a DLCO <40% predicted. Subjects in strata with DLCO ≤ 70% had significantly better survival if they were on augmentation (p<0.01, HR 0.44 (CI 0.30–0.58)).
A multivariable regression was performed to examine the impact of baseline differences in demographics using age, sex, smoking pack-years, education, FEV1% predicted, DLCO % predicted, TLC % predicted and BMI between augmented and non-augmented populations. The analysis for the entire population showed that survival was influenced by age, augmentation, FEV1% predicted, and DLCO %predicted. Replicate analysis limited to the population with FEV1 <50% predicted showed identical independent correlates.
Discussion
In rare diseases, the ability to undertake large and definitive randomized control trials is limited due to lack of patients and cost of studies. Longitudinal studies in rare diseases such as the NHLBI Registry of Individuals Severely Deficient in Alpha-1 Antitrypsin have historically provided great insight into the natural history of disease. Although this dataset suffers from lack of randomization as regards allocation of augmentation therapy, the observations regarding lung function decline on or off therapy have stood the test of time and have allowed powering of more recent prospective, randomized clinical trials. However, no study since has prospectively examined the mortality signal in AATD because of the slow progression of COPD and the reticence to prospectively enroll individuals with severely impaired lung function in registration studies.
Although many researchers have given recognition to the mortality signal in the NHLBI database, the type of analysis performed on the data may have led to some misinterpretation by physicians and by some regulatory authorities as showing no differences in mortality for those with low FEV1 values. Our analysis shows that participants had improved survival in augmentation therapy cohorts in all deciles of baseline FEV1 from 10–60% predicted and suggests a larger population who would benefit from augmentation.
Air trapping, as measured by RV > 120% predicted, and low DLCO occurs with advanced emphysema in AATD. Although the Registry did not collect computed tomography scans, the subgroups with air trapping and low diffusion that likely had advanced emphysema had improved survival with augmentation therapy. In short, there was no cohort of advanced emphysema participants in whom we could not detect a mortality difference associated with infusion of augmentation therapy except for the 6 individuals with baseline FEV1 <10% predicted in which no therapy was given.
The improved power in our analysis was due to the use of all sources available (visits, spirometry appointments, telephone reports) rather than only visits to establish date of last contact. When individuals who made a visit to a study center 6 months after enrollment were the only subjects included in analysis, we still found significantly better survival in augmented subjects with a baseline FEV1% predicted 10–50% when measured by 10% increments (all p<0.05). The change in significance in the 50–60% baseline FEV1 group was due to the removal of a single non-augmented subject who died within the 180 days after first spirometry.
Although the NHLBI protocol was written at study initiation, the analysis plan that added the 6-month augmentation therapy requirement before becoming an evaluable patient was not in the initial protocol. The later statistical analysis plan added this requirement after noting the incomplete follow-up associated with this pragmatic study. The original statistical plan to exclude these participants will always stand as the primary analysis of mortality since it was pre-hoc.
Historically, COPD mortality has been known to correlate with FEV1.12 Since this NHLBI registry data was originally collected, the understanding of risk factors for COPD mortality has progressed. Increased mortality is seen in COPD patients with a low body mass index (BMI).13,14 When BMI is combined with airflow obstruction, dyspnea and exercise capacity in the BODE score, the ability to predict mortality is enhanced.15 Six minute walk was not performed in this historic NHLBI dataset precluding an analysis of survival in similarly matched patients on parameters other than FEV1. Also, it is now established that FEV1 ceases to linearly decline at lower FEV1 levels and may not be the best way to assess disease severity or follow patients serially.16
This is an observational, retrospective analysis of a NHLBI database. As such, there are many possible limitations to the conclusions drawn from this effort. The analysis of a registry population is not universally generalizable to all patients with AATD deficiency, as this is not a population-based analysis. In the registry the decision of therapy was made by treating physicians and subjects. Therefore, there was systematic bias between those that received augmentation therapy and those that did not around medical insurance coverage in the US healthcare system. Bias might also be present associated with severity of illness, since some patients may be deemed too sick for augmentation therapy to likely help. Weekly dates of augmentation therapy were not available and compliance with therapy was subject to recall bias. As such, duration of time on therapy was not felt to be sufficiently robust to become a study variable. Therefore, a combined analysis of those always or sometimes on therapy (usually beginning in the first few months after baseline visit) allowed sufficient power for the current analyses, but do not help define the intensity of therapy needed to get mortality improvement from augmentation therapy.
Other studies have since looked at survival and mortality in AATD. McElvaney et al extrapolated data from RAPID open label extension suggesting an improvement of 5.6 years in the life of a patient receiving AAT replacement therapy.17 Recently Ellis et al presented a study of comparison made between augmentation naive AATD patients from the AATD UK registry (United Kingdom) and United States patients on augmentation therapy followed in AlphaNet’s Disease Management and Prevention Program (ADMAPP). Propensity-matched patients had longer mean survival in the US treatment group with augmentation (20.3 years, 95% CI 19.4 to 21.2), compared to the UK control (13.7 years, 95% CI 13.1 to 14.3) p<0.001.18 These studies extend our knowledge concerning the effect of augmentation therapy on mortality.
This analysis of the data will also be important to regulatory authorities and payers who may not see any advantage to testing and treating advanced or elderly patients.
Conclusion
In summary, the previous report of this dataset showed a lack of improvement in FEV1 decline in those with severe obstruction with FEV1<35% predicted and reported on mortality reduction in individuals with FEV1 baseline <50% predicted. Assumptions have followed that the mortality signal was driven by individuals with higher ranges of FEV1 values. We find that a more complete analysis shows the differential effect of augmentation therapy on mortality is most significant in the most severely obstructed phenotypes of AATD. Withholding augmentation therapy and AATD screening from those with severe airflow obstruction appears to place a higher priority on a physiologic measurement (FEV1) that has a floor to further decline, than on survival. We should revise this practice and pay greater attention to screening and provision of therapy to more advanced patients.
Acknowledgments
This study was supported by contract number N01-HR-86036 from the National Heart, Lung, and Blood Institute, National Institutes of Health.
Disclosure
TB reports grants with Grifols, outside the submitted work. CS is employed in part by AlphaNet, a disease management company for Alpha-1 antitrypsin deficiency. He reports grants from Adverum, Arrowhead, CSL Behring, Grifols, MatRx, Takeda, and Vertex. He has received personal fees from CSL Behring, Dicerna, and Vertex, all outside the submitted work. FR reports grants and personal fees, from Grifols, and Takeda outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Laurell CB, Eriksson S. The electrophoretic alpha1-globulin pattern of serum in alpha1-antitrypsin deficiency. 1963. Copd. 2013;10(Suppl 1):3–8. doi:10.3109/15412555.2013.771956
2. Wewers MD, Casolaro MA, Sellers SE, et al. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N Engl J Med. 1987;316(17):1055–1062. doi:10.1056/NEJM198704233161704
3. Stockley RA, Miravitlles M, Vogelmeier C. Augmentation therapy for alpha-1 antitrypsin deficiency: towards a personalised approach. Orphanet J Rare Dis. 2013;8:149. doi:10.1186/1750-1172-8-149
4. Sandhaus RA, Stoller JK. Introduction to the 50th anniversary of the description of alpha-1 antitrypsin deficiency. COPD. 2013;10(Suppl sup1):1–2. doi:10.3109/15412555.2013.765840
5. Chapman KR, Stockley RA, Dawkins C, Wilkes MM, Navickis RJ. Augmentation therapy for alpha1 antitrypsin deficiency: a meta-analysis. Copd. 2009;6(3):177–184. doi:10.1080/15412550902905961
6. McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of alpha 1-antitrypsin deficiency. Chest. 1997;111(2):394–403. doi:10.1378/chest.111.2.394
7. Stoller JK, Tomashefski J, Crystal RG, et al. Mortality in individuals with severe deficiency of alpha1-antitrypsin: findings from the National Heart, Lung, and Blood Institute Registry. Chest. 2005;127(4):1196–1204.
8. The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Respir Crit Care Med. 1998;158(1):49–59. doi:10.1164/ajrccm.158.1.9712017
9. Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999;159(1):179–187. doi:10.1164/ajrccm.159.1.9712108
10. Crapo RO, Morris AH, Clayton PD, Nixon CR. Lung volumes in healthy nonsmoking adults. Bull Eur Physiopathol Respir. 1982;8(3):419–425.
11. Crapo RO, Morris AH. Standardized single breath normal values for carbon monoxide diffusing capacity. Am Rev Respir Dis. 1981;123(2):185–189.
12. Fletcher C, Peto R. The natural history of chronic airflow obstruction. Br Med J. 1977;1(6077):1645–1648. doi:10.1136/bmj.1.6077.1645
13. Bhavani S, Tsai CL, Perusich S, et al. Clinical and immunological factors in emphysema progression: 5-year prospective LES-COPD study. Am J Respir Crit Care Med. 2015;192(10):1171–1178. doi:10.1164/rccm.201504-0736OC
14. Budweiser S, Harlacher M, Pfeifer M, Jorres RA. Co-morbidities and hyperinflation are independent risk factors of all-cause mortality in very severe COPD. Copd. 2014;11(4):388–400. doi:10.3109/15412555.2013.836174
15. Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(10):1005–1012. doi:10.1056/NEJMoa021322
16. Parr DG, Stoel BC, Stolk J, Stockley RA. Validation of computed tomographic lung densitometry for monitoring emphysema in alpha1-antitrypsin deficiency. Thorax. 2006;61(6):485–490. doi:10.1136/thx.2005.054890
17. McElvaney NG, Burdon J, Holmes M, et al. Long-term efficacy and safety of α1 proteinase inhibitor treatment for emphysema caused by severe α1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE). Lancet Respir Med. 2017;5(1):51–60. doi:10.1016/S2213-2600(16)30430-1
18. Ellis P, Holm K, Choate R, et al. Comparison of outcomes in augmentation naïve and augmented patients with alpha-1 antitrypsin deficiency related lung disease. Eur Respir J. 2019;54:PA3383.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.