Back to Journals » Journal of Inflammation Research » Volume 19
Alopecia Areata and Atopic Dermatitis: Common Mechanisms and Emerging Therapeutics
Authors Tang A, Rachko G ![]() , Manzo Margiotta F, Levine J, Del Duca E, Ungar B
, Manzo Margiotta F, Levine J, Del Duca E, Ungar B ![]()
Received 22 January 2026
Accepted for publication 31 March 2026
Published 7 April 2026 Volume 2026:19 549496
DOI https://doi.org/10.2147/JIR.S549496
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Anish R. Maskey
Alice Tang,1 Grace Rachko,2 Flavia Manzo Margiotta,1,3,4 Jasmine Levine,1 Ester Del Duca,1,5 Benjamin Ungar1
1Department of Dermatology, Icahn School of Medicine at Mount Sinai, New York, NY, USA; 2Rutgers New Jersey Medical School, Newark, NJ, USA; 3Department of Dermatology, University of Pisa, Pisa, Italy; 4Health Science Interdisciplinary Center, Sant’Anna School of Advanced Studies, Pisa, Italy; 5Department of Medical and Cardiovascular Sciences, Sapienza University of Rome, Rome, Italy
Correspondence: Benjamin Ungar, Department of Dermatology, Icahn School of Medicine at Mount Sinai, New York, NY, USA, Tel +1 212-241-9728, Fax +1 212-987-1197, Email [email protected]
Abstract: Alopecia areata (AA) and atopic dermatitis (AD) have historically been classified as immunologically distinct—AA as a T helper 1 (Th 1) driven autoimmune condition and AD as a T helper 2 (Th 2) polarized inflammatory disease. However, emerging evidence demonstrates substantial immunological overlap, with both conditions exhibiting multi-axis cytokine dysregulation converging on Janus kinase-signal transducer and activator of transcription (JAK-STAT) dependent signaling and regulatory T cell insufficiency/dysfunction. Epidemiologically, these conditions frequently co-occur: up to 40% of AA patients have atopic comorbidities. Both diseases share genetic susceptibility loci, including interleukin 13 (IL-13) and human leukocyte antigen (HLA) associations, further supporting convergent pathogenic mechanisms. Key shared biomarkers include elevated interleukin 4 (IL-4), IL-13, and IgE, alongside dysregulated interferon-gamma and interleukin 15 signaling through the JAK-STAT pathway. These mechanistic insights have catalyzed the development of therapeutics for both conditions. JAK inhibitors—including baricitinib, ritlecitinib, upadacitinib, and abrocitinib—have demonstrated efficacy in AA and/or AD by interrupting cytokine signaling central to both diseases. Dupilumab, an interleukin − 4 receptor alpha antagonist FDA-approved for AD, has demonstrated benefit in a specific subset of AA patients with concurrent atopic disease; these findings highlight pathophysiologic heterogeneity within the disease and support the need for endotype-stratified treatment strategies. Investigational therapies targeting the OX40/OX40 ligand costimulatory pathway, interleukin 2 receptor agonists (rezpegaldesleukin), interleukin 7 receptor antagonists (bempikibart), and tri-specific cytokine inhibitors (EQ101) represent the next generation of agents that may treat both conditions by restoring immune homeostasis rather than broadly suppressing immunity. In this narrative review, we synthesize current understanding of the shared immunopathogenesis of AA and AD and highlight approved and investigational therapeutics with dual-disease relevance, pointing toward an era of mechanistically targeted, potentially disease-modifying interventions.
Keywords: alopecia areata, atopic dermatitis, JAK inhibitors, dupilumab, Th2
Introduction
Alopecia areata (AA) and atopic dermatitis (AD) are common inflammatory skin disorders historically viewed as immunologically distinct, with AA being a cell-mediated autoimmune hair loss condition driven by T helper type 1 (Th1) inflammation,1,2 and AD dominated by T helper type 2 (Th2)-type pathways,3,4 a dichotomy that has increasingly been challenged as advances in immune-mediated inflammatory disease research reveal shared pathogenic mechanisms across conditions once considered unrelated.5 Clinically, up to 15–20% of AA patients may have concurrent AD, and AA has been linked to other atopic conditions like asthma and allergic rhinitis, with up to 40% demonstrating atopic disease overall.6–8 The primary trigger for AA is loss of immune privilege (IP), leading to an autoimmune attack on the hair follicle, driven by CD8⁺ T cells that target the follicular bulb. AD pathogenesis is multifactorial, with type 2 inflammation now recognized as a central driver that both arises from and perpetuates epidermal barrier dysfunction.9,10 Both diseases exhibit multi-axis cytokine dysregulation with many shared features of both Th1 and Th2 dysregulation, converging on Janus kinase-signal transducer and activator of transcription (JAK-STAT) dependent cytokine signaling and regulatory T cell (Treg) insufficiency/dysfunction as shared pathogenic denominators.11
The management of patients with comorbid AA and AD presents a particular clinical challenge: comorbid AD has been identified as a poor prognostic factor for AA treatment response and a predictor of relapse risk, and therapies effective for one condition may have limited or uncertain efficacy for the other — IL-13 inhibitor monotherapy, for example, is approved for AD but has not shown efficacy in AA thus far, while topical therapies that are central to AD management are largely ineffective in AA.
Notably, immunomodulators like Janus kinase (JAK) inhibitors and biologics such as dupilumab have transformed treatment of both AA and AD, and offer further therapeutic promise across both diseases by intervening on shared cytokine signaling pathways.12,13 In this narrative review, we discuss the immunological background of AA and AD with a focus on shared immune axes, highlight key biomarkers of disease activity, and review approved and investigative therapeutics that may successfully treat both diseases.
Epidemiology
The global prevalence of AA is approximately 2%,14 with roughly 5–10% of AA cases being alopecia totalis (AT) or alopecia universalis (AU), complete scalp and body hair loss, respectively.14,15 Across races, a cross-sectional study found a significantly higher odds of AA in Black patients compared with White patients and a lower odds of AA in Asian patients compared with White patients.16 Individuals aged 25–40 have the highest likelihood of developing AA.17 The incidence appears similar between males and females.18 AA is also the third most common dermatologic condition in children, and the annual number of new pediatric cases continues to rise.19 Although the underlying pathogenesis of AA has yet to be fully elucidated, studies have identified genetic susceptibility loci centered around innate and adaptive immunity, including human leukocyte antigen (HLA-DR), cytotoxic T-lymphocyte-associated protein 4 (CTLA4), interleukin-2 (IL-2)/IL-21, and interleukin-2 receptor alpha (IL-2RA).20,21 Of note, one genome wide association study identified interleukin-13 (IL-13), a central cytokine in AA pathogenesis, as one of the most strongly associated genes.20 Family history of AA is a risk factor, as AA tends to present in multiple members within the same family and presents more often among siblings than among parents and offspring.18 Other risk factors for AA include having an autoimmune or atopic background such as atopic dermatitis, psoriasis, or inflammatory bowel disease.6
AD is a chronic inflammatory skin disease that can present at any age with recurrent eczematous lesions and intense itching. The global prevalence rate over one year of AD is approximately 2.6%, approximately 2.0% in adults and 4.0% in children, and the lifetime prevalence rate is up to 10% globally in adults, and up to 20% globally in children.22–24 AD is slightly more common in females (2.8%) than in males (2.4%).22 Although it may develop throughout life, between 60% and 73% of affected children present with AD within the first one to two years of age.22,25
AD and AA are epidemiologically linked, with multiple studies demonstrating bidirectional increase in risk; providing support for AD as the most common comorbidity in AA patients.26,27 In several meta-analyses, AA patients were found to have an increased risk of AD, and conversely, AD patients were found to have a significantly increased risk of AA.26,28 A Danish population-based study reported this association to have as high an adjusted odds ratio as 26.31 for AD among patients with AA.27 This genetic link is further supported by multiple Mendelian randomization studies looking at AD and AA, strengthening a potential underlying shared etiology in some patients.29,30 Clinically, the significance of this association extends beyond epidemiology, as comorbid AD has been identified as a poor prognostic factor for treatment response and a predictor of relapse risk in AA across several studies.6,31,32
Biomarkers of Alopecia Areata
AA is believed to arise from the collapse of immune privilege (IP) in the hair follicle.33 Under normal conditions, the anagen hair follicle maintains a distinct state of IP, characterized by minimal expression of major histocompatibility complex (MHC) proteins in its inferior segment, reduced levels of stress-induced ligands, local production of anti-inflammatory cytokines/hormones (eg. transforming growth factor-beta (TGFβ), interleukin-10 (IL-10), alpha-melanocyte-stimulating hormone (α-MSH)), and a relative scarcity of immune cell infiltrates.34,35 For as yet unclear reasons, disruption of this protective environment leads to upregulated expression of self-antigenic peptides presented by MHC molecules along the follicular epithelium, thus facilitating infiltration of self-reactive T cells and a dense immune cell infiltrate which histopathologically appears as a distinctive “bee-swarm pattern,” marked by dense lymphocytic infiltrates surrounding the bulbar region of anagen hair follicles.34
In AA, peribulbar immune cell infiltrates are predominantly composed of CD4+ and CD8+ T cells, accompanied by increased numbers of natural killer (NK) cells, Langerhans cells, dendritic cells, and mast cells.36 Among these, CD8+ T cells closely associated with the hair follicle were found to express NKG2D, an activating receptor typically associated with the NK cell lineage.37 Notably, CD8+ NKG2D+ T cells are sufficient to induce disease in a cell-transfer murine model of AA, highlighting a potential pathogenic role.12 Furthermore, activated CD8+ T cells produce high levels of pro-inflammatory cytokines, such as Interferon-gamma (IFN- γ), which skew the hair follicle microenvironment towards a proinflammatory state.38
The primary Th1 cytokine IFN-γ is hypothesized to be a key mediator of IP collapse within the hair follicle. Its ability to disrupt IP has been demonstrated ex-vivo: anagen-stage hair follicles collected from healthy scalps cultured in the presence of IFN-γ resulted in increased expression of MHC I molecules.35 IL-2 is another key cytokine released by CD4+ cells which signals through JAK1 and JAK3 receptors, driving gene transcription programs that promote cellular expansion, survival, and maintenance.39 In AA patients, elevated IL-2 expression is detected within the deep dermis, where the follicular bulb resides, and is significantly increased compared to levels of the upper dermis.39 Furthermore, active lesions of AA patients also show higher expression of IL-2 than inactive areas.39 IL-15, structurally related to IL-2 and essential for the survival of CD8⁺ memory T cells, further contributes to disease pathogenesis by supporting the expansion of T cells and NK cells and enhancing CD8⁺ T-cell production of granzymes, IFN-γ, and TNF-α.40 Circulating IL-15 levels correlate with disease severity,40 and both patients with AA and murine models exhibit increased IL-15 and IL-15Rα expression in the outer root sheath of the hair follicle compared with controls.12
Beyond IFN-γ, IL-2, and IL-15, Th2-associated cytokines, particularly IL-4 and IL-13, have more recently been implicated in AA. Genetic susceptibility involving IL-13 was identified in a large genome-wide study that included patients with and without atopy, while IL-4 polymorphisms were similarly associated with increased AA risk.11,20 Transcriptomic profiling of AA scalp tissues revealed IL-13 as one of the most highly upregulated cytokines, with significant increases in lesional versus nonlesional scalp, and serum IL-13 levels were also elevated in a cohort of 51 AA patients compared with controls.11 Alongside Th2 activation, elevations in Th1-related and cardiovascular- associated proteins (MMP9, AGER, IL1RL1/ST2/IL33R) were detected in both scalp and serum, with the scalp exhibiting a greater degree of immune dysregulation than serum.41 Additional evidence for Th2 involvement comes from elevated circulating levels of IL-4, IL-13 and IgE in AA patients.41–43 Furthermore, increased IL-13+/ cutaneous lymphocyte-associated antigen (CLA)+ skin-homing CD4+ T and CD8+ T cells have been identified in the peripheral blood of AA patients, with IL-13+CD4+CLA− T cell frequencies correlating with disease severity and IFN-γ+CD8+ T-cell frequencies correlating with disease duration.44
Biomarkers of Atopic Dermatitis
Immune dysregulation is central to the pathogenesis of AD, driving skin barrier disruption and the clinical manifestations of eczematous lesions and itch. AD has long been characterized as a Th2-polarized disease, with elevated expression of IL-13 and IL-4, and numerous additional Th2-associated markers.45 Increasing evidence, however, supports its heterogeneity, with distinct endotypes demonstrating variable contributions from Th1, Th17, and Th22 pathways. For example, Asian patients exhibit greater Th17 activation, Black patients display fewer FLG loss-of-function mutations, and pediatric patients show higher expression of TSLP receptor, among other immunologic differences.46 Several Th2-related biomarkers, including TARC/CCL17, periostin, and serum IgE, correlate with disease activity and therapeutic response.23
Epidermal barrier dysfunction is another hallmark of AD,47 characterized by dysregulated expression of key structural proteins such as filaggrin (FLG), transglutaminases, keratins, and intercellular structural proteins. FLG, a major structural protein in the stratum corneum, is critical for maintaining epidermal integrity; FLG mutations are associated with an increased risk of severe AD with earlier onset, longer persistence, and skin infection.48,49 Immune activation may also influence FLG gene expression, as IL-4 and IL-13 downregulate FLG and contribute to impaired barrier function.50 Critically, IL-4/JAK signaling–mediated suppression of antimicrobial peptides has direct clinical consequences in AD, which is associated with cutaneous infections including Staphylococcus aureus colonization and eczema herpeticum, with no corresponding increased risk in AA. Beyond suppressing antimicrobial peptides (AMP), these cytokines promote allergic inflammation and chemokine production, underscoring their central roles in AD pathophysiology.50 The clinical success of targeted therapies such as dupilumab (IL-4Rα blockade) and the IL-13 inhibitors tralokinumab and lebrikizumab further highlights the pathogenic relevance of these cytokines.3,51–53
Interleukin-22 (IL-22) is another proinflammatory cytokine implicated in AD due to its critical role in wound healing and immune stimulation. Produced primarily by Th22 with additional production from Th17 cells, IL-22 plays a central role in stimulating the proliferation, migration, and differentiation of the cells involved in tissue repair.54 IL-22 can directly stimulate the production of the extracellular matrix components in fibroblasts via binding to the IL-22 receptor and triggering downstream JAK/ STAT dimerization.55 In keratinocytes, IL-22 induces hyperproliferation and decreases FLG expression, contributing to epidermal thickening and barrier dysfunction.54 IL-22 may also modulate pruritus, as gastrin-related peptide (GRP), a mediator of non-histaminergic itch, is increased in AD and correlates with itch severity, with IL-22 upregulating GRP receptor expression in keratinocytes.56 Although anti-IL-22 therapies have not shown broad clinical efficacy, additional analysis suggested that inhibiting IL-22 in the subset of patients with high expression of IL-22 may have clinical utility.57 In this context, therapeutically engaging the IL-22 receptor itself may represent a more promising strategy, as emerging data from a Phase 2b clinical trial (NCT05923099) on temtokibart, a monoclonal antibody directed against the interleukin-22 receptor subunit alpha-1 (IL-22RA1), indicate encouraging early signals of efficacy.58–60
CCL17, also known as thymus and activation-regulated chemokine (TARC), is a chemokine produced by dendritic cells, endothelial cells, keratinocytes, and fibroblasts that acts as a chemoattractant for CCR4+ Th2 lymphocytes, playing a central role in the pathogenesis of AD by promoting Th2 cell migration into inflamed skin.61 CCL17/TARC is considered one of the most robust and specific biomarkers for monitoring AD activity and treatment response, and is used clinically in several countries to guide management.45,62 Its specificity extends beyond general atopic diseases, with elevated CCL17/TARC levels are more strongly associated with AD than with asthma or allergic rhinitis.63 CCL17/TARC is markedly overexpressed in lesional AD skin and elevated in the serum, correlating closely with disease activity and severity as measured by clinical scores such as SCORing Atopic Dermatitis (SCORAD).45,62 Elevated CCL17/TARC can be detected even before clinical onset of AD, supporting a role in early disease pathogenesis and as a predictive biomarker.64 Other Th2 chemokines, including CCL18, CCL22, and CCL26, are likewise elevated in AD and contribute to the self-perpetuating cycle of Th2-driven inflammation.45
Interleukin-31 (IL-31), a key pruritogenic cytokine produced primarily by Th2 cells, acts directly on sensory neurons and keratinocytes to induce itch, impair barrier function, and drive inflammation.65–67 Elevated IL-31 levels correlate strongly with disease severity and propagate the itch-scratch cycle, worsening skin lesions and quality of life.65 Nemolizumab, a humanized monoclonal antibody targeting the IL-31 receptor α (IL-31Rα), has demonstrated rapid and sustained reduction in pruritus, improvement in skin signs, and enhanced quality of life in patients with moderate-to-severe AD inadequately controlled by topical therapies.67
Thymic stromal lymphopoietin (TSLP) is highly expressed in the epidermis of patients with AD, with significantly elevated serum levels in both adults and children.68 Its production is induced by environmental triggers such as allergens, microbes, diesel exhaust, cigarette smoke, and chemical irritants.69 Notably, in a Korean birth cohort, skin tape stripping revealed increased TSLP expression in the skin of 2 month-old infants who later developed AD at 24 months, suggesting a pathogenic role early in disease development.70 TSLP overexpression is specific to AD lesions, with no significant elevations detected in nonlesional AD skin or in lesions from nickel-induced allergic contact dermatitis.71
Shared Pathways
Having outlined the distinct biomarker landscapes of AA and AD, collectively, current biomarker and genetic data suggest that AA and AD, though clinically distinct, share several key pathogenic pathways, with JAK-STAT dependent cytokine dysregulation and impaired Treg function emerging as central convergent mechanisms (Figure 1). These common mechanisms help to explain both their epidemiologic association and their emerging overlap in therapeutic strategies.
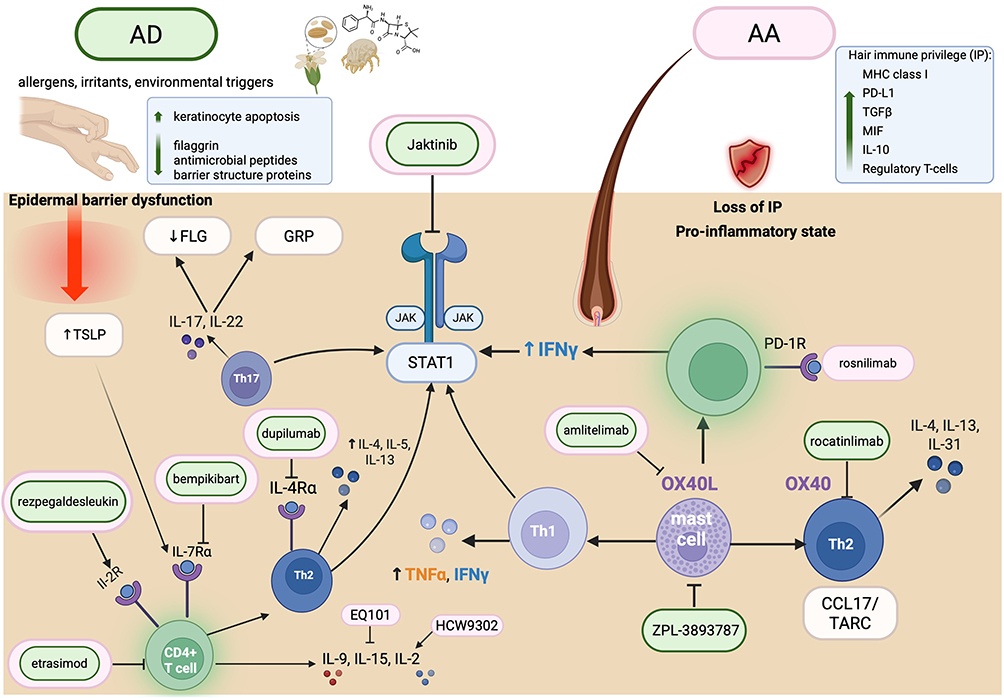 |
Figure 1 Convergent and divergent immunopathogenic pathways in atopic dermatitis (AD) and alopecia areata (AA), with therapeutic targets. AD (left) is characterized by a bidirectional interplay between type 2 inflammation and epidermal barrier dysfunction: Th2 cytokines (IL-4, IL-13) suppress epidermal differentiation proteins — including filaggrin (FLG) and loricrin — and disrupt stratum corneum lipid composition, while barrier impairment in turn facilitates allergen penetration and keratinocyte-derived TSLP release, amplifying the inflammatory cycle. AA (right) arises from collapse of hair follicle immune privilege (IP), characterized by loss of local immunosuppressive mediators (TGFβ, IL-10, PD-L1, MIF) and CD8+ T cell–mediated autoimmune attack on the follicular bulb. Both diseases converge on shared JAK-STAT signaling, Th1/Th2 cytokine dysregulation (IFN-γ, IL-4, IL-5, IL-13), mast cell activation, and OX40/OX40L costimulatory signaling. Therapeutic agents are color-coded by primary indication: green (AD) and pink (AA), shown with their respective molecular targets. Created in BioRender. Abbreviations: FLG, filaggrin; GRP, gastrin-releasing peptide; IP, immune privilege; TSLP, thymic stromal lymphopoietin; CCL17/TARC, thymus and activation-regulated chemokine. |
IFN-γ has been studied as a biomarker of disease severity in AA and disease chronicity in AD. IFN-γ and its downstream chemokines, particularly CXCL9 and CXCL10, are essential mediators of inflammatory amplification.72 In AA, IFN-γ is produced primarily by CD8+ T cells and other lymphocyte subsets, contributing to the collapse of hair follicle IP, upregulation of MHC class I, and recruitment of additional inflammatory cells, which collectively drive follicular damage and hair loss.38,73 Elevated serum IFN-γ levels and increased IFN-γ gene expression in lesional tissue correlate with disease severity, disease activity, the extent and chronicity of hair loss, and the degree of CD8+ T -cell perifollicular infiltration.74,75 Animal models further support its pathogenic contributions, as exogenous IFN-γ administration can induce AA-like lesions.76 In AD, IFN-γ sustains Th1- and Th22-cell activation, induces keratinocyte apoptosis, and impairs the epidermal barrier, thereby increasing transepidermal water loss.72 Chronic AD is characterized by progressive enrichment of IFN-γ driven Th1 pathways, paralleling the expansion of multiple immune pathways.77–79
Although TNF-α is not considered a primary driver of AA or AD pathogenesis, it plays an important homeostatic role in skin immunity, helping to maintain the balance between pro- and anti-inflammatory signals that preserves both hair follicle immune privilege and epidermal barrier integrity. Reports of TNF-α inhibitor-induced AA highlight this role,80 leading Perez et al to propose that TNF-α may contribute to the maintenance of hair follicle IP.81 Similarly, AD has been observed in patients receiving TNF-α inhibitors for inflammatory bowel disease.82 One proposed mechanism is that TNF-α blockade may shift the immunologic environment toward Th2 upregulation, with personal or familial atopic history predisposing individuals to anti-TNF-α–induced AD.82
IL-2 is another Th1 cytokine responsible for T-cell proliferation, including Treg survival and expansion, which positions it as a key homeostatic regulator across diverse inflammatory conditions.83 As discussed above, in AA, IL-2 promotes CD8+NKG2D+ T cell-mediated hair follicle destruction.12 In AD, impaired IL-2 production contributes to Treg dysfunction and loss of immune tolerance.83 This dual role as a pathogenic effector in AA versus a Treg sustainer in AD depends on dose and receptor context: low doses of IL-2 preferentially expand Tregs, whereas higher doses of IL-2 promotes effector T cell activation.84
The Th2 axis, composed of CD4+ T cells producing IL-4, IL-5, and IL-13, drives type 2 immunity, including eosinophil activation, IgE production, and barrier dysfunction.85 This pathway represents a shared immunologic mechanism in both AA and AD (Figure 1). Both diseases feature expansion of circulating Th2 and Th22 cells, and reduced regulatory T cells, with disease severity correlating with the degree of Th1/Th2/Th17 activation and keratin dysregulation.85 In AA, suppression of Th2 biomarkers with dupilumab correlates with clinical improvement and restoration of hair keratin expression, further supporting a pathogenic role of the Th2 axis in AA.86 Even less specific JAK pathway inhibitors such as ritlecitinib (JAK3/TEC) and brepocitinib (JAK1/TYK2) demonstrate that modulation of Th2 responses is strongly associated with improvements in keratin expression and clinical outcomes.87 In comorbid AA and AD, immune signatures differ based on atopic status. Kageyama et al demonstrated that atopic and non-atopic AA patients exhibit distinct transcriptomic profiles.5 Non-atopic AA is characterized by a more pronounced Th1-skewed immune activation, with higher ratios of CD4+ IFN-γ+ cells to IL-4+ or IL-13+ CD4+ cells.5 In contrast, patients with co-incident AD exhibit dense infiltration of both CXCR3+ (Th1-associated) and CCR4+ (Th2-associated) cells around the hair bulb.5 Furthermore, IgE levels are associated with dupilumab response in both atopic and non-atopic AA, suggesting that IgE is not exclusive to atopic disease.5 Dupilumab therapy significantly reduces CCR4+ cells (Th2) cell infiltration in atopic AA, whereas CXCR3+ cell numbers (Th1) remain unchanged. These findings underscore the immunologic heterogeneity of atopic and non-atopic AA and suggest that type 2 inflammation plays a particularly important role in AA associated with extrinsic AD, whereas non-atopic AA has more prominently type 1 inflammation.5
While IL-15 plays a well-characterized pathogenic role in AA — supporting CD8+ T cell expansion and correlating with disease severity — its contribution to AD pathogenesis remains poorly understood, representing a point of mechanistic divergence between the two diseases and raising uncertainty about whether IL-15–targeting strategies will have therapeutic relevance in AD.
The OX40 costimulatory pathway further contributes to T-cell–mediated inflammation and the development of tissue resident memory (TRM) T cells. OX40 is an inducible receptor predominantly expressed on activated T cells and is upregulated by cytokines such as IL-1, IL-2, IL-4, and TNF-α.88 Engagement of OX40 by OX40L promotes T-cell proliferation and survival, TRM cell generation, and proinflammatory cytokine production.88 Persistent antigen exposure and prolonged expression of IFN-γ, IL-4, IL-17, and IL-22 sustain OX40 signaling, perpetuating a self-amplifying inflammatory loop.89 In AD, OX40+ T cells and OX40L+ dendritic cells are enriched in lesional skin compared with nonlesional or psoriatic skin.89
In AA, mast cells in the perifollicular region also exhibit increased OX40L expression.36 As described above, OX40L+ mast cells activate CD8+ T cells through the OX40-OX40L interaction, enhancing IFN-γ production and promoting Th2 cell maturation, while downregulating signals required for Treg induction.36 These findings suggest that OX40L+ mast cells undermine Treg–mediated immune tolerance and thereby contribute directly to AA pathogenesis.90 Transcriptomic studies show robust upregulation of Th1 and Th2-associated products in AA scalp, with OX40 signaling playing a particularly important role in AA comorbid with atopy.91 Both lesional and nonlesional AA tissue exhibit increased perifollicular infiltration by OX40+ and OX40L+ cells, which strongly correlate with hair-keratin downregulation.91 Notably, current hair loss episode duration correlates positively with OX40+ infiltrates in atopic AA, but negatively in non-atopic AA, suggesting distinct immunologic trajectories.91 Finally, cutaneous OX40/OX40L upregulation is mirrored by elevations in circulating OX40+ and OX40L+ leukocytes, and OX40 is highly expressed on non-skin-homing Tregs in the context of increased circulating OX40L+ antigen presenting cells, further suggesting that Treg dysfunction in AA may originate systemically.91
Early mechanistic studies established that IFN-γ and interleukin-15, key cytokines in AA, signal through JAKs, and clinical case series demonstrated hair regrowth with oral JAK inhibitors in recalcitrant disease.2 More than 50 cytokines signal via the JAK – signal transducer and activator of transcription (STAT) pathway, wherein cytokines binding to transmembrane receptors trigger JAK dimerization, which then activate STATs to translocate to the nucleus and regulate transcription of target genes.92,93 The JAK–STAT pathway plays a central role in immune dysregulation in AD, contributing to exaggerated Th2 polarization, eosinophil activation, B-cell maturation, and suppression of Tregs.94 Additionally, IL-4–mediated activation of JAK–STAT signaling promotes AD pathogenesis by inducing epidermal chemokines, pro-inflammatory cytokines, and pro-angiogenic factors, while simultaneously suppressing antimicrobial peptide expression and key mediators of skin barrier integrity.94
Despite these convergent inflammatory axes, the site of immune activation in AA and AD imposes important therapeutic constraints that limit the interchangeability of treatment strategies. In AA, the principal target of immune attack is the hair follicle bulb, an anatomically deep structure residing in the mid-to-lower dermis or sometimes even subcutis, rendering topical therapies largely ineffective due to presumed inadequate drug penetration, an observation consistent with failed trials of topical JAK inhibitors in AA.95–97 By contrast, AD pathogenesis involves a bidirectional interplay between type 2 inflammation and epidermal barrier dysfunction,51,98 positioning the skin surface as a site of immune-driven pathology and a pharmacologically accessible target. Thus, barrier-directed therapies remain central to AD management but have little therapeutic rationale in AA. Furthermore, the regulatory T-cell landscape diverges between the two diseases. AA patients exhibit significantly depleted Tregs compared to both controls and AD patients, and this Treg deficiency correlates with activated CD8+ memory T cells,44 suggesting a fundamental collapse of peripheral immune tolerance coupled with cytotoxic effector activation — in contrast to AD, which is instead driven by CD4+ Th2 cells with compensatory Treg expansion.
Traditional Therapies for Alopecia Areata
Traditional treatments for AA include intralesional and topical corticosteroids, systemic steroids, contact immunotherapy, and traditional immunosuppressants, although these approaches are limited by variable efficacy and side effects.99 Intralesional corticosteroids remain the first-line treatment for adults with one to two small patches of hair loss, but their use is associated with pitting scars and limited effectiveness in more extensive disease.99 Systemic corticosteroids, such as prednisolone (with dosing regimens commonly in the range of 0.5 mg/kg/day tapering over 6–12 weeks),100 may be used in severe AA, but their utility is tempered by suboptimal durability of response and considerable adverse effects. Risks include weight gain, worsening diabetes or hypertension, and suppression of the hypothalamic–pituitary–adrenal axis, along with high relapse rates.100 Other traditional modalities include contact immunotherapy which stimulates an allergic contact dermatitis reaction using agents such as diphencyclopropenone (DPCP) and squaric acid dibutylester (SADBE) to promote hair regrowth.101 Adverse effects include urticaria, regional lymphadenopathy. This strategy is generally contraindicated in patients with acute, rapidly progressive AA.101 For more severe forms of AA, systemic immunosuppressants such as methotrexate and cyclosporine have been used, although their efficacy is inferior to that of newer targeted therapies and they carry risks of renal, hepatic and other toxicities.102,103 Topical and low dose oral minoxidil are also commonly used, typically as adjunctive therapy in combination with immune-modulating approaches.104 Limited discussion has centered around topical phosphodiesterase-4 inhibitors such as apremilast, which decrease proinflammatory cytokines and have shown moderate efficacy in smaller studies. However, evidence supporting their use in AA remains insufficient.99
Traditional Therapies for Atopic Dermatitis
In patients with mild AD, treatment focuses on reducing exposure to aggravating factors and minimizing flare frequency. For mild-to-moderate AD, topical corticosteroids have long been the cornerstone of therapy, but the emergence of non-steroidal topical therapies has expanded treatment options. These include topical calcineurin inhibitors (pimecrolimus and tacrolimus), topical phosphodiesterase inhibitors (crisaborole and roflumilast), topical JAK inhibitors (ruxolitinib), and topical aryl hydrocarbon receptor modulators (tapinarof). Phototherapy with narrowband UVB is another therapeutic option,105 while historically, systemic immunosuppressants (eg. methotrexate, cyclosporine, azathioprine, mycophenolate mofetil) were used for moderate-to-severe AD,106,107 though the therapeutic landscape has shifted substantially with the development of targeted biologics and small-molecule inhibitors.
JAK Inhibitors
The clinical efficacy of JAK inhibitors across both AA and AD provides perhaps the strongest validation of JAK-STAT signaling as a key shared pathogenic axis underlying both conditions. JAK inhibitors are an established class of drugs with significant therapeutic overlap for both AA and AD. JAKs are a family of non-receptor tyrosine kinases that transmit signals from the cell membrane to the cell nucleus via STAT proteins.93 Binding of cytokines to extracellular receptor domains induces JAK activation, making these kinases central regulators of cytokine-induced signal transport.93 Unlike biologics which are often subcutaneous or IV injections, most JAK inhibitors are administered orally.93 While delgocitinib 2% cream was recently approved for moderate to severe chronic hand eczema (CHE),108 large clinical trials of delgocitinib ointment,95 ruxolitinib 1.5% cream,96 and tofacitinib 2% ointment97 in AA failed to demonstrate marked clinical efficacy. All JAK inhibitors carry a black box warning for increased risk of major adverse cardiovascular events (MACE), venous thromboembolism, blood clots, severe infection, malignancy, and overall mortality as a result of the ORAL Surveillance trial.109 This randomized post-authorization safety study evaluated the side effects and safety profile of tofacitinib in rheumatoid arthritis patients ≥50 years old with at least one additional cardiovascular risk factor.109 It is worth noting that AA and AD patients treated with JAK inhibitors tend to be younger, often lack the cardiovascular comorbidities required for ORAL Surveillance enrollment, and are frequently not on concurrent methotrexate — factors that substantially limit direct extrapolation of these safety signals to dermatologic practice. When compared to dupilumab in AD patients, JAK inhibitors were associated with a greater risk of skin infections, herpes simplex, herpes zoster, anemia, neutropenia, thrombocytopenia, hyperlipidemia, and acneiform eruptions.110 However, risk of severe adverse events such as malignancy, VTE, or major adverse cardiovascular events were comparable between JAK inhibitor and dupilumab groups.110 Accordingly, the American Academy of Dermatology recommends reserving JAK inhibitors for patients who have failed or are not candidates for other systemic therapies and advises caution in patients with cardiovascular risk factors, history of malignancy, or chronic/recurrent infections.111
JAK inhibitors differ in their selectivity profiles. JAK1-selective agents (upadacitinib, abrocitinib) and JAK3-selective agents (ritlecitinib) primarily suppress inflammatory cytokine signaling, while additional JAK2 blockade may contribute to impaired hematopoiesis, providing a mechanistic basis for anemia which has been reported with pan-JAK inhibitors (tofacitinib, baricitinib).112,113 Importantly, however, JAK selectivity is relative rather than absolute: at therapeutic doses, “selective” inhibitors retain meaningful cross-JAK activity, and both clinical efficacy and adverse events likely reflect suppression of multiple JAK-dependent signaling pathways rather than blockade of a single pathway.114 This may partly explain why both JAK1-selective and less-selective agents demonstrate efficacy across AA and AD despite their nominally distinct cytokine targets.
For moderate-to-severe AD, the American Academy of Dermatology recommends the JAK1-selective inhibitors upadacitinib and abrocitinib as second-line systemic therapy for patients who have failed or are not candidates for other systemic agents, including biologics such as dupilumab.115 In the JADE COMPARE trial, a Phase 3 head to head comparing 100 or 200mg abrocitinib to standard dupilumab dosing and placebo in adults with moderate to severe AD, abrocitinib 200 mg once daily demonstrated significantly higher response rates than placebo at week 12: 70.3% of patients achieved EASI-75 compared with 27.1% on placebo, and 48.4% reached an investigator global response (IGA) 0/1 response versus ~4.0% with placebo.116 Neither abrocitinib dose differed significantly from dupilumab for most secondary outcomes at later time points.116 The 200 mg dose showed superior itch relief at week 2 compared with dupilumab.116 A different head to head trial comparing upadacitinib 30mg to dupilumab (HEADS UP) showed that 72.4% of upadacitinib treated patients achieved the week 16 primary endpoint of EASI-75 compared to 62.6% of the dupilumab treated cohort.117 All ranked secondary end points also demonstrated the superiority of upadacitinib vs dupilumab at that time point.117
Focusing on the use of JAK inhibitors in AA, meta-analyses of randomized trials confirm that they are associated with significantly greater odds of achieving ≥50% and ≥90% improvement in SALT scores compared to placebo, with a favorable safety profile and no increase in severe adverse events over short-term follow-up.118,119 Real-world cohort studies corroborate these findings, showing substantial regrowth in most patients over 6–12 months, although relapse is common after discontinuation.120 In particular, the first FDA-approved treatment for AA was baricitinib, a JAK1/JAK2 inhibitor approved in 2022 for adult patients with severe AA.2 Ritlecitinib, a JAK3/TEC inhibitor, and deuruxolitinib, JAK1/JAK2-selective, were approved in 2023 and 2024, respectively, with ritlecitinib currently being the only FDA approved JAK inhibitor for patients ≥12 years.121 In the pivotal phase 3 BRAVE-AA1 and BRAVE-AA2 trials, baricitinib (2 mg and 4 mg daily) was evaluated in adults with a Severity of Alopecia Tool (SALT) score ≥ 50 (≥ 50% scalp hair loss) and a current AA episode lasting > 6 months to < 8 years without spontaneous improvement. The primary outcome was a SALT≤20 at week 36. At week 36, the estimated proportion of patients achieving a SALT ≤20 in BRAVE-AA1 was 38.8% with baricitinib 4 mg, 22.8% with 2 mg, and 6.2% with placebo; corresponding rates in BRAVE-AA2 were 35.9%, 19.4%, and 3.3%, respectively. In BRAVE-AA1, baricitinib 4 mg demonstrated a 32.6% improvement over placebo (95% CI, 25.6–39.5), while the 2-mg dose showed a 16.6% improvement (95% CI, 9.5–23.8), with both doses significantly outperforming placebo (p<0.001 for each comparison).13 Findings from the BRAVE-AA1 randomized withdrawal study highlight the chronicity of AA: after 52 weeks of baricitinib, approximately 80% of patients who discontinued treatment relapsed by week 152, compared to only 7% of those who continued therapy.13 Relapse typically began to manifest around 8 weeks after stopping the drug.13 Expert consensus currently recommends considering discontinuation of systemic therapy only after complete and stable regrowth has been maintained for at least 6 months, or when disease activity is sufficiently controlled to allow transition to topical therapy. However, for most patients with severe AA, long-term maintenance therapy is required to sustain regrowth.13 A new development in JAK inhibition for both AA (NCT05051761)122 and AD (NCT05526222)123 is represented by the compound jaktinib (also known as gecacitinib), a broad JAKi.
Dupilumab
Treatment of moderate-to-severe AD was revolutionized with the approval of dupilumab, the first targeted therapy shown to be effective for AD in 2017.3 Dupilumab is a fully human monoclonal antibody that targets the IL-4Rα subunit, thereby inhibiting signaling of both IL-4 and IL-13, which are central drivers of Th2 inflammation, implicated in AD and other allergic diseases.3,4,124,125 By blocking the shared IL-4Rα subunit, dupilumab disrupts the downstream inflammatory cascade, reducing Th2-associated cytokines, chemokines, and markers of epidermal hyperplasia, while promoting restoration of skin barrier function.125
Robust evidence from phase 3 randomized controlled trials demonstrates that dupilumab significantly improves clinical signs, symptoms, and quality of life in adults with moderate-to-severe AD inadequately controlled by topical therapies.3 In the SOLO 1 (NCT02277743) and SOLO 2 (NCT02277769) trials, 16 weeks of dupilumab (300 mg subcutaneously weekly or every other week) resulted in a significantly higher proportion of patients achieving clear or almost clear skin and at least 75% improvement in Eczema Area and Severity Index (EASI-75) compared to placebo, with additional reduction in pruritus (an improvement of at least 4 points in patient-reported pruritus score by week 16) and improvement in mental health outcomes (a reduction of at least 4 points in Dermatology Quality of Life Index surveys).3,4 Long-term safety of dupilumab is favorable with FDA approval as young as age 6 months.126 Dupilumab was also effective in treating anti-TNF-α–induced AD, suggesting a potential role for Th2 inhibition in patients with AD induced during anti-TNF-α therapies.127 Molecular studies confirm that dupilumab progressively normalizes lesional skin transcriptomes and suppresses systemic type 2 biomarkers,51 a finding that provides a biological rationale for its potential utility in AA. Indeed, initial case series showed that AD patients with AA treated with dupilumab not only experienced improvement in their skin, but also significant hair regrowth.128 This paved the way for current clinical trials investigating the potential benefit of dupilumab in treating AA in both adult and pediatric patients (NCT03359356).129 Thus far, AA patients with and without concomitant AD have shown improvement in SALT scores in response to dupilumab.129 Interestingly, responders were more likely to have personal or familial atopy and/or high serum total IgE levels compared to non-responders. A possible explanation to these observations may be that increased levels of serum IgE are indicative of Th2-associated inflammation in the hair follicle, which may respond better to Th2-targeting approaches.129
Dupilumab paved the way for more biologics in AD targeting the type 2 inflammatory axis that may also show some potential in treating AA. Two anti-IL-13 antibodies, tralokinumab and lebrikizumab, have shown efficacy in AD and were recently FDA approved in June 2021 and September 2024 for tralokinumab and lebrikizumab, respectively.52,53,130 In phase 3 clinical trials, 16–22% of patients on tralokinumab achieved an IGA of 0–1 (indicating clear or almost clear skin) compared to 7–10% in placebo,131 while 33–43% of patients on lebrikizumab achieved an IGA of 0–1 compared to placebo rates of 10–12%.132 Although a few reports have been published describing cases of AA successfully treated with tralokinumab,133,134 a Phase 2 randomized, double-blind, placebo-controlled pilot study (NCT02684097) on adults affected by moderate to severe AA did not show significant clinical efficacy.135 While the underlying mechanistic explanation for the failure of tralokinumab in AA remains unclear, one possibility is that IL-13 blockade alone is insufficient in AA, and that dupilumab’s broader dual IL-4/IL-13 blockade, combined with patient enrichment for atopic background and elevated IgE, may represent necessary conditions for type 2–targeted efficacy in AA. Alternatively, the dose that demonstrated efficacy in AD (300 mg q2weeks) may be insufficient to adequately suppress the more complex inflammatory milieu of AA.
Investigative Treatments
Building on the approved therapies discussed above, and given the overlapping immunologic mechanisms underlying AD and AA pathogenesis, the therapeutic landscape for agents capable of treating both conditions is highly promising. Anti-OX40/OX40L agents are among the most advanced investigational systemic therapies for moderate-to-severe AD (Table 1).136,137 A phase 2b trial of amlitelimab (anti-OX40L) demonstrated significant reductions in Eczema Area and Severity Index (EASI) scores, with durable clinical responses persisting for months after treatment cessation, suggesting potential disease-modifying effects,136 and a recent phase 3 trial (COAST 1) met all primary (IGA of 0–1) and key secondary endpoints (at least a 4 point reduction in patient reported pruritus score).138 Phase 3 trials for rocatinlimab (anti-OX40) in moderate-to-severe AD (ROCKET, ROCKET HORIZON) also recently showed statistically significant improvement in IGA and EASI-75 at 24 weeks compared to placebo.137,139 Since it has been established that blocking OX40 signaling may dampen autoreactive T-cell expansion, reduce inflammatory cytokine production, and restore immune tolerance at the hair follicle,140 amlitelimab is also currently in phase 2 trials for severe AA (NCT06444451) as the concept of tempering pathogenic T cell responses could also apply to AA.141
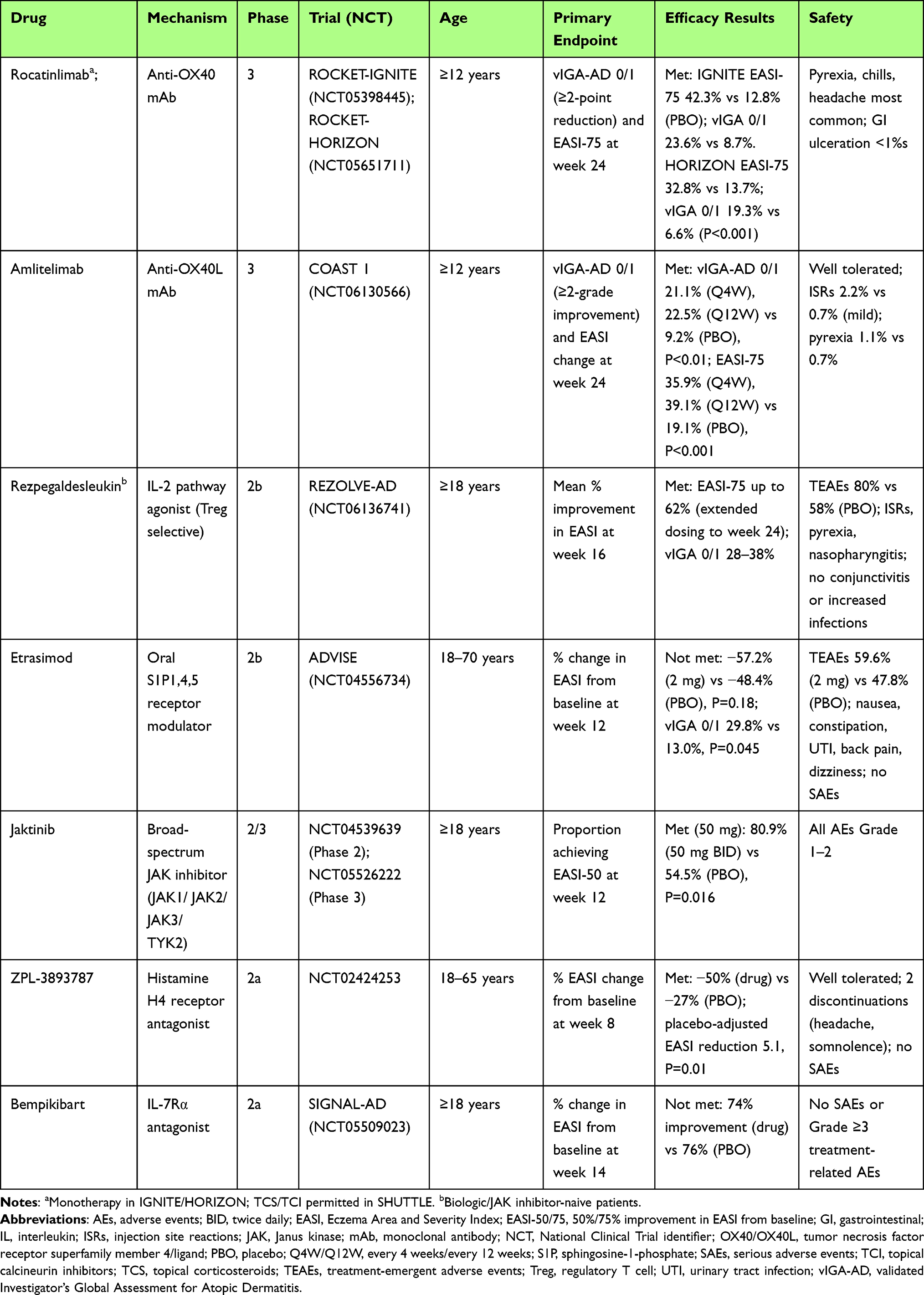 |
Table 1 Investigational Therapies for Atopic Dermatitis |
As discussed above, the role of IL-2 in the immunopathogenesis of both AA and AD makes this cytokine an appealing therapeutic target, supporting the development of novel IL-2–modulating agents for both conditions (Table 2). The Treg-selective IL-2 receptor agonist rezpegaldesleukin is currently the most extensively studied molecule within this pharmacologic class. While current biologics like dupilumab focus on neutralizing the effects of IL-4 and IL-13, rezpegaldesleukin is an agonist that stimulates the expansion and activity of regulatory Tregs. Rezpegaldesleukin demonstrated a favorable safety and tolerability profile, with consistent pharmacokinetics across dosing cohorts (AD, psoriasis).83 In the AD cohort, patients receiving the higher dose achieved a mean 83% improvement in EASI score after 12 weeks.83 Notably, EASI scores were sustained for up to 36 weeks post-treatment discontinuation in a majority of week 12 responders (71% and 80%, respectively).83 A phase 2b trial for this drug in AD (REZOLVE-AD) was recently completed with significant reductions in EASI scores after 16 weeks of therapy.142
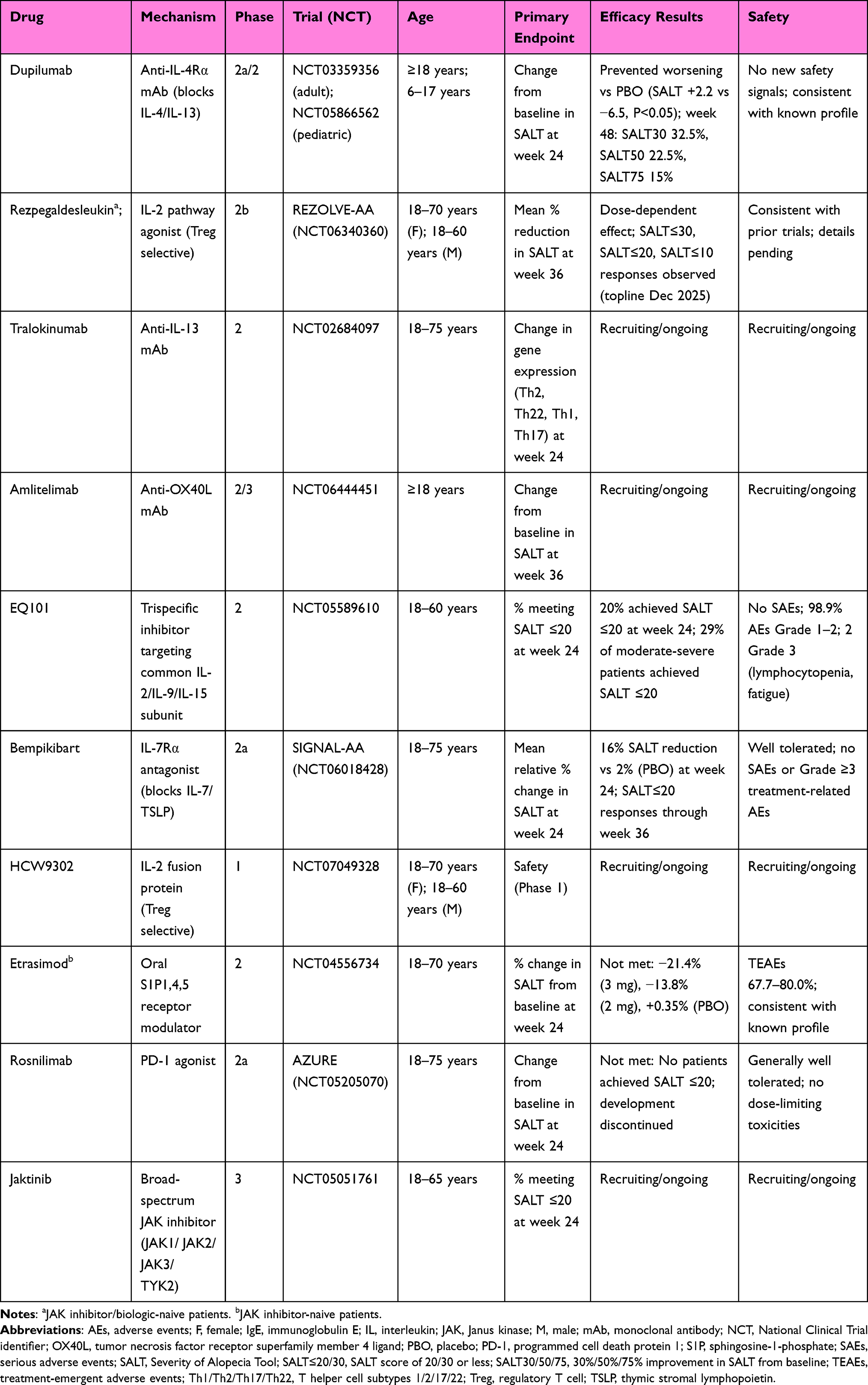 |
Table 2 Investigational Therapies for Alopecia Areata |
Rezpegaldesleukin is also currently being studied in a phase 2b trial (NCT06340360) for severe to very severe AA in patients aged 12 years and older.143 After 36 weeks of therapy, patients on rezpegaldesleukin achieved a mean reduction in SALT of 28.2% - 30.3% compared to 11.2% in placebo, providing promising results for future phase 3 trials.144 A novel IL-2 fusion protein (HCW9302) has entered Phase 1 trial for moderate-to-severe AA as of August 2025 (NCT07049328).145 This molecule selectively engages IL-2 receptors predominantly expressed on regulatory T cells, functioning similarly to rezpegaldesleukin to enhance Treg-mediated immune regulation. Notably, IL-2 has been incorporated as one of the targets in a tri-specific antibody currently under investigation for AA, reflecting how the simultaneous blockade of multiple immunologic pathways is emerging as a new therapeutic frontier. EQ101 is a first-in-class tri-specific inhibitor for AA that targets the common receptor subunit shared by IL-2, IL-9, and IL-15, thereby selectively blunting those cytokines’ activity without broad JAK inhibition.146 This strategy aims to more precisely inhibit pathogenic T cell signals in AA, particularly IL-15–driven cytotoxic T cells, with IL-2 and IL-9 contributing to T cell and mast cell activation. Preclinical data showed EQ101 reversed alopecia in a mouse model more effectively than ruxolitinib, and phase 2 data has demonstrated favorable safety, tolerability, and efficacy in adults.146 However, the efficacy of EQ101 for AD has yet to be examined and its specific targets may not address the pathogenic mechanisms underlying AD.
Another compound of interest is bempikibart (ADX-194), a monoclonal antagonist of the IL-7 receptor α-chain. IL-7 expression is critical for T cell survival and expansion, and blockade of IL-7Rα suppresses inflammatory T cell responses while relatively sparing Treg populations.147 In AA, IL-7Rα inhibition halted disease progression and even reversed early-stage disease in murine models, underscoring the relevance of IL-7 signaling in AA pathogenesis.147 Although IL-7 is not considered a primary driver of AD, IL-7 and TSLP share structural similarities and both signal through the IL-7Rα subunit.148 This overlap provides a mechanistic rationale suggesting that IL-7Rα-mediated pathways may influence immune dysregulation in AD as well. Thus, IL-7 signaling may have pathogenic relevance in both AA and AD by modulating the balance between inflammatory T-cell activity and regulatory control. For these reasons, bempikibart has advanced into Phase 2 development for both AA and AD. In AA, early efficacy was observed in the Phase 2a SIGNAL-AA trial (NCT06018428), where adults treated over 24 weeks, with follow-up through 36 weeks, registered mean SALT reductions of 16% at Weeks 24 versus 2% with placebo, and 9% of treated patients achieved SALT ≤20 compared with 0% in the placebo group.149 In AD, the two-part Phase 2a SIGNAL-AD study (NCT05509023) evaluated subcutaneous dosing every two weeks in adults with moderate-to-severe AD. In Part A (n=15), mean EASI improvements at Week 14 were 58% with 2 mg/kg, 84% with 3 mg/kg, and 72% pooled, versus 38% for placebo. However, in Part B (n=106), bempikibart 200 mg Q2W produced a 74% mean EASI improvement at Week 14, essentially identical to placebo (76%), and thus the primary endpoint was not met. This failure may be due to inadequate study design given the high placebo response rate in Part B. Post-hoc analysis suggested some efficacy in the bempikibart compared to control group, but further studies will be needed to prove efficacy or lack thereof. The drug was well-tolerated with no serious or Grade≥3 treatment-related adverse events. As a result, current development is now primarily focused on AA rather than AD.150
Antihistamines and histamine receptor antagonists may also play a dual role in AA/ AD. The histamine H4 receptor on eosinophils and T cells mediates itch and inflammation. A drug candidate, ZPL-3893787, an H4 antagonist, reached Phase 2 in AD and showed a reduction in eczema severity and itch compared to placebo.151 There is some clinical evidence for using antihistamines in alopecia areata as an adjunctive therapy, consistent with the concept of an allergic or atopic immune component in AA.152–154
Finally, several early (Phase 1 and Phase 2) investigational agents with novel mechanisms of action are also being explored. Although these molecules have yet to establish clinical efficacy, they may help refine future therapeutic targets. Among them, etrasimod, a sphingosine 1-phosphate (S1P) receptor modulator that retains lymphocyte subsets within skin-draining lymph nodes to limit migration into inflamed tissue,155 showed a statistically significant decrease in IGA (29.8%) compared to placebo (13%) in a 12-week phase 2b trial for AD.156 On the other hand, it failed to meet primary and secondary endpoints at week 24 in a Phase 2, randomized, double-blind, placebo-controlled trial evaluating the efficacy and safety of etrasimod in adults with moderate-to-severe AA (NCT04556734).157 While any mechanistic explanations remain speculative given lack of published data, this failure may reflect the inability of S1P-mediated lymphocyte retention in lymph nodes to adequately address the established tissue-resident memory T cell infiltrate in AA, which by definition is not recirculating and therefore not susceptible to this mechanism of action. Similarly, rosnilimab, a programmed death (PD) 1 agonist that was examined recently in phase 2 studies for AA, failed to meet primary or secondary endpoints in both groups.158 Rosnilimab’s failure may reflect downregulation or dysfunction of PD-1 on chronically activated perifollicular CD8+ T cells in established AA, limiting the efficacy of checkpoint-based suppression in this context, or may alternatively reflect insufficient trial power. Nevertheless, its mechanism of action as an immune checkpoint agonist, potentially broadly decreasing T cell overactivity, is a novel approach to temper the T-cell mediated immune response seen in AA and AD.
Conclusion
Once regarded as immunologically distinct disorders, AD and AA are now understood to share convergent inflammatory pathways — centrally, JAK-STAT dependent cytokine dysregulation and Treg insufficiency/dysfunction — that together explain both their epidemiologic association and their emerging therapeutic overlap. There are several important limitations of the current evidence base. Much of the data supporting shared mechanisms derives from transcriptomic data. Agents effective in one disease may not adequately address the full pathogenic architecture of the other. Critically, the shared mechanism framework appears most robust for the atopic-associated AA endotype, wherein Th2 dysregulation plays a prominent pathogenic role, and less so for non-atopic AA, which is characterized by predominantly Th1-skewed inflammation. As the field advances, endotype-stratified approaches, selecting therapies based on atopic background, serum IgE, and transcriptomic immune signatures, represent the most promising path toward personalized, potentially disease-modifying interventions for both AA and AD. Continued advances in our understanding of hair follicle immune privilege, T-cell dysregulation, and type 1 and type 2 immune circuits are rapidly reshaping the treatment landscape. Emerging shared therapeutic strategies, ranging from JAK inhibitors to OX40/OX40L blockade and IL-2–modulating compounds, point toward a future in which interventions are not merely immunosuppressive but restore immune homeostasis and potentially modify disease course.159
Data Sharing Policy
Data sharing is not applicable to this article as no data were created or analyzed in this study.
Author Contributions
A.T.: Conceptualization, Data curation, Writing – original draft, Writing – review & editing, Visualization. G.R.: Data curation, Writing – original draft. F.M.M.: Conceptualization, Validation, Writing – review & editing. J.L.: Data curation, Writing – original draft. E.D.D.: Conceptualization, Validation, Writing – review & editing, Visualization. B.U.: Conceptualization, Validation, Writing – review & editing, Visualization, Supervision, Project administration.
All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding was utilized in the preparation of this article.
Disclosure
BU is an employee of Mount Sinai and has received research funds (grants paid to the institution) from: Bristol Myers Squibb, Incyte, Rapt Therapeutics, Pfizer, and Sanofi. He is also a consultant for AbbVie, Arcutis Biotherapeutics, Apogee, Therapeutics Bristol Myers Squibb, Botanix Pharmaceuticals, Castle Biosciences, Ebla Holdco, Fresenius Kabi, Galderma, J&J, Leo Pharma, Lilly, Nektar Therapeutics, Pfizer, Primus Pharmaceuticals, Sanofi, Sun Pharma, UCB, Veradermics, VRG Therapeutics.
FMM declares consulting fees and speaking fees from Abbvie, Eli Lilly, and Sanofi, and consulting fees from Almirall, Leopharma, and Pfizer.
The authors declare no other conflicts of interest with this study.
References
1. Islam N, Leung PS, Huntley AC, Gershwin ME. The autoimmune basis of alopecia areata: a comprehensive review. Autoimmun Rev. 2015;14(2):81–20. doi:10.1016/j.autrev.2014.10.014
2. Lensing M, Jabbari A. An overview of JAK/STAT pathways and JAK inhibition in alopecia areata. Front Immunol. 2022;13:955035. doi:10.3389/fimmu.2022.955035
3. Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371(2):130–139. doi:10.1056/NEJMoa1314768
4. Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375(24):2335–2348. doi:10.1056/NEJMoa1610020
5. Kageyama R, Ito T, Hanai S, et al. Immunological properties of atopic dermatitis-associated alopecia areata. Int J Mol Sci. 2021;22(5):2618. doi:10.3390/ijms22052618
6. Dainichi T, Iwata M, Kaku Y. Alopecia areata: what’s new in the epidemiology, comorbidities, and pathogenesis? J Dermatol Sci. 2023;112(3):120–127. doi:10.1016/j.jdermsci.2023.09.008
7. Krajewski PK, Jastrząb B, Szepietowski JC, Jankowska-Konsur A, Saceda Corralo D. Immune-mediated disorders in patients with alopecia areata: systematic review and meta-analysis. Int J Dermatol. 2025;64(11):2029–2037. doi:10.1111/ijd.17895
8. Huang KP, Mullangi S, Guo Y, Qureshi AA. Autoimmune, atopic, and mental health comorbid conditions associated with alopecia areata in the United States. JAMA Dermatol. 2013;149(7):789–794. doi:10.1001/jamadermatol.2013.3049
9. Brunner PM, Leung DYM, Guttman-Yassky E. Immunologic, microbial, and epithelial interactions in atopic dermatitis. Ann Allergy Asthma Immunol. 2018;120(1):34–41. doi:10.1016/j.anai.2017.09.055
10. Leung DY, Guttman-Yassky E. Deciphering the complexities of atopic dermatitis: shifting paradigms in treatment approaches. J Allergy Clin Immunol. 2014;134(4):769–779. doi:10.1016/j.jaci.2014.08.008
11. Suárez-Fariñas M, Ungar B, Noda S, et al. Alopecia areata profiling shows TH1, TH2, and IL-23 cytokine activation without parallel TH17/TH22 skewing. J Allergy Clin Immunol. 2015;136(5):1277–1287. doi:10.1016/j.jaci.2015.06.032
12. Xing L, Dai Z, Jabbari A, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med. 2014;20(9):1043–1049. doi:10.1038/nm.3645
13. King B, Ohyama M, Kwon O, et al. Two phase 3 trials of baricitinib for alopecia areata. N Engl J Med. 2022;386(18):1687–1699. doi:10.1056/NEJMoa2110343
14. Lee HH, Gwillim E, Patel KR, et al. Epidemiology of alopecia areata, ophiasis, totalis, and universalis: a systematic review and meta-analysis. J Am Acad Dermatol. 2020;82(3):675–682. doi:10.1016/j.jaad.2019.08.032
15. Mostaghimi A, Gao W, Ray M, et al. Trends in prevalence and incidence of alopecia areata, alopecia totalis, and alopecia universalis among adults and children in a us employer-sponsored insured population. JAMA Dermatol. 2023;159(4):411–418. doi:10.1001/jamadermatol.2023.0002
16. Lee H, Jung SJ, Patel AB, Thompson JM, Qureshi A, Cho E. Racial characteristics of alopecia areata in the United States. J Am Acad Dermatol. 2020;83(4):1064–1070. doi:10.1016/j.jaad.2019.06.1300
17. Zhou J, Liang L, Zhang H, et al. Global burden of alopecia areata and associated diseases: a trend analysis from 1990 to 2021. J Cosmet Dermatol. 2025;24(3):e70076. doi:10.1111/jocd.70076
18. Ho C-Y, Wu C-Y, Chen JY-F, Wu C-Y. Clinical and genetic aspects of alopecia areata: a cutting edge review. Genes. 2023;14(7):1362. doi:10.3390/genes14071362
19. Wohlmuth-Wieser I, Osei JS, Norris D, et al. Childhood alopecia areata—data from the national alopecia areata registry. Pediatric Dermatol. 2018;35(2):164–169. doi:10.1111/pde.13387
20. Jagielska D, Redler S, Brockschmidt FF, et al. Follow-up study of the first genome-wide association scan in alopecia areata: IL13 and KIAA0350 as susceptibility loci supported with genome-wide significance. J Invest Dermatol. 2012;132(9):2192–2197. doi:10.1038/jid.2012.129
21. Betz RC, Petukhova L, Ripke S, et al. Genome-wide meta-analysis in alopecia areata resolves HLA associations and reveals two new susceptibility loci. Nat Commun. 2015;6(1):5966. doi:10.1038/ncomms6966
22. Tian J, Zhang D, Yang Y, et al. Global epidemiology of atopic dermatitis: a comprehensive systematic analysis and modelling study. Br J Dermatol. 2023;190(1):55–61. doi:10.1093/bjd/ljad339
23. Liu D, Patel D, Lau M, et al. A translational approach to improve therapeutics in atopic dermatitis and beyond. J Immunol. 2025;214(9):2165–2179. doi:10.1093/jimmun/vkaf049
24. Bylund S, Kobyletzki LB, Svalstedt M, Svensson Å. Prevalence and Incidence of Atopic Dermatitis: a Systematic Review. Acta Derm Venereol. 2020;100(12):adv00160. doi:10.2340/00015555-3510
25. Narla S, Silverberg JI. Current updates in the epidemiology and comorbidities of atopic dermatitis. Ann Allergy Asthma Immunol. doi:10.1016/j.anai.2025.05.003
26. Wei Y-H, Tai Y-H, Dai Y-X, Chang Y-T, Chen T-J, Chen M-H. Bidirectional association between alopecia areata and atopic dermatitis: a population-based cohort study in Taiwan. Clin Exp Immunol. 2020;50(12):1406–1414. doi:10.1111/cea.13729
27. Davis DMR, Drucker AM, Alikhan A, et al. American academy of dermatology guidelines: awareness of comorbidities associated with atopic dermatitis in adults. J Am Acad Dermatol. 2022;86(6):1335–1336.e18. doi:10.1016/j.jaad.2022.01.009
28. Mohan GC, Silverberg JI. Association of vitiligo and alopecia areata with atopic dermatitis: a systematic review and meta-analysis. JAMA dermatol. 2015;151(5):522–528. doi:10.1001/jamadermatol.2014.3324
29. Zhou W, Cai J, Li Z, Lin Y. Association of atopic dermatitis with autoimmune diseases: a bidirectional and multivariable two-sample mendelian randomization study. Front Immunol. 2023;14:1132719. doi:10.3389/fimmu.2023.1132719
30. O’Hagan R, Caldas SA, Correa da Rosa JM, Guttman-Yassky E, Ungar B. The impact of atopic dermatitis on alopecia areata: a 2-sample Mendelian randomization study. J Am Acad Dermatol. 2023;89(3):600–602. doi:10.1016/j.jaad.2023.05.023
31. Hitaka T, Haruyama S, Ohmori S, et al. Treatment outcomes and considerations for topical immunotherapy in patients with alopecia totalis and alopecia universalis. Front Med Lausanne. 2025;12:1573929. doi:10.3389/fmed.2025.1573929
32. Ucak H, Cicek D, Demir B, Erden I, Ozturk S. Prognostic factors that affect the response to topical treatment in patchy alopecia areata. J Eur Acad Dermatol Venereol. 2014;28(1):34–40. doi:10.1111/jdv.12043
33. Bertolini M, McElwee K, Gilhar A, Bulfone-Paus S, Paus R. Hair follicle immune privilege and its collapse in alopecia areata. Exp Dermatol. 2020;29(8):703–725. doi:10.1111/exd.14155
34. Paus R, Ito N, Takigawa M, Ito T. The hair follicle and immune privilege. J Investig Dermatol Symp Proc. 2003;8(2):188–194. doi:10.1046/j.1087-0024.2003.00807.x
35. Ito T, Ito N, Bettermann A, Tokura Y, Takigawa M, Paus R. Collapse and restoration of MHC class-I-dependent immune privilege: exploiting the human hair follicle as a model. Am J Pathol. 2004;164(2):623–634. doi:10.1016/s0002-9440(10)63151-3
36. Bertolini M, Zilio F, Rossi A, et al. Abnormal interactions between perifollicular mast cells and CD8+ T-cells may contribute to the pathogenesis of alopecia areata. PLoS One. 2014;9(5):e94260. doi:10.1371/journal.pone.0094260
37. Ito T, Ito N, Saatoff M, et al. Maintenance of hair follicle immune privilege is linked to prevention of NK cell attack. J Invest Dermatol. 2008;128(5):1196–1206. doi:10.1038/sj.jid.5701183
38. Gilhar A, Laufer-Britva R, Keren A, Paus R. Frontiers in alopecia areata pathobiology research. J Allergy Clin Immunol. 2019;144(6):1478–1489. doi:10.1016/j.jaci.2019.08.035
39. Zhang X, Zhao Y, Ye Y, et al. Lesional infiltration of mast cells, Langerhans cells, T cells and local cytokine profiles in alopecia areata. Arch Dermatol Res. 2015;307(4):319–331. doi:10.1007/s00403-015-1539-1
40. El Aziz Ragab MA, Hassan EM, El Niely D, Mohamed MM. Serum level of interleukin-15 in active alopecia areata patients and its relation to age, sex, and disease severity. Postepy Dermatol Alergol. 2020;37(6):904–908. doi:10.5114/ada.2020.102103
41. Glickman JW, Dubin C, Dahabreh D, et al. An integrated scalp and blood biomarker approach suggests the systemic nature of alopecia areata. Allergy. 2021;76(10):3053–3065. doi:10.1111/all.14814
42. Song T, Pavel AB, Wen HC, et al. An integrated model of alopecia areata biomarkers highlights both T(H)1 and T(H)2 upregulation. J Allergy Clin Immunol. 2018;142(5):1631–1634.e13. doi:10.1016/j.jaci.2018.06.029
43. Attia EA, El Shennawy D, Sefin A. Serum Interleukin-4 and total immunoglobulin e in nonatopic alopecia areata patients and HLA-DRB1 Typing. Dermatol Res Pract. 2010;2010:503587. doi:10.1155/2010/503587
44. Czarnowicki T, He HY, Wen H-C, et al. Alopecia areata is characterized by expansion of circulating Th2/Tc2/Th22, within the skin-homing and systemic T-cell populations. Allergy. 2018;73(3):713–723. doi:10.1111/all.13346
45. Renert-Yuval Y, Thyssen JP, Bissonnette R, et al. Biomarkers in atopic dermatitis-a review on behalf of the International Eczema Council. J Allergy Clin Immunol. 2021;147(4):1174–1190.e1. doi:10.1016/j.jaci.2021.01.013
46. Czarnowicki T, He H, Krueger JG, Guttman-Yassky E. Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol. 2019;143(1):1–11. doi:10.1016/j.jaci.2018.10.032
47. Schleimer RP, Berdnikovs S. Etiology of epithelial barrier dysfunction in patients with type 2 inflammatory diseases. J Allergy Clin Immunol. 2017;139(6):1752–1761. doi:10.1016/j.jaci.2017.04.010
48. Irvine AD, McLean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med. 2011;365(14):1315–1327. doi:10.1056/NEJMra1011040
49. Sandilands A, Sutherland C, Irvine AD, McLean WH. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci. 2009;122(Pt 9):1285–1294. doi:10.1242/jcs.033969
50. Hussein YM, Shalaby SM, Nassar A, Alzahrani SS, Alharbi AS, Nouh M. Association between genes encoding components of the IL-4/IL-4 receptor pathway and dermatitis in children. Gene. 2014;545(2):276–281. doi:10.1016/j.gene.2014.04.024
51. Guttman-Yassky E, Bissonnette R, Ungar B, et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J Allergy Clin Immunol. 2019;143(1):155–172. doi:10.1016/j.jaci.2018.08.022
52. Guttman-Yassky E, Sun Z, Mena LR, et al. Lebrikizumab rapidly lowers inflammatory biomarkers with clinical correlations in moderate-to-severe atopic dermatitis. Dermatol Ther. 2025;15(9):2595–2614. doi:10.1007/s13555-025-01481-4
53. Bieber T. Atopic dermatitis: an expanding therapeutic pipeline for a complex disease. Nat Rev Drug Discov. 2022;21(1):21–40. doi:10.1038/s41573-021-00266-6
54. Zaharie RD, Popa C, Schlanger D, Vălean D, Zaharie F. The role of il-22 in wound healing. potential implications in clinical practice. Int J Mol Sci. 2022;23(7):3693. doi:10.3390/ijms23073693
55. McGee HM, Schmidt BA, Booth CJ, et al. IL-22 promotes fibroblast-mediated wound repair in the skin. J Invest Dermatol. 2013;133(5):1321–1329. doi:10.1038/jid.2012.463
56. Lou H, Lu J, Choi EB, et al. Expression of IL-22 in the skin causes th2-biased immunity, epidermal barrier dysfunction, and pruritus via stimulating epithelial th2 cytokines and the grp pathway. J Immunol. 2017;198(7):2543–2555. doi:10.4049/jimmunol.1600126
57. Guttman-Yassky E, Brunner PM, Neumann AU, et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: a randomized, double-blind, phase 2a trial. J Am Acad Dermatol. 2018;78(5):872–881.e6. doi:10.1016/j.jaad.2018.01.016
58. Del Duca E. Temtokibart, an IL-22RA1 monoclonal antibody broadly dampens gene expression markers of activated immune pathways in atopic dermatitis: results from a phase 2b trial subgroup analysis. 2025.
59. Weidinger S. IL-22RA1 antagonism with temtokibart provides significant early and sustained improvements in atopic dermatitis: results from a phase 2b dose-finding trial. 2025.
60. Pharma L. LEO Pharma presents two late-breaking abstracts for temtokibart reporting positive Phase 2b efficacy, safety and biomarker results in moderate-to-severe atopic dermatitis at the 2025 EADV Annual Meeting in Paris. Available from: https://www.leo-pharma.com/media-center/news/2025-temtokibart-late-breaker-press-release.
61. Kakinuma T, Nakamura K, Wakugawa M, et al. Thymus and activation-regulated chemokine in atopic dermatitis: serum thymus and activation-regulated chemokine level is closely related with disease activity. J Allergy Clin Immunol. 2001;107(3):535–541. doi:10.1067/mai.2001.113237
62. Kataoka Y. Thymus and activation-regulated chemokine as a clinical biomarker in atopic dermatitis. J Dermatol. 2014;41(3):221–229. doi:10.1111/1346-8138.12440
63. Hijnen D, De Bruin-Weller M, Oosting B, et al. Serum thymus and activation-regulated chemokine (TARC) and cutaneous T cell- attracting chemokine (CTACK) levels in allergic diseases: TARC and CTACK are disease-specific markers for atopic dermatitis. J Allergy Clin Immunol. 2004;113(2):334–340. doi:10.1016/j.jaci.2003.12.007
64. D’Erme AM, Fidanzi C, Bevilacqua M, et al. Cord blood serum levels of IL-31 and CCL17, cutaneous markers, and development of atopic dermatitis. JAMA dermatol. 2024;160(10):1112–1115. doi:10.1001/jamadermatol.2024.3178
65. Łacwik J, Kraik K, Laska J, Tota M, Sędek Ł, Gomułka K. IL-31/33 axis in atopic dermatitis. Int J Mol Sci. 2025;26(20):10162. doi:10.3390/ijms262010162
66. Kabashima K, Matsumura T, Komazaki H, Kawashima M. Nemolizumab plus topical agents in patients with atopic dermatitis (AD) and moderate-to-severe pruritus provide improvement in pruritus and signs of AD for up to 68 weeks: results from two Phase III, long-term studies. Br J Dermatol. 2022;186(4):642–651. doi:10.1111/bjd.20873
67. Ruzicka T, Hanifin JM, Furue M, et al. Anti–interleukin-31 receptor a antibody for atopic dermatitis. N Engl J Med. 2017;376(9):826–835. doi:10.1056/NEJMoa1606490
68. Nygaard U, Hvid M, Johansen C, et al. TSLP, IL-31, IL-33 and sST2 are new biomarkers in endophenotypic profiling of adult and childhood atopic dermatitis. J Eur Acad Dermatol Venereol. 2016;30(11):1930–1938. doi:10.1111/jdv.13679
69. Thyssen JP, Kezic S. Causes of epidermal filaggrin reduction and their role in the pathogenesis of atopic dermatitis. J Allergy Clin Immunol. 2014;134(4):792–799. doi:10.1016/j.jaci.2014.06.014
70. Kim J, Kim BE, Lee J, et al. Epidermal thymic stromal lymphopoietin predicts the development of atopic dermatitis during infancy. J Allergy Clin Immunol. 2016;137(4):1282–1285.e4. doi:10.1016/j.jaci.2015.12.1306
71. Soumelis V, Reche PA, Kanzler H, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3(7):673–680. doi:10.1038/ni805
72. Tordjman L, Mashoudy KD, Czarnowicki T. Converging paths toward unified therapeutic approaches in atopic dermatitis, vitiligo, and alopecia areata. J Allergy Clin Immunol. 2025;156(2):237–251. doi:10.1016/j.jaci.2025.04.016
73. Gregoriou S, Papafragkaki D, Kontochristopoulos G, Rallis E, Kalogeromitros D, Rigopoulos D. Cytokines and other mediators in alopecia areata. Mediators Inflamm. 2010;2010:928030. doi:10.1155/2010/928030
74. Yang X, Zhang W, Zhao X, et al. Changes and significance of Th1/Th2 and Treg/Th17 cells and their cytokines in patients with alopecia areata. Exp Cell Res. 2024;442(2):114259. doi:10.1016/j.yexcr.2024.114259
75. Omar SI, Hamza AM, Eldabah N, Habiba DA. IFN-α and TNF-α serum levels and their association with disease severity in Egyptian children and adults with alopecia areata. Int J Dermatol. 2021;60(11):1397–1404. doi:10.1111/ijd.15658
76. Shin JM, Choi DK, Sohn KC, et al. Induction of alopecia areata in C3H/HeJ mice using polyinosinic-polycytidylic acid (poly[I:C]) and interferon-gamma. Sci Rep. 2018;8(1):12518. doi:10.1038/s41598-018-30997-3
77. Pandey R, Jangid A, Vinjamuri RG, Ramaswamy R. Modelling of indirect cell-cell interaction networks mediated by IFNγ/IL-4 cytokine involved in atopic dermatitis. J Theor Biol. 2023;556:111291. doi:10.1016/j.jtbi.2022.111291
78. Wasserer S, Jargosch M, Mayer KE, et al. Characterization of high and low IFNG-expressing subgroups in atopic dermatitis. Int J Mol Sci. 2024;25(11):6158. doi:10.3390/ijms25116158
79. Gittler JK, Shemer A, Suárez-Fariñas M, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. 2012;130(6):1344–1354. doi:10.1016/j.jaci.2012.07.012
80. Tauber M, Buche S, Reygagne P, et al. Alopecia areata occurring during anti-TNF therapy: a national multicenter prospective study. J Am Acad Dermatol. 2014;70(6):1146–1149. doi:10.1016/j.jaad.2014.03.005
81. Perez SM, Kabashima K, Reich K, Gilliet M, Gilhar A, Paus R. Lessons from TNF-α inhibitor-induced alopecia areata: does tnf-α operate as a hair follicle immune privilege guardian under inflammatory conditions? J Invest Dermatol. 2025;145(10):2410–2416.e3. doi:10.1016/j.jid.2025.04.024
82. Nakamura M, Lee K, Singh R, et al. Eczema as an adverse effect of anti-TNFα therapy in psoriasis and other Th1-mediated diseases: a review. J Dermatol Treat. 2017;28(3):237–241. doi:10.1080/09546634.2016.1230173
83. Silverberg JI, Rosmarin D, Chovatiya R, et al. The regulatory T cell-selective interleukin-2 receptor agonist rezpegaldesleukin in the treatment of inflammatory skin diseases: two randomized, double-blind, placebo-controlled phase 1b trials. Nat Commun. 2024;15(1):9230. doi:10.1038/s41467-024-53384-1
84. Ross SH, Cantrell DA. Signaling and function of interleukin-2 in t lymphocytes. Annu Rev Immunol. 2018;36(1):411–433. doi:10.1146/annurev-immunol-042617-053352
85. Zaaroura H, Gilding AJ, Sibbald C. Biomarkers in alopecia Areata: a systematic review and meta-analysis. Autoimmun Rev. 2023;22(7):103339. doi:10.1016/j.autrev.2023.103339
86. Renert-Yuval Y, Pavel AB, Del Duca E, et al. Scalp biomarkers during dupilumab treatment support Th2 pathway pathogenicity in alopecia areata. Allergy. 2023;78(4):1047–1059. doi:10.1111/all.15561
87. Guttman-Yassky E, Pavel AB, Diaz A, et al. Ritlecitinib and brepocitinib demonstrate significant improvement in scalp alopecia areata biomarkers. J Allergy Clin Immunol. 2022;149(4):1318–1328. doi:10.1016/j.jaci.2021.10.036
88. Redmond WL, Triplett T, Floyd K, Weinberg AD. Dual anti-OX40/IL-2 therapy augments tumor immunotherapy via IL-2R-mediated regulation of OX40 expression. PLoS One. 2012;7(4):e34467. doi:10.1371/journal.pone.0034467
89. Croft M, Esfandiari E, Chong C, et al. OX40 in the pathogenesis of atopic dermatitis-a new therapeutic target. Am J Clin Dermatol. 2024;25(3):447–461. doi:10.1007/s40257-023-00838-9
90. Iriki H, Takahashi H, Amagai M. Diverse role of ox40 on t cells as a therapeutic target for skin diseases. J Invest Dermatol. 2023;143(4):545–553. doi:10.1016/j.jid.2022.11.009
91. Kim M, Del Duca E, Dahabreh D, et al. Alopecia areata exhibits cutaneous and systemic OX40 activation across atopic backgrounds. Allergy. 2024;79(12):3401–3414. doi:10.1111/all.16268
92. Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018;27(12):1984–2009. doi:10.1002/pro.3519
93. O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66(1):311–328. doi:10.1146/annurev-med-051113-024537
94. Guttman-Yassky E, Irvine AD, Brunner PM, et al. The role of Janus kinase signaling in the pathology of atopic dermatitis. J Allergy Clin Immunol. 2023;152(6):1394–1404. doi:10.1016/j.jaci.2023.07.010
95. Mikhaylov D, Glickman JW, Del Duca E, et al. A phase 2a randomized vehicle-controlled multi-center study of the safety and efficacy of delgocitinib in subjects with moderate-to-severe alopecia areata. Arch Dermatol Res. 2023;315(2):181–189. doi:10.1007/s00403-022-02336-0
96. Olsen EA, Kornacki D, Sun K, Hordinsky MK. Ruxolitinib cream for the treatment of patients with alopecia areata: a 2-part, double-blind, randomized, vehicle-controlled phase 2 study. J Am Acad Dermatol. 2020;82(2):412–419. doi:10.1016/j.jaad.2019.10.016
97. Liu LY, Craiglow BG, King BA. Tofacitinib 2% ointment, a topical Janus kinase inhibitor, for the treatment of alopecia areata: a pilot study of 10 patients. J Am Acad Dermatol. 2018;78(2):403–404.e1. doi:10.1016/j.jaad.2017.10.043
98. Berdyshev E, Goleva E, Bissonnette R, et al. Dupilumab significantly improves skin barrier function in patients with moderate-to-severe atopic dermatitis. Allergy. 2022;77(11):3388–3397. doi:10.1111/all.15432
99. Dahabreh D, Jung S, Renert-Yuval Y, Bar J, Del Duca E, Guttman-Yassky E. Alopecia areata: current treatments and new directions. Am J Clin Dermatol. 2023;24(6):895–912. doi:10.1007/s40257-023-00808-1
100. Harries MJ, Ascott A, Asfour L, et al. British association of dermatologists living guideline for managing people with alopecia areata 2024. Br J Dermatol. 2025;192(2):190–205. doi:10.1093/bjd/ljae385
101. Zhou C, Li X, Wang C, Zhang J. alopecia areata: an update on etiopathogenesis, diagnosis, and management. Clin Rev Allergy Immunol. 2021;61(3):403–423. doi:10.1007/s12016-021-08883-0
102. Phan K, Ramachandran V, Sebaratnam DF. Methotrexate for alopecia areata: a systematic review and meta-analysis. J Am Acad Dermatol. 2019;80(1):120–127.e2. doi:10.1016/j.jaad.2018.06.064
103. Acikgoz G, Caliskan E, Tunca M, Yeniay Y, Akar A. The effect of oral cyclosporine in the treatment of severe alopecia areata. Cutaneous Ocular Toxicol. 2014;33(3):247–252. doi:10.3109/15569527.2013.839997
104. Kalil L, Valido K, Peterson D, King B. Oral minoxidil treatment of alopecia areata. J Am Acad Dermatol. 2025;93(4):1099–1101. doi:10.1016/j.jaad.2025.05.1446
105. Rodenbeck DL, Silverberg JI, Silverberg NB. Phototherapy for atopic dermatitis. Clin Dermatol. 2016;34(5):607–613. doi:10.1016/j.clindermatol.2016.05.011
106. Ross G. Treatments for atopic dermatitis. Austr Prescr. 2023;46(1):9–12. doi:10.18773/austprescr.2023.002
107. Sidbury R, Tom WL, Bergman JN, et al. Guidelines of care for the management of atopic dermatitis: section 4. Prevention of disease flares and use of adjunctive therapies and approaches. J Am Acad Dermatol. 2014;71(6):1218–1233. doi:10.1016/j.jaad.2014.08.038
108. Bissonnette R, Warren RB, Pinter A, et al. Efficacy and safety of delgocitinib cream in adults with moderate to severe chronic hand eczema (DELTA 1 and DELTA 2): results from multicentre, randomised, controlled, double-blind, phase 3 trials. Lancet. 2024;404(10451):461–473. doi:10.1016/S0140-6736(24)01027-4
109. Ytterberg SR, Bhatt DL, Mikuls TR, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. 2022;386(4):316–326. doi:10.1056/NEJMoa2109927
110. Tsai SY, Phipatanakul W, Hawryluk EB, Oyoshi MK, Schneider LC, Ma KS. Comparative safety of oral Janus kinase inhibitors versus dupilumab in patients with atopic dermatitis: a population-based cohort study. J Allergy Clin Immunol. 2024;154(5):1195–1203.e3. doi:10.1016/j.jaci.2024.07.019
111. Davis DMR, Drucker AM, Alikhan A, et al. Guidelines of care for the management of atopic dermatitis in adults with phototherapy and systemic therapies. J Am Acad Dermatol. 2024;90(2):e43–e56. doi:10.1016/j.jaad.2023.08.102
112. Keystone EC, Taylor PC, Drescher E, et al. Safety and efficacy of baricitinib at 24 weeks in patients with rheumatoid arthritis who have had an inadequate response to methotrexate. Ann Rheum Dis. 2015;74(2):333–340. doi:10.1136/annrheumdis-2014-206478
113. Parmentier JM, Voss J, Graff C, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. 2018;2(1):23. doi:10.1186/s41927-018-0031-x
114. Dowty ME, Lin TH, Jesson MI, et al. Janus kinase inhibitors for the treatment of rheumatoid arthritis demonstrate similar profiles of in vitro cytokine receptor inhibition. Pharmacol Res Perspect. 2019;7(6):e00537. doi:10.1002/prp2.537
115. Chu AWL, Wong MM, Rayner DG, et al. Systemic treatments for atopic dermatitis (eczema): systematic review and network meta-analysis of randomized trials. J Allergy Clin Immunol. 2023;152(6):1470–1492. doi:10.1016/j.jaci.2023.08.029
116. Bieber T, Simpson EL, Silverberg JI, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med. 2021;384(12):1101–1112. doi:10.1056/NEJMoa2019380
117. Blauvelt A, Teixeira HD, Simpson EL, et al. Efficacy and safety of upadacitinib vs dupilumab in adults with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA dermatol. 2021;157(9):1047–1055. doi:10.1001/jamadermatol.2021.3023
118. Liu M, Gao Y, Yuan Y, et al. Janus kinase inhibitors for alopecia areata: a systematic review and meta-analysis. JAMA Network Open. 2023;6(6):e2320351–e2320351. doi:10.1001/jamanetworkopen.2023.20351
119. Husein-ElAhmed H, Husein-ElAhmed S. Comparative efficacy of oral Janus kinase inhibitors and biologics in adult alopecia areata: a systematic review and Bayesian network meta-analysis. J Eur Acad Dermatol Venereol. 2024;38(5):835–843. doi:10.1111/jdv.19797
120. Van Helmond SC, Willaert M, Nguyen VH, Nijsten T, Waalboer-Spuij R, Hijnen D. Real-world effectiveness and safety of janus kinase inhibitors in alopecia areata: a retrospective cohort study of 72 patients. Acta Derm Venereol. 2025;105:adv42990. doi:10.2340/actadv.v105.42990
121. King B, Zhang X, Harcha WG, et al. Efficacy and safety of ritlecitinib in adults and adolescents with alopecia areata: a randomised, double-blind, multicentre, phase 2b–3 trial. Lancet. 2023;401(10387):1518–1529. doi:10.1016/S0140-6736(23)00222-2
122. Study to evaluate the safety and efficacy of jaktinib in adults with alopecia areata (AA)(ClinicalTrials.gov (US). 2025.
123. A phase iii study of jaktinib in adults with moderate and severe atopic dermatitis. 2024.
124. Gooderham MJ, Hong HC, Eshtiaghi P, Papp KA. Dupilumab: a review of its use in the treatment of atopic dermatitis. J Am Acad Dermatol. 2018;78(3 Suppl 1):S28–s36. doi:10.1016/j.jaad.2017.12.022
125. Harb H, Chatila TA. Mechanisms of Dupilumab. Clin Exp Allergy. 2020;50(1):5–14. doi:10.1111/cea.13491
126. Paller AS, Siegfried EC, Simpson EL, et al. Dupilumab safety and efficacy up to 1 year in children aged 6 months to 5 years with atopic dermatitis: results from a phase 3 open-label extension study. Am J Clin Dermatol. 2024;25(4):655–668. doi:10.1007/s40257-024-00859-y
127. Pagan AD, Ghalili S, Cices A, et al. Atopic dermatitis induced during anti-TNF-α therapy for inflammatory bowel disease: potential for Th2 inhibition with dupilumab. J Allergy Clin Immunol Pract. 2023;11(7):2235–2238.e1. doi:10.1016/j.jaip.2023.03.054
128. McKenzie PL, Castelo-Soccio L. Dupilumab therapy for alopecia areata in pediatric patients with concomitant atopic dermatitis. J Am Acad Dermatol. 2021;84(6):1691–1694. doi:10.1016/j.jaad.2021.01.046
129. Guttman-Yassky E, Renert-Yuval Y, Bares J, et al. Phase 2a randomized clinical trial of dupilumab (anti-IL-4Rα) for alopecia areata patients. Allergy. 2022;77(3):897–906. doi:10.1111/all.15071
130. Müller S, Maintz L, Bieber T. Treatment of atopic dermatitis: recently approved drugs and advanced clinical development programs. Allergy. 2024;79(6):1501–1515. doi:10.1111/all.16009
131. Wollenberg A, Blauvelt A, Guttman‐Yassky E, et al. Tralokinumab for moderate‐to‐severe atopic dermatitis: results from two 52‐week, randomized, double‐blind, multicentre, placebo‐controlled phase III trials (ECZTRA 1 and ECZTRA 2)*. Br J Dermatol. 2021;184(3):437–449. doi:10.1111/bjd.19574
132. Silverberg JI, Guttman-Yassky E, Thaçi D, et al. Two phase 3 trials of lebrikizumab for moderate-to-severe atopic dermatitis. N Engl J Med. 2023;388(12):1080–1091. doi:10.1056/NEJMoa2206714
133. Kussini J, Pfützner W, Mühlenbein S. A case of unexpected successful treatment of alopecia areata with tralokinumab in a patient with atopic dermatitis. Int J Dermatol. 2025;64(10):1905–1906. doi:10.1111/ijd.17755
134. Tavoletti G, Chiei-Gallo A, Barei F, Marzano AV, Ferrucci SM. Tralokinumab as a therapeutic option for patients with concurrent atopic dermatitis and alopecia areata. Int J Dermatol. 2024;63(3):374–375. doi:10.1111/ijd.17010
135. Gaumond SI, Kamholtz I, Opstal M, Jimenez JJ. 0479 Biologics for the treatment of alopecia areata: a comprehensive review of clinical trials. J Invest Dermatol. 2025;145(8):S82. doi:10.1016/j.jid.2025.06.484
136. Weidinger S, Blauvelt A, Papp KA, et al. Phase 2b randomized clinical trial of amlitelimab, an anti-OX40 ligand antibody, in patients with moderate-to-severe atopic dermatitis. J Allergy Clin Immunol. 2025;155(4):1264–1275. doi:10.1016/j.jaci.2024.10.031
137. Guttman-Yassky E, Kabashima K, Worm M, et al. Efficacy and safety of rocatinlimab for the treatment of moderate-to-severe atopic dermatitis in ROCKET-IGNITE and ROCKET-HORIZON: two global, double-blind, placebo-controlled, randomised phase 3 clinical trials. Lancet. doi:10.1016/S0140-6736(25)01865-3
138. Press Release: sanofi’s amlitelimab met all primary and key secondary endpoints in the COAST 1 phase 3 study in adults and adolescents with atopic dermatitis. 2025. Available from: https://www.sanofi.com/en/media-room/press-releases/2025/2025-09-04-05-00-00-3144170.
139. Guttman-Yassky E, Simpson E, Bissonnette R, et al. ROCKET: a phase 3 program evaluating the efficacy and safety of rocatinlimab in moderate-to-severe atopic dermatitis. Immunotherapy. 2025;17(2):83–94. doi:10.1080/1750743x.2025.2464528
140. Passeron T, King B, Seneschal J, et al. Inhibition of T-cell activity in alopecia areata: recent developments and new directions. Front Immunol. 2023;14:1243556. doi:10.3389/fimmu.2023.1243556
141. A study to evaluate the efficacy and safety of subcutaneous amlitelimab monotherapy compared with placebo in adult participants with severe alopecia areata. 2024.
142. Rezolve-AD phase 2b study of rezpegaldesleukin meets primary and key secondary endpoints in patients with moderate-to-severe atopic dermatitis. 2025. Available from: https://ir.nektar.com/news-releases/news-release-details/rezolve-ad-phase-2b-study-rezpegaldesleukin-meets-primary-and.
143. Therapeutics N. A phase 2b study to evaluate rezpegaldesleukin (rezpeg) in the treatment of severe to very severe alopecia areata in adult patients (Rezolve AA). 2024. NCT06340360.
144. REZOLVE-AA phase 2b study of rezpegaldesleukin establishes proof-of-concept in patients with severe-to-very-severe alopecia areata. 2025. Available from: https://ir.nektar.com/news-releases/news-release-details/rezolve-aa-phase-2b-study-rezpegaldesleukin-establishes-proof.
145. HCW9302 (Interleukin-2 Fusion Protein) for Alopecia Areata (IL-2) (ClinicalTrials.gov). 2025.
146. Equillium reports positive phase 2 results for eq101 in alopecia areata. 2024. Available from: https://synapse.patsnap.com/article/equillium-reports-positive-phase-2-results-for-eq101-in-alopecia-areata.
147. Dai Z, Wang EHC, Petukhova L, Chang Y, Lee EY, Christiano AM. Blockade of IL-7 signaling suppresses inflammatory responses and reverses alopecia areata in C3H/HeJ mice. Sci Adv. 2021;7(14). doi:10.1126/sciadv.abd1866
148. Marković I, Savvides SN. Modulation of signaling mediated by tslp and il-7 in inflammation, autoimmune diseases, and cancer. Front Immunol. 2020;11:1557. doi:10.3389/fimmu.2020.01557
149. Q32 bio presents results from SIGNAL-AA part a clinical trial evaluating bempikibart in patients with alopecia areata at the 2025 American academy of dermatology meeting. 2025. Available from: https://ir.q32bio.com/news-releases/news-release-details/q32-bio-presents-results-signal-aa-part-clinical-trial/.
150. Q32 bio provides bempikibart program update, including next steps for advancing alopecia areata development program. 2024. Available from: https://ir.q32bio.com/news-releases/news-release-details/q32-bio-provides-bempikibart-program-update-including-next-steps.
151. Werfel T, Layton G, Yeadon M, et al. Efficacy and safety of the histamine H(4) receptor antagonist ZPL-3893787 in patients with atopic dermatitis. J Allergy Clin Immunol. 2019;143(5):1830–1837.e4. doi:10.1016/j.jaci.2018.07.047
152. Lee YB, Lee WS. Efficacy of antihistamines in combination with topical corticosteroid and superficial cryotherapy for treatment of alopecia areata: a retrospective cohort study. J Am Acad Dermatol. 2021;84(4):1152–1154. doi:10.1016/j.jaad.2020.06.1026
153. Pham C, Sung C, Juhasz M, et al. The Role of antihistamines and dupilumab in the management of alopecia areata: a systematic review. J Drugs Dermatol. 2022;21(10):1070–1083. doi:10.36849/jdd.6553
154. Ohyama M, Shimizu A, Tanaka K, Amagai M. Experimental evaluation of ebastine, a second-generation anti-histamine, as a supportive medication for alopecia areata. J Dermatological Sci. 2010;58(2):154–157. doi:10.1016/j.jdermsci.2010.03.009
155. Silverberg JI, Bissonnette R, Kircik L, et al. Efficacy and safety of etrasimod, a sphingosine 1-phosphate receptor modulator, in adults with moderate-to-severe atopic dermatitis (ADVISE). J Eur Acad Dermatol Venereol. 2023;37(7):1366–1374. doi:10.1111/jdv.18914
156. Etrasimod significantly improves atopic dermatitis in phase 2b trial April 24 2021. 2021. Available from: https://www.healio.com/news/dermatology/20210423/etrasimod-significantly-improves-atopic-dermatitis-in-phase-2b-trial.
157. King B, Mesinkovska N, Senna M, Luo X, Minkiewicz J, Selfridge A. Efficacy and safety of etrasimod in alopecia areata: a multicentre, randomized, double-blind, placebo-controlled, Phase 2 study. J Eur Acad Dermatol Venereol. 2025;39(6):1174–1184. doi:10.1111/jdv.20605
158. A study to evaluate the efficacy and safety of rosnilimab (ANB030) in treatment of subjects with moderate to severe alopecia areata (AZURE). 2021.
159. Lai HJ, Ye ZM, Chen SQ, McElwee KJ, Guo HW. Immune therapies for alopecia areata: evidence and new perspectives. Expert Rev Clin Immunol. 2025;21(10):1421–1446. doi:10.1080/1744666x.2025.2575365
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.