")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Alisertib, an Aurora kinase A inhibitor, induces apoptosis and autophagy but inhibits epithelial to mesenchymal transition in human epithelial ovarian cancer cells
Authors Ding Y, Zhou Z, Ha C, Zhang X, Pan S, He Z, Edelman J, Wang D, Yang Y, Zhang X, Duan W, Yang T, Qiu J, Zhou S
Received 10 September 2014
Accepted for publication 3 October 2014
Published 9 January 2015 Volume 2015:9 Pages 425—464
DOI https://doi.org/10.2147/DDDT.S74062
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Min Li
Yong-Hui Ding,1,2 Zhi-Wei Zhou,2,3 Chun-Fang Ha,1 Xue-Yu Zhang,1 Shu-Ting Pan,4 Zhi-Xu He,3 Jeffrey L Edelman,2 Dong Wang,5 Yin-Xue Yang,6 Xueji Zhang,7 Wei Duan,8 Tianxin Yang,9 Jia-Xuan Qiu,4 Shu-Feng Zhou2,3
1Department of Gynecology, General Hospital of Ningxia Medical University, Yinchuan, People’s Republic of China; 2Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL, USA ; 3Guizhou Provincial Key Laboratory for Regenerative Medicine, Stem Cell and Tissue Engineering Research Center and Sino-US Joint Laboratory for Medical Sciences, Guiyang Medical University, Guiyang, 4Department of Oral and Maxillofacial Surgery, The First Affiliated Hospital of Nanchang University, Nanchang, 5Cancer Center, Daping Hospital and Research Institute of Surgery, Third Military Medical University, Chongqing, 6Department of Colorectal Surgery, General Hospital of Ningxia Medical University, Yinchuan, 7Research Center for Bioengineering and Sensing Technology, University of Science and Technology Beijing, Beijing, People’s Republic of China; 8School of Medicine, Deakin University, Waurn Ponds, Australia; 9Department of Internal Medicine, University of Utah and Salt Lake Veterans Affairs Medical Center, Salt Lake City, UT, USA
Abstract: Ovarian cancer is a leading killer of women, and no cure for advanced ovarian cancer is available. Alisertib (ALS), a selective Aurora kinase A (AURKA) inhibitor, has shown potent anticancer effects, and is under clinical investigation for the treatment of advanced solid tumor and hematologic malignancies. However, the role of ALS in the treatment of ovarian cancer remains unclear. This study investigated the effects of ALS on cell growth, apoptosis, autophagy, and epithelial to mesenchymal transition (EMT), and the underlying mechanisms in human epithelial ovarian cancer SKOV3 and OVCAR4 cells. Our docking study showed that ALS, MLN8054, and VX-680 preferentially bound to AURKA over AURKB via hydrogen bond formation, charge interaction, and π-π stacking. ALS had potent growth-inhibitory, proapoptotic, proautophagic, and EMT-inhibitory effects on SKOV3 and OVCAR4 cells. ALS arrested SKOV3 and OVCAR4 cells in G2/M phase and induced mitochondria-mediated apoptosis and autophagy in both SKOV3 and OVCAR4 cell lines in a concentration-dependent manner. ALS suppressed phosphatidylinositol 3-kinase/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) and p38 mitogen-activated protein kinase pathways but activated 5′-AMP-dependent kinase, as indicated by their altered phosphorylation, contributing to the proautophagic activity of ALS. Modulation of autophagy altered basal and ALS-induced apoptosis in SKOV3 and OVCAR4 cells. Further, ALS suppressed the EMT-like phenotype in both cell lines by restoring the balance between E-cadherin and N-cadherin. ALS downregulated sirtuin 1 and pre-B cell colony enhancing factor (PBEF/visfatin) expression levels and inhibited phosphorylation of AURKA in both cell lines. These findings indicate that ALS blocks the cell cycle by G2/M phase arrest and promotes cellular apoptosis and autophagy, but inhibits EMT via phosphatidylinositol 3-kinase/Akt/mTOR-mediated and sirtuin 1-mediated pathways in human epithelial ovarian cancer cells. Further studies are warranted to validate the efficacy and safety of ALS in the treatment of ovarian cancer.
Keywords: alisertib, Aurora kinase A, epithelial ovarian cancer, cell cycle, apoptosis, autophagy, epithelial to mesenchymal transition, sirtuin 1
Introduction
Ovarian cancer is the eighth most common type of cancer and the seventh most common cause of cancer-related death in women worldwide, with 239,000 new cases and around 152,000 deaths in 2012.1 In 2010, 19,959 women in the USA were diagnosed with ovarian cancer, and 14,572 women in the USA died from the disease. In 2014, it is estimated that there will be 21,980 new cases of ovarian cancer and that an estimated 14,270 people will die of the disease in the USA.2 In Europe, there were around 65,600 new cases of ovarian cancer and around 42,700 deaths due to the disease in 2012.1 In 2011, there were around 7,100 new cases of ovarian cancer diagnosed in the UK, and 4,300 women died from the disease.3 The estimated incidence of ovarian cancer is increasing among Chinese women; it has increased by 30% in the last 10 years and the mortality rate has increased by 18%, with 15,000 deaths each year.1,4 Age and genetics are the most well established risk factors for ovarian cancer. The majority of women with epithelial ovarian cancer (EOC) are diagnosed at an advanced stage when the disease has spread beyond the ovary, which substantially increases the risk of recurrence and early death.5 Peritoneal dissemination and ascites are the major causes of patient morbidity and mortality. Platinum-based and taxane-based chemotherapy is standard of care for the first-line treatment of patients with advanced ovarian, fallopian tube, or peritoneal cancer.5 Responses are observed in up to ~80% of patients; however, 70%–80% of responding patients will relapse, requiring further systemic chemotherapy.6 The 5-year survival rate ranges from ~30% to 60%. Patients who relapse more than 6–12 months after completing platinum-based chemotherapy may respond again to platinum-based therapy, but repeated use of platinum agents can be limited due to acquired resistance or intolerance.6 Hence, there is an urgent need to develop new, safe, and effective therapeutics for advanced EOC.
The Aurora kinases are a family of serine/threonine kinases consisting of three members, Aurora kinase A (AURKA), AURKB, and AURKC.7 The three members of the Aurora kinases of varying peptide lengths share similar catalytic domains located in the C-terminus, but their N-terminal extensions are of variable length and display little or no similarity. AURKA and AURKB are essential regulators of mitosis and centrosome function and act by polymerizing microfilaments and controlling chromatid segregation, but the function of AURKC is less clear.7 All three Aurora kinases have a conserved adenosine triphosphate binding pocket but different amino acid sequences at the N-terminal domain. AURKA has critical roles in the regulation of mitotic entry, centrosome function, bipolar spindle assembly, and segregation of chromosomes.8,9 AURKA is critical for correct formation of the mitotic spindle. It is required for the recruitment of several different proteins important to spindle formation. Among these target proteins is TACC, a microtubule-associated protein that stabilizes centrosomal microtubules and kinesin 5, a motor protein involved in formation of the bipolar mitotic spindle.7
As a key regulator of the cell cycle, AURKA regulates G2/M transition. AURKA is regulated by phosphorylation in a cell cycle-dependent manner, occurring on a conserved residue, Thr288, within the activation loop of the catalytic domain of the kinase and resulting in a significant increase in enzymatic activity.10 Inhibition of AURKA expression leads to G2/M arrest and apoptosis, whereas ectopic expression allows cells to bypass the G2/M DNA damage checkpoint.11 AURKA maps to human chromosome 20q13 and AURKB to 17q13.1, which are loci frequently altered in human cancers. AURKC is located on chromosome 19q13.2 to 13.4, a region associated with loss of heterozygosity in ovarian cancer and pancreatic carcinomas. The expression and activity of Aurora kinases are tightly regulated, and dysregulation results in genetic instability, aneuploidy, and tumorigenesis.7,12 The AURKA gene is frequently amplified and/or overexpressed in a number of malignancies, including cancers of the bladder, breast, colon, liver, ovary, pancreas, stomach, and esophagus, and aberrant AURKA signaling is associated with malignant tumor behavior such as invasion and metastasis, advanced stage, and poor prognosis.11,13,14 Overexpression of AURKA is common in ovarian cancer, which is associated with supernumerary centrosomes, a poor response to chemotherapy, and reduced overall survival.10,15–17
AURKA has become a target of interest for the treatment of cancer, and a number of Aurora kinase inhibitors that have dual specificity for AURKA and AURKB, including MK-0457 and PHA-739358, have been developed.11,14,18 Alisertib (MLN8237, ALS, Figure 1) is an investigational small-molecule inhibitor developed by Millennium Pharmaceuticals Inc (Boston, MA, USA) which selectively inhibits AURKA and has been shown in preclinical studies to induce cell cycle arrest, polyploidy, and mitotic catastrophe in various tumor cells, and to induce tumor regression in vivo.19–21 Currently, ALS is being tested in various Phase I and Phase II clinical trials for advanced solid tumors and hematologic malignancies.22–27 In the present study, we aimed to uncover the underlying mechanisms for the anticancer effects of ALS in human EOC cells. Before we performed our benchmarking experiments, we ran molecular docking assays to check how ALS bound to AURKA and AURKB and to compare the differences in the binding mode with those of other Aurora kinase inhibitors, including AMG-900, barasertib, CYC116, danusertib, MLN8054, and VX-680 (also called MK-0457), which are selective or nonselective inhibitors for AURKA.11,28
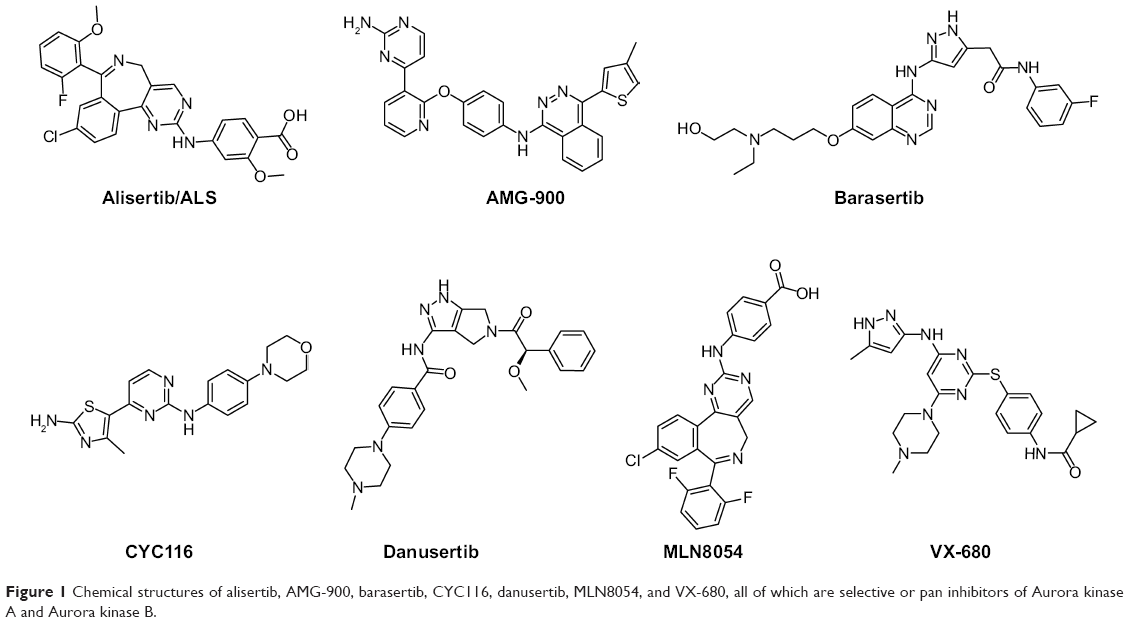 | Figure 1 Chemical structures of alisertib, AMG-900, barasertib, CYC116, danusertib, MLN8054, and VX-680, all of which are selective or pan inhibitors of Aurora kinase A and Aurora kinase B. |
Materials and methods
Molecular docking
In order to determine the molecular interactions between AURKA and AURKB and their inhibitors, the Discovery Studio program 3.1 designed by Accelrys Inc (San Diego, CA, USA) was used to dock ALS, AMG-900 (a potent and highly selective pan-AURKA, AURKB, and AURKC inhibitor29), barasertib (a highly selective AURKB inhibitor30), CYC116 (a potent inhibitor of AURKA and AURKB31), danusertib (an AURKA, AURKB, and AURKC inhibitor31), MLN8054 (a potent and selective inhibitor of AURKA32), and VX-680 (a pan-AURKA, AURKB, and AURKC, mostly against AURKA33) (Figure 1) into the active sites of human AURKA (Protein Data Bank [PDB] identification [ID]: 2DWB) and AURKB (PDB ID: 4AF3) as previously described.34–36 The crystal structures of human AURKA and AURKB were obtained from the PDB (http://www.rcsb.org/pdb/). The protein and ligand were prepared prior to the docking. For protein preparation, AURKA and AURKB were cleaned, modified, and prepared for defining and editing the binding site. During preparation for ALS, AMG-900, barasertib, CYC116, danusertib, MLN8054, and VX-680, the duplicate structures were deleted and ionization change, tautomer or isomer generation, Lipinski filter, and three-dimensional generator were all set true. A harmonic potential with the force constant of 300 kcal/mol was applied outside the grid boundary. Following preparation of protein, ligand, and grid setting, ALS, AMG-900, barasertib, CYC116, danusertib, MLN8054, and VX-680 were docked into binding sites of AURKA and AURKB. Electrostatic energy and van der Waals forces were considered during the docking process. For each defined van der Waals force or electrostatic probe, the interactions with all protein atoms were stored at predetermined grid points. For ligand atoms located between grid points, a trilinear interpolation was used to approximate the energies.
Chemicals and reagents
ALS, Dulbecco’s phosphate-buffered saline (PBS), thiazolyl blue tetrazolium bromide (MTT), 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), ethylenediamine tetraacetic acid, RNase A, propidium iodide, and fetal bovine serum were purchased from Sigma-Aldrich Inc (St Louis, MO, USA). 4′ 6-Diamidino-2-phenylindole and 5-(and 6)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) were sourced from Invitrogen (Carlsbad, CA, USA). Dulbecco’s Modified Eagle’s Medium and McCoy’s 5A medium were purchased from Corning Cellgro Inc (Herndon, VA, USA). The Annexin V:phycoerythrin apoptosis was purchased from BD Biosciences Inc (San Jose, CA, USA). A Cyto-ID® autophagy detection kit was obtained from Enzo Life Sciences Inc (Farmingdale, NY, USA). The polyvinylidene difluoride membrane was sourced from Millipore Inc (Bedford, MA, USA). Western blotting substrate was obtained from Thermo Scientific Inc (Hudson, NH, USA). The autophagy inhibitors chloroquine, bafilomycin A, and wortmannin (an irreversible and selective phosphatidylinositol 3-kinase [PI3K] inhibitor and a blocker of autophagosome formation), and the apoptosis inhibitor Z-VAD(OMe)-FMK were purchased from Invivogen (Carlsbad, CA, USA). SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl) 1H-imidazole; an autophagy inducer and specific p38 mitogen-activated protein kinase inhibitor (MAPK)] was purchased from Santa Cruz Biotechnology Inc (Santa Cruz, CA, USA). Primary antibodies against human silent mating type information regulation 1 (sirtuin 1 [Sirt1]), p53, acetylated (Ac)-p53 (Lys379), p21 Wafl/Cipl, p27 Kipl, cyclin B1, cell division cycle protein 2 homolog (CDC2), the p53-upregulated modulator of apoptosis (PUMA), B-cell lymphoma 2 (Bcl-2), Bcl-2-like protein4/Bcl-2-associated X protein (Bax), B-cell lymphoma-extra-large (Bcl-xl), cytochrome c, cleaved caspase 3, cleaved caspase 9, microtubule-associated protein 1A/1B-light chain 3 (LC3-I), LC3-II, p38 MAPK, phosphorylated (p)-38 MAPK at Thr180/Tyr182, protein kinase B (Akt), p-Akt at Ser473, mammalian target of rapamycin (mTOR), p-mTOR at Ser2448, PI3K, p-PI3K at Tyr458, 5′ AMP-activated protein kinase (AMPK), p-AMPK at Thr172, beclin1, AURKA, p-AURKA at Thr288, and epithelial to mesenchymal transition (EMT) antibody sampler kit (#9782) were all purchased from Cell Signaling Technology Inc (Beverly, MA, USA). The EMT antibody sampler kit contains primary antibodies to N-cadherin, E-cadherin, zona occludens protein 1 (ZO-1), vimentin, slug, snail, zinc finger E-box-binding homeobox 1 (TCF8/ZEB1), and β-catenin. The antibody against human β-actin and pre-B-cell colony-enhancing factor (PBEF/visfatin, also called nicotinamide phosphoribosyltransferase) were obtained from Santa Cruz Biotechnology Inc.
Cell lines and cell culture
SKOV3 and OVCAR4 cells are human EOC cell lines, and T80 cells are a normal epithelial ovarian cell line. SKOV3, OVCAR4, and T80 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in McCoy’s 5A medium. All medium contained L-glutamine, phenol red, L-cysteine, L-methionine, and sodium bicarbonate, and was supplemented with 10% heat-inactivated fetal bovine serum. The cells were maintained at 37°C in a 5% CO2/95% air humidified incubator. ALS was dissolved in dimethyl sulfoxide (DMSO) with a stock concentration of 50 mM, and was freshly diluted to the desired concentration with culture medium. The final concentration of DMSO was 0.05% (v/v).
Cell viability assay
The MTT assay was performed to examine the effect of ALS on cell viability. Briefly, cells were seeded in 96-well culture plates at a density of 8×l03 per well. After the cells were attached, the medium was changed with freshly prepared medium containing ALS at different concentrations (from 0.1 to 100 μM). The control cells received the vehicle only. After 24 hours of incubation, 10 μL MTT (5 g/L) was added to each well and cultured for another 4 hours. The medium was then removed and 150 μL of DMSO was added. It was shaken for 15 minutes for crystal dissolution. Absorbance at the 490 nm wavelength was measured with a Synergy H4 Hybrid microplate reader (BioTek Inc, Winooski, VT, USA). IC50 values were determined using the relative viability over ALS concentration curve.
Cell cycle analysis using flow cytometry
Propidium iodide is used as a DNA stain to determine DNA content in cell cycle analysis. The effect of ALS on cell cycle distribution was determined by flow cytometry as described previously.37 Briefly, SKOV3 and OVCAR4 cells were treated with ALS at different concentrations (0.1, 1, and 5 μM) for 24 hours. In separate experiments, SKOV3 and OVCAR4 cells were treated with ALS 5 μM for 4, 8, 12, 24, 48, or 72 hours. The cells were suspended, washed with PBS, centrifuged, and fixed in 70% ethanol at −20°C overnight. The cells were then resuspended in 1 mL of PBS containing 1 mg/mL RNase A and 50 μg/mL propidium iodide. The cells were incubated in the dark for 30 minutes at room temperature. In total, 1×104 cells were subject to cell cycle analysis using a flow cytometer (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA).
Simultaneous determination of apoptosis and autophagy using flow cytometry
To explore the potential crosstalk between ALS-induced apoptosis and autophagy, we further determined the two modes of programmed cell death simultaneously. SKOV3 and OVCAR4 cells were plated in six-well cell culture plates (Corning Incorporated, Corning, NY, USA) at a density of 1×105 cells per well. After incubation overnight, the cells were incubated with fresh medium, vehicle alone (0.05% DMSO, v/v), or ALS at 0.1, 1, and 5 μM for 24 hours. For comparison purposes, the autophagy inducer SB203580 at 10 μM, autophagy inhibitor wortmannin at 10 μM, chloroquine at 30 μM, bafilomycin A at 10 μM, and the apoptosis inhibitor Z-VAD(OMe)-FMK at 20 μM were used alone or in combination with 5 μM ALS to treat the cells for 24 hours. At the end of treatment, the cells were trypsinized and centrifuged at 1,000× g for 5 minutes to pellet the cells. The cells were divided into two samples of equal volume and washed with PBS. For detection of apoptosis using Annexin V, each sample was resuspended in 1 mL of binding buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2; filtered through a 0.2 μm filter). An aliquot of cell suspension (195 μL) was mixed with 5 μL of Annexin-ATTO 647N (Enzo Life Sciences Inc; ALX-209-259-T100), and incubated for 10 minutes in the dark. The cells were washed once using PBS and resuspended in 190 μL of binding buffer; 10 μL of 20 μg/mL propidium iodide was then added, and the cells were subjected to apoptotic analysis using flow cytometry. For detection of autophagy, each sample was washed by resuspending the cell pellet in 1× assay buffer (Enzo Life Sciences Inc; ENZ-51031-K200) and collected by centrifugation. Cells were resuspended in 250 μL of phenol red-free culture medium (Invitrogen; #1294895) containing 5% fetal bovine serum, and 250 μL of diluted Cyto-ID green stain solution (Enzo Life Sciences Inc; #ENZ-51031-K200) was added to each sample and mixed well. Cells were incubated for 30 minutes at 37°C in the dark, collected by centrifugation, washed with 1× assay buffer, and resuspended in 500 μL of fresh 1× assay buffer. Cells were analyzed using the green (FL1) channel of a flow cytometer.
Confocal fluorescence microscopy
The cellular autophagy level was examined using a Cyto-ID autophagy detection kit according to the manufacturer’s instructions. The SKOV3 and OVCAR4 cells were seeded into an eight-well chamber slide at 30% confluence. After incubation overnight, the cells were treated with ALS 0.1, 1, and 5 μM for 24 hours. In separate experiments, cells were pretreated with the autophagy inducer SB203580 (10 μM), autophagy inhibitor wortmannin (10 μM), chloroquine (30 μM), bafilomycin A (10 μM), and the apoptosis inhibitor Z-VAD(OMe)-FMK (20 μM) for 1 hour, and then treated with ALS 5 μM. After incubation for 24 hours, the cells reached ~60% confluence, were washed three times with 1× assay buffer, and then fixed by paraformaldehyde. A 40 μL aliquot of assay buffer with 4′ 6-diamidino-2-phenylindole was used to stain DNA molecules in the cells. The slides were covered with glass coverslips and sealed with polish oil. Samples were observed using a TCS SP2 laser scanning confocal microscope (Leica, Wetzlar, Germany) at a 405/488 nm wavelength.
Western blotting analysis
Expression levels of the cellular proteins of interest were determined using Western blotting assays.37 SKOV3 and OVCAR4 cells were washed with pre-cold PBS after 24 hours of treatment with ALS 0.1, 1, and 5 μM, lysed with radioimmunoprecipitation assay (RIPA) buffer (50 mmol HEPES at pH 7.5, 150 mmol NaCl, 10% glycerol, 1.5 mmol MgCl2, 1% Triton-X 100, 1 mmol ethylenediamine tetraacetic acid at pH 8.0, 10 mmol sodium pyrophosphate, 10 mmol sodium fluoride) containing the protease inhibitor and phosphatase inhibitor cocktails, and centrifuged at 3,000× g for 10 minutes at 4°C. The protein concentrations were measured using a bicinchoninic acid protein assay kit (Pierce, Rockford, IL, USA). An equal amount of protein sample (30 μg) was resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer and electrophoresed on 7%-12% SDS-PAGE minigel after thermal denaturation at 95°C for 5 minutes. The proteins were transferred onto an Immobilon® polyvinylidene difluoride membrane at 400 mA for 2 hours at 4°C. The membranes were probed with the primary antibodies indicated in the “Chemicals and reagents” section overnight at 4°C and then blotted with respective secondary antimouse or antirabbit antibody. Visualization was performed using a ChemiDoc™ XRS system (Bio-Rad, Hercules, CA, USA) with an enhanced chemiluminescence substrate, and the blots were analyzed using Image Lab 3.0 (BioRad). The protein level was normalized to the matching densitometry values of the internal control, β-actin.
Statistical analysis
The data are presented as the mean ± standard deviation. Comparisons of multiple groups were evaluated by one-way analysis of variance followed by Tukey’s multiple comparison procedure. P<0.05 was considered to be statistically significant. Assays were performed at least three times independently.
Results
Molecular interactions between ALS and other inhibitors, and AURKA and AURKB
ALS is known to be a potent selective AURKA inhibitor with an IC50 of 1.2 nM, and also inhibits the activity of AURKB with an IC50 of 396.5 nM.21,38 To explore how ALS interacts with AURKA and AURKB and to compare the differences in molecular interactions between ALS and other Aurora kinase inhibitors, we first carried out docking experiments using the Discovery Studio program 3.1.
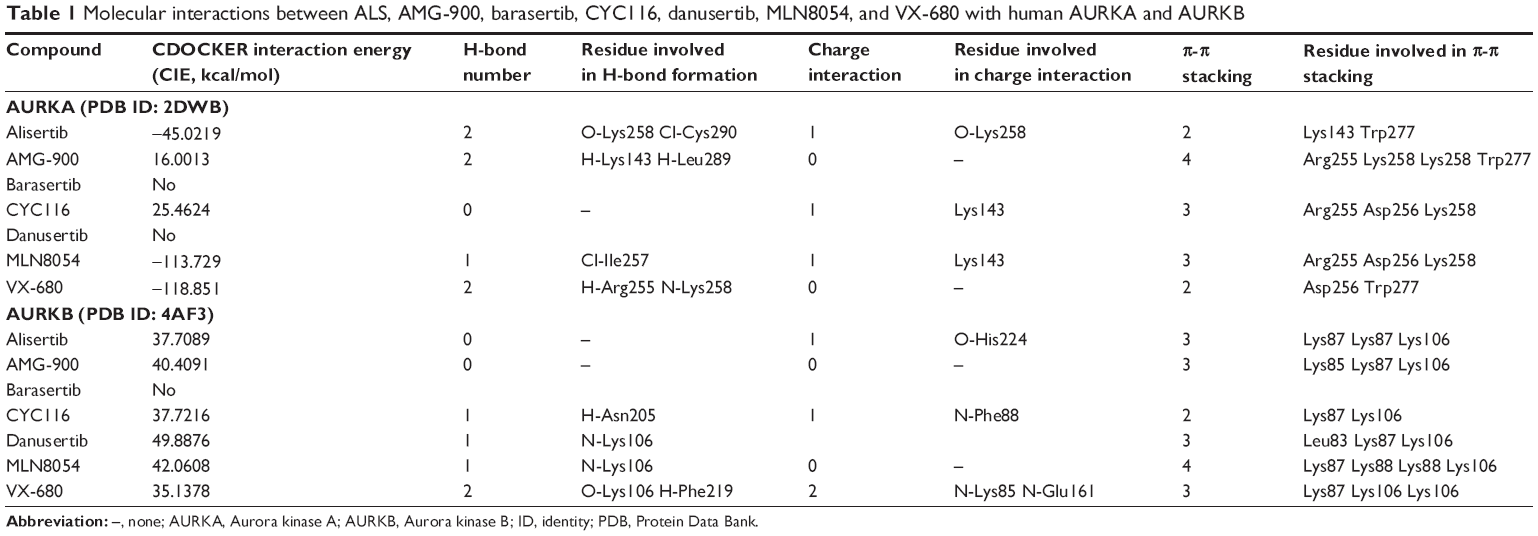
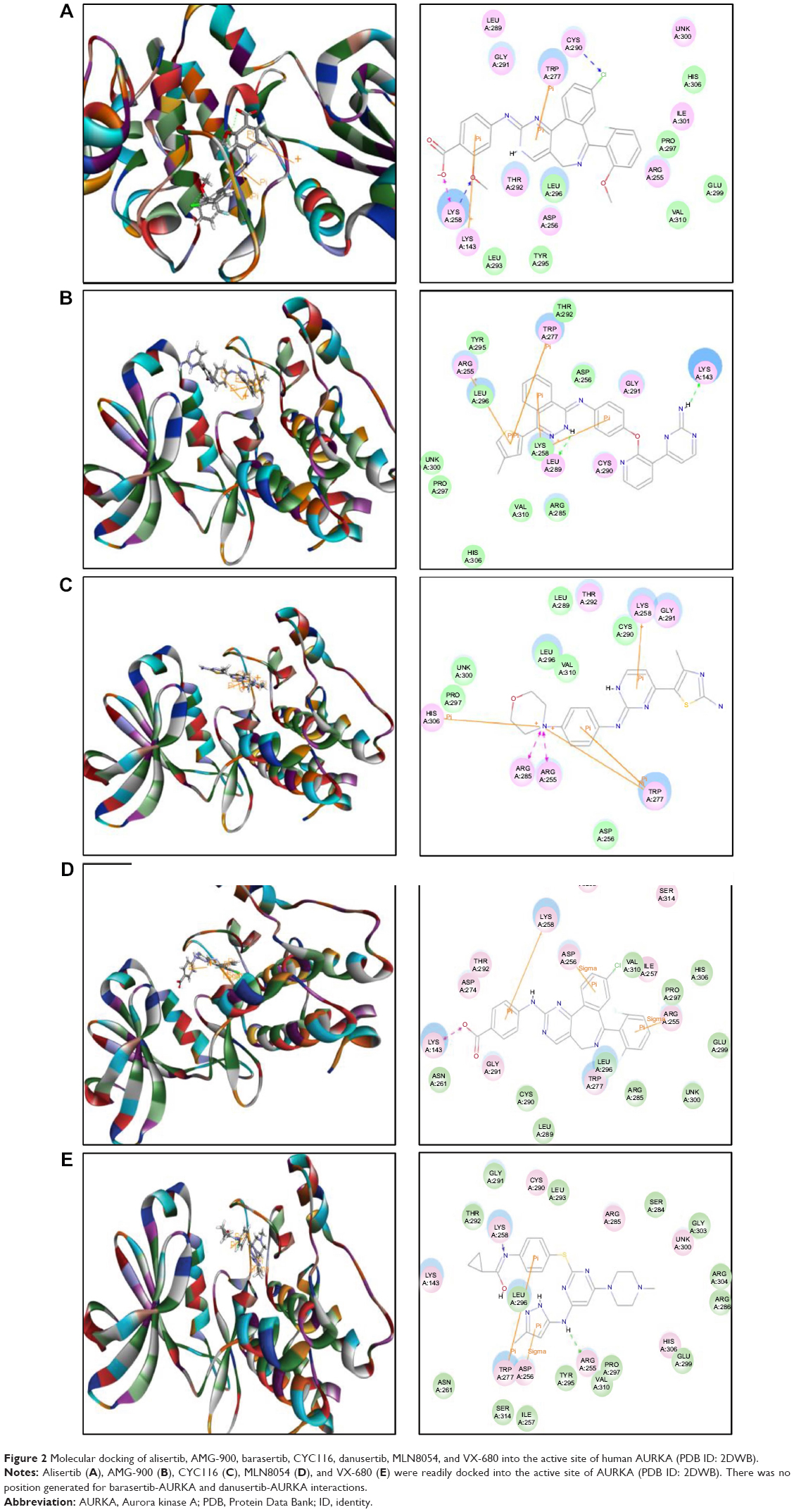
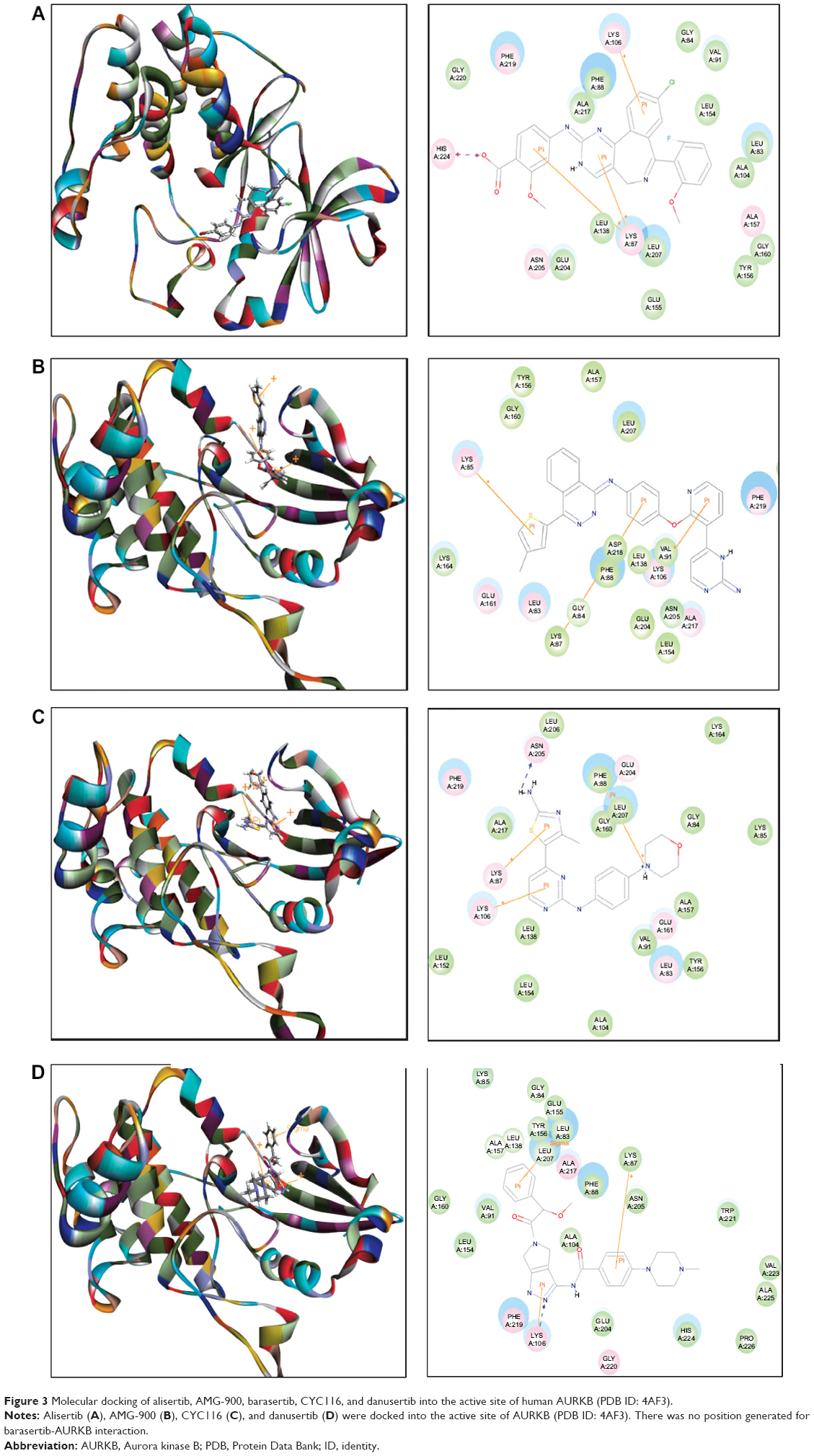
The docking results are shown in Figures 2–4. After docking ALS into the active sites of AURKA and AURKB, there were one and 50 positions generated for ALS-AURKA and ALS-AURKB interactions, respectively (Figures 2A and 3A). The CDOCKER interaction energy ranged from around −45 to 58 kcal/mol. Each compound-enzyme complex with the highest CDOCKER interaction energy was selected, and two-dimensional and three- dimensional pictures of these complexes were collected. The data showed that ALS bound to AURKA through formation of a hydrogen bond at Lys258 and Cys290, a charge interaction at Lys258, and π-π stacking at Lys143 and Trp277 (Figure 2A and Table 1). ALS also bound to AURKB via charge interaction at the site of His224 and π-π stacking at Lys87 and Lys106 (Figure 3A). There was a remarkable difference in CDOCKER interaction energy for the interactions between ALS and AURKA (–45.0219 kcal/mol) and AURKB (37.7089 kcal/mol), which may partly explain the difference in inhibitory effect of ALS on these two Aurora kinases.
 | Table 1 Molecular interactions between ALS, AMG-900, barasertib, CYC116, danusertib, MLN8054, and VX-680 with human AURKA and AURKB |
 | Figure 2 Molecular docking of alisertib, AMG-900, barasertib, CYC116, danusertib, MLN8054, and VX-680 into the active site of human AURKA (PDB ID: 2DWB). |
 | Figure 3 Molecular docking of alisertib, AMG-900, barasertib, CYC116, and danusertib into the active site of human AURKB (PDB ID: 4AF3). |
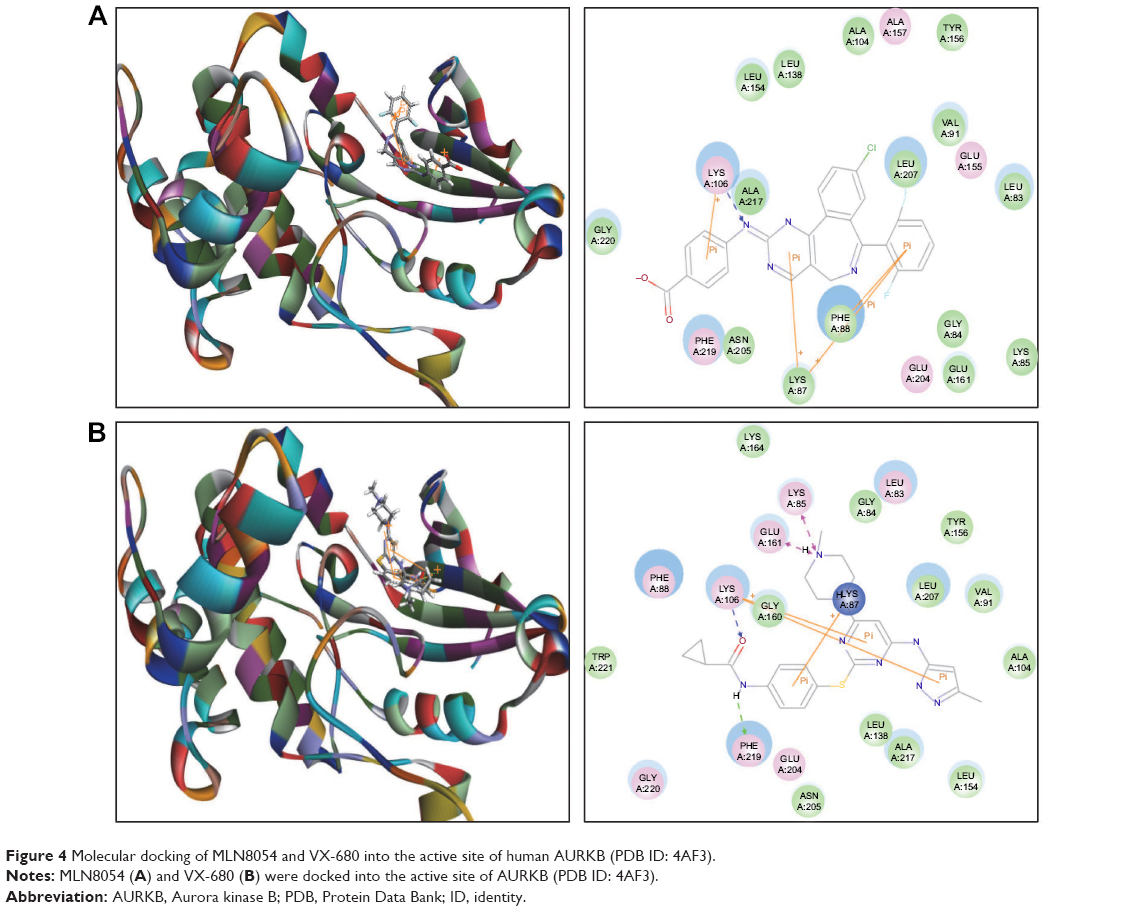 | Figure 4 Molecular docking of MLN8054 and VX-680 into the active site of human AURKB (PDB ID: 4AF3). |
Further, we docked AMG-900, barasertib, CYC116, danusertib, MLN8054, and VX-680 into the active sites of AURKA and AURKB. There were one and 90 positions generated for AMG-900-AURKA and AMG-900-AURKB interactions, respectively. AMG-900 interacted with AURKA via hydrogen bond formation at Lys143 and Leu289 and π-π stacking with Arg255, Lys258, and Trp277 (Figure 2B). AMG-900 also bound to AURKB through π-π stacking at Lys85, Lys87, and Lys106 (Figure 3B). The CDOCKER interaction energy was 16.0013 kcal/mol and 40.4091 kcal/mol for AMG-900-AURKA and AMG-900-AURKB, respectively (Table 1).
CYC116 bound to AURKA via charge interaction at Lys143 and π-π stacking at Arg255, Asp256, and Lys258, with a CDOCKER interaction energy of 25.4624 kcal/mol (Figure 2C and Table 1), and bound to AURKB through hydrogen bond formation at Asn205, charge interaction at Phe88, and π-π stacking at Lys87 and Lys106, with a CDOCKER interaction energy of 37.7216 kcal/mol (Figure 3C and Table 1). There were 360 positions for both CYC116-AURKA and CYC116-AURKB interactions.
The MLN8054-AURKA interactions had 12 positions involving hydrogen bond formation at Ile257, charge interaction at Lys143, and π-π stacking at Arg255, Asp256, and Lys258 (Figure 2D), and the MLN8054-AURKB interactions had 50 positions involving hydrogen bond formation at Lys106 and π-π stacking at Lys87, Lys88, and Lys106 (Figure 4A). There was a marked difference in the CDOCKER interaction energy for the interactions between MLN8054 and AURKA (–113.729 kcal/mol) and AURKB (42.0608 kcal/mol; Table 1), which may explain the much higher binding affinity to AURKA.
There were 18 and 400 positions produced for VX-680- AURKA and VX-680-AURKB interactions, respectively. VX-680 interacted with AURKA via hydrogen bond formation at Arg255 and Lys258 and via π-π stacking at Asp256 and Trp277 (Figure 2E). VX-680 bound to AURKB through hydrogen bond formation at Lys106 and Phe219, charge interaction at Lys85 and Glu161, and π-π stacking at Lys87 and Lys106 (Figure 4B). The CDOCKER interaction energy between VX-680-AURKA and VX-680-AURKB interactions was markedly different, ie, –113.729 kcal/mol and 35.1378 kcal/mol, respectively (Table 1). This marked difference in CDOCKER interaction energy may be associated with the preference of VX-680 toward AURKA.
Danusertib did not bind to AURKA and there was no position generated for the danusertib-AURKA interactions, whereas danusertib bound to AURKB with 158 positions via hydrogen bond formation at Lys106 and π-π stacking at Leu93, Lys87, and Lys106, with a CDOCKER interaction energy of 49.8876 kcal/mol (Figure 3D). In addition, there was no position generated for the barasertib-AURKA/AURKB interactions. Taken together, the docking results show that danusertib binds to AURKB, but not to AURKA, while barasertib does not bind to either AURKA or AURKB.
ALS inhibits proliferation of SKOV3 and OVCAR4 cells
To determine whether ALS inhibited the proliferation of SKOV3 and OVCAR4 cells and whether ALS was toxic toward normal human epithelial ovarian (T80) cells, we first examined the effect of ALS on cell proliferation in the T80 cell line and in the SKOV3 and OVCAR4 EOC cell lines using the MTT assay. The concentration-dependent inhibitory effects of ALS on growth of T80, SKOV3, and OVCAR4 cells are shown in Figure 5. When T80 cells were treated with ALS at 0.1, 0.5, 2.5, 10, 25, and 100 μM for 24 hours, the percentage cell viability over the control cells (100%) was 94.3%, 78.7%, 80.0%, 75.1%, 59.5%, and 54.5%, respectively (Figure 5A). The percentage of live SKOV3 cells over the control cells (100%) was 86.8%, 78.7%, 67.6%, 62.1%, 52.8%, and 28.3%, respectively (Figure 5B). The percentage of live OVCAR4 cells over the control cells (100%) was 88.4%, 81.4%, 79.1%, 59.2%, 53.0%, and 31.2%, respectively (Figure 5C). The IC50 values for ALS were 164.7 μM in T80 cells, 20.48 μM in SKOV3 cells, and 22.13 μM in OVCAR4 cells. These results demonstrate that ALS induces a concentration-dependent inhibitory effect on the growth of T80, SKOV3, and OVCAR4 cells, but the inhibitory effect on normal T80 cells is significantly lower than the inhibitory effect on EOC cells. This suggests that ALS has no or low toxicity toward normal human ovarian epithelial cells.
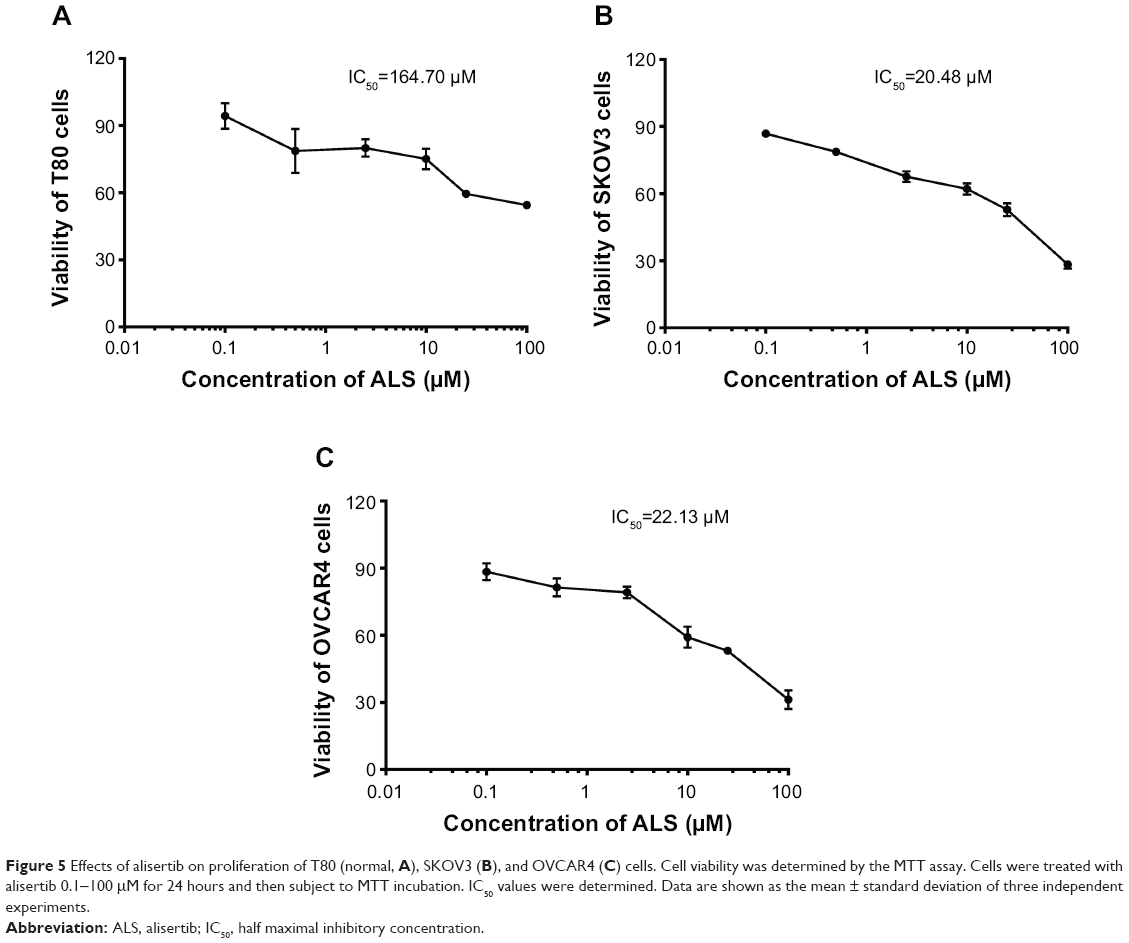 | Figure 5 Effects of alisertib on proliferation of T80 (normal, A), SKOV3 (B), and OVCAR4 (C) cells. Cell viability was determined by the MTT assay. Cells were treated with alisertib 0.1–100 μM for 24 hours and then subject to MTT incubation. IC50 values were determined. Data are shown as the mean ± standard deviation of three independent experiments. |
ALS induces G2/M arrest, downregulates cyclin B1 and CDC2, and upregulates p27 Kip1 and p53 in SKOV3 and OVCAR4 cells
We examined the effect of ALS on cell cycle distribution using a flow cytometer in both cell lines. ALS significantly induced G2/M phase arrest (Figure 6A). Compared with the control cells, the percentage of SKOV3 and OVCAR4 cells in G2/M phase were all increased in a concentration-dependent manner after treatment with ALS (Figures 6A and 6B). At basal levels, the percentage of SKOV3 and OVCAR4 cells in G2/M phase was 23.5% and 14.9%, respectively. When treated with ALS at 0.1, 1, and 5 μM for 24 hours, the percentage of SKOV3 cells was 51.9%, 86.5%, and 85.7%, respectively, and the percentage of OVCAR4 cells was 33%, 87.9%, and 81.3%, respectively. We conducted further separate experiments to evaluate the effect of ALS 5 μM on cell cycle distribution in SKOV3 and OVCAR4 cells over 72 hours (Figure 6C). Compared with control cells, the percentage of SKOV3 cells in G2/M phase was increased from 6.11% at the basal level to 11.9%, 24.3%, 55.2%, and 88.1% after 4, 8, 12, and 24 hours of treatment with ALS 5 μM, and declined to 86.8% and 52.2% after 48 and 72 hours of treatment with ALS 5 μM, respectively (Figure 6D). Similarly, compared with control cells, the percentage of OVCAR4 cells in G2/M phase increased from 25.6% at the basal level to 31.5%, 44.7%, 53.7%, and 81.4% after 4, 8, 12, and 24 hours of treatment with 5 μM ALS and declined to 79.0% and 19.3%, after 48 and 72 hours of treatment with 5 μM ALS, respectively (Figure 6D).
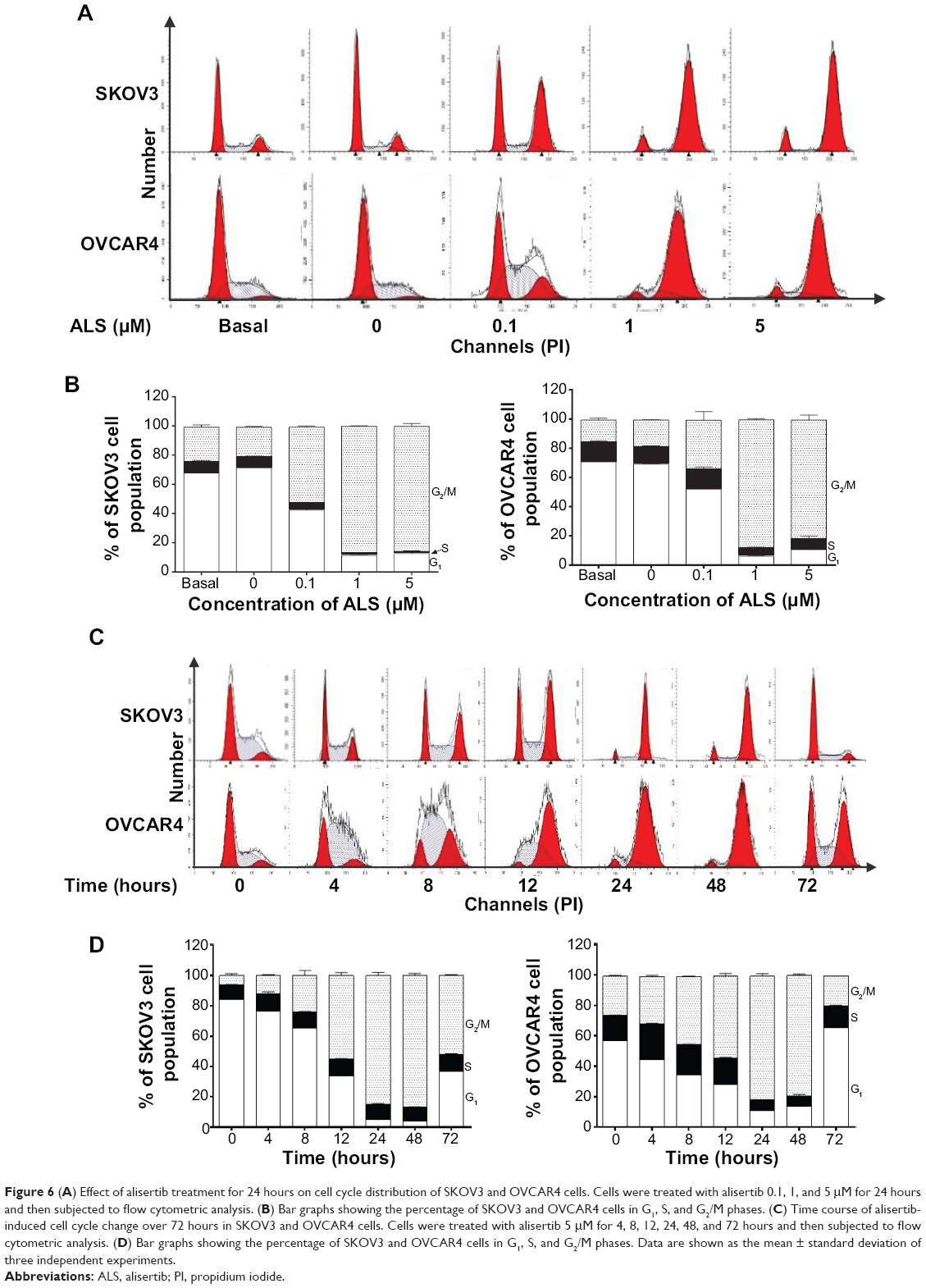 | Figure 6 (A) Effect of alisertib treatment for 24 hours on cell cycle distribution of SKOV3 and OVCAR4 cells. Cells were treated with alisertib 0.1, 1, and 5 μM for 24 hours and then subjected to flow cytometric analysis. (B) Bar graphs showing the percentage of SKOV3 and OVCAR4 cells in G1, S, and G2/M phases. (C) Time course of alisertib-induced cell cycle change over 72 hours in SKOV3 and OVCAR4 cells. Cells were treated with alisertib 5 μM for 4, 8, 12, 24, 48, and 72 hours and then subjected to flow cytometric analysis. (D) Bar graphs showing the percentage of SKOV3 and OVCAR4 cells in G1, S, and G2/M phases. Data are shown as the mean ± standard deviation of three independent experiments. |
On the contrary, treatment of SKOV3 and OVCAR4 cells with ALS expressed a significantly reduced G1 phase at 0.1, 1, and 5 μM for 24 hours. The number of cells in G1 phase was significantly reduced by 40.5%, 83.3%, and 82.5% when SKOV3 was treated in ALS for 24 hours. The number of OVCAR4 cells was also significantly reduced by 26.3%, 91.3%, and 85.1% in G1 phase when treated with ALS for 24 hours (Figure 6B). Similarly, there was a decrease in G1 phase when SKOV3 and OVCAR4 cells were treated with ALS 5 μM for 72 hours. Compared with the control cells, the percentage of SKOV3 cells in G1 phase was decreased to 52.8%, 82.2%, 92.3%, 94.4%, 89.3%, and 36.8% after 4, 8, 12, 24, 48, and 72 hours, respectively (Figures 6C and 6D). Compared with control cells, the percentages of OVCAR4 cells were decreased to 21.6%, 39.7%, 50.1%, 80.9%, 76.2%, and 36.8% after 4, 8, 12, 24, and 48 hours, respectively, and increased to 15.7% after 72 hours of treatment with 5 μM ALS.
In addition, compared with control cells, treatment of SKOV3 and OVCAR4 cells with ALS 0.1, 1, and 5 μM for 24 hours significantly decreased the percentage of cells in S phase to 36.4%, 76.4%, and 79.8%, respectively, and to 0.3%, 58.3%, and 45.7%, respectively (Figure 6B). Compared with control cells, treatment of SKOV3 cells with ALS 5 μM for 4, 8, 12, 24, 48, and 72 hours increased the percentage of cells in S phase to 22.9%, 10%, 15.4%, 13.4%, 1.3%, and 16.3%, respectively (Figure 6C), and in OVCAR4 cells, the percentage of cells in S phase increased to 37.9% and 20.8% at 4 and 8 hours and decreased to 57.3%, 57.4%, and 14.6% at 24, 48, and 72 hours, respectively (Figure 6C).
To explore the mechanism for the ALS-induced effects on cell cycle arrest in SKOV3 and OVCAR4 cells, the expression levels of key regulators responsible for G2 checkpoints were examined using the Western blotting assay (Figure 7A). Cyclin B1 and CDK1/CDC2 are two key regulators for the G2 to M phase transition,39 so their expression level was determined in SKOV3 and OVCAR4 cells. The expression of cyclin B1 was significantly suppressed in SKOV3 and OVCAR4 cells treated with ALS 0.1, 1, and 5 μM for 24 hours (P<0.05 or P<0.01). In comparison with control cells, the expression level of cyclin B1 in SKOV3 cells was decreased 61.7%, 83.7%, and 58.8% when treated with ALS 0.1, 1, and 5 μM for 24 hours, respectively. The expression level of cyclin B1 in OVCAR4 cells was suppressed 15.6%, 44.5%, and 61.7% when treated with ALS 0.1, 1, and 5 μM for 24 hours, respectively. There was a 19.9%, 29.1%, and 44.6% reduction in the expression level of CDK1/CDC2 in SKOV3 cells incubated with ALS 0.1, 1, and 5 μM for 24 hours, respectively (P<0.05 or P<0.01). There was also a 19.1%, 43.7%, and 59.2% reduction in the expression level of CDK1/CDC2 in OVCAR4 cells incubated with ALS 0.1, 1, and 5 μM for 24 hours, respectively (P<0.05 or P<0.01; Figure 7B).
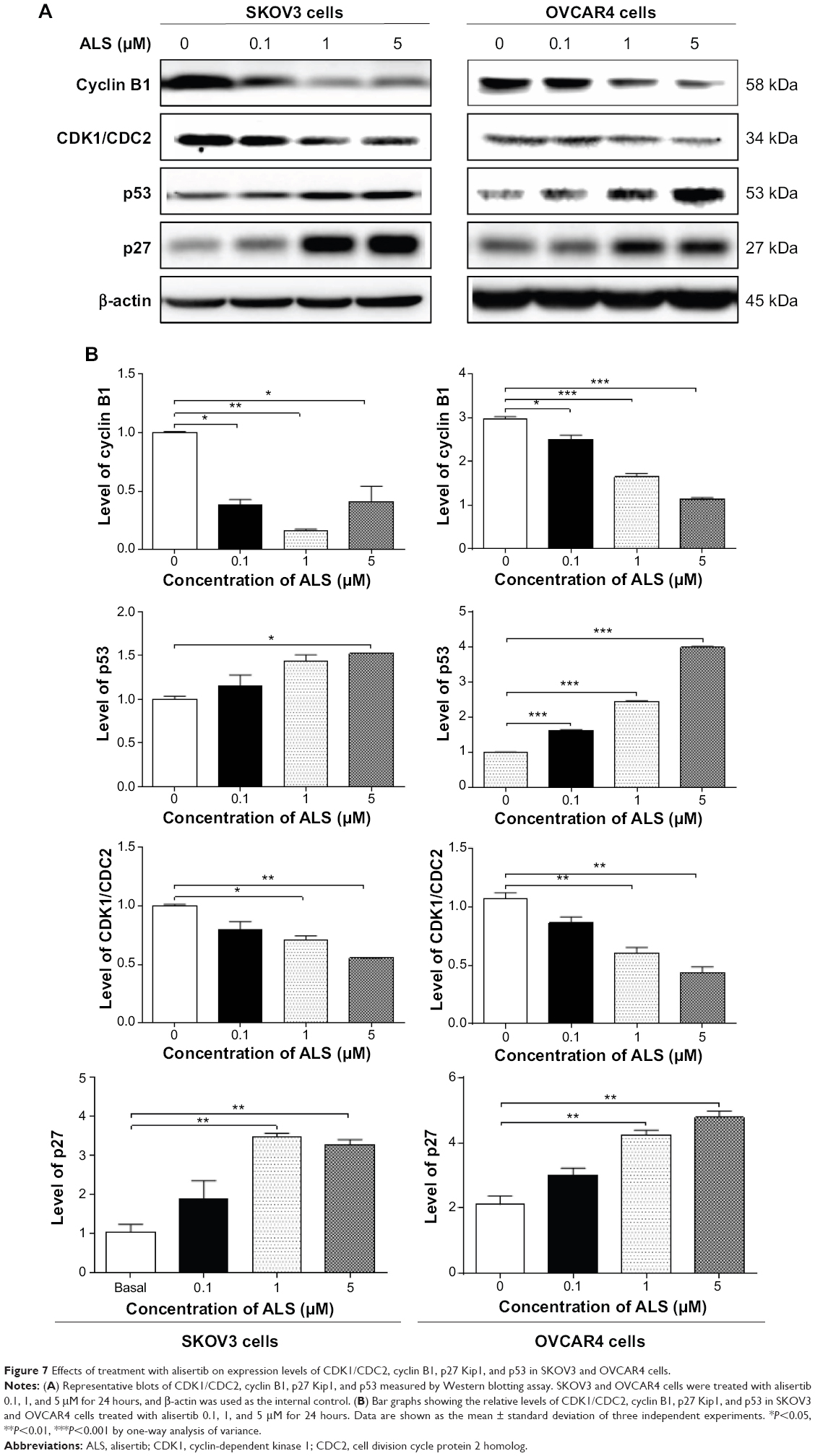 | Figure 7 Effects of treatment with alisertib on expression levels of CDK1/CDC2, cyclin B1, p27 Kip1, and p53 in SKOV3 and OVCAR4 cells. |
These results demonstrate that ALS can downregulate CDK1/CDC2 and cyclin B1 in SKOV3 and OVCAR4 cells with differential effects. This would contribute to cell cycle arrest in both cell lines when exposed to ALS. p21 Waf1/Cip1, p27 Kip1, and p53 in SKOV3 and OVCAR4 cells treated with ALS were determined using Western blotting assay. The tumor suppressor protein p21 Waf1/Cip1 acts as an inhibitor of cell cycle progression, and serves to inhibit kinase activity and block progression through G1/S in association with CDK2 complexes.40 During cell cycle stages when cell division cycle protein 2 homolog (CDC2)/cyclin B or cyclin-dependent kinase 2 (CDK2)/cyclin A is active, p53 is phosphorylated and upregulates p21 Waf1/Cip1 transcription via a p53-responsive element. p27 Kip1 is a member of the Cip/Kip family of cyclin-dependent kinase inhibitors.41 Like p57 Kip2 and p21 Waf1/Cip1, p27 Kip1 enforces the G1 restriction point via its inhibitory binding to CDK2/cyclin E and other CDK/cyclin complexes.41 p53 is a tumor suppressor protein that plays a major role in the cellular response to DNA damage and other genomic aberrations.42 Activation of p53 can lead to either cell cycle arrest and DNA repair or apoptosis. p53 is phosphorylated at multiple sites and by several different protein kinases. DNA damage induces phosphorylation of p53 at Ser15 and Ser20, which leads to a reduced interaction between p53 and its negative regulator, mouse double minute 2 homologue.42 As shown in Figure 7A, the expression level of p27 Kip1 was concentration-dependently increased in SKOV3 cells and OVCAR4 cells treated with ALS for 24 hours. In comparison with control cells, there was a 2.0-fold and 2.3-fold increase in expression of p27 Kip1 in SKOV3 cells treated with ALS at 1 and 5 μM for 24 hours (P<0.05; Figure 7B), and a 1.4-fold increase in expression of p27 Kip1 in SKOV3 cells treated with ALS at 0.1 μM for 24 hours. However, these changes were not statistically significant. When OVCAR4 cells were treated with ALS 1 μM and 5 μM for 24 hours, the expression level of p27 Kip1 was increased 2.0-fold and 2.3-fold, respectively (P<0.05; Figure 7B). When OVCAR4 cells were treated with ALS 0.1 μM for 24 hours, the expression of p27 Kip1 only increased 1.4-fold, but had no statistical significance in comparison with control cells.
Moreover, there was a 1.5-fold increase in the expression level of p53 in SKOV3 cells treated with ALS 5 μM for 24 hours (P<0.05; Figure 7B). There was a 1.6-fold, 2.4-fold, and 3.9-fold increase in expression of p53 in OVCAR4 cells treated with ALS 0.1, 1, and 5 μM for 24 hours, respectively, compared with control cells (P<0.05; Figure 7B). ALS did not affect the expression of p21 Waf1/Cip1 in SKOV3 and OVCAR4 cells.
ALS induces apoptosis of SKOV3 and OVCAR4 cells via activation of the mitochondria-dependent pathway
Flow cytometric analysis was used to examine the effect of ALS on apoptosis in SKOV3 and OVCAR4 cells. The number of apoptotic cells was first quantified and the results are shown in Figure 8A. The number of apoptotic cells at the basal level was 6.4% and was 6.2% in SKOV3 cells treated with the control vehicle only (0.05% DMSO, v/v). When SKOV3 cells were treated with ALS 0.1, 1, and 5 μM for 24 hours, the total percentages of apoptotic cells (early and late apoptosis) were 9.8%, 9.0%, and 10.4%, respectively, with a 1.6-fold, 1.4-fold, and 1.7-fold increase, respectively, compared with control cells. When treated with ALS 0.1 and 5 μM, the results showed a statistically significant difference (P<0.05; Figure 8B). In OVCAR4 cells, the number of apoptotic cells was 4.5% at the basal level and 4.1% in cells treated with the control vehicle only (0.05% DMSO, v/v). When OVCAR4 cells were treated with ALS 1 and 5 μM for 24 hours, the total percentage of apoptotic cells was increased 3.4-fold and 2.0-fold compared with control cells (P<0.05; Figure 8B).
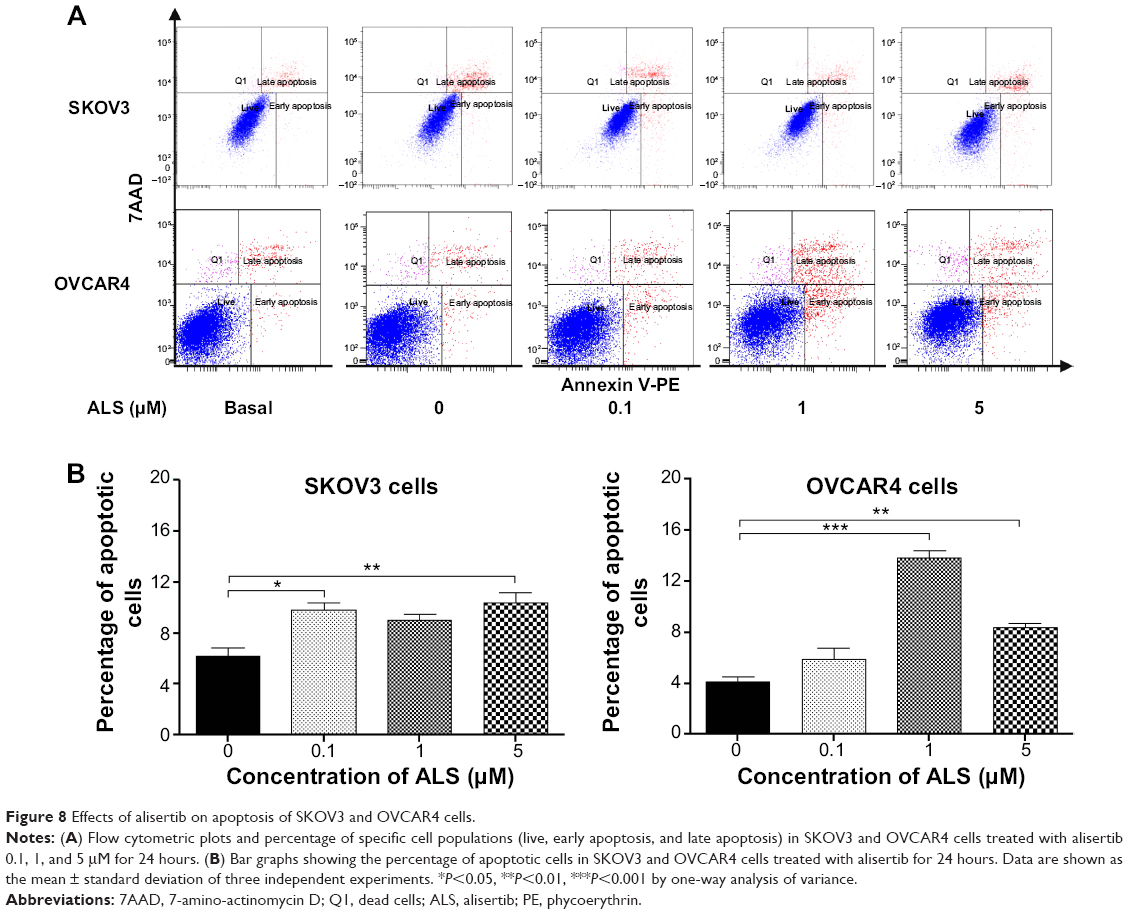 | Figure 8 Effects of alisertib on apoptosis of SKOV3 and OVCAR4 cells. |
The results of flow cytometric analysis clearly showed that ALS induced apoptotic cell death of both SKOV3 and OVCAR4 cells in a concentration-dependent manner. In order to elucidate the mechanism for the proapoptotic effect of ALS in SKOV3 and OVCAR4 cells, we first examined the effects of treatment with ALS on expression levels of the proapoptotic protein Bax and the antiapoptotic proteins Bcl-2 and Bcl-xl (Figure 9A). In the mitochondrial/cytochrome c-mediated apoptotic pathway, various stimuli converge at the mitochondria and activate one or more members of the BH3-only protein family, which are tightly regulated by Bcl-2 family members. This promotes assembly of Bcl-2-antagonist/killer-1 (Bak) and Bax oligomers within the outer membranes of the mitochondria, leading to release of cytochrome c into the cytosol, which in turn induces apoptotic protease-activating factor-1 to generate apoptosome and activates caspase 9, and subsequently activates the downstream caspases 2, 3, 6–8, and 10.43 SKOV3 and OVCAR4 cells were treated with ALS 0.1, 1, and 5 μM for 24 hours. The expression level of Bax was concentration-dependently increased in both cell lines (P<0.05; Figures 9B and 9C). Incubation of SKOV3 cells with ALS 0.1, 1, and 5 μM significantly increased Bax expression (4.5-fold, 6.1-fold, and 6.3-fold, respectively, compared with control cells), and treatment of OVCAR4 cells with ALS 5 μM for 24 hours resulted in a remarkable increase in expression of Bax (2.0-fold compared with control cells). In contrast, the expression level of Bcl-2 was decreased by 40.7% and by 51.7% in SKOV3 cells treated with ALS 1 and 5 μM, and by 27.9%, 36.8%, and 47.6% in OVCAR4 cells treated with ALS at 0.1, 1, and 5 μM, respectively (P<0.05; Figures 9B and 9C). A significant increase in the expression level of Bax and reduction of Bcl-2 resulted in a significant decrease in the Bcl-2/Bax ratio in both cell lines, with a 6.7%, 6.6%, and 8.2% reduction in SKOV3 cells and a 25.7%, 27.8%, and 24.2% reduction in OVCAR4 cells when treated with ALS 0.1, 1, and 5 μM for 24 hours, respectively (P<0.01; Figures 9B and 9C). In addition, the expression level of Bcl-xl was suppressed after treatment with ALS 5 μM in both cell lines in a concentration-dependent manner. Compared with control cells, the expression level of Bcl-xl was reduced by 54.6% and 85.5% in SKOV3 and OVCAR4 cells, respectively, when treated with ALS 5 μM for 24 hours. The effect of ALS on expression of PUMA was also examined due to the important role of PUMA in the regulation of antiapoptotic proteins. Incubation of SKOV3 and OVCAR4 cells with ALS increased the expression level of PUMA in a concentration-dependent manner. Treatment of SKOV3 cells with ALS 0.1, 1, and 5 μM for 24 hours significantly increased the expression level of PUMA 2.4-fold, 2.6-fold, and 4.1-fold (P<0.05; Figure 9B), and treatment of OVCAR4 cells with ALS 1 and 5 μM for 24 hours significantly increased the expression level of PUMA 1.6-fold and 1.8-fold, respectively (P<0.05; Figure 9C).
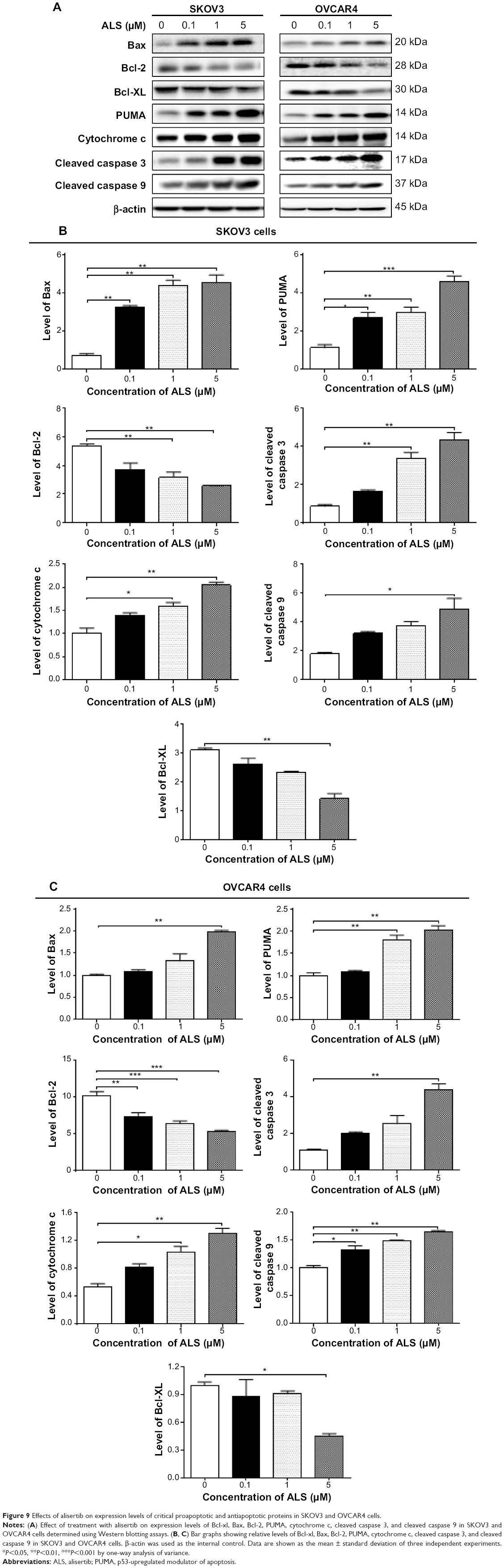 | Figure 9 Effects of alisertib on expression levels of critical proapoptotic and antiapoptotic proteins in SKOV3 and OVCAR4 cells. |
The effect of treatment with ALS on the mitochondria-related apoptotic pathway was further examined in SKOV3 and OVCAR4 cells. The release of cytochrome c from the mitochondria into the cytosol initiates the caspase-dependent apoptotic cascade. The release of cytochrome c from the mitochondrial intermembrane space is the early event during apoptotic cell death.44 Upon apoptotic stimulation, cytochrome c released from the mitochondria associates with procaspase-9/Apaf 1 to form a complex that processes caspase 9 from its inactive proenzyme to its active form, eventually triggering activation of caspase 3 and apoptosis.44
We observed a significant increase in the activation of caspases 9 and 3 in SKOV3 and OVCAR4 cells treated with ALS. Compared with control cells, treatment of SKOV3 and OVCAR4 cells with ALS 0.1, 1, and 5 μM for 24 hours significantly increased the level of cleaved caspase 3 and caspase 9. Treatment of SKOV3 cells with ALS 1 and 5 μM for 24 hours significantly increased the expression level of cleaved caspase 3 by 3.9-fold and 5.0-fold, respectively, and treatment with ALS 5 μM increased the expression level of cleaved caspase 9 by 2.7-fold (P<0.01; Figure 9B). Treatment of OVCAR4 cells with ALS 5 μM for 24 hours significantly increased the expression level of cleaved caspase 3 by 5.1-fold and treatment of OVCAR4 cells with ALS 0.1, 1, and 5 μM for 24 hours significantly increased the expression level of cleaved caspase 9 by 1.3-fold, 1.5-fold, and 1.6-fold, respectively (P<0.05; Figure 9C). These results indicate that ALS induced marked activation of caspase 9 and caspase 3 and eventually led to apoptotic death of both SKOV3 and OVCAR4 cells.
Further, we observed a significant increase in activation of cytochrome c in SKOV3 and OVCAR4 cells treated with ALS. Compared with control cells, treatment of SKOV3 and OVCAR4 cells with ALS 1 and 5 μM for 24 hours significantly increased the level of cytochrome c. Treatment of SKOV3 cells with ALS 1 and 5 μM for 24 hours significantly increased the expression level of cytochrome c 1.6-fold and 2.0-fold, respectively (P<0.05 and P<0.01; Figure 9B), and treatment of OVCAR4 cells with ALS 1 and 5 μM increased the expression level of cytochrome c 1.0-fold and 1.3-fold, respectively (P<0.05 and P<0.01; Figure 9C).
ALS induces autophagy via inhibition of the PI3K/Akt/mTOR pathway in SKOV3 and OVCAR4 cells
After observing that ALS induced apoptosis in SKOV3 and OVCAR4 cells, we examined the effect of ALS on autophagy of SKOV3 and OVCAR4 cells using flow cytometric analysis and confocal microscopy (Figures 10 and 11). The percent of autophagic cells at the basal level was 5.0% and 4.43% for SKOV3 and OVCAR4 cells, respectively (Figure 10B). Incubation of SKOV3 and OVCAR4 cells with ALS for 24 hours significantly increased the percentage of autophagic cells (Figure 10A). In SKOV3 cells, there was a 10.7-fold and 11.5-fold increase in the percentage of autophagic cells treated with ALS 0.1 and 1 μM for 24 hours compared with control cells (11.0% versus 1.0% and 11.9% versus 1.0%, respectively, P<0.05; Figure 10B). When SKOV3 cells were treated with ALS 5 μM for 24 hours, the percentage of autophagic cells was only slightly increased compared with control cells, but the difference was not statistically significant (5.5% versus 1.0%, P>0.05; Figure 10B). In addition, treatment of OVCAR4 cells with ALS 5 μM for 24 hours resulted in a 3.24-fold increase in the percentage of autophagic cells (17.97% versus 5.53%; P<0.001; Figure 10B). Treatment of OVCAR4 cells with ALS 0.1 and 1 μM for 24 hours only slightly increased autophagic death.
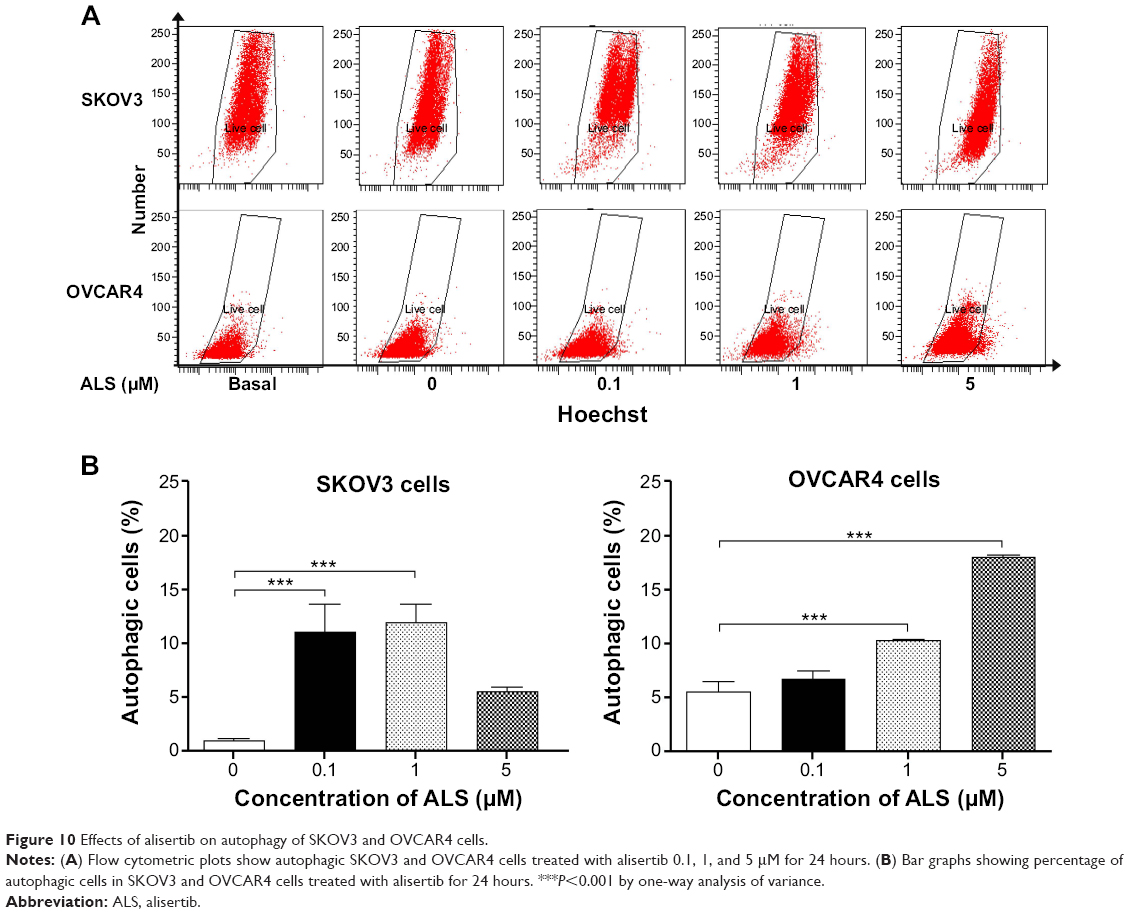 | Figure 10 Effects of alisertib on autophagy of SKOV3 and OVCAR4 cells. |
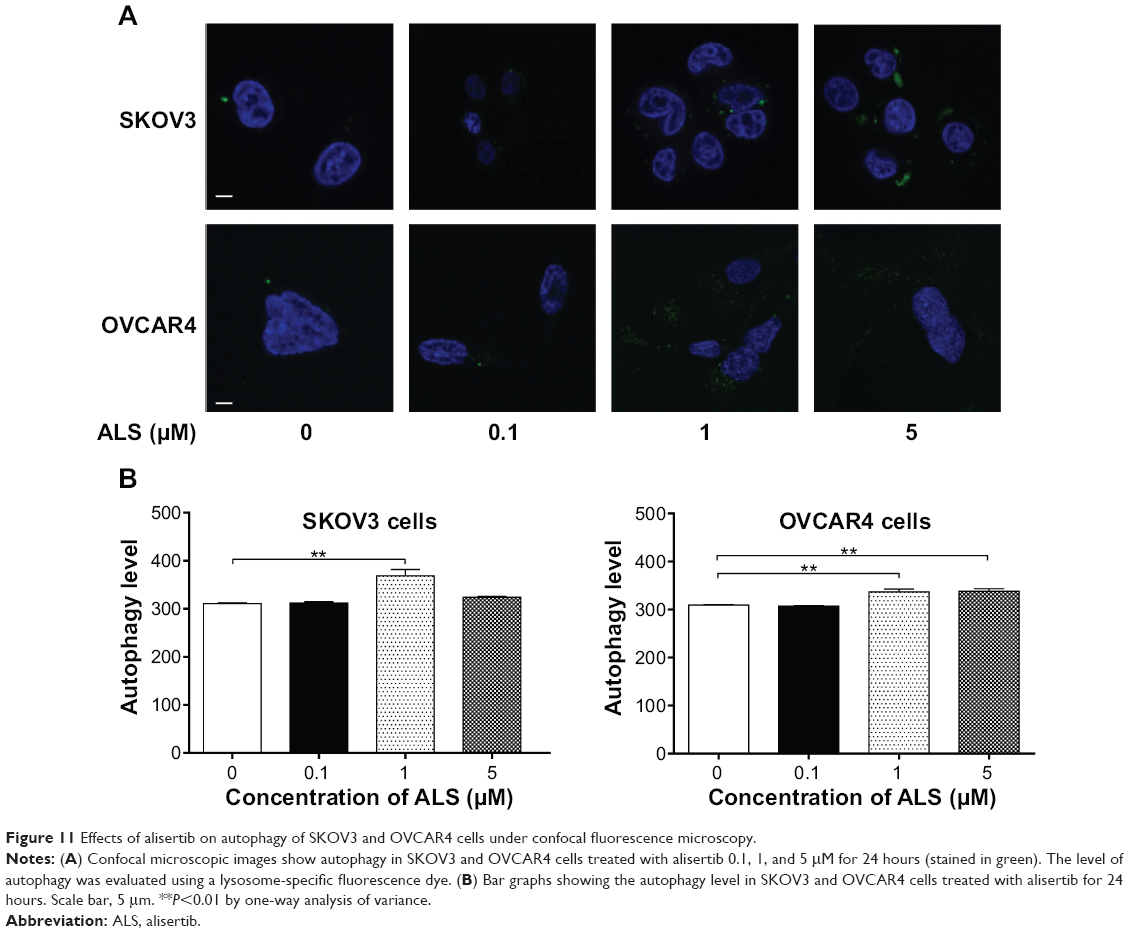 | Figure 11 Effects of alisertib on autophagy of SKOV3 and OVCAR4 cells under confocal fluorescence microscopy. |
We further confirmed the autophagy-inducing effects of ALS on SKOV3 and OVCAR4 cells using confocal microscopic examination (Figure 11A). In comparison with control cells, treatment with ALS caused an increase in autophagy in a concentration-dependent manner in SKOV3 and OVCAR4 cells (Figure 11B). There was a 1.2-fold increase in autophagic death of SKOV3 cells when treated with ALS 1 μM for 24 hours, (P<0.05; Figure 11B), but the lower concentration (0.1 μM) and higher concentration (5 μM) of ALS did not significantly affect autophagy of SKOV3 cells. For OVCAR4 cells, there was a 1.1-fold and 1.1-fold increase in autophagy when treated with ALS 1 and 5 μM (P<0.05; Figure 11B). A lower concentration of ALS (0.1 μM) did not significantly affect autophagy in OVCAR4 cells. These results demonstrate that ALS induces autophagy in both SKOV3 and OVCAR4 cells.
Next, we investigated the mechanisms for the autophagy-inducing effect of ALS in SKOV3 and OVCAR4 cells. First, we examined the phosphorylation levels of PI3K at Tyr199 and AMPK at Thr172; these are upstream signaling molecules in the Akt/mTOR pathway and play an important role in regulation of cell proliferation and death.45 PI3K catalyzes the formation of phosphatidylinositol-3,4,5-triphosphate via phosphorylation of phosphatidylinositol, phosphatidylinositol- 4-phosphate, and phosphatidylinositol-4,5 bisphosphate.46 Growth factors and hormones trigger this phosphorylation event, which in turn coordinates cell growth, cell cycle, cell migration, and cell survival. In this study, ALS significantly inhibited phosphorylation of PI3K at Tyr199 in both cell lines in a concentration-dependent manner compared with control cells (Figure 12A). Exposure of SKOV3 and OVCAR4 cells to ALS 0.1, 1, and 5 μM for 24 hours decreased the phosphorylation level of PI3K at Tyr199 (Figure 12B). However, incubation of each cell line with ALS did not significantly affect the expression level of total PI3K. The ratio of p-PI3K over PI3K was concentration-dependently decreased by ALS in both cell lines compared with control cells. In SKOV3 cells, the p-PI3K/PI3K ratio was decreased from 4.9 at the basal level to 3.5, 2.7, and 2.6 when SKOV3 cells were treated with ALS 0.1, 1, and 5 μM, respectively (P<0.05 or P<0.01; Figure 12B). In OVCAR4 cells, ALS 0.1, 1, and 5 μM decreased the ratio of p-PI3K/PI3K from 4.4 at the basal level to 2.2, 1.0, and 2.9, respectively (P<0.05 or P<0.01; Figure 12B).
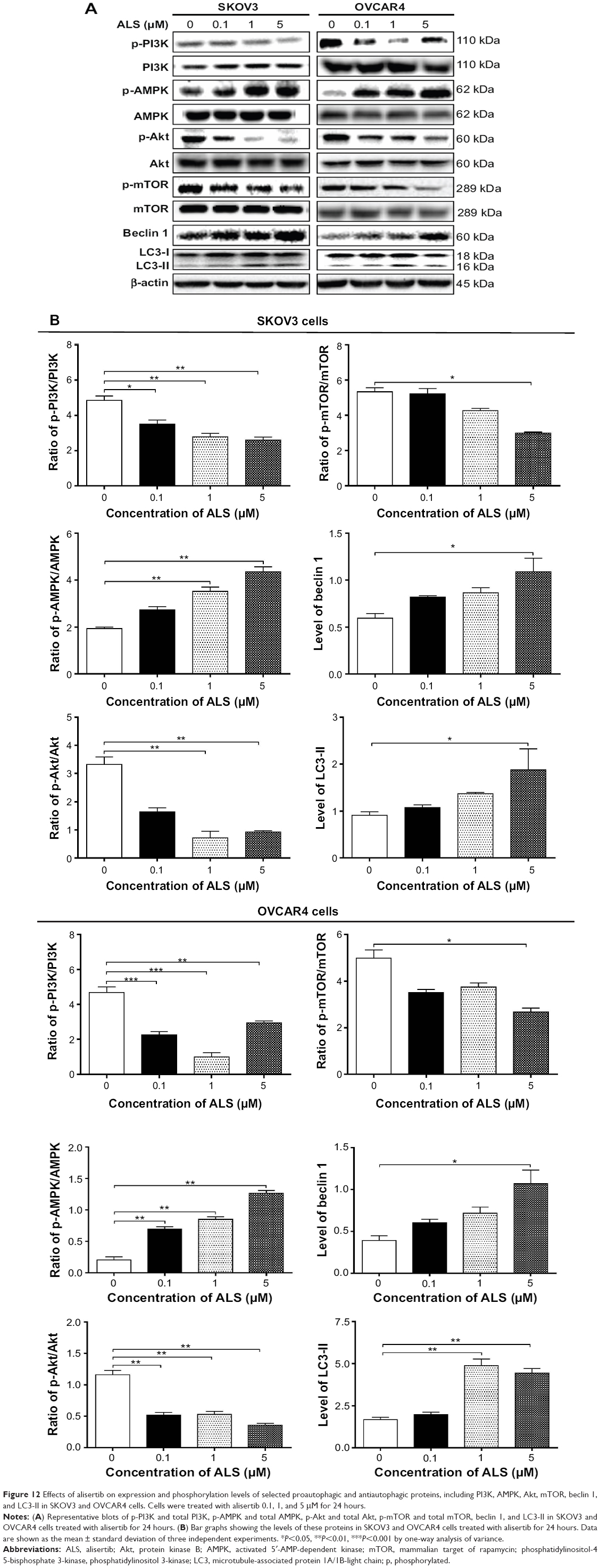 | Figure 12 Effects of alisertib on expression and phosphorylation levels of selected proautophagic and antiautophagic proteins, including PI3K, AMPK, Akt, mTOR, beclin 1, and LC3-II in SKOV3 and OVCAR4 cells. Cells were treated with alisertib 0.1, 1, and 5 μM for 24 hours. |
AMPK is highly conserved from yeast to plants and animals and plays a key role in the regulation of energy homeostasis, cell survival, and cell death.47 AMPK is a heterotrimeric complex composed of a catalytic α subunit and regulatory β and γ subunits. The kinase is activated by an elevated AMP/adenosine triphosphate ratio due to cellular and environmental stress, such as heat shock, hypoxia, or ischemia. The tumor suppressor LKB1, in association with accessory proteins STRAD and MO25, phosphorylates AMPK at Thr172 in the activation loop, and this phosphorylation is required for AMPK activation. Under glucose starvation, AMPK promotes autophagy by directly activating Unc-51-like kinase 1 (ULK1, a homologue of yeast Atg1) through phosphorylation of Ser317 and Ser777.48 Under nutrient sufficiency, high mTOR activity prevents ULK1 activation by phosphorylating ULK1 Ser757 and disrupting the interaction between ULK1 and AMPK.48
ALS showed a promoting effect on phosphorylation of AMPK at Thr172 in both cell lines (Figure 12B). Incubation of SKOV3 cells with ALS for 24 hours activated phosphorylation of AMPK at Thr172 in a concentration-dependent manner. In comparison with control cells, there was an increase in the AMPK phosphorylation level at Thr172 in SKOV3 and OVCAR4 cells treated with ALS 0.1, 1, and 5 μM for 24 hours (Figure 12B). However, there was no significant change in the expression of total AMPK compared with control cells. The ratio of p-AMPK over AMPK was concentration-dependently increased by ALS in both cell lines compared with control cells. In SKOV3 cells, the p-AMPK/AMPK ratio was increased from 1.9 at the basal level to 2.7, 3.5, and 4.3, when SKOV3 cells were treated with ALS 0.1, 1, and 5 μM, respectively (P<0.05 or P<0.01; Figure 12B). In OVCAR4 cells, ALS 0.1, 1, and 5 μM increased the ratio of p-AMPK/AMPK from 0.2 at the basal level to 0.7, 0.8, and 1.3, respectively (P<0.05 or P<0.01; Figure 12B).
We examined further the regulatory effect of ALS on phosphorylation of Akt at Ser473 and mTOR at Ser2448 in SKOV3 and OVCAR4 cells (Figure 12A). Akt is involved in the regulation of various signaling downstream pathways, including metabolism, cell proliferation, survival, growth, and angiogenesis.49 As a downstream effector of PI3K, Akt can activate mTOR, while mTORC2 phosphorylates Akt at Ser473. Thus, stimulation of Akt phosphorylation at Thr308 by 3-phosphoinositide-dependent protein kinase-1 leads to full Akt activation.49 mTOR plays a key role in cell growth, autophagic cell death, and homeostasis.50 mTOR is phosphorylated at Ser2448 via the PI3 kinase/Akt signaling pathway and is autophosphorylated at Ser2481.50 mTOR inhibition promotes dissociation of mTOR from the complex of Atg13 with ULK1 and ULK2. The dissociation releases ULK1/2 to activate FIP200, a protein critical for autophagosome formation and autophagy initiation.50 In comparison with control cells, the phosphorylation level of Akt at Ser473 was decreased in SKOV3 and OVCAR4 cells on exposure to ALS 0.1, 1, and 5 μM for 24 hours, respectively (Figure 12B). However, there was no significant alteration in expression of Akt in either cell line (Figure 12B). However, the p-Akt/Akt ratio was significantly decreased in both cell lines treated with ALS. In SKOV3 cells, the p-Akt/Akt ratio was decreased from 3.3 at the basal level to 1.6, 0.7, and 0.9 when cells were treated with ALS 0.1, 1, and 5 μM for 24 hours, respectively. In OVCAR4 cells, the p-Akt/Akt ratio decreased from 1.14 at the basal level to 0.5, 0.5, and 0.3 on exposure to ALS 0.1, 1, and 5 μM, respectively (Figure 12B).
Exposure of SKOV3 and OVCAR4 cells to ALS 5 μM resulted in a decrease in the phosphorylation level of mTOR at Ser2448 (Figure 12B). Treatment of SKOV3 and OVCAR4 cells with ALS 0.1 and 1 μM for 24 hours slightly reduced mTOR phosphorylation. There was no significant change in expression of total mTOR in either cell line when treated with ALS for 24 hours. Similarly, a decreased p-mTOR/mTOR ratio was observed in both cell lines when treated with ALS 5 μM (Figure 12B). In SKOV3 cells, the p-mTOR/mTOR ratio was decreased from 5.3 at the basal level to 2.9 when treated with ALS 5 μM (Figure 12B), but treatment of SKOV3 cells with ALS 0.1 and 1 μM for 24 hours reduced mTOR phosphorylation by 5.2% and 4.2%, respectively; however, the difference did not reach statistical significance. In OVCAR4 cells, the p-mTOR/mTOR ratio decreased from 5.0 at the basal level to 2.7 on exposure to ALS 5 μM, (P<0.05 or P<0.01; Figure 12B). However, treatment of OVCAR4 cells with ALS 0.1 and 1 μM for 24 hours reduced mTOR phosphorylation by only 3.5% and 3.5%, respectively (P>0.05).
Finally, we examined the effect of ALS on the expression levels of beclin 1 and LC3-I/II. Autophagy is tightly regulated by beclin 1 (a mammalian homologue of yeast Atg6), which forms a complex with vacuolar sorting protein 34 (Vps34, also called class III PI3K), and serves as a platform for recruitment of other Atgs that are critical for autophagosome formation.51,52 Upon initiation of autophagy, LC3 is cleaved at the C-terminus by Atg4 to form cytosolic LC3-I.53 LC3-I is consequently proteolytically cleaved and lipidated by Atg3 and Atg7 to form LC3-II, which localizes to the autophagosome membrane. Treatment of SKOV3 and OVCAR4 with ALS5 μM for 24 hours significantly increased the expression of beclin 1 (Figure 12B). There was a 1.8-fold and 2.7-fold increase of beclin 1 in SKOV3 and OVCAR4 cells, respectively, when treated with ALS 5 μM for 24 hours (P<0.05; Figure 12B). However, treatment of SKOV3 and OVCAR4 cells with ALS 0.1 and 1 μM for 24 hours increased beclin 1 by 1.3-fold and 1.4-fold and by 1.5-fold and 1.8-fold, respectively, but none of these increases were statistically significant.
After 24 hours of treatment with ALS, our Western blotting analysis revealed two clear bands of LC3-I and LC3-II in SKOV3 and OVCAR4 cells (Figure 12A). LC3-II migrated more rapidly than LC3-I on SDS-PAGE, leading to the appearance of two bands after immunoblotting, ie, LC3-I with an apparent mobility of about 18 kDa and LC3-II with an apparent mobility of 16 kDa. In both SKOV3 and OVCAR4 cells, there was a concentration-dependent increase in expression of LC3-II (Figure 12A). Compared with control cells, there was an increase in the LC3-II level in SKOV3 cells treated with ALS 5 μM for 24 hours and in the LC3-II level in OVCAR4 cells treated with ALS 1 and 5 μM for 24 hours, respectively. Treatment of SKOV3 and OVCAR4 cells with ALS had no significant effect on expression of LC3-I. However, the ratio of LC3-II to LC3-I was remarkably increased 2.1-fold in SKOV3 cells treated by ALS 5 μM for 24 hours and was significantly increased 2.9-fold and 2.7-fold in OVCAR4 cells on exposure to ALS 1 and 5 μM, respectively (Figure 12B). These findings indicate that ALS had a strong autophagy-inducing effect on SKOV3 and OVCAR4 cells via inhibition of the PI3K/Akt/mTOR pathway.
ALS-induced apoptosis and autophagy crosstalk in SKOV3 and OVCAR4 cells
Flow cytometry and confocal fluorescence microscopy were used to evaluate the effect of inducers and inhibitors of autophagy on basal and ALS-induced apoptosis and autophagy in SKOV3 and OVCAR4 cells. Treatment of SKOV3 cells with SB203580 10 μM (a selective p38 inhibitor and autophagy inducer), bafilomycin A10 μM, wortmannin 10 μM (a PI3K inhibitor and autophagy blocker), Z-VAD(OMe)-FMK (a pan-caspase inhibitor) 20 μM, and chloroquine (an autophagy inhibitor) 30 μM alone for 24 hours had no significant effect on apoptosis of SKOV3 cells (Figure 13A). In addition, the above inducers and inhibitors coincubated with ALS5 μM; only wortmannin and Z-VAD(OMe)-FMK significantly enhanced the apoptosis-inducing effect of ALS compared with cells treated with ALS alone (12.3% versus 6.3% and 12.2% versus 6.3%, respectively, P<0.05; Figure 13B). Others chemical regulators had no effect on apoptosis induced by ALS. In OVCAR4 cells, only Z-VAD(OMe)-FMK significantly enhanced the apoptosis-inducing effect of ALS when compared with cells treated with ALS alone (21.9% versus 5%, respectively, P<0.001; Figure 13B).
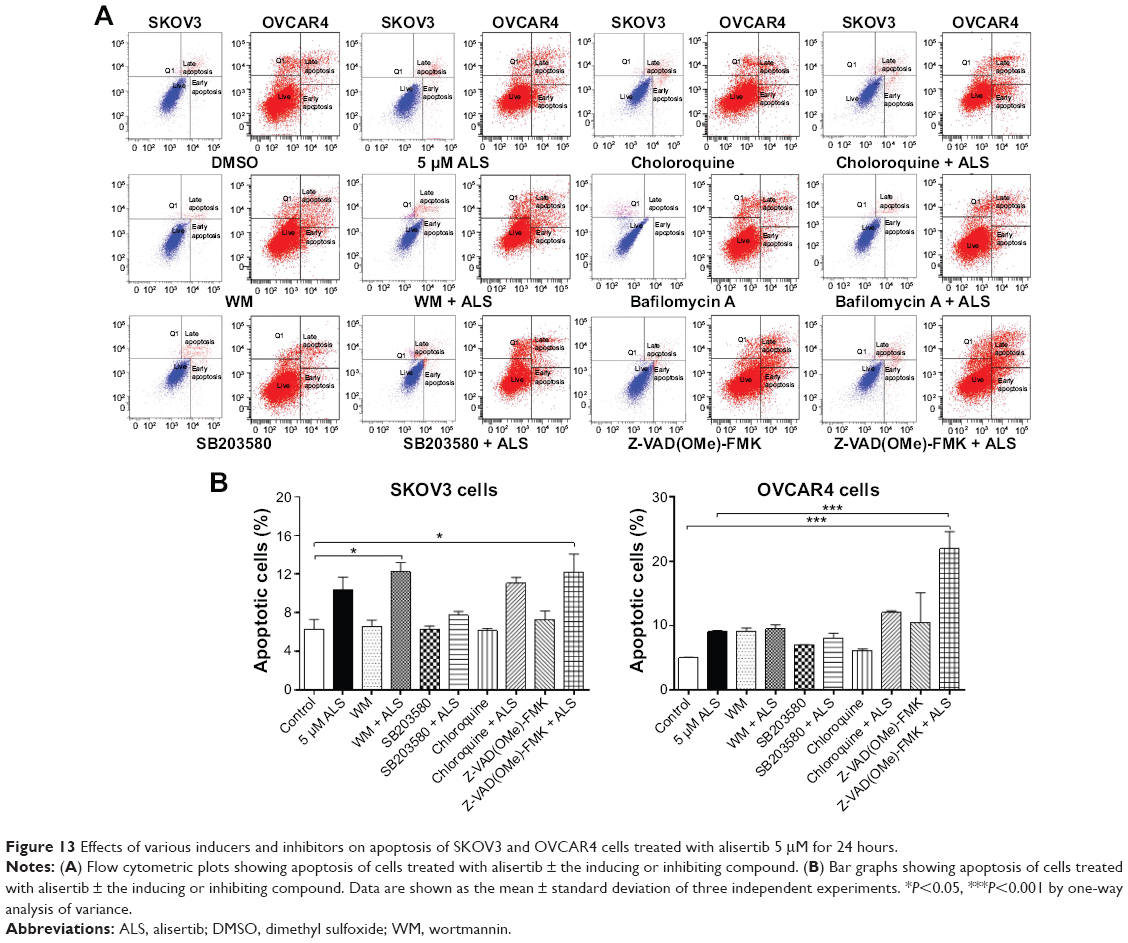 | Figure 13 Effects of various inducers and inhibitors on apoptosis of SKOV3 and OVCAR4 cells treated with alisertib 5 μM for 24 hours. |
Next, we evaluated the effect of induction or inhibition of autophagy on basal and ALS-induced autophagy in SKOV3 and OVCAR4 cells (Figure 14A). In SKOV3 cells, treatment with ALS 5 μM slightly increased autophagy, but the difference was not statistically different. SKOV3 cells treated with SB203580 10 μM, wortmannin 10 μM, or chloroquine 30 μM alone for 24 hours showed a 39.0-fold, 18.1-fold, and 18.5-fold increase in basal autophagy compared with control cells, respectively (P<0.05 or P<0.01; Figure 14B). SB203580 10 μM combined with ALS 5 μM increased ALS-induced autophagy by 10.9-fold. Wortmannin 10 μM coincubated with ALS 5 μM then increased autophagy 5.7-fold, while chloroquine 30 μM and ALS 5 μM increased autophagy 6.8-fold compared with ALS 5 μM alone (P<0.001; Figure 14B). Z-VAD(OMe)-FMK 20 μM and bafilomycin A 10 μM significantly decreased autophagy at the basal level, but when they were combined with ALS 5 μM, Z-VAD(OMe)-FMK 20 μM promoted a 2.6-fold increase of the autophagy induced by ALS compared with ALS 5 μM alone (P>0.05; Figure 14B). However, bafilomycin A 10 μM did not affect the ALS-induced autophagy effect.
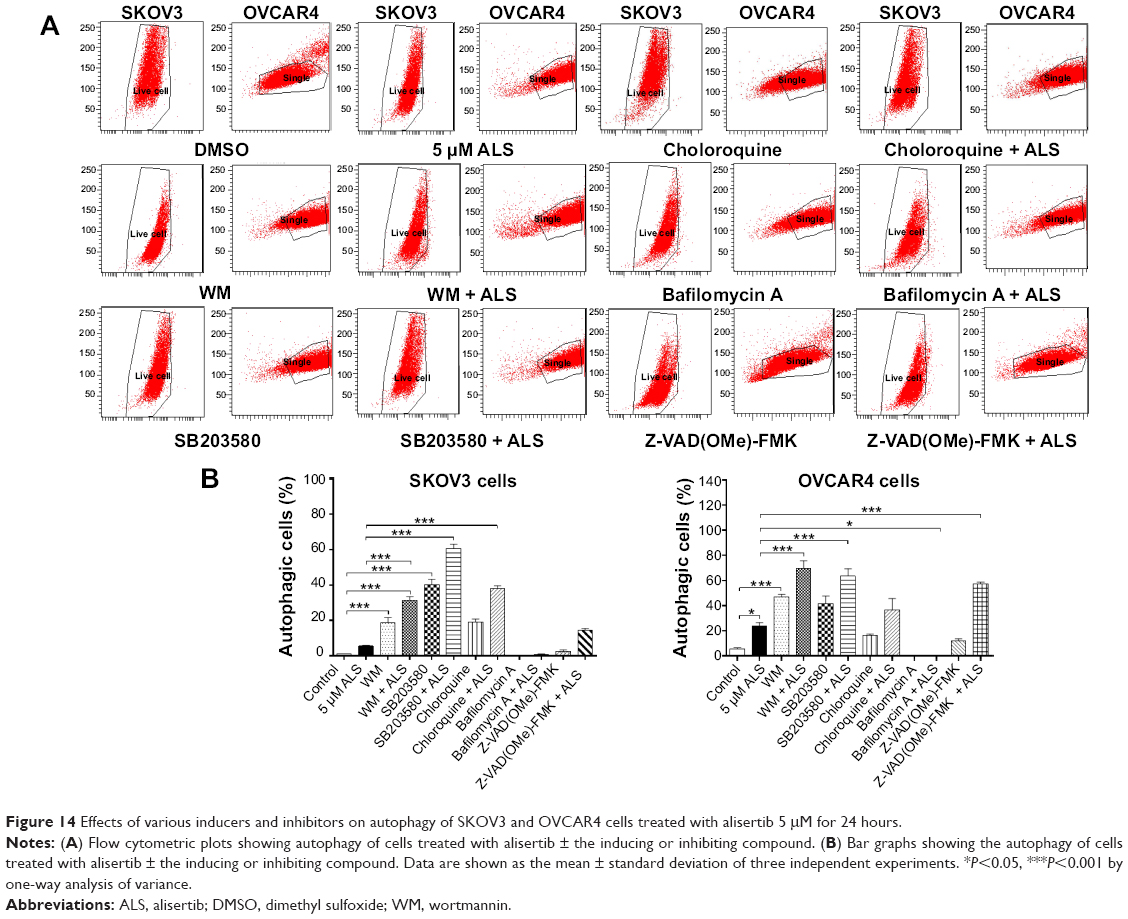 | Figure 14 Effects of various inducers and inhibitors on autophagy of SKOV3 and OVCAR4 cells treated with alisertib 5 μM for 24 hours. |
In OVCAR4 cells, treatment with ALS 5 μM increased autophagy 4.3-fold (P<0.05; Figure 14B). When OVCAR4 cells were treated with SB203580 10 μM or wortmannin 10 μM alone for 24 hours there was a 7.5-fold and 8.5-fold increase in basal autophagy compared with control cells, respectively (P<0.05 or P<0.01; Figure 14B). OVCAR4 cells treated with chloroquine 30 μM showed slightly increased autophagy, but the differences were not statistically significant (Figure 14B). SB203580 10 μM combined with ALS 5 μM increased autophagy 2.7-fold. Wortmannin 10 μM and ALS 5 μM increased autophagy 2.9-fold (P<0.001; Figure 14B), but chloroquine 30 μM with ALS 5 μM only increased autophagy 1.5-fold compared with ALS 5 μM alone (P>0.05; Figure 14B). Z-VAD(OMe)-FMK 20 μM slightly increased autophagy by 2.2-fold compared with basal levels, and the difference was not statistically significant. However, when Z-VAD(OMe)-FMK 20 μM was combined with ALS 5 μM, the effect of autophagy increased to 2.4-fold compared with ALS 5 μM (P<0.001; Figure 14B). Bafilomycin A 10 μM significantly decreased autophagy compared with basal levels; even when ALS 5 μM was combined, the effect of autophagy did not significantly increase (P>0.05; Figure 14B).
We tested the effect of inducers and inhibitors of autophagy on basal and ALS-induced autophagy in SKOV3 and OVCAR4 cells further, using confocal fluorescence microscopy (Figure 15A). In SKOV3 cells, we observed a significant increase in autophagy from the basal level when cells were treated with wortmannin 10 μM, which increased basal autophagy 2.0-fold; and chloroquine 30 μM, which increased the basal autophagy 1.6-fold. When wortmannin 10 μM or chloroquine 30 μM was combined with ALS 5 μM, the autophagic effect was markedly enhanced. Compared with ALS alone, wortmannin 10 μM increased autophagy 2.7-fold and chloroquine 30 μM increased it 1.9-fold (Figure 15B). Z-VAD(OMe)-FMK 20 μM did not affect basal autophagy, but increased ALS-induced autophagy 2.2-fold (P<0.05; Figure 15B). Treatment with bafilomycin A 10 μM reduced basal autophagy 3.9-fold and decreased ALS-induced autophagy 2.8-fold (P<0.05; Figure 15B).
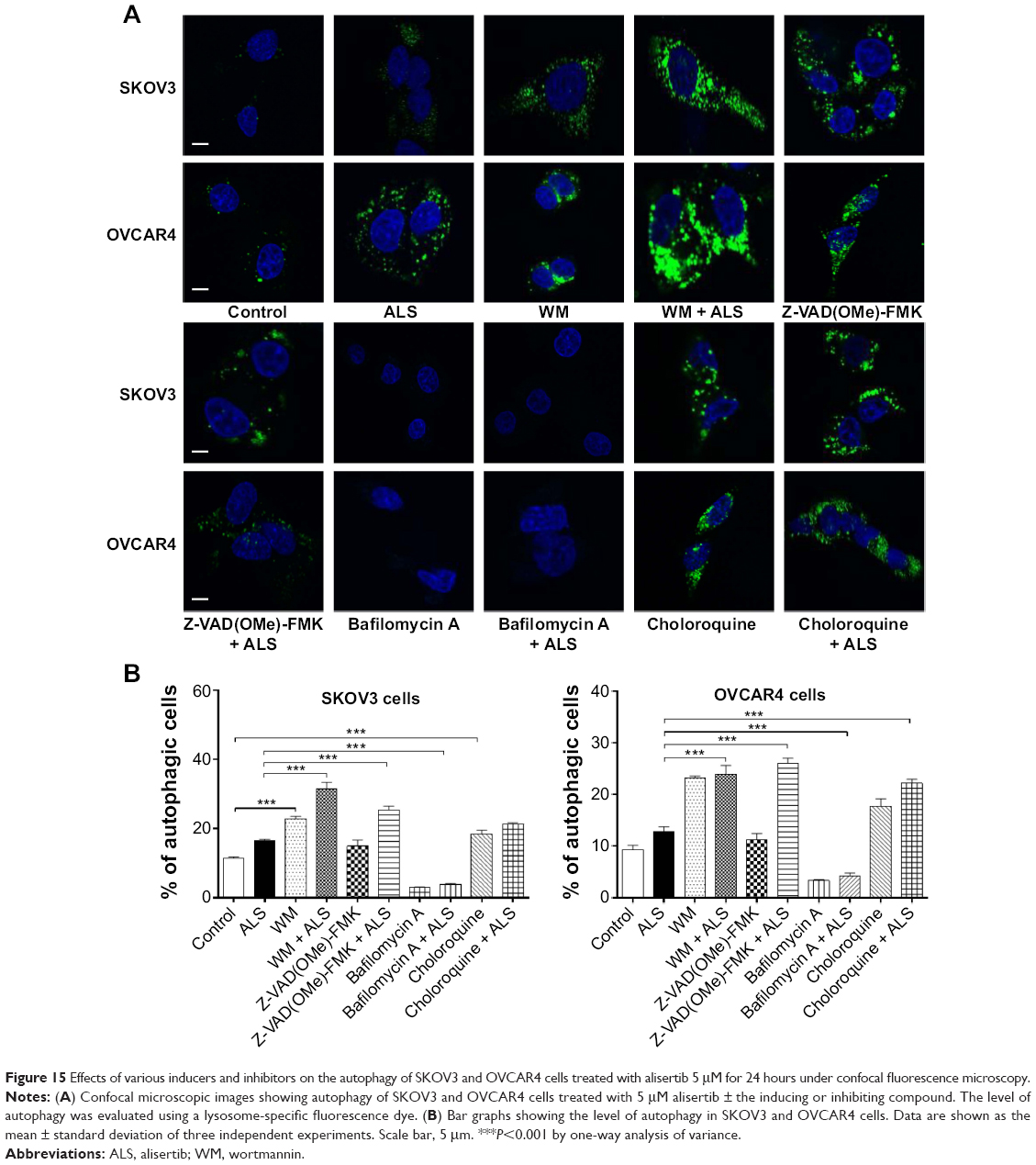 | Figure 15 Effects of various inducers and inhibitors on the autophagy of SKOV3 and OVCAR4 cells treated with alisertib 5 μM for 24 hours under confocal fluorescence microscopy. |
In OVCAR4 cells, a similar effect of wortmannin 10 μM increased autophagy 2.5-fold, and chloroquine 30 μM increased autophagy 1.9-fold from basal levels (P<0.001). Z-VAD(OMe)-FMK 20 μM did not affect basal autophagy. Bafilomycin A 10 μM diminished the basal autophagy 2.7-fold (Figure 15B). When cotreated with ALS 5 μM, autophagy was significantly increased in all groups. Wortmannin 10 μM increased ALS-induced autophagy 2.6-fold, chloroquine 30 μM increased it 2.4-fold, Z-VAD(OMe)-FMK 20 μM increased it 2.8-fold, and bafilomycin A 10 μM increased it 1.2-fold (Figure 15B).
ALS inhibits EMT in SKOV3 and OVCAR4 cells
EMT is a critical process involving the initiation, growth, invasion, and metastasis of cancer.54 EMT depends on a reduction in expression of cell adhesion molecules. E-cadherin is considered an active suppressor of invasion and growth of many epithelial cancers. Tight junctions, or zonula occludens, form a continuous barrier to fluids across the epithelium and endothelium. They function in the regulation of paracellular permeability and in the maintenance of cell polarity, blocking the movement of transmembrane proteins between the apical and basolateral cell surfaces. Tight junctions are composed of claudin and occludin proteins, which join the junctions to the cytoskeleton. ZO-1, 2, and 3 are peripheral membrane adaptor proteins that link junctional transmembrane proteins such as occludin and claudin to the actin cytoskeleton.55 Herein, we examined the effect of treatment with on EMT-associated markers in SKOV3 and OVCAR4 cells using the Western blotting assay (Figure 16A).
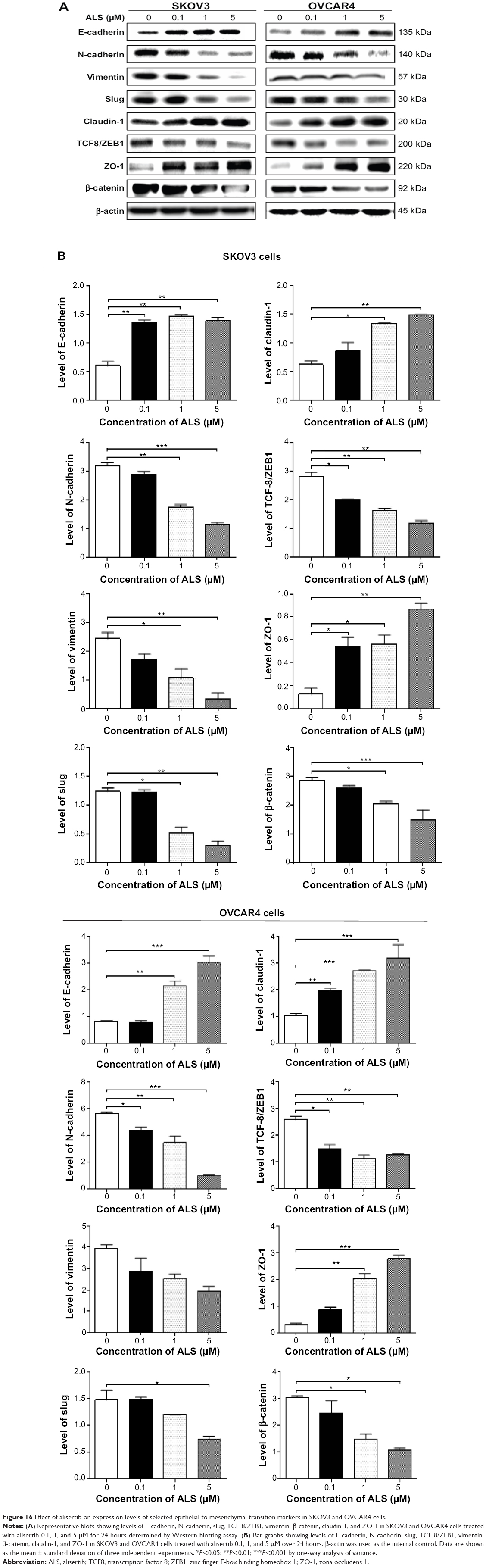 | Figure 16 Effect of alisertib on expression levels of selected epithelial to mesenchymal transition markers in SKOV3 and OVCAR4 cells. |
Cadherins are a superfamily of transmembrane glycoproteins that contain cadherin repeats of approximately 100 residues in their extracellular domain. They mediate calcium-dependent cell–cell adhesion, and the classic cadherin subfamily includes N-cadherin, P-cadherin, R-cadherin, B-cadherin, and E-cadherin. The cytoplasmic domain of classical cadherins interacts with β-catenin, γ-catenin, and p120 catenin. Cancer cells often show upregulated N-cadherin in addition to loss of E-cadherin.55 As shown in Figure 16A, incubation of SKOV3 cells with ALS resulted in a concentration-dependent increase in the expression level of E-cadherin and a decrease in the expression level of N-cadherin. In SKOV3 cells, there was a 2.1-fold, 2.2-fold, and 1.9-fold increase in expression of E-cadherin when treated with ALS 0.1, 1, and 5 μM for 24 hours, respectively, whereas ALS 1 and 5 μM suppressed the expression level of N-cadherin 1.8-fold and 2.8-fold, respectively (P<0.05; Figure 16B). Consequently, with increasing concentrations of ALS, an increased ratio of E-cadherin over N-cadherin was observed. The E-cadherin/N-cadherin ratio was increased from 0.18 at the basal level to 0.5, 0.8, and 1.2 when SKOV3 cells were treated with ALS 0.1, 1, and 5 μM for 24 hours, respectively (P<0.05; Figure 16B). In OVCAR4 cells, there was a 2.7-fold and 3.5-fold increase in expression of E-cadherin when cells were treated with ALS 1 and 5 μM, respectively (Figure 16B). Meanwhile, ALS decreased the expression of N-cadherin 1.3-fold, 1.8-fold, and 5.8-fold, respectively, compared with control cells when OVCAR4 cells were treated with ALS 0.1, 1, and 5 μM for 24 hours, respectively. The E-cadherin/N-cadherin ratio was increased from 0.15 at the basal level to 0.5, 0.8, and 1.2 when OVCAR4 cells were treated with ALS 0.1, 1, and 5 μM for 24 hours, respectively (P<0.05; Figure 16B).
In order to examine the effect of ALS on EMT in SKOV3 and OVCAR4 cells further, we tested the expression levels of several key regulators of E-cadherin (Figure 16B). Slug (a zinc finger transcriptional factor) together with TCF8/ZEB1 are suppressors of E-cadherin in EMT.55 In addition, slug protects damaged cells from apoptosis by repressing P53-induced transcription of the proapoptotic Bcl-2 family protein, PUMA.55 ALS significantly reduced the expression level of slug in both cell lines (Figure 16B). In SKOV3 cells, the expression level of slug was significantly suppressed 1.9-fold and 3.1-fold when treated with ALS 1 and 5 μM for 24 hours, respectively (P<0.01; Figure 16B). In OVCAR4 cells, treatment with ALS 5 μM for 24 hours, respectively significantly reduced the expression level of slug by 2.1-fold (P<0.05; Figure 16B). Further, ALS induced a concentration-dependent reduction in the expression level of TCF-8/ZEB1 in SKOV3 and OVCAR4 cells. In SKOV3 cells, ALS 0.1, 1, and 5 μM significantly suppressed the expression level of TCF-8/ZEB1 by 1.4-fold, 1.7-fold, and 2.4-fold, respectively (P<0.05; Figure 16B). Similarly, there was a 1.8-fold, 2.4-fold, and 2.3-fold reduction in the expression of TCF-8/ZEB1 in OVCAR4 cells treated with ALS 0.1, 1, and 5 μM, respectively (P<0.001; Figure 16B). Vimentin is a type III intermediate filament protein that is expressed in mesenchymal cells.55 β-catenin can act as an integral component of a protein complex in adherens junctions, helps cells maintain epithelial layers, and participates in the Wnt signaling pathway as a downstream target.55 Treatment of cells with ALS 1 and 5 μM significantly suppressed the expression level of vimentin by 2.8-fold and 7.4-fold in SKOV3 cells (P<0.05; Figure 16B). ALS 0.1 μM reduced the vimentin level by 1.4-fold, but this change was not statistically significant. In OVCAR4 cells, treatment with ALS 0.1, 1, and 5 μM for 24 hours resulted in a 1.4-fold, 1.6-fold, and 2.0-fold reduction in vimentin expression levels, but these changes were not statistically significant (Figure 16B). There was a significant reduction in the expression level of β-catenin in both cell lines treated with ALS 0.1, 1, and 5 μM for 24 hours. ALS 1 and 5 μM significantly decreased the expression level of β-catenin by 1.4-fold and 2.6-fold in SKOV3 cells, and by 2.0-fold and 2.8-fold in OVCAR4 cells, respectively (Figure 16B).
Finally, expression of ZO-1 was examined in SKOV3 and OVCAR4 cells exposed to ALS. ZO-1 and ZO-2 are required for the formation and function of tight junctions. In subconfluent proliferating cells, ZO-1 and ZO-2 have been shown to colocalize to the nucleus and play a role in transcriptional regulation, possibly by facilitating nuclear import/export of transcriptional regulators.55 A significant effect of ALS on the expression of ZO-1 was observed in both cell lines (Figure 16A). Treatment of SKOV3 cells with ALS 0.1, 1, and 5 μM for 24 hours increased 6.4-fold, 6.7-fold, and 10.3-fold in the expression of ZO-1 and 1 and 5 μM ALS resulted in a 7.1-fold and 9.7-fold increase in the expression level of ZO-1 in OVCAR4 cells (P<0.05; Figure 16B).
Sirt1 plays a role in ALS-induced autophagy and EMT inhibition in SKOV3 and OVCAR4 cells
Sirt1 plays an important role in the regulation of cellular autophagy through two major mechanisms. Sirt1 can influence autophagy directly via its deacetylation of key components of the autophagy induction network, such as Atgs 5, 7, and 8. Nucleus-localized Sirt1 is also known to induce the expression of components of the autophagy pathway via activation of members of the FoxO transcription factor family.56 Therefore, we speculated that ALS may regulate expression of Sirt1 in SKOV3 and OVCAR4 cells and examined the effect of ALS on the expression level of Sirt1 in both cell lines. As shown in Figure 17A, there was a concentration-dependent reduction in the expression of Sirt1 in SKOV3 and OVCAR4 cells. Incubation of SKOV3 cells with ALS 1 and 5 μM for 24 hours reduced the expression of Sirt1 by 58.9% and 64.1%, respectively; in OVCAR4 cells, treatment with ALS 5 μM for 24 hours significantly suppressed the expression level of Sirt1 by 69.5% (Figure 17B).
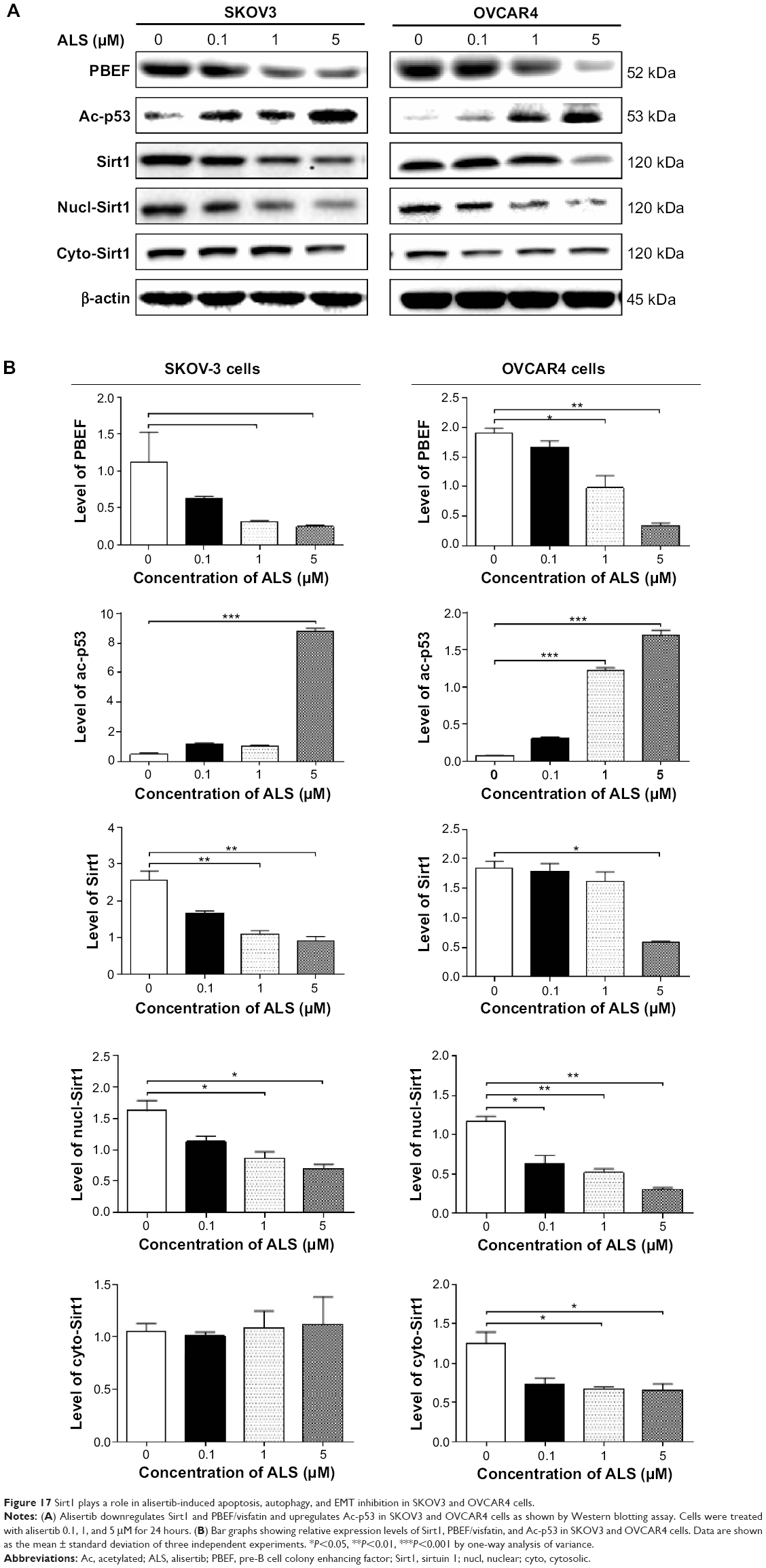 | Figure 17 Sirt1 plays a role in alisertib-induced apoptosis, autophagy, and EMT inhibition in SKOV3 and OVCAR4 cells. |
To further verify the expression of Sirt1, we extracted cytoplasmic protein and nuclear protein from SKOV3 and OVCAR4 cells treated with ALS 0.1, 1, and 5 μM. In the nuclear protein of SKOV3, exposure to ALS 1 and 5 μM for 24 hours reduced the expression of Sirt1 by 46.6% and 56.9%, respectively (Figure 17B). However, in the cytosol of SKOV3, expression of Sirt1 was not significantly different. In OVCAR4 cells, expression of Sirt1 was significantly decreased in both the nucleus and cytosol. In the nucleus, expression of Sirt1 was suppressed by 47.1%, 55.7%, and 74.5% when OVCAR4 cells were treated with ALS 0.1, 1, and 5 μM, respectively; in the cytosol, expression of Sirt1 was suppressed by 45.8% and 48.5% when these cells were treated with ALS 1 and 5 μM, respectively (P<0.05; Figure 17B). Downregulation of Sirt1 by ALS may partially contribute to ALS’ autophagy-inducing and EMT-inhibitory effects.
ALS inhibits phosphorylation of AURKA in SKOV3 and OVCAR4 cells
To verify further the anticancer effect of ALS on the treatment of EOC by suppressing AURKA, we investigated the expression of AURKA and phosphorylated AURKA in SKOV3 and OVCAR4 cell lines exposed to ALS. When SKOV3 cells were treated with ALS 0.1 and 5 μM for 24 hours, expression of AURKA increased 1.8-fold and 1.9-fold, respectively (P<0.05; Figure 18B). Expression of p-AURKA increased 1.1-fold, 0.9-fold, and 1.4-fold when SKOV3 cells were treated with ALS 0.1, 1, and 5 μM for 24 hours, respectively, but these changes were not statistically significant. However, the p-AURKA/AURKA ratio was suppressed by 49.2%, 48.5%, and 40.8% when SKOV3 cells were treated with ALS 0.1, 1, and 5 μM, respectively (P<0.01 or P<0.05; Figure 18B).
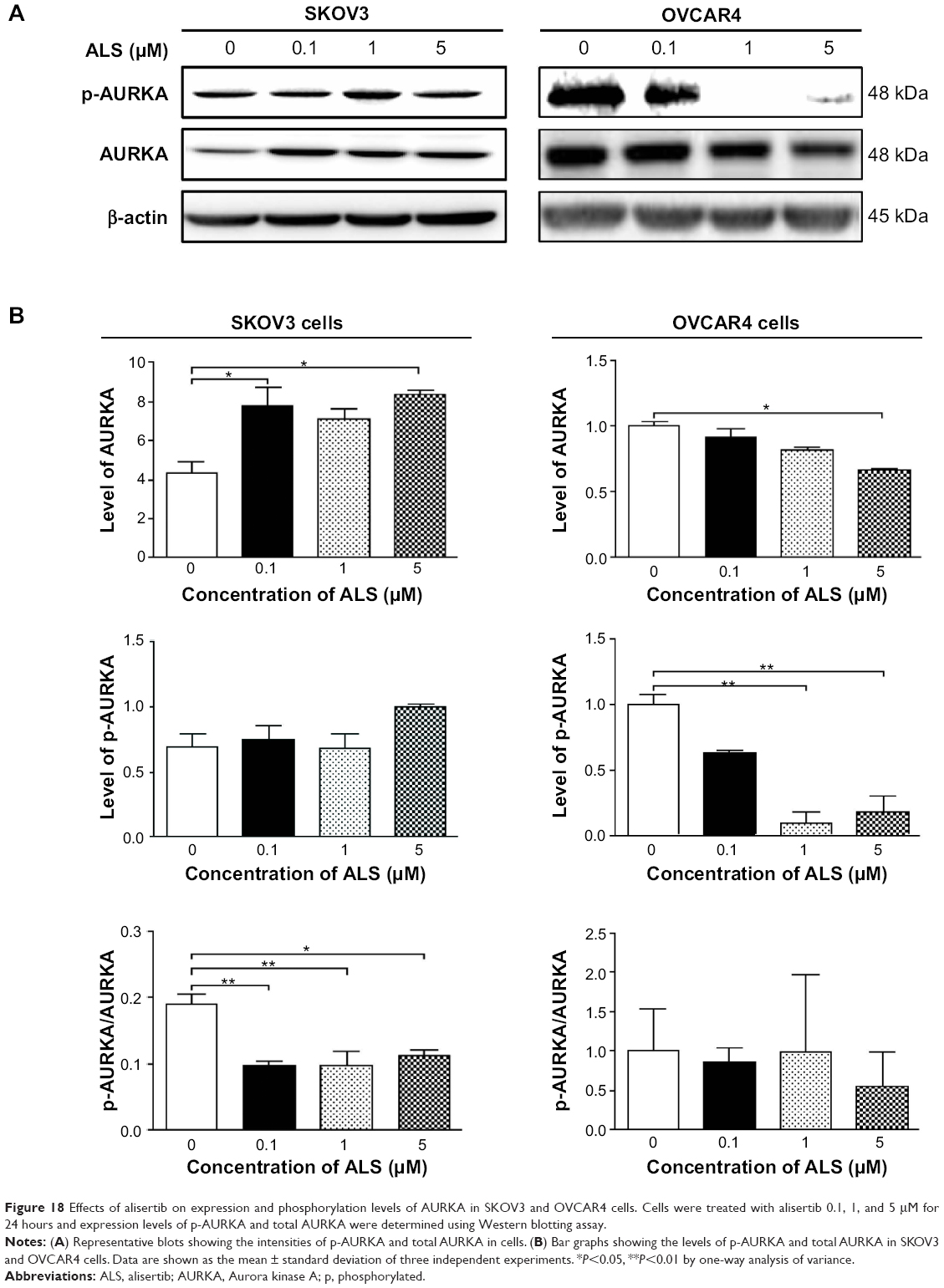 | Figure 18 Effects of alisertib on expression and phosphorylation levels of AURKA in SKOV3 and OVCAR4 cells. Cells were treated with alisertib 0.1, 1, and 5 μM for 24 hours and expression levels of p-AURKA and total AURKA were determined using Western blotting assay. |
In OVCAR4 cells, expression of AURKA decreased significantly by 63.9% when these cells were treated with ALS 5 μM for 24 hours (Figure 18B). There was no statistically significant difference in AURKA expression when OVCAR4 cells were treated with ALS 0.1 or 1 μM for 24 hours. Similarly, expression of p-AURKA and the ratio of p-AURKA/AURKA were not significantly different when OVCAR4 cells were treated with different concentrations of ALS.
Discussion
Treatment of advanced ovarian cancer remains a major challenge because of the poor efficacy of current therapies and chemotherapy. Despite novel therapeutic advances, improvement in the survival rate of patients with EOC has been marginal, suggesting the presence of unique, active, intrinsic mechanisms that impart resistance to chemotherapeutic agents in EOC. Recently, there has been an increased interest in seeking new effective drugs for EOC from new compounds. AURKA is expressed in late S phase, peaking at G2/M phase and declining at G1 phase, and is involved in centrosome maturation and separation, bipolar spindle assembly, and mitotic entry. AURKB, known as the chromosomal passenger protein, is essential for accurate chromosome segregation and cytokinesis,57 and AURKC complements the function of AURKB, which is expressed in the testes.58 With more and more studies focusing on the effect of the Aurora kinases in formation of solid tumors, AURKA has become more important in cancer therapy. Both overexpression and gene amplification of AURKA have been characterized in human tumors, and have been shown to correlate with tumor proliferation rates and prognostic markers.59 Recent reports suggest that AURKA can induce chemotherapeutic resistance and regulate several key signaling pathways related to the cell cycle, apoptosis, autophagy, and EMT 8,59,60 in cancer cells, suggesting AURKA’s role as a central node in cancer cell signaling.61 With the goal of exploiting this pathway in anticancer therapy, we evaluated the impact of the AURKA inhibitor ALS on EOC cells. Based on our docking studies, ALS, MLN8054, and VX-680 showed a binding preference for AURKA mainly via hydrogen bond formation, charge interactions, and π-π stacking interactions, which are all known AURKA inhibitors. Other compounds including AMG-900, CYC116, and danusertib could all bind to AURKA or AURKB with higher binding energy. ALS,38 AMG-900,29 barasertib,30 CYC116,31 danusertib,31 MLN8054,32 and VX-68033 are all known inhibitors of Aurora kinases with distinct binding affinity and preference. ALS is a selective AURKA inhibitor with an IC50 of 1.2 nM. For AURKB, the IC50 value is more than 200-fold higher than that of AURKA.38 MLN8054 is a potent and selective inhibitor of AURKA, with an IC50 of 4 nM. It is more than 40-fold selective for AURKA over AURKB.32 In addition, VX-680 is a pan-Aurora kinase inhibitor, mostly targeting AURKA, with an IC50 of 0.6 nM. VX-680 is less potent to AURKB and AURKC, with IC50 values of 18 nM and 4.6 nM, respectively.33 Our docking results showed that ALS, MLN8045, and VX-680 docked easily into the active site of AURKA and with low CDOCKER interaction energy when compared with their interactions with AURKB. The docking results show that the low CDOCKER interaction energy may explain their high potency and preference for AURKA over AURKB.
The Aurora kinases are related to the AGC (protein kinase A, protein kinase G, and protein kinase C) branch of the protein kinases. Part of the activation mechanism for most AGC kinases is mediated by binding of the phosphorylated C-terminal “hydrophobic motif”, FXXF(T/S)F, to the N-terminal lobe of the kinase catalytic domain.9,58 However, Aurora kinases lack canonical C-terminal hydrophobic motifs and are activated in trans through binding of regulatory proteins to their N-terminal lobe. The catalytic domain of AURKA has the typical bilobal kinase fold, comprised of an N-terminal β-strand domain (residues 127–215) and a C-terminal α-helical domain (residues 216–385).62 These domains are linked together by a hinge region (residues 210–216) that plays an important part in forming the catalytic active site. Residues 273–292 are located at the N-terminal end of the activation loop of AURKA which adopt a unique conformation. Trp277 in the activation loop is stabilized by interactions with residue Phe144 in the flexible glycine-rich loop and residue Phe275.62 Trp277 in AURKA corresponds to Phe187 in cAMP-dependent kinase and Leu407 in SRC proto-oncogene kinase, which is conserved in all three human Aurora kinases and all known Aurora-like sequences. In our docking study, we observed that Trp277 bound to ALS, VX-680, and AMG-900, indicating the important role of Trp277 in ligand binding. Notably, Lys87 and Lys106 in AURKB bind to ALS, AMG-900, danusertib, MLN8054, and VX-68 via hydrogen bond formation and/or π-π stacking, suggesting their important role in ligand binding of AURKB.
Next, we explored the effects of ALS on growth, proliferation, apoptosis, and autophagy in SKOV3 and OVCAR4 cells. We observed a potent inhibitory effect of ALS on cell proliferation in the two EOC cell lines. We found that ALS promoted cell apoptosis and autophagy, and induced cell cycle arrest in SKOV3 and OVCAR4 cells. Further, we observed that ALS inhibited EMT (Figure 16) in both cell lines and Sirt1 was involved in the inhibitory effects of ALS on the growth of EOC cells (Figure 17).
Recent studies indicate that AURKA may enable cancer cells to become chemoresistant and radioresistant through dysregulation of cell cycle progression and the DNA damage response. Direct evidence suggests that AURKA inhibits p53, p21 Waf1/Cip1, and p27 Kip1, but enhances cyclin B1 to repeal cell cycle checkpoints and to promote cell cycle progression.63 In previous studies, the effect of ALS on cell cycle distribution has been confirmed in various cancer cell lines.59 In the present study, we found that ALS arrested SKOV3 and OVCAR4 cells in G2/M phase in a concentration-dependent and time-dependent manner (Figure 6). We further explored the effect of ALS on key regulators of cell cycle checkpoints, including CDC2 and cyclin B1, in both cell lines. The CDC2-cyclin B1 complex is vital in regulation of the G2/M phase transition and mitosis. We observed a significant decrease in the expression level of cyclin B1 and CDC2 in SKOV3 and OVCAR4 cells treated with ALS, providing an explanation for the effect of ALS on G2/M phase arrest in SKOV3 and OVCAR4 cells (Figure 7). It has been reported that p27 Kip1, a cyclin-dependent kinase inhibitor regulated by p53, can bind to the CDK1/CDC2-cyclin B1 complex, thereby inducing cell cycle arrest.63 We observed that expression of p53 and p27 Kip1 was increased in a concentration-dependent and time-dependent manner in SKOV3 and OVCAR4 cells treated with ALS, which probably contributes to the inhibitory effect of ALS on cell proliferation and its inducing effect on cell cycle arrest in SKOV3 and OVCAR4 cells (Figure 7).
Programmed cell death, referring to apoptosis, autophagy, and programmed necrosis, is proposed to be death of a cell in any pathological format when mediated by an intracellular program. These three forms of programmed cell death may jointly decide the fate of malignant neoplastic cells; apoptosis and programmed necrosis invariably contribute to cell death, whereas autophagy can have either prosurvival or prodeath roles.64
Apoptosis, or type I programmed cell death, is characterized by specific morphological and biochemical changes of dying cells, including cell shrinkage, nuclear condensation and fragmentation, dynamic membrane blebbing, and loss of adhesion to neighboring cells or to the extracellular matrix. Biochemical changes include chromosomal DNA cleavage into internucleosomal fragments, phosphatidylserine externalization, and a number of intracellular substrate cleavages by specific proteolysis.64 Apoptosis is executed by members of the caspase family of cysteine proteases, which can be activated by two main pathways, ie, the extrinsic death receptor pathway and the intrinsic mitochondria/cytochrome c-mediated pathway. The two pathways are linked and both trigger the activation of caspases 3, 6, and 7. In the death receptor pathway, binding of extracellular death ligands to members of the tumor necrosis factor and nerve growth factor receptor superfamily induces activation of caspase 8, which in turn activates caspases 3 and 7, resulting in further caspase activation events and finally cell death.65,66 It has been reported that suppression of AURKA increased apoptosis of small cell lung cancer cells. A potential mechanism for the increase of apoptosis is the downregulation of Bcl-2 and upregulation of Bax.67 Another study indicated AURKA plays a crucial role in growth by inhibiting cell apoptosis and propelling cell cycle.8 The proapoptotic effect of ALS has been reported in various cancer cell lines, including Burkitt lymphoma, breast cancer, melanoma, and leukemia.68–70 In the present study, we observed concentration-dependent ALS-induced apoptosis in SKOV3 and OVCAR4 cells (Figure 8). It is known that mitochondrial disruption and the subsequent release of cytochrome c initiate the process of apoptosis.66 The release of cytochrome c from the mitochondria was initiated by proapoptotic members of the Bcl-2 family but antagonized by antiapoptotic members of this family. Antiapoptotic members of the Bcl-2 family can be inhibited by post-translational modification and/or by increased expression of PUMA, which is an essential regulator of p53-mediated cell apoptosis.66 In addition, the cytosolic cytochrome c released from the mitochondria following an apoptotic stimulus serves as a main trigger for caspase enzyme activation.66 In our study, we found that the cytosolic level of cytochrome c was significantly increased after treatment with ALS, which subsequently activated caspase 9. Activated caspase 9 in turn activated caspase 3. Activated caspase 3 ultimately induced apoptosis, with a decrease in the Bcl-2 level (Figure 9). Moreover, we noted a concentration-dependent increase in expression of PUMA in SKOV3 and OVCAR4 cells. These results indicate that ALS induces mitochondria-dependent apoptosis in both SKOV3 and OVCAR4 cells, with the involvement of p53 (Figure 9).
Autophagy (also known as type II programmed cell death), the process of degradation of unwanted or damaged cell elements, is extremely important for a variety of human diseases, especially cancers. This process influences various stages of initiation and progression of cancer, which is caused by overlapping signaling pathways of autophagy and carcinogenesis. However, due to the complexity of cancer as a systemic disease, the fate of tumor cells is not determined by one signaling pathway.71 Recently, the PI3K/AKT/mTOR signaling pathway has come to be regarded as the key regulator of a series of cell processes, as it can be deregulated by various genetic and epigenetic mechanisms in a wide range of cancer cells.64 PI3K activates the serine/threonine kinase Akt, which, in turn, through a cascade of regulators, results in phosphorylation and activation of the serine/threonine kinase mTOR.72 Targeting apoptosis and autophagy concurrently has emerged as a potential approach in the treatment of prostate cancer.73 Overexpression of AURKA induces growth-promoting and survival-promoting oncogenic signaling pathways, such as the PI3K/Akt and β-catenin pathways in UGC cancer cells.74
In this study, ALS induced autophagy in both SKOV3 and OVCAR4 cells, which may contribute to its anticancer activity. In fact, previous studies have shown the autophagy-inducing effect of ALS in various cancer cell lines via regulation of the PI3K/Akt/mTOR axis.75 In the present study, we also observed that the autophagy-inducing effect of ALS outweighed its apoptosis-inducing effect. Autophagy has a multifaceted role in cancer; it functions as a tumor suppressor at an early stage of tumor development, but can also be used by cancer cells as a cytoprotection mechanism to promote survival of established tumors.64
In general, cancer cells proliferate depending on glycolysis, whereas supply of glucose and other nutrients may not always be sufficient. Therefore, cancer cells can often be exposed to an intracellular energy and/or nutrient imbalance and various forms of stress associated with energetic insufficiency. To cope with such metabolic stresses, cells have various mechanisms to sense nutrient status and to adapt to stressful cell conditions. These include the nutrient-signaling pathways regulated by AMPK and mechanistic/mTOR and also cellular stress responses, such as mitochondrial and endoplasmic reticulum stress responses, as well as autophagy. AMPK, the master energy sensor, is an important regulator of cell death under various conditions, through activation of JNK and p53 and inhibition of mTOR.76 In the present study, there was a significant activation of AMPK in response to treatment with ALS, which may contribute to inhibition of mTOR.
Sirt1 is the best characterized member of the sirtuin family and functions as a longevity-promoting protein, playing a role in lifespan extension induced by caloric restriction. Sirt1 is an nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylase overexpressed in prostate, colon, breast, gastric, liver, and pancreatic tumors.77 Sirt1 also attenuates p53 and PTEN, promotes EMT, and increases cell migration, all of which promote carcinogenesis.77
Recently, studies have shown that Sirt1 has an important role in the regulation of apoptosis and autophagy. Sirt1 deacetylates both histone and nonhistone proteins, such as p53 and FoxOs.78 Deacetylation of p53 and inhibition of p53-regulated cell death indicate that Sirt1 is a negative regulator of p53.78 In our study, we found that ALS significantly increased intracellular levels of deacetylated p53 and decreased the expression levels of Sirt1 and PBEF. PBEF is a rate-limiting enzyme in the NAD+ biosynthesis pathway and plays an essential role in regulation of Sirt1 activation.79 Taken together, ALS-induced cell apoptosis and autophagy in EOC cells may occur via a Sirt1-mediated pathway (Figure 17).
Autophagy and apoptosis are connected both positively and negatively, with extensive crosstalk between the two processes. We have found that ALS induces both apoptosis and autophagy. Inhibition or induction of autophagy inhibited ALS-induced apoptosis in SKOV3 and OVCAR4 cells. We speculate that these two key processes of cell death initiated by ALS can be coordinated by certain important molecules, including p53, beclin 1, and Akt (Figure 12). ALS can induce apoptosis and autophagy in a coordinated manner.
EMT is an important step in the developmental process. Recent evidence indicates that EMT allows tumor cells to acquire invasive properties and to develop metastatic growth characteristics. Some of the transcription factors that are actively involved in EMT have a significant role in the EMT-metastasis linkage. A number of studies have reported that EMT-inducing transcription factors, such as Twist, snail, slug, and ZEB, are directly or indirectly involved in cancer cell metastasis through a different signaling cascades, including the Akt, signal transducer and activator of transcription 3, MAPK, and Wnt pathways, with the ultimate consequence of downregulating E-cadherin and upregulating metastatic proteins, such as N-cadherin, vimentin, and matrix metalloproteinase-2.80
In the present study, ALS significantly increased the ratio of E-cadherin over N-cadherin, which would result in EMT inhibition in EOC. Further, ALS suppressed the expression of slug and TCF-8 in SKOV3 and OVCAR4 cells. Slug is a master regulator of EMT, downregulating E-cadherin by silencing gene expression.55 TCF8/ZEB1 functions as an activator of the EMT process through downregulation of epithelial genes.55 Moreover, ALS significantly increased the expression level of ZO-1 but decreased the expression level of vimentin. ZO-1 is required for tight junction formation and function, involving the regulation of paracellular permeability and maintenance of cell polarity, blocking the movement of transmembrane proteins between the apical and basolateral cell surface,81 whereas vimentin is a marker expressed in mesenchymal cells.55
Sirtuins are a highly conserved family of NAD+-dependent protein lysine-modifying enzymes, with deacetylase, adenosine diphosphateribosyltransferase, and other deacetylase activities. Mammals have seven sirtuins, namely Sirts1–7. They are key regulators for a wide variety of cellular and physiological processes, such as cell proliferation, differentiation, DNA damage and stress response, genome stability, cell survival, metabolism, energy homeostasis, organ development, aging, and cancer. Here we present an extensive literature review of the roles of mammalian sirtuins, particularly Sirt1, given that it is the most studied sirtuin, being involved in human epithelial, neuronal, hematopoietic, and mesenchymal malignancies, covering breast, prostate, lung, thyroid, liver, colon, gastric, pancreatic, ovarian, and cervical cancers, tumors of the central nervous system, leukemia and lymphoma, and soft tissue sarcomas. The collective evidence suggests that sirtuins are involved in both promoting and suppressing tumorigenesis depending on cellular and molecular contexts. We discuss the potential use of sirtuin modulators, especially sirtuin inhibitors, in cancer treatment.82
It was reported that reduction of Sirt1 promoted metastasis of breast epithelial cells in an orthotopic model of breast cancer and promoted EMT and cell motility in these cells in vitro. Further, EMT could be induced in both breast epithelial and kidney epithelial cells in vitro, and this was also repressed by Sirt1.83 In our study, we found that ALS suppressed Sirt1 in SKOV3 and OVCAR4 cells in a concentration-dependent manner (Figure 17). This result indicates that Sirt1 has a role in the oncogenesis of EOC and that ALS might inhibit EMT through a Sirt1-mediated pathway. In our study, ALS downregulated expression of PBEF/visfatin in SKOV3 and OVCAR4 cells. PBEF/visfatin has been proposed to be involved in preventing apoptosis in cancer cells, and as such has received a great deal of attention in recent years and stimulated the development of specific inhibitors for treating cancer.84
In summary, ALS preferentially bound to AURKA over AURKB via hydrogen bond formation, charge interactions, and π-π stacking interactions. ALS induced inhibition of the PI3K/Akt/mTOR and p38 MAPK pathways and activation of AMPK, contributing to the autophagy-inducing activities of ALS. Modulation of autophagy altered basal and ALS-induced apoptosis in both cell lines. In addition, ALS suppressed EMT via regulation of E-cadherin suppressor with involvement of Sirt1. ALS also downregulated PBEF/visfatin, and inhibition of PBEF/visfatin significantly enhanced basal and ALS-induced apoptosis and autophagy in both cell lines. ALS may represent a new anticancer drug that can kill EOC cells and prevent EMT. More studies are needed to reveal the underlying mechanisms and other potential targets of ALS in the treatment of EOC.
Acknowledgments
The authors appreciate the financial support from the Startup Fund of the College of Pharmacy, University of South Florida, Tampa, Florida, USA. Dr Zhi-Wei Zhou is a holder of a postdoctoral scholarship from the College of Pharmacy, University of South Florida, Tampa, Florida, USA.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Soerjomataram I, Ervik M, et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. Lyon, France: International Agency for Research on Cancer; 2013. Available from: http://globocan.iarc.fr. Accessed October 21, 2014. | ||
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Doufekas K, Olaitan A. Clinical epidemiology of epithelial ovarian cancer in the UK. Int J Womens Health. 2014;6:537–545. | ||
Lowe KA, Chia VM, Taylor A, et al. An international assessment of ovarian cancer incidence and mortality. Gynecol Oncol. 2013;130(1):107–114. | ||
Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014;384(9951):1376–1388. | ||
Davis A, Tinker AV, Friedlander M. “Platinum resistant” ovarian cancer: what is it, who to treat and how to measure benefit? Gynecol Oncol. 2014; 133(3):624–631. | ||
Bolanos-Garcia VM. Aurora kinases. Int J Biochem Cell Biol. 2005;37(8): 1572–1577. | ||
Do TV, Xiao F, Bickel LE, et al. Aurora kinase A mediates epithelial ovarian cancer cell migration and adhesion. Oncogene. 2014;33(5): 539–549. | ||
Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL Jr, Golemis EA. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci. 2013;70(4):661–687. | ||
Hsu LC, Kapali M, DeLoia JA, Gallion HH. Centrosome abnormalities in ovarian cancer. Int J Cancer. 2005;113(5):746–751. | ||
Hilton JF, Shapiro GI. Aurora kinase inhibition as an anticancer strategy. J Clin Oncol. 2014;32(1):57–59. | ||
Zhou H, Kuang J, Zhong L, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20(2):189–193. | ||
Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN Jr, Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res. 2008;14(6):1639–1648. | ||
Mountzios G, Terpos E, Dimopoulos MA. Aurora kinases as targets for cancer therapy. Cancer Treat Rev. 2008;34(2):175–182. | ||
Landen CN Jr, Lin YG, Immaneni A, et al. Overexpression of the centrosomal protein Aurora-A kinase is associated with poor prognosis in epithelial ovarian cancer patients. Clin Cancer Res. 2007; 13(14):4098–4104. | ||
Lassmann S, Shen Y, Jutting U, et al. Predictive value of Aurora-A/STK15 expression for late stage epithelial ovarian cancer patients treated by adjuvant chemotherapy. Clin Cancer Res. 2007;13(14):4083–4091. | ||
Gritsko TM, Coppola D, Paciga JE, et al. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin Cancer Res. 2003;9(4):1420–1426. | ||
Kollareddy M, Zheleva D, Dzubak P, Brahmkshatriya PS, Lepsik M, Hajduch M. Aurora kinase inhibitors: progress towards the clinic. Invest New Drugs. 2012;30(6):2411–2432. | ||
Maris JM, Morton CL, Gorlick R, et al. Initial testing of the Aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Pediatr Blood Cancer. 2010;55(1):26–34. | ||
Tomita M, Mori N. Aurora A selective inhibitor MLN8237 suppresses the growth and survival of HTLV-1-infected T-cells in vitro. Cancer Sci. 2010;101(5):1204–1211. | ||
Gorgun G, Calabrese E, Hideshima T, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. 2010;115(25):5202–5213. | ||
Cervantes A, Elez E, Roda D, et al. Phase I pharmacokinetic/pharmacodynamic study of MLN8237, an investigational, oral, selective Aurora A kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2012;18(17):4764–4774. | ||
Dees EC, Cohen RB, von Mehren M, et al. Phase I study of Aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin Cancer Res. 2012;18(17):4775–4784. | ||
Matulonis UA, Sharma S, Ghamande S, et al. Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol Oncol. 2012;127(1): 63–69. | ||
Friedberg JW, Mahadevan D, Cebula E, et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol. 2014;32(1):44–50. | ||
Goldberg SL, Fenaux P, Craig MD, et al. An exploratory phase 2 study of investigational Aurora A kinase inhibitor alisertib (MLN8237) in acute myelogenous leukemia and myelodysplastic syndromes. Leuk Res Rep. 2014;3(2):58–61. | ||
Macarulla T, Cervantes A, Elez E, et al. Phase I study of the selective Aurora A kinase inhibitor MLN8054 in patients with advanced solid tumors: safety, pharmacokinetics, and pharmacodynamics. Mol Cancer Ther. 2010;9(10):2844–2852. | ||
Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer. 2004;4(12):927–936. | ||
Uitdehaag JC, Verkaar F, Alwan H, de Man J, Buijsman RC, Zaman GJ. A guide to picking the most selective kinase inhibitor tool compounds for pharmacological validation of drug targets. Br J Pharmacol. 2012;166(3):858–876. | ||
Thoresen SB, Campsteijn C, Vietri M, et al. ANCHR mediates Aurora-B-dependent abscission checkpoint control through retention of VPS4. Nat Cell Biol. 2014;16(6):550–560. | ||
Markant SL, Esparza LA, Sun J, et al. Targeting sonic hedgehog-associated medulloblastoma through inhibition of Aurora and Polo-like kinases. Cancer Res. 2013;73(20):6310–6322. | ||
Kozyreva VK, McLaughlin SL, Livengood RH, et al. NEDD9 regulates actin dynamics through cortactin deacetylation in an AURKA/HDAC6-dependent manner. Mol Cancer Res. 2014;12(5):681–693. | ||
Kim Y, Holland AJ, Lan W, Cleveland DW. Aurora kinases and protein phosphatase 1 mediate chromosome congression through regulation of CENP-E. Cell. 2010;142(3):444–455. | ||
Wang ZX, Sun J, Howell CE, et al. Prediction of the likelihood of drug interactions with kinase inhibitors based on in vitro and computational studies. Fundam Clin Pharmacol. 2014;28(5):551–582. | ||
Qiu JY, Zhou ZW, He ZX, et al. Plumbagin elicits differential proteomic responses mainly involving cell cycle, apoptosis, autophagy, and epithelial to mesenchymal transition pathways in human prostate cancer PC-3 and DU145 cells. Drug Des Devel Ther. In press. | ||
Yin JJ, Sharma S, Shumyak SP, et al. Synthesis and biological evaluation of novel folic acid receptor-targeted, beta-cyclodextrin-based drug complexes for cancer treatment. PLoS One. 2013;8(5):e62289. | ||
Li YC, He SM, He ZX, et al. Plumbagin induces apoptotic and autophagic cell death through inhibition of the PI3K/Akt/mTOR pathway in human non-small cell lung cancer cells. Cancer Lett. 2014; 344(2):239–259. | ||
Manfredi MG, Ecsedy JA, Chakravarty A, et al. Characterization of alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res. 2011;17(24):7614–7624. | ||
Hu X, Moscinski LC. Cdc2: a monopotent or pluripotent CDK? Cell Prolif. 2011;44(3):205–211. | ||
Warfel NA, El-Deiry WS. p21WAF1 and tumourigenesis: 20 years after. Curr Opin Oncol. 2013;25(1):52–58. | ||
Yoon MK, Mitrea DM, Ou L, Kriwacki RW. Cell cycle regulation by the intrinsically disordered proteins p21 and p27. Biochem Soc Trans. 2012;40(5):981–988. | ||
Carvajal LA, Manfredi JJ. Another fork in the road – life or death decisions by the tumour suppressor p53. EMBO Rep. 2013;14(5):414–421. | ||
Estaquier J, Vallette F, Vayssiere JL, Mignotte B. The mitochondrial pathways of apoptosis. Adv Exp Med Biol. 2012;942:157–183. | ||
Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–489. | ||
Neri LM, Cani A, Martelli AM, et al. Targeting the PI3K/Akt/mTOR signaling pathway in B-precursor acute lymphoblastic leukemia and its therapeutic potential. Leukemia. 2014;28(4):739–748. | ||
Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. 2014;4:64. | ||
Wang X, Wang XL, Chen HL, et al. Ghrelin inhibits doxorubicin cardiotoxicity by inhibiting excessive autophagy through AMPK and p38-MAPK. Biochem Pharmacol. 2014;88(3):334–350. | ||
Dunlop EA, Tee AR. The kinase triad, AMPK, mTORC1 and ULK1, maintains energy and nutrient homoeostasis. Biochem Soc Trans. 2013;41(4):939–943. | ||
Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 2011;1813(11):1978–1986. | ||
Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330(6009):1344–1348. | ||
Kang R, Zeh HJ, Lotze MT, Tang D. The beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. | ||
Maiuri MC, Criollo A, Kroemer G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 2010;29(3): 515–516. | ||
Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–5728. | ||
Cannito S, Novo E, di Bonzo LV, Busletta C, Colombatto S, Parola M. Epithelial-mesenchymal transition: from molecular mechanisms, redox regulation to implications in human health and disease. Antioxid Redox Signal. 2010;12(12):1383–1430. | ||
Nauseef JT, Henry MD. Epithelial-to-mesenchymal transition in prostate cancer: paradigm or puzzle? Nat Rev Urol. 2011;8(8):428–439. | ||
Ng F, Tang BL. Sirtuins’ modulation of autophagy. J Cell Physiol. 2013; 228(12):2262–2270. | ||
Ruchaud S, Carmena M, Earnshaw WC. The chromosomal passenger complex: one for all and all for one. Cell. 2007;131(2):230–231. | ||
Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 2003;4(11):842–854. | ||
Zhou N, Singh K, Mir MC, et al. The investigational Aurora kinase A inhibitor MLN8237 induces defects in cell viability and cell-cycle progression in malignant bladder cancer cells in vitro and in vivo. Clin Cancer Res. 2013;19(7):1717–1728. | ||
D’Assoro AB, Liu T, Quatraro C, et al. The mitotic kinase Aurora – a promotes distant metastases by inducing epithelial-to-mesenchymal transition in ERα+ breast cancer cells. Oncogene. 2014;33(5): 599–610. | ||
Melaiu O, Cristaudo A, Melissari E, et al. A review of transcriptome studies combined with data mining reveals novel potential markers of malignant pleural mesothelioma. Mutat Res. 2012;750(2):132–140. | ||
Cheetham GM, Knegtel RM, Coll JT, et al. Crystal structure of Aurora-2, an oncogenic serine/threonine kinase. J Biol Chem. 2002;277(45): 42419–42422. | ||
Wang Y, Sun H, Wang Z, et al. Aurora-A: a potential DNA repair modulator. Tumour Biol. 2014;35(4):2831–2836. | ||
Ouyang L, Shi Z, Zhao S, et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45(6):487–498. | ||
Xu DR, Huang S, Long ZJ, et al. Inhibition of mitotic kinase Aurora suppresses Akt-1 activation and induces apoptotic cell death in all-trans retinoid acid-resistant acute promyelocytic leukemia cells. J Transl Med. 2011;9:74. | ||
Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. | ||
Lu Y, Liu Y, Jiang J, et al. Knocking down the expression of Aurora-A gene inhibits cell proliferation and induces G2/M phase arrest in human small cell lung cancer cells. Oncol Rep. 2014;32(1):243–249. | ||
Ierano C, Chakraborty AR, Nicolae A, et al. Loss of the proteins Bak and Bax prevents apoptosis mediated by histone deacetylase inhibitors. Cell Cycle. 2013;12(17):2829–2838. | ||
Liu Y, Hawkins OE, Su Y, et al. Targeting aurora kinases limits tumour growth through DNA damage-mediated senescence and blockade of NF-kappaB impairs this drug-induced senescence. EMBO Mol Med. 2013;5(1):149–166. | ||
Yang J, Ikezoe T, Nishioka C, Nobumoto A, Udaka K, Yokoyama A. CD34(+)/CD38(−) acute myelogenous leukemia cells aberrantly express Aurora kinase A. Int J Cancer. 2013;133(11):2706–2719. | ||
Lisiak N, Toton E, Rybczynska M. [Autophagy, new perspectives in anticancer therapy]. Postepy Hig Med Dosw (Online). 2014;68(0): 925–935. Polish. | ||
Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10(3):143–153. | ||
Zielinski RR, Eigl BJ, Chi KN. Targeting the apoptosis pathway in prostate cancer. Cancer J. 2013;19(1):79–89. | ||
Sehdev V, Katsha A, Ecsedy J, Zaika A, Belkhiri A, El-Rifai W. The combination of alisertib, an investigational Aurora kinase A inhibitor, and docetaxel promotes cell death and reduces tumor growth in preclinical cell models of upper gastrointestinal adenocarcinomas. Cancer. 2013;119(4):904–914. | ||
Brewer Savannah KJ, Demicco EG, Lusby K, et al. Dual targeting of mTOR and aurora-A kinase for the treatment of uterine leiomyosarcoma. Clin Cancer Res. 2012;18(17):4633–4645. | ||
Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19(1):87–95. | ||
Shackelford RE, Mayhall K, Maxwell NM, Kandil E, Coppola D. Nicotinamide phosphoribosyltransferase in malignancy: a review. Genes Cancer. 2013;4(11–12):447–456. | ||
Preyat N, Leo O. Sirtuin deacylases: a molecular link between metabolism and immunity. J Leukoc Biol. 2013;93(5):669–680. | ||
Skokowa J, Lan D, Thakur BK, et al. NAMPT is essential for the G-CSF-induced myeloid differentiation via a NAD(+)-sirtuin-1-dependent pathway. Nat Med. 2009;15(2):151–158. | ||
Tania M, Khan MA, Fu J. Epithelial to mesenchymal transition inducing transcription factors and metastatic cancer. Tumour Biol. 2014; 35(8):7335–7342. | ||
Shin K, Margolis B. ZOning out tight junctions. Cell. 2006;126(4): 647–649. | ||
Yuan H, Su L, Chen WY. The emerging and diverse roles of sirtuins in cancer: a clinical perspective. Onco Targets Ther. 2013;6:1399–1416. | ||
Simic P, Williams EO, Bell EL, Gong JJ, Bonkowski M, Guarente L. SIRT1 suppresses the epithelial-to-mesenchymal transition in cancer metastasis and organ fibrosis. Cell Rep. 2013;3(4):1175–1186. | ||
Nowell M, Evans L, Williams A. PBEF/NAMPT/visfatin: a promising drug target for treating rheumatoid arthritis? Future Med Chem. 2012;4(6):751–769. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.