Back to Journals » Drug Design, Development and Therapy » Volume 16
Allosteric Binding Sites of the SARS-CoV-2 Main Protease: Potential Targets for Broad-Spectrum Anti-Coronavirus Agents
Authors Alzyoud L ![]() , Ghattas MA
, Ghattas MA ![]() , Atatreh N
, Atatreh N
Received 28 April 2022
Accepted for publication 23 July 2022
Published 2 August 2022 Volume 2022:16 Pages 2463—2478
DOI https://doi.org/10.2147/DDDT.S370574
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Lara Alzyoud,1 Mohammad A Ghattas,1,2 Noor Atatreh1,2
1College of Pharmacy, Al Ain University, Abu Dhabi, United Arab Emirates; 2AAU Health and Biomedical Research Center, Al Ain University, Abu Dhabi, United Arab Emirates
Correspondence: Mohammad A Ghattas; Noor Atatreh, Email [email protected]; [email protected]
Abstract: The current pandemic caused by the COVID-19 disease has reached everywhere in the world and has affected every aspect of our lives. As of the current data, the World Health Organization (WHO) has reported more than 300 million confirmed COVID-19 cases worldwide and more than 5 million deaths. Mpro is an enzyme that plays a key role in the life cycle of the SARS-CoV-2 virus, and it is vital for the disease progression. The Mpro enzyme seems to have several allosteric sites that can hinder the enzyme catalytic activity. Furthermore, some of these allosteric sites are located at or nearby the dimerization interface which is essential for the overall Mpro activity. In this review paper, we investigate the potential of the Mpro allosteric site to act as a drug target, especially since they interestingly appear to be resistant to mutation. The work is illustrated through three subsequent sections: First, the two main categories of Mpro allosteric sites have been explained and discussed. Second, a total of six pockets have been studied and evaluated for their druggability and cavity characteristics. Third, the experimental and computational attempts for the discovery of new allosteric inhibitors have been illustrated and discussed. To sum up, this review paper gives a detailed insight into the feasibility of developing new Mpro inhibitors to act as a potential treatment for the COVID-19 disease.
Keywords: COVID-19, Mpro, SARS-CoV-2, allosteric sites, druggability, antiviral
Graphical Abstract:

Introduction
Since January 2020, the world has been afflicted by an unprecedented and emergent pandemic of coronavirus disease 2019 (COVID-19), which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).1 The newly discovered coronavirus has spread to every country, with cases reported in Asia, Europe, North and South America, Australia, and Africa.2 As of February 01, 2022, The World Health Organization (WHO) has reported 373,229,380 confirmed COVID-19 cases worldwide and 5,658,702 deaths (https://covid19.who.int/table). Besides SARS-CoV-2, coronaviruses from the betacoronavirus genus such as the severe acute respiratory syndrome coronavirus (SARS-CoV) and the Middle East respiratory syndrome coronavirus (MERS-CoV), have several cases reported previously in China and the Middle East, respectively.3,4 However, COVID19 consequences on global health and the economy have made it an unprecedented public health crisis, prompting pharmaceutical companies and research institutes to devote significant time and resources to developing antiviral medications and vaccines as quickly as possible.5
The SARS-CoV-2 virus is a single-stranded positive-sense RNA genome enclosed within a membrane envelope belonging to the Coronaviridae family of viruses.6,7 Similar to other viral infections, the entry of the genetic material into the host cell for replication and release of virions is a critical stage in the virus life cycle (Figure 1).8 The viral RNA is translated once the SARS-CoV-2 is internalized in the host cell resulting in virus-encoded proteins of various open reading frames (ORFs). Subsequently, the ORFs are translated into two viral replicase polyproteins (pp1a and pp1ab).9 The cleavage of these viral polyproteins into individual nonstructural proteins is essential for viral genome replication. With papain-like protease (PLpro) cleaving two non-structural proteins at the N-terminus and main protease or 3C-Like protease (Mpro or 3CLpro) recognizing 11 separate cleavage sites at the C-terminus, this cleaving process generates 16 mature non-structural proteins.6,8–10 The remaining ORFs encode four structural proteins (spike, membrane, envelope, and nucleocapsid) and several accessory proteins.11,12
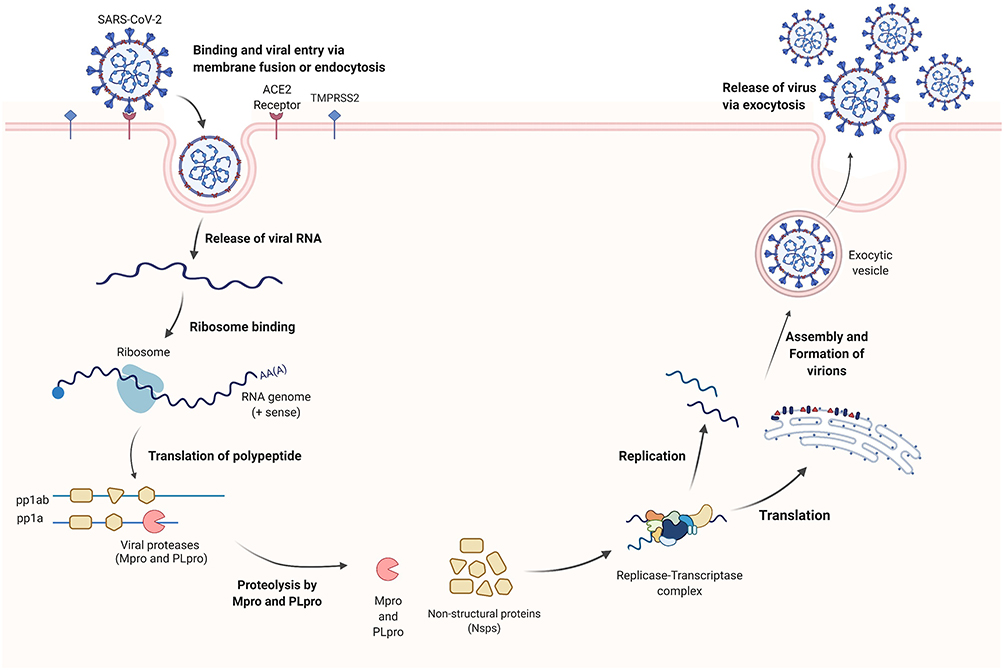 |
Figure 1 The life cycle of the SARS-CoV-2 coronavirus inside the host cell. Created with BioRender.com. |
Various important enzymes and proteins are involved in the viral replication and infectious capacity of the SARS-CoV-2 virus.13 Among them, two key proteases, such as main protease (Mpro) and papain-like protease (PLpro), are required for viral replication.14,15 PLpro is known to cleave nsp1, nsp2, and nsp3, whereas Mpro cleaves the remaining 13 non-structural proteins (Figure 1).16 In addition, PLpro is involved in antagonizing the host’s immune response upon viral infection through its deubiquitinating and deISGylating activities.17,18 However, because both PLpro and human deubiquitinases (DUBs) bind ubiquitin at the extended C-terminus with the consensus sequence Leu-X-Gly-Gly, PLpro inhibitors may exhibit off-target effects on human DUBs.19 Hence, the issue of target selection must be addressed early on in the developmental process. The Mpro enzyme, on the other hand, is unique in that it exclusively cleaves polypeptides after a glutamine (Gln) residue, and no known human protease possesses the same cleavage specificity, making it a viable target for the development of highly selective antiviral agents.20–23 With new COVID-19 variants and the susceptibility of RNA viruses for genetic reshuffling, mutations, and interspecies transmission, it has become more challenging to rely only on vaccines to effectively protect individuals and limit down the community spread of the infection.24,25 Thus, small molecules that target conserved viral proteases like the main protease could lock key steps of the SARS-CoV-2 life cycle providing a broad-spectrum antiviral effect with minimal side effects.20,26 In December 2021, the FDA issued emergency use authorization (EUA) for SARS-CoV-2 main protease (Mpro) inhibitor PAXLOVID (nirmatrelvir and ritonavir).27 This is the first antiviral drug to be approved as an oral treatment of mild to moderate COVID-19 in patients who are at high risk of developing severe illness.27
The Mpro enzyme encompasses several pockets on its surface that were proven to be important for its catalytic activity; some of them exist in distal areas from the main catalytic pocket, and others have been identified on the dimerization interface. The latter type, in particular, has shown promising results and seems to hinder the overall catalytic activity of the Mpro enzyme if a suitable organic molecule can successfully bind to it. Hence, in this review, we showcase all allosteric sites identified so far and illustrate all drug discovery efforts to target them.
Mpro Structure and Dimerization
The SARS-CoV-2 Mpro is a homodimer made up of two monomers, arranged almost perpendicular to one another.23 Each monomer consists of three domains – domains I, II, and III. Domains I (residues 10–99) and II (residues 100–182) assume a chymotrypsin-like structure with a six-stranded antiparallel b-barrel fold.23 Domains II and III are linked by a loop (residues 183–198) that takes on two different conformations: a long helix shape or a short helix shape.6
The active site is located in a cleft between the two N-terminal domains (Domain I and II) of the three-domain structure of the monomer.23 The C-terminal helical domain III (residues 198–303) is involved in the regulation and dimerization of the enzyme via an intermolecular salt-bridge interaction between Glu290 and Arg4.28,29 Upon dimerization and activation (Figure 2), both monomers orient themselves in the appropriate conformation to perform the catalytic function, and the N finger of each monomer interacts with Glu166 of the other monomer.23 This step is vital for the enzyme’s catalytic activity as it shapes the S1 pocket of the substrate-binding site, which contains the catalytic dyad, His41, and Cys145.25 Given the importance of dimerization, targeting the dimer site might be used to attenuate the catalytic activity.30 Earlier MD simulations and mutational analyses studies revealed key residues involved in stabilizing the catalytically active dimeric structure of the SARS-CoV-2 Mpro enzyme, including Arg4, Ser10, Gly11, Glu14, Asn28, Ser139, Phe140, Ser147, Glu290, and Arg298.25 These residues operate cooperatively to control dimerization and maintain the integrity of the dimer interface which is important for dimer stability.31 Several inhibitors have been identified to bind to the dimer site, inducing weak to moderate antiviral activity.32,33 Thus, interfering with the residues on the dimer site is likely to deter the enzymes` catalytic activity and tendency to dimerize.34
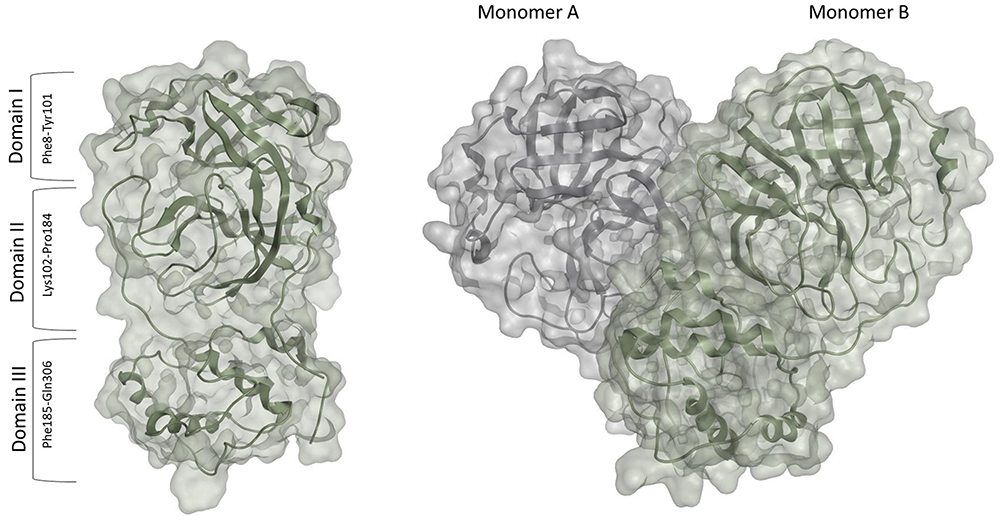 |
Figure 2 The Monomer (PDB: 6LU723) and Dimer (PDB:7CAM79) forms of the SARS-CoV-2 Mpro. |
Allosteric Binding Sites on the Mpro Enzyme
Over the past two years, there have been extensive research efforts worldwide to develop new Mpro inhibitors, most of which targeted the enzyme catalytic pocket.23 However, several inhibitors appeared to bind at sites distant from the catalytic pocket, which opens the door for an alternative approach to target the Mpro enzyme via an allosteric mechanism.32 Nevertheless, there are limited data on this area so far, which hinders the search for allosteric inhibitors.
Currently, two pockets of varying size and druggability properties have been identified as possible therapeutic targets: distal site and dimerization sites.25,35,36 Computational studies have predicted that targeting either sites will result in allosteric modulation by inducing either conformational changes or thermal fluctuations around a fixed, mean conformation.37–42 Given the difficulty associated with targeting the active site, investigating distal and dimer, allosteric sites will provide a better understanding of their characteristics and potential as molecular targets, ultimately assisting in the development of novel broad-spectrum inhibitors of SARS-CoV-2 Mpro.
Allosteric Sites on the Distal Region of the Mpro Enzyme
Analysis of the key contacts in the SARS-CoV-2 Mpro, which include Arg131-Thr199, Arg131-Asp289, Pro132-Thr196, Asp197, and Thr198-Asn238, Tyr239-Leu287, shows the presence of a pocket at the interface between domains II and III.43 This pocket, which is located far from the active site, is known as the distal site. The distal site contains a number of residues surrounding the beta-sheet and alpha-helices. Due to the position of these residues, allosteric inhibitors targeting this site operate via reversible non-competitive inhibition.44
Up to this point, no inhibitors of the distal site have been reported, suggesting that a druggability analysis would be necessary to determine the viability of the cavities distant from the catalytic pocket. When evaluated using the PockDrug45 web server, these pockets druggability scores ranged from 0.37 to 1.00, 60% of which possess scores greater than 0.90.36 As a result of its ability to bind drug-like molecules, the distal site provides a promising molecular target for potential allosteric inhibitors of the Mpro enzyme.
Further studies have looked into the coupled movement of residues from aggregate 1 μs MD simulations of the apo and dimer forms of Mpro to assess the effect of small molecules binding to the distal site on catalytic activity.36 Regardless of whether the correlation is positive or negative, the distal and active sites appear to be dynamically dependent on one another in both the apo and dimer forms, demonstrating long-range communication between the distal and active sites.36,46
Allosteric Sites on the Mpro Dimerization Interface
Located between domains II and III, the dimer site encompasses residues from the N finger and the C-terminal helix. These residues are essential for the dimerization and subsequent activation and stabilization of the protease.47,48 The SARS-CoV-2 Mpro, similar to other viral proteases, is made up of two monomers that dimerize to produce an active homodimer. The dimerization process is vital for enzyme activity, as the monomer form of Mpro cannot fulfill the enzymatic role in catalysis.49
Due to the complexities of dimer dynamics, it is frequently assumed that one monomer is active in the dimer while the other is inactive.50 Protease activity can be disrupted by mutating key residues in the dimer interface.25,51 These residues operate cooperatively to control dimerization and maintain dimer stability.31 Mutations of Arg298 in the dimer interface, in particular, are detrimental to viral activity because they prevent dimerization and inactivate the enzyme irreversibly.47 Hence, compounds that block dimerization may operate as allosteric inhibitors of protease activity by impairing its catalytic activity, inducing weak to moderate antiviral activity.52,53
Given the allosteric linkage between the dimerization site and the catalytic region, targeting protease dimerization could potentially impact the substrate pocket and thus inhibit Mpro activity.54,55 x1187 and x1086 are two fragments that disrupt dimer formation by binding to hydrophobic pockets at the dimer site, as illustrated in Figure 3.32,44,51 By disrupting dimerization, these fragments can be used to develop more potent drugs that allosterically modulate Mpro activity. Such fragments can induce antiviral activity by disrupting the dimer interface and conserving Mpro in its inactive monomer state.44,56 Jiménez-Avalos et al53 have found that the binding of CHEMBL2171598 at the dimer site prevents subsequent dimerization and catalytic activity, mirroring the effect of the detrimental mutation Arg298Ala.
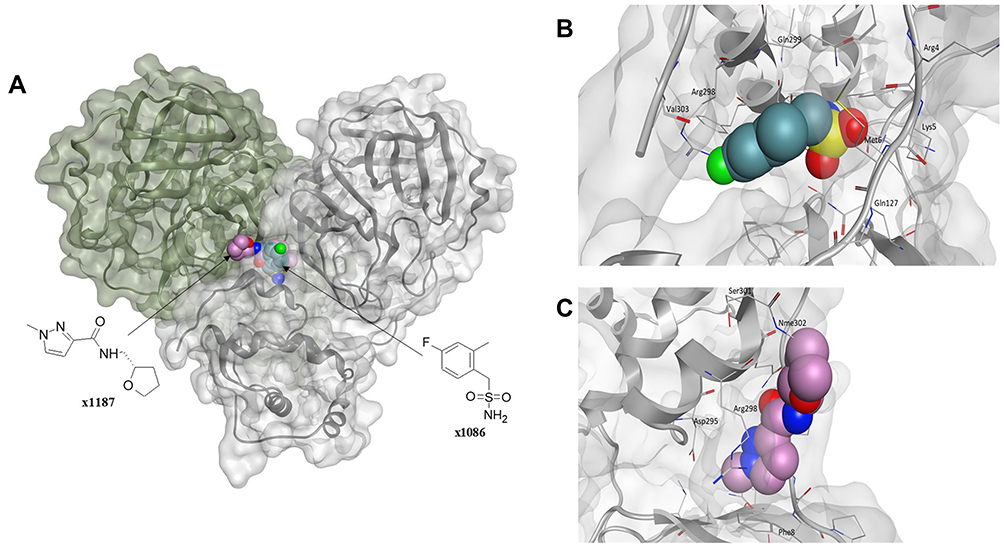 |
Figure 3 (A) The Mpro enzyme in the dimer form with co-crystallized ligands, (B) x1086 (PDB:5RGQ51) and (C) x1187 (PDB:5RFA51) at the dimer interface indicates opportunities for allosteric modulation. For clarity, superimposition on the dimer form of Mpro (PDB:7CAM79) was performed to demonstrate binding onto the dimerization site. |
Due to the fact that dimerization stabilizes both the protein and its active site, apo-structures tend to lack any distinctive pockets at the dimer interface.53 However, when preserved in the monomer state, the protease features a well-defined pocket with desirable size and polarity that may bind ligand-like molecules. As a result, ligand-bound structures that maintain the monomer state are more likely to produce a druggable pocket at the dimer interface than apo-structures. Investigating these pockets represents a promising strategy for inhibiting the Mpro via allosteric means.
Druggability Assessment of Allosteric Sites
As multiple allosteric pockets have been identified experimentally,32,51 it was necessary to perform a proper evaluation of these sites and assess them in terms of druggability or the ability of the target site to recruit and bind drug-like ligands. At the time of writing this review, six allosteric sites were experimentally published along with eight co-crystallized ligands or fragments, as shown in Figure 4.32,51 Accordingly, we run a well-established module in the Schrodinger57 software that is called SiteMap.58 In this tool, we can identify the co-crystallized ligand as the center of the pocket that has to be assessed. Druggability scores (Dscore) will be then generated for each of these pockets; the higher the scoring, the better the druggability (typically pockets with a Dscore ≥1.0 are considered very druggable, whereas sites with a Dscore less than 0.8 are classified as difficult non-drug binding sites).59 Such scoring is calculated according to three major factors: pocket size, hydrophobicity, and enclosure.59
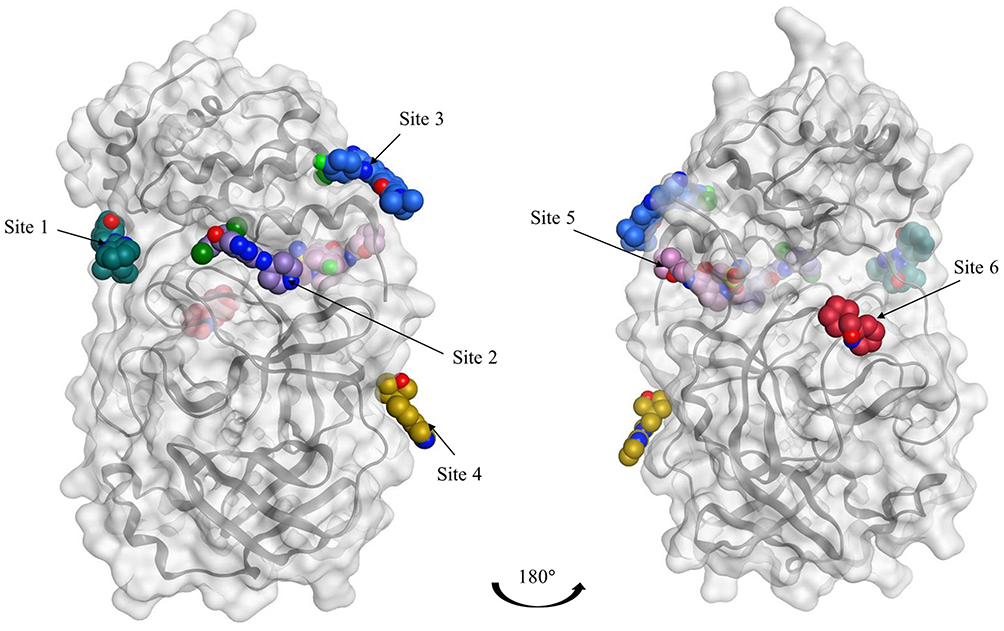 |
Figure 4 Representation of the Mpro enzyme in the monomer form labeled with its six experimentally proven allosteric sites, along with their co-crystallized ligands and Dscores. |
Interestingly, we found that only two sites have shown very druggable pockets (ie, site #2 and site #5), while one showed poor druggability (ie, site #3), and the other three sites could not be detected at all by SiteMap (ie, site #1, site #4 and site #6), obviously because of their very shallow nature.
From Table 1, it can be seen that the highest-ranking site, site #2, possesses a large pocket (n = 102 spheres) which is well-defined (e = 0.71) with high hydrophilicity (p = 1.083), as illustrated in Figure 5. Next to site 2, also at the dimer interface, is another “druggable” site, site #5, which has comparable features as it is a large (n = 100 spheres), less defined cavity (e = 0.71), and lower hydrophilicity (p = 0.92). Similarly, site #3 is also located at the dimer interface; however, it is considered a difficult target due to its very small (n = 14 spheres), and shallow cavity (e = 0.46) that is significantly less hydrophilic (p = 0.433). Finally, no well-defined pocket was detected by SiteMap for sites #1, #4, and #6, as they showed a relatively flat surface. Visual inspection of these sites confirms their superficial character as they appear on the interface (Figure 5). With protease movement, these sites may become more exposed; nevertheless, further dynamic analysis is required. Hence, out of the experimentally found allosteric sites, only sites #2 and #5 seem to be promising and able to bind druglike inhibitors.
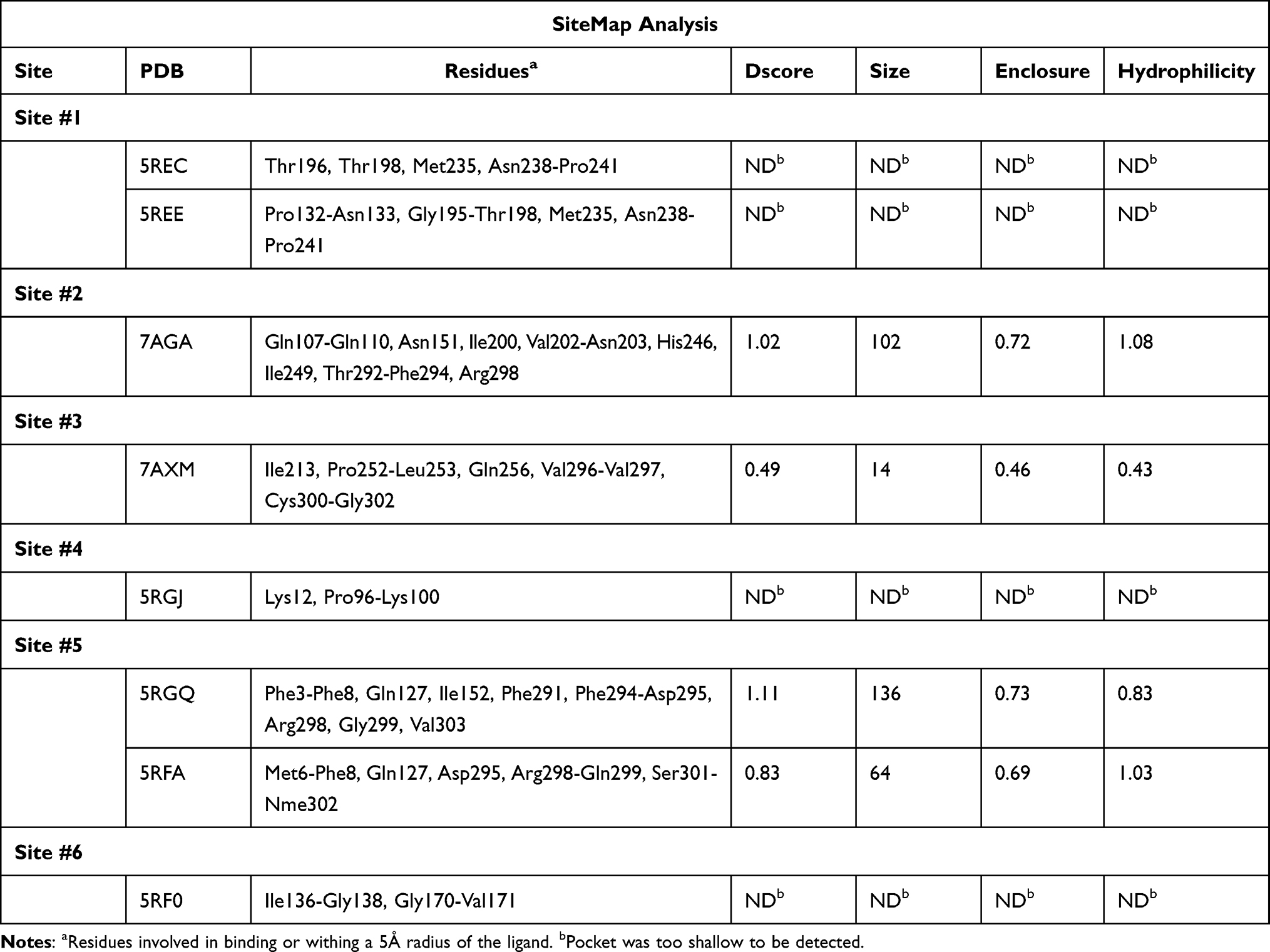 |
Table 1 Druggability Assessment of the Six Experimentally Proven Allosteric Sites, Along with Their Co-Crystallized Ligands and Dscores. (ND: Not Detected) |
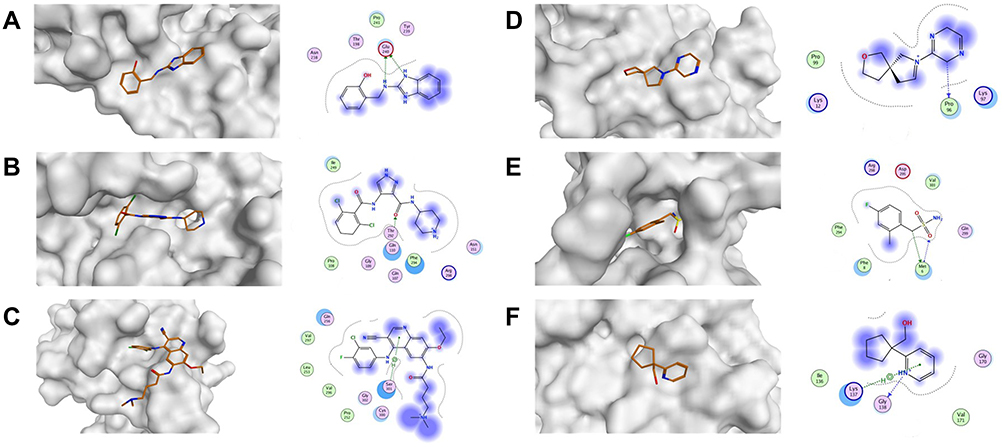 |
Figure 5 The overview shows the surface of the Mpro and ligand-receptor interaction diagrams across all reported allosteric sites. (A) Site 1 (x0390; PDB:5REC51); (B) Site 2 (AT7519; PDB:7AGA32); (C) Site 3 (Pelitinib; PDB:7AXM32); (D) Site 4 (x0425; PDB:5RGJ51); (E) Site 5 (x1086; PDB:5RGQ51); (F) Site 6 (x0887; PDB:5RF051). |
Recent Advances in the Development of Allosteric SARS-CoV-2 Mpro Inhibitors
Several strategies were employed to find therapeutic agents targeting the Mpro enzyme, which involve screening approaches, such as high throughput virtual screening, mass spectrometry, X-ray crystallographic, electrophilic, and NMR fragment screening.32,44,51,60 Most discovered inhibitors block the active site and bind covalently to its nucleophilic cysteine residue. Interestingly, few appeared to bind at sites distant to the catalytic pocket, opening the door for an alternative approach that possibly targets the Mpro via an allosteric mechanism. It is important to note that only one attempt has currently been made to target the allosteric site through both computational and experimental means, which is discussed in the section.
Experimental Studies
Limited work has been done to evaluate reported Mpro allosteric inhibitors in vitro as shown in Figure 6. Gunther et al32 used X-Ray screening to identify drugs that bind at the Mpro active site. Surprisingly, they have identified five compounds that bind at two distinct allosteric sites. The first allosteric site (site #3 in our paper) binds five compounds: pelitinib, RS-102895, Ifenprodil, PD-168568, and tofogliflozin. Pelitinib, an anti-cancer drug, has shown considerable anti-viral activity with EC50 of 1.25 μM, outperforming other inhibitors in this study as the top-ranking allosteric inhibitor. The remaining four drugs had little to no anti-viral activity. The second allosteric site is found near the dimerization site (site #5 in our analysis), binds CDK inhibitor AT-7519, and has exhibited weak anti-viral activity with EC50 of 25.2 μM. Although this validates their anti-viral activity and affinity for allosteric sites, they have not been experimentally tested for their enzyme inhibition property. When evaluating the binding mode of the co-crystallized ligands AT-7519 (Figure 7), this ligand shows a reasonable fitting; however, many of its functional groups seem to have no role in binding (in line with its experimentally proven weak activity). Similarly, Pelitinib (Figure 7) exhibits a similar binding pattern and weak interactions. Moreover, both described sites are in the dimer site, which is essential for protease dimerization and activation.
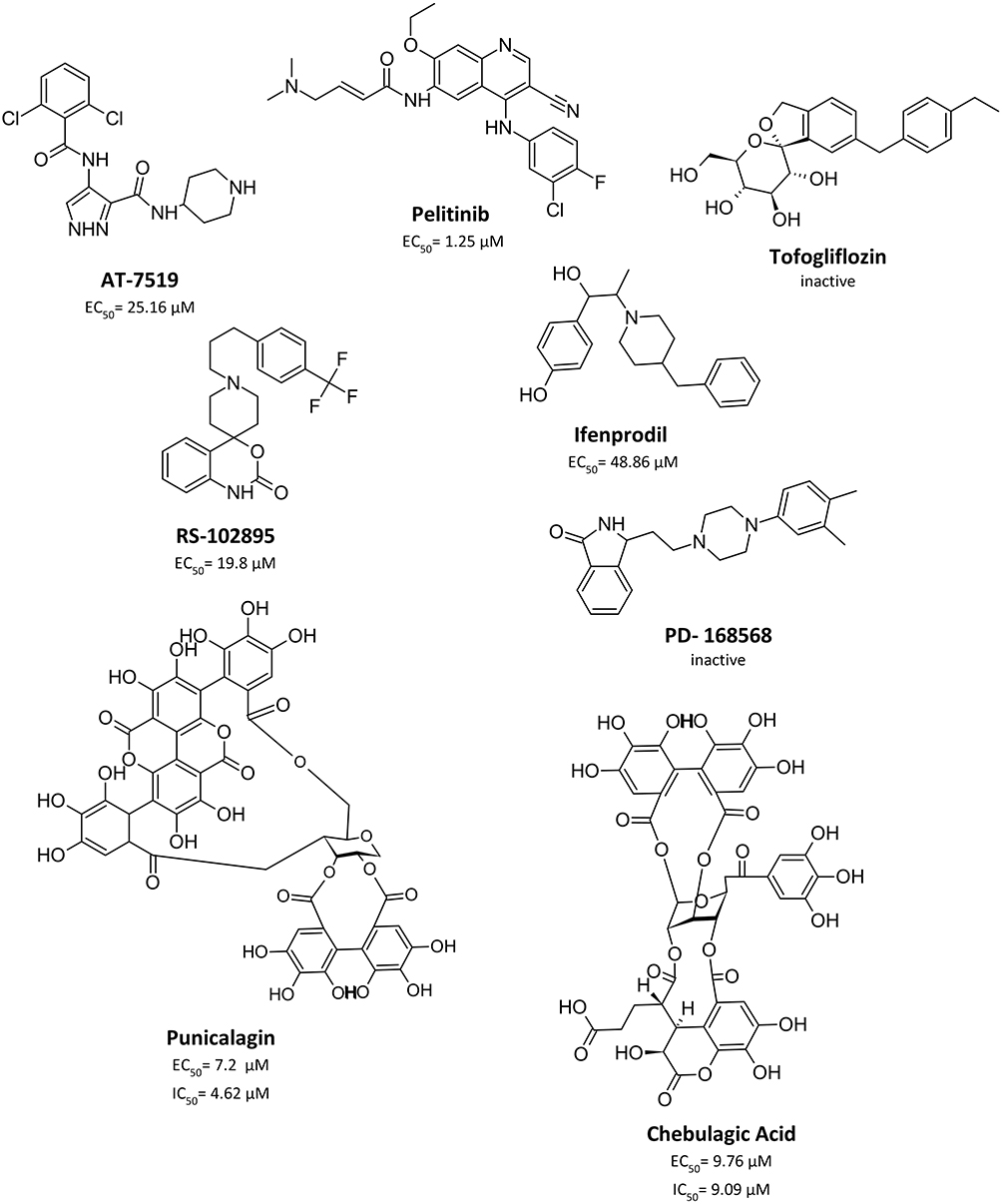 |
Figure 6 Chemical structure of experimentally evaluated allosteric Mpro inhibitor. |
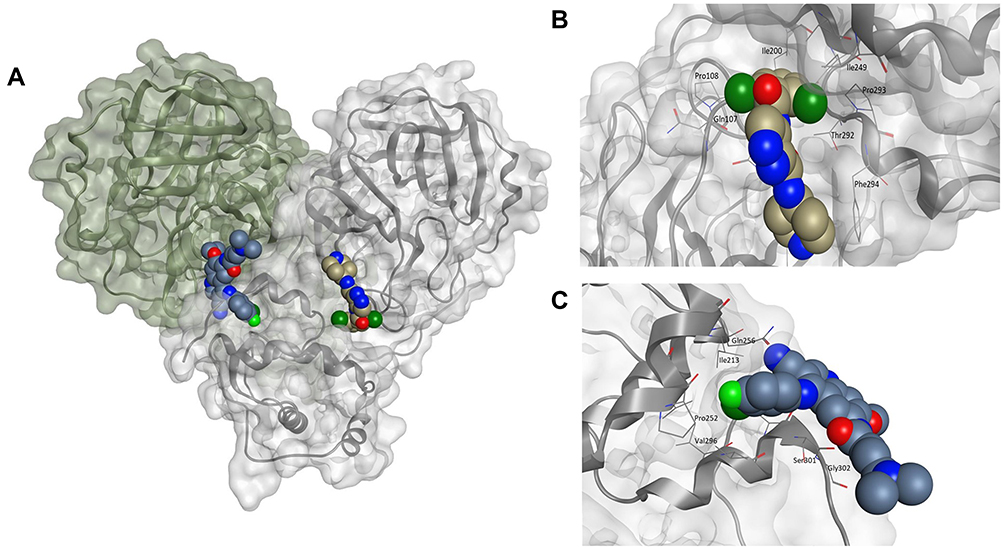 |
Figure 7 (A) The Mpro enzyme in the dimer form with co-crystallized ligands, (B) AT-7519 (PDB:7AGA32) and (C) Pelitinib (PDB:7AXM32) at the dimer interface indicates opportunities for allosteric modulation. For clarity, superimposition on the dimer form of Mpro (PDB:7CAM79) was performed to demonstrate binding onto the dimerization site. |
Broad-spectrum antiviral agents, chebulagic acid and punicalagin are another set of compounds that were previously identified as non-competitive Mpro inhibitors. Such compounds demonstrated a non-druglike character as they exhibit large sizes with EC50 and IC50 values in the micromolar range.33 There is no information about their binding site, but they are suspected to bind near the dimer site. Nonetheless, these results demonstrate that chebulagic acid and pelitinib are reversible inhibitors that bind non-competitively at distinct allosteric sites on the Mpro.32,33
Another notable drug discovery strategy is fragment screening, which uses small molecular fragments to probe new cavities on the SARS-CoV-2 Mpro interface. Depending on their structural features, these cavities serve as potential targets for therapeutic development. So far, electrophilic and noncovalent fragment screening appears to be the most effective and informative strategy for defining these sites.32,51,60
Douangamath et al51 used a combination of mass spectrometry and crystallographic techniques, resulting in six Mpro complexes with co-crystallized allosteric fragments (Figure 8), two of which bind at the dimerization site (x1086 and x1087), and four of which bind in distinct hydrophilic allosteric cavities (x0390, x0464, x0425, and x0887). Recently, NMR Fragment-based screening found fragment F15 to bind at the Mpro dimer interface, where it is predicted to mimic the binding of fragments x1086 and x1187 in the hydrophobic pocket.60
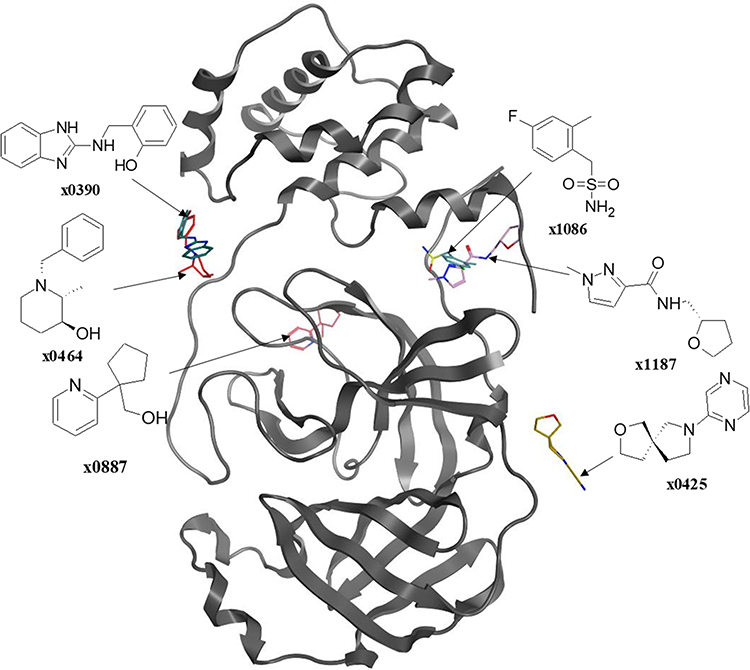 |
Figure 8 The so far known co-crystallized fragments bound to different allosteric sites after superposing them onto a single crystal structure of the SARS-CoV-2 Mpro. |
Since there is no testing performed, it is highly predictable that these small molecular fragments have little to no antiviral activity; yet, they form multiple interactions with their respective binding site, shown in Figure 7, which boosts the binding energy. Researchers can benefit from these interactions by using the fragment as the lead compound, which could be later optimized to derive a series of small molecules with higher affinity and in-vitro and in-vivo antiviral activity.
Computational Studies
Given the similarities between the SARS-CoV-2 Mpro and other viral cysteine proteases, repurposing FDA-approved drugs as anti-COVID-19 therapeutics provides a time and cost-effective alternative to novel drug design.20 The initial concept was to re-propose current antiviral medications used in the treatment of HIV, hepatitis B and C, influenza, and the common cold, as they have shown to be effective in treating past coronavirus infections.61 A few of these drugs, listed in Table 2, appear to bind at high binding energies to sites distant from the active site, indicating a preference for allosteric sites.
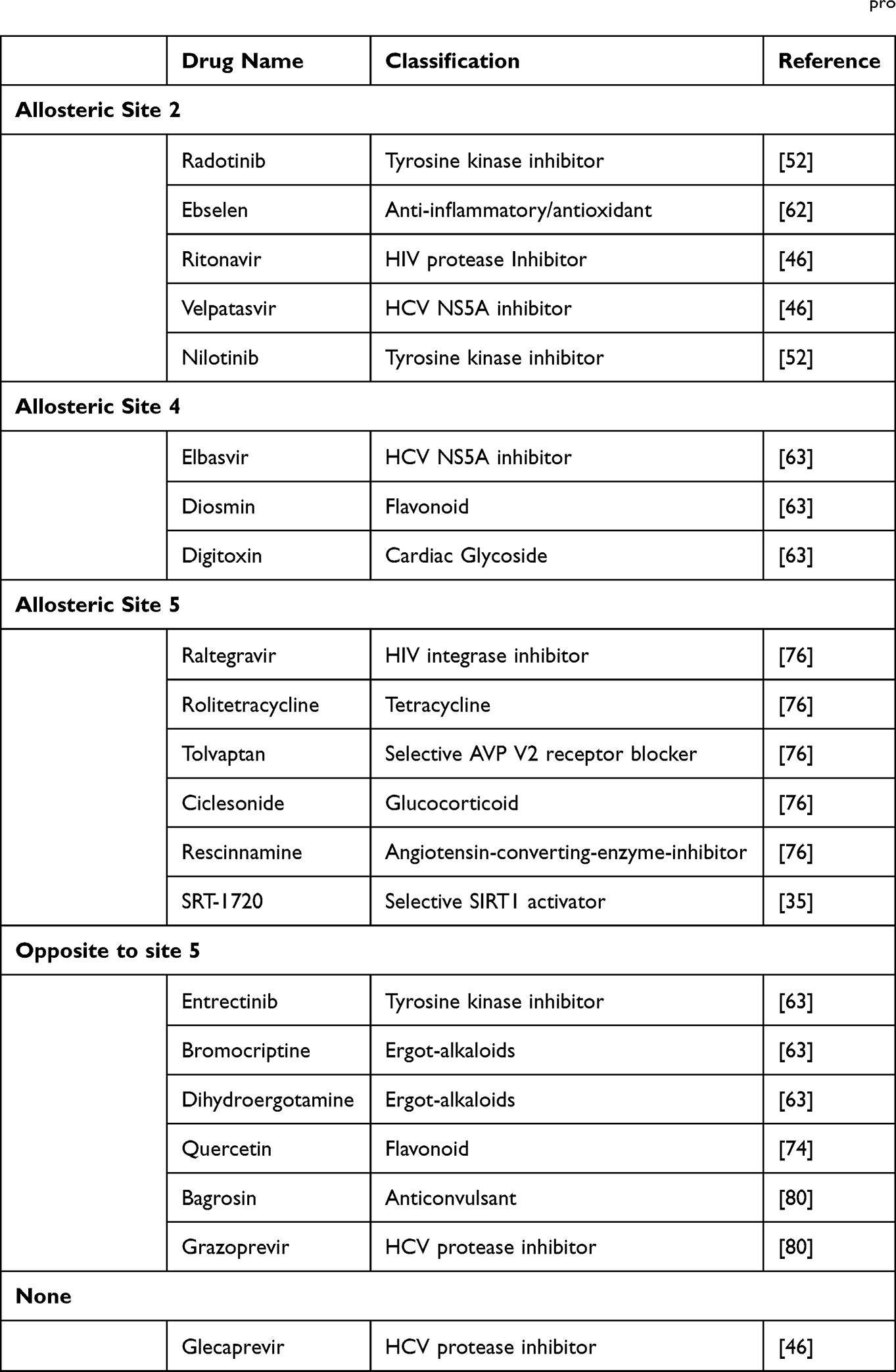 |
Table 2 List of Repurposed Drugs That Bind to Allosteric Sites on the SARS-CoV-2 Mpro |
Ebselen, a synthetic organoselenium molecule, exhibits higher binding energy for the potential allosteric site (−8.87 kcal/mol) compared to the active site (−5.55 kcal/mol). When ebselen binds to the site between the II and III domains, it exerts a significant allosteric effect that limits catalytic site access via surface-loop interactions.62
The antiviral drug elbasvir has a higher docking score at both potential allosteric sites (−10.8 kcal/mol and −11.1 kcal/mol) compared to the active site (−8.8 kcal/mol), as well as a higher binding affinity towards both allosteric sites (−51.08 kcal/mol and −42.37 kcal/mol), making it an ideal candidate for clinical evaluation.63 However, it is important to remember that binding at higher energy does not necessarily mean a higher binding affinity for the allosteric sites. Some drugs, such as dihydroergotamine, bromocriptine, and diosmin, have higher binding energies for potential allosteric sites, but a higher binding affinity for the active site.63
Although drug repurposing has frequently shown to be a successful approach, the majority of these studies were carried out computationally through virtual screening of existing drugs, meaning that they require additional in-vitro and in-vivo testing for viral activity. The lack of in-vitro and in-vivo testing data makes it difficult to draw firm conclusions about whether the presupposing drug approach acts on the Mpro or other biological pathways involved with disease progression and symptoms.
Advantages of Targeting Allosteric Sites on the SARS-CoV-2 Mpro
Allosteric modulation has proven to be a viable alternative to orthosteric ligands, contributing significantly to the development of GPCR and protein kinase modulators.64 As targeting the SARS-CoV-2 active site appears to be a challenging endeavor, allosteric sites have emerged as attractive sites for the design of small-molecule inhibitors, offering several advantages over their orthosteric counterpart.25,35,36 All allosteric inhibitors identified thus far act via reversible means, making them potentially safer than many of the previously discovered Mpro inhibitors, which contain reactive groups and form a covalent bond with the enzyme, and hence possibly with other enzymes.65,66 Therefore, the Mpro allosteric sites appear to be an interesting target for the discovery of broad-spectrum anti-corona viral agents that can act alone or in combination with the other Mpro competitive inhibitors.
Dimerization-Targeted Allosteric Inhibition
Acting on the dimerization site seems to be rather promising as this strategy has previously been reported to modulate several viral enzymes, such as HIV reverse transcriptase, integrase, and herpes simplex virus.67 The dimeric nature of SARS-CoV-2 Mpro allows targeting dimerization at certain hot spots by small molecule inhibitors, which seems to disrupt and inactivate the enzyme.68 The dimerization interface, located between domains II and III, is densely packed with high-energy hot spots.69 A few of these residues are essential for Mpro dimerization (Ser1, Arg4, His172, and Glu290), as they form salt bridges between the two monomers.28,69,70
Beyond promoting dimer dissociation, the mechanism by which Mpro dimerization inhibitors interfere with the catalytic activity is not entirely clear.30 Some studies have linked enzyme inhibition to the compound’s tendency to induce dimer dissociation.25 Others have proposed the presence of an allosteric connection between the sizes of the catalytic and allosteric dimerization sites.71 AMOEBA trajectory analysis verifies this, revealing that the studied structures had an open catalytic site and a small allosteric dimerization site, and vice versa.71 It also implies that although inhibiting Mpro dimerization disrupts its activation, it does not necessarily block its interaction with the substrate. Nonetheless, current findings provide encouraging data on dimerization-targeted Mpro allosteric inhibition.
Maximizing Antiviral Activity Through Synergy
As previously stated, inhibiting Mpro dimerization does not necessarily hamper its interaction with the substrate,30 as there will always be other dimeric molecules that will exert their enzymatic activity upon substrate binding. This, however, hints at the possibility of synergism, in which a combination of active and allosteric site inhibitors can be employed to induce maximum inhibition of Mpro activity.30,72
In fact, a previous study investigated the potential synergistic effect of combining allosteric and active site inhibitors.72 When tested alone, Quinacrine demonstrated competitive inhibition with an IC50 of 6.3 μM, while suramin exhibited non-competitive inhibition with an IC50 of 7.8 μM. Interestingly, combining them together has successfully boosted the Mpro inhibition activity by more than ten folds (new IC50 = 0.46 μM).72 Thus, Mpro allosteric inhibitors seem to have the potential to act alone or in conjunction with their competitive peers, to allow lower doses and hence greater safety for the COVID19 patients.30,72
Conservation of Allosteric Sites
Targeting conserved regions on the protease can vastly enhance treatment efficiency.73,74 The Mpro is distinct from other viral proteins that it is highly conserved across the coronavirus family, as any mutation can be detrimental to the virus and its activity.75,76 However, the active site is of high plasticity and is regulated by a flexible C44-P52 loop, raising concerns that any mutations to its amino acid sequence may impede small-molecule entry.77 Accordingly, resistance mutations to these active site inhibitors should be expected. Structural changes that confer resistance may occur in any site throughout the protease and disrupt the complex interactions required for enzymatic function.45 Hence, targeting multiple regions on the protease, including allosteric sites, might be a better strategy than using a single compound that targets the active site.
Most of the residues implicated in allosteric sites were reported to be conserved in most SARS-CoV-2 structures studies. This could be interpreted as a reassuring sign of the viability of the newly identified allosteric targets, as potential drugs targeting these sites are assumed to be robust against mutations.46,78 Moreover, hydrophobic residues at the allosteric sites are substantially conserved throughout the Mpro of all human coronaviruses, indicating that mutations are unlikely.43 As a result, the likelihood of mutation-mediated drug resistance is quite low, and inhibitors will have broad antiviral activity.30
Conclusions and Future Prospective
This review paper summarizes the work done so far on the Mpro allosteric site and it illustrates the potential of such pockets to act as a drug target (Figure 5). There have been several attempts to target the Mpro allosteric sites; however, many of these are computational and do not provide us with very conclusive data. Having said that, a few experimental works have confirmed the presence of multiple pockets, at least two of them appear to have a reasonable druggability potential (as per an in-house computational assessment). Both pockets are located at the dimerization site or in its vicinity, offering more potential to interfere with the Mpro catalytic site through a dimerization-based allosteric mechanism.
As a future perspective, computer-aided drug design seems to be in a good position for the discovery of new inhibitors, especially since several crystal structures have been already revealed, some of them co-crystallized with small ligands in their allosteric sites. For instance, compounds x1086 and x1187 both together exhibited a complementary binding mode in the allosteric site #5, having their sulfonamide and the hetero aromatic ring sitting in two distinct subpockets (Figure 9). Based on the aforementioned observations, Site #5 can be targeted by docking-guided derivatization through synthesizing a series of compounds designed based on both x1086 and x1187, where distinguishingly bound moieties will be joined together. To sum up, druglike allosteric inhibitors seem to be achievable and can provide us with an extra weapon in our war against the COVID-19 disease, and possibly against other coronaviruses-caused pandemics.
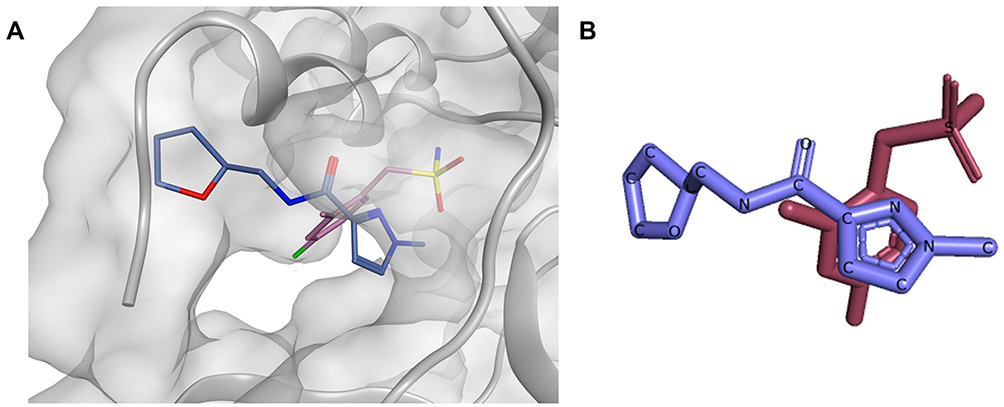 |
Figure 9 (A) The binding mode of compounds x1086 (pink) and x1187 (blue) aligned on top of each other inside the allosteric site #5, (B) 2D representation of compounds x1086 (pink) and x1187 (blue) aligned on top of each other. |
Acknowledgments
We would like to acknowledge the generous financial support from Al Jalila Foundation, Dubai, UAE.
Funding
This work was funded by Al Jalila Foundation, Dubai, UAE (Seed Grant Reference no: AJF202073).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chams N, Chams S, Badran R., et al. COVID-19: a multidisciplinary review. Front Public Heal. 2020;8. doi:10.3389/fpubh.2020.00383
2. Jang WD, Jeon S, Kim S, Lee SY. Drugs repurposed for COVID-19 by virtual screening of 6218 drugs and cell-based assay. Proc Natl Acad Sci USA. 2021;118(30). doi:10.1073/pnas.2024302118
3. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. 2020;26(4):450–452. doi:10.1038/s41591-020-0820-9
4. Zhou P, Lou YX, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi:10.1038/s41586-020-2012-7
5. Jebril N. World Health Organization declared a pandemic public health menace: a systematic review of the coronavirus disease 2019 “COVID-19.”. SSRN Electron J. 2020. doi:10.2139/ssrn.3566298
6. Arya R, Kumari S, Pandey B, et al. Structural insights into SARS-CoV-2 proteins. J Mol Biol. 2021;433(2). doi:10.1016/j.jmb.2020.11.024
7. Pal M, Berhanu G, Desalegn C, Kandi V. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): an update. Cureus. 2020;12(3). doi:10.7759/cureus.7423
8. Gioia M, Ciaccio C, Calligari P, et al. Role of proteolytic enzymes in the COVID-19 infection and promising therapeutic approaches. Biochem Pharmacol. 2020;182:114225. doi:10.1016/j.bcp.2020.114225
9. Romano M, Ruggiero A, Squeglia F, Maga G, Berisio R, Structural A. View of SARS-CoV-2 RNA replication machinery: RNA synthesis, proofreading and final capping. Cells. 2020;9(5). doi:10.3390/cells9051267
10. Razali R, Asis H, Budiman C. Structure-function characteristics of SARS-CoV-2 proteases and their potential inhibitors from microbial sources. Microorganisms. 2021;9(12). doi:10.3390/MICROORGANISMS9122481
11. Alanagreh L, Alzoughool F, Atoum M. The human coronavirus disease covid-19: its origin, characteristics, and insights into potential drugs and its mechanisms. Pathogens. 2020;9(5). doi:10.3390/pathogens9050331
12. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020;92(4):418–423. doi:10.1002/jmv.25681
13. Shagufta AI. The race to treat COVID-19: potential therapeutic agents for the prevention and treatment of SARS-CoV-2. Eur J Med Chem. 2021;213. doi:10.1016/j.ejmech.2021.113157
14. Peele KA, Potla Durthi C, Srihansa T, et al. Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: a computational study. Informatics Med Unlocked. 2020;19:100345. doi:10.1016/j.imu.2020.100345
15. Das S, Sarmah S, Lyndem S, Singha Roy A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J Biomol Struct Dyn. 2021;39(9):3347–3357. doi:10.1080/07391102.2020.1763201/SUPPL_FILE/TBSD_A_1763201_SM9561.PDF
16. Klemm T, Ebert G, Calleja DJ, et al. Mechanism and inhibition of the papain‐like protease, PLpro, of SARS‐CoV‐2. EMBO J. 2020;39(18). doi:10.15252/embj.2020106275
17. Klemm T, Ebert G, Calleja DJ, et al. Mechanism and inhibition of SARS-CoV-2 PLpro. bioRxiv. 2020. doi:10.1101/2020.06.18.160614
18. Gao X, Qin B, Chen P, et al. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm Sin B. 2021;11(1):237–245. doi:10.1016/j.apsb.2020.08.014
19. Tan H, Hu Y, Jadhav P, Tan B, Wang J. Progress and challenges in targeting the SARS-CoV-2 papain-like protease. Cite This J Med Chem. 2022;2022. doi:10.1021/acs.jmedchem.2c00303
20. Mengist HM, Dilnessa T, Jin T. Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Front Chem. 2021;9:7. doi:10.3389/fchem.2021.622898
21. Cui W, Yang K, Yang H. Recent progress in the drug development targeting SARS-CoV-2 main protease as treatment for COVID-19. Front Mol Biosci. 2020;7:398. doi:10.3389/FMOLB.2020.616341/BIBTEX
22. Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem. 2016;59(14):6595–6628. doi:10.1021/acs.jmedchem.5b01461
23. Zhang L, Lin D, Sun X, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science (80-). 2020;368(6489):409–412. doi:10.1126/science.abb3405
24. Citarella A, Scala A, Piperno A, Micale N. Sars-cov-2 mpro: a potential target for peptidomimetics and small-molecule inhibitors. Biomolecules. 2021;11(4). doi:10.3390/biom11040607
25. Goyal B, Goyal D. Targeting the dimerization of the main protease of coronaviruses: a potential broad-spectrum therapeutic strategy. ACS Comb Sci. 2020;22(6):297–305. doi:10.1021/acscombsci.0c00058
26. Zumla A, Chan JFW, Azhar EI, Hui DSC, Yuen KY. Coronaviruses-drug discovery and therapeutic options. Nat Rev Drug Discov. 2016;15(5):327–347. doi:10.1038/nrd.2015.37
27. Coronavirus (COVID-19) update: FDA authorizes first oral antiviral for treatment of COVID-19 | FDA. Available from: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-first-oral-antiviral-treatment-covid-19.
28. Shi J, Song J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006;273(5):1035–1045. doi:10.1111/j.1742-4658.2006.05130.x
29. Shi J, Wei Z, Song J. Dissection study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme. Defining the extra domain as a new target for design of highly specific protease inhibitors. J Biol Chem. 2004;279(23):24765–24773. doi:10.1074/jbc.M311744200
30. Silvestrini L, Belhaj N, Comez L, et al. The dimer-monomer equilibrium of SARS-CoV-2 main protease is affected by small molecule inhibitors. Sci Rep. 2021;11(1):1–16. doi:10.1038/s41598-021-88630-9
31. Hu T, Zhang Y, Li L, et al. Two adjacent mutations on the dimer interface of SARS coronavirus 3C-like protease cause different conformational changes in crystal structure. Virology. 2009;388(2):324–334. doi:10.1016/j.virol.2009.03.034
32. Günther S, Reinke PYA, Fernández-Garciá Y, et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science (80-). 2021;372(6542):642–646. doi:10.1126/science.abf7945
33. Du R, Cooper L, Chen Z, Lee H, Rong L, Cui Q. Discovery of chebulagic acid and punicalagin as novel allosteric inhibitors of SARS-CoV-2 3CLpro. Antiviral Res. 2021;190. doi:10.1016/j.antiviral.2021.105075
34. Lin PY, Chou CY, Chang HC, Hsu WC, Chang GG. Correlation between dissociation and catalysis of SARS-CoV main protease. Arch Biochem Biophys. 2008;472(1):34–42. doi:10.1016/j.abb.2008.01.023
35. Liang J, Karagiannis C, Pitsillou E, et al. Site mapping and small molecule blind docking reveal a possible target site on the SARS-CoV-2 main protease dimer interface. Comput Biol Chem. 2020;89. doi:10.1016/j.compbiolchem.2020.107372
36. Sztain T, Amaro R, McCammon JA. Elucidation of cryptic and allosteric pockets within the SARS-CoV-2 main protease. J Chem Inf Model. 2021;61(7):3495–3501. doi:10.1021/acs.jcim.1c00140
37. Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12(1):88–118. doi:10.1016/S0022-2836(65)80285-6
38. Cooper A, Dryden DTF. Allostery without conformational change - a plausible model. Eur Biophys J. 1984;11(2):103–109. doi:10.1007/BF00276625
39. Hawkins RJ, McLeish TCB. Dynamic allostery of protein alpha helical coiled-coils. J R Soc Interface. 2006;3(6):125–138. doi:10.1098/rsif.2005.0068
40. McLeish TCB, Rodgers TL, Wilson MR. Allostery without conformation change: modelling protein dynamics at multiple scales. Phys Biol. 2013;10(5). doi:10.1088/1478-3975/10/5/056004
41. McLeish T, Schaefer C, von der Heydt AC. The “allosteron” model for entropic allostery of self-assembly. Philos Trans R Soc B Biol Sci. 2018;373(1749). doi:10.1098/rstb.2017.0186
42. Dubanevics I, McLeish TCB. Computational analysis of dynamic allostery and control in the SARS-CoV-2 main protease: computational analysis of dynamic allostery and control in the SARS-CoV-2 main protease. J R Soc Interface. 2021;18(174):20200591. doi:10.1098/rsif.2020.0591
43. Carli M, Sormani G, Rodriguez A, Laio A. Candidate binding sites for allosteric inhibition of the SARS-CoV-2 main protease from the analysis of large-scale molecular dynamics simulations. J Phys Chem Lett. 2020;12(1):65–72. doi:10.1021/ACS.JPCLETT.0C03182
44. El-Baba TJ, Lutomski CA, Kantsadi AL, et al. Allosteric inhibition of the SARS-CoV-2 main protease: insights from mass spectrometry based assays**. Angew Chemie Int Ed. 2020;59(52):23544–23548. doi:10.1002/ANIE.202010316
45. Roe MK, Junod NA, Young AR, Beachboard DC, Stobart CC. Targeting novel structural and functional features of coronavirus protease nsp5 (3CLpro, Mpro) in the age of COVID-19. J Gen Virol. 2021;102(3):001558. doi:10.1099/JGV.0.001558
46. Bhat ZA, Chitara D, Iqbal J, Sanjeev BS, Madhumalar A. Targeting allosteric pockets of SARS-CoV-2 main protease Mpro. J Biomol Struct Dyn. 2021. doi:10.1080/07391102.2021.1891141
47. Shi J, Sivaraman J, Song J. Mechanism for controlling the dimer-monomer switch and coupling dimerization to catalysis of the severe acute respiratory syndrome coronavirus 3C-like protease. J Virol. 2008;82(9):4620–4629. doi:10.1128/JVI.02680-07
48. Sang P, Tian S-H, Meng Z-H, Yang L-Q. Anti-HIV drug repurposing against SARS-CoV-2. RSC Adv. 2020;10(27):15775–15783. doi:10.1039/D0RA01899F
49. Chen H, Wei P, Huang C, Tan L, Liu Y, Lai L. Only one protomer is active in the dimer of SARS 3C-like proteinase. J Biol Chem. 2006;281(20):13894–13898. doi:10.1074/jbc.M510745200
50. Grottesi A, Bešker N, Emerson A, et al. Computational studies of SARS-CoV-2 3CLpro: insights from MD simulations. Int J Mol Sci. 2020;21(15):1–18. doi:10.3390/IJMS21155346
51. Douangamath A, Fearon D, Gehrtz P, et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat Commun. 2020;11(1):1–11. doi:10.1038/s41467-020-18709-w
52. Novak J, Rimac H, Kandagalla S, et al. Proposition of a new allosteric binding site for potential SARS-CoV-2 3CL protease inhibitors by utilizing molecular dynamics simulations and ensemble docking. J Biomol Struct Dyn. 2021. doi:10.1080/07391102.2021.1927845
53. Jiménez-Avalos G, Vargas-Ruiz AP, Delgado-Pease NE, et al. Comprehensive virtual screening of 4.8 k flavonoids reveals novel insights into allosteric inhibition of SARS-CoV-2 MPRO. Sci Rep. 2021;11(1):1–19. doi:10.1038/s41598-021-94951-6
54. Wei P, Fan K, Chen H, et al. The N-terminal octapeptide acts as a dimerization inhibitor of SARS coronavirus 3C-like proteinase. Biochem Biophys Res Commun. 2006;339(3):865–872. doi:10.1016/j.bbrc.2005.11.102
55. Ding L, Zhang XX, Wei P, Fan K, Lai L. The interaction between severe acute respiratory syndrome coronavirus 3C-like proteinase and a dimeric inhibitor by capillary electrophoresis. Anal Biochem. 2005;343(1):159–165. doi:10.1016/j.ab.2005.04.027
56. Chen YW, Yiu CPB, Wong KY. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research. 2020;9. doi:10.12688/f1000research.22457.2
57. Maestro, Schrödinger, LLC. New York, NY; 2021. Available from: http://www.schrodinger.com/.
58. SiteMap, Schrödinger, LLC. New York, NY; 2021. Available from: http://www.schrodinger.com/.
59. Halgren TA. Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model. 2009;49(2):377–389. doi:10.1021/ci800324m
60. Cantrelle F-X, Boll E, Brier L, et al. NMR spectroscopy of the main protease of SARS‐CoV‐2 and fragment‐based screening identify three protein hotspots and an antiviral fragment. Angew Chemie Int Ed. 2021. doi:10.1002/ANIE.202109965
61. Pinzi L, Tinivella A, Caporuscio F, Rastelli G. Drug repurposing and polypharmacology to fight SARS-CoV-2 through inhibition of the main protease. Front Pharmacol. 2021;12:84. doi:10.3389/fphar.2021.636989
62. Menéndez CA, Byléhn F, Perez-Lemus GR, Alvarado W, de Pablo JJ. Molecular characterization of ebselen binding activity to SARS-CoV-2 main protease. Sci Adv. 2020;6(37). doi:10.1126/sciadv.abd0345
63. Yuce M, Cicek E, Inan T, Dag AB, Kurkcuoglu O, Sungur FA. Repurposing of FDA-approved drugs against active site and potential allosteric drug-binding sites of COVID-19 main protease. Proteins Struct Funct Bioinforma. 2021. doi:10.1002/PROT.26164
64. Gregory KJ, Sexton PM. Overview of receptor allosterism. Curr Protoc Pharmacol. 2010;Chapter 1(SUPPL.51). doi:10.1002/0471141755.PH0121S51
65. Owen DR, Allerton CMN, Anderson AS, et al. An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19. Science (80-). 2021. doi:10.1126/science.abl4784
66. Hoffman RL, Kania RS, Brothers MA, et al. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J Med Chem. 2020;63(21):12725–12747. doi:10.1021/acs.jmedchem.0c01063
67. Boggetto N, Reboud-Ravaux M. Dimerization inhibitors of HIV-1 protease. Biol Chem. 2002;383(9):1321–1324. doi:10.1515/BC.2002.150
68. Tekpinar M, Yildirim A. Impact of dimerization and N3 binding on molecular dynamics of SARS-CoV and SARS-CoV-2 main proteases. J Biomol Struct Dyn. 2021. doi:10.1080/07391102.2021.1880481
69. Strömich L, Wu N, Barahona M, Yaliraki SN. Allosteric hotspots in the main protease of SARS-CoV-2. bioRxiv. 2020. doi:10.1101/2020.11.06.369439
70. Chou CY, Chang HC, Hsu WC, Lin TZ, Lin CH, Chang GG. Quaternary structure of the severe acute respiratory syndrome (SARS) coronavirus main protease. Biochemistry. 2004;43(47):14958–14970. doi:10.1021/bi0490237
71. El AD, Lagardère L, Inizan TJ, et al. Interfacial water many-body effects drive structural dynamics and allosteric interactions in SARS-CoV-2 main protease dimerization interface. J Phys Chem Lett. 2021;12(26):6218–6226. doi:10.1021/ACS.JPCLETT.1C01460
72. Eberle RJ, Olivier DS, Amaral MS, et al. The repurposed drugs suramin and quinacrine cooperatively inhibit sars-cov-2 3clpro in vitro. Viruses. 2021;13(5). doi:10.3390/v13050873
73. Wu C, Liu Y, Yang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10(5):766–788. doi:10.1016/j.apsb.2020.02.008
74. Verma S, Pandey AK. Factual insights of the allosteric inhibition mechanism of SARS-CoV-2 main protease by quercetin: an in silico analysis. 3 Biotech. 2021;11(2):67. doi:10.1007/s13205-020-02630-6
75. Olubiy OO, Olagunju M, Keutmann M, Loschwitz J, Strodel B. High throughput virtual screening to discover inhibitors of the main protease of the coronavirus SARS-CoV-2. Molecules. 2020;25(14). doi:10.3390/molecules25143193
76. Sencanski M, Perovic V, Pajovic SB, Adzic M, Paessler S, Glisic S. Drug repurposing for candidate SARS-CoV-2 main protease inhibitors by a novel in silico method. Molecules. 2020;25(17). doi:10.3390/molecules25173830
77. Kneller DW, Phillips G, O’Neill HM, et al. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat Commun. 2020;11(1). doi:10.1038/s41467-020-16954-7
78. Jaffrelot Inizan T, Célerse F, Adjoua O, et al. High-resolution mining of the SARS-CoV-2 main protease conformational space: supercomputer-driven unsupervised adaptive sampling. Chem Sci. 2021;12(13):4889–4907. doi:10.1039/d1sc00145k
79. Sharun K, Tiwari R, Dhama K. Protease inhibitor GC376 for COVID-19: lessons learned from feline infectious peritonitis. Ann Med Surg. 2021;61:122–125. doi:10.1016/j.amsu.2020.12.030
80. Iftikhar H, Ali HN, Farooq S, Naveed H, Shahzad-ul-Hussan S. Identification of potential inhibitors of three key enzymes of SARS-CoV2 using computational approach. Comput Biol Med. 2020;122:103848. doi:10.1016/j.compbiomed.2020.103848
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.