Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Aldehyde dehydrogenase-2 protects against myocardial infarction-related cardiac fibrosis through modulation of the Wnt/β-catenin signaling pathway
Authors Zhao X, Hua Y, Chen H, Yang H, Zhang T, Huang G, Fan H, Tan Z, Huang X, Liu B, Zhou Y
Received 9 May 2015
Accepted for publication 3 June 2015
Published 11 September 2015 Volume 2015:11 Pages 1371—1381
DOI https://doi.org/10.2147/TCRM.S88297
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor De Yun Wang
Xinjun Zhao,1,2,* Yue Hua,1,2,* Hongmei Chen,1,2,* Haiyu Yang,3,* Tao Zhang,1,2,* Guiqiong Huang,4,* Huijie Fan,1,2 Zhangbin Tan,1,2 Xiaofang Huang,1,2 Bin Liu,5 Yingchun Zhou1,2
1The Key Laboratory of Molecular Biology, State Administration of Traditional Chinese Medicine, School of Traditional Chinese Medicine, Southern Medical University, Guangdong, Guangzhou, People’s Republic of China; 2Nanfang Hospital, Southern Medical University, Guangdong, Guangzhou, People’s Republic of China; 3Jiangmen Wuyi Traditional Chinese Medicine Hospital, Guangdong, Jiangmen, People’s Republic of China; 4Huizhou Hospital of Traditional Chinese Medicine, Huizhou, People’s Republic of China; 5The Second Affiliated Hospital of Guangzhou Medical University, Guangdong, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Background: Aldehyde dehydrogenase-2 (ALDH2) has a protective effect on ischemic heart disease. Here, we examined the protective effects of ALDH2 on cardiac fibrosis through modulation of the Wnt/ß-catenin signaling pathway in a rat model of myocardial infarction (MI).
Methods: Wistar rats were divided into the sham (control), MI (model), and ALDH2 activator (Alda-1) groups. After 10 days of treatment, the left ventricular (LV) remodeling parameters of each animal were evaluated by echocardiography. Myocardial fibrosis was evaluated by Masson’s trichrome staining and Sirius Red staining. Expression levels of collagen types I and III and β-smooth muscle actin (α-SMA) were examined. Finally, the expression and activity of ALDH2 and the levels of several Wnt-related proteins and genes, such as phospho-glycogen synthase kinase (GSK)-3β, GSK-3β, β-catenin, Wnt-1, WNT1-inducible signaling-pathway protein 1, and tumor necrosis factor (TNF)-α, were also analyzed.
Results: After MI, the heart weight/body weight ratio, LV dimension at end diastole, and LV dimension at end systole were decreased, while the LV ejection fraction and LV fractional shortening were increased in the Alda-1 group. Myocardial fibrosis was also reduced in the Alda-1 group, accompanied by decreased expression collagen types I and III and α-SMA. β-Catenin, phosphorylated GSK-3β, and Wnt-1 levels were significantly increased in the model group. Interestingly, this alteration was partly reversed by Alda-1 treatment. Immunohistochemical staining showed that numerous WNT1-inducible signaling-pathway protein 1 (WISP-1)- and TNF-α-positive cells were found in the model group. However, few WISP-1- and TNF-α-positive cells were detected in the Alda-1 group.
Conclusion: The reduction of cardiac fibrosis and the down-regulation of β-catenin, phosphorylated GSK-3β, Wnt-1, and WISP-1 may be mediated by increased ALDH2 activity, leading to reduction of MI-related cardiac fibrosis.
Keywords: myocardial infarction, aldehyde dehydrogenase, cardiac fibrosis, Wnt signaling
Introduction
Cardiovascular disease is the leading cause of mortality and morbidity in developed countries.1,2 All over the world, the incidence of death from cardiovascular disease has risen by one-third between 1990 and 2010.3 Furthermore, according to the report of WHO, the amount of cardiovascular disease has been achieved to 17 million in 2008.4
The Wnt/β-catenin signaling pathway is a conserved pathway that plays a critical role in most physiological and pathological processes, including tissue and organ formation, maintenance of intracellular homeostasis,5 wound repair and regeneration, and disease progression.6 The Wnt/β-catenin signaling pathway is inactive under physiological conditions and can be activated after stimulation by a variety of endogenous and exogenous pathogens. Recently, studies have shown that the occurrence and progression of kidney, lung, liver, heart, and skin fibrosis are closely related to abnormal activation of the Wnt signaling pathway.7,8
After myocardial infarction (MI), the canonical Wnt signaling pathway is activated, and epicardial Wnt signaling is markedly enhanced, inducing the epithelial–mesenchymal transition into fibroblasts. Wnt-1 is upregulated in cardiac fibroblasts after MI, suggesting increased proliferation and extracellular matrix secretion.9 Appropriate increases in Wnt signaling can promote the repair of necrotic myocardial tissue after MI, reduce MI size, and improve ventricular function.10 However, continuous and excessive activation of the Wnt signaling pathway can lead to serious myocardial fibrosis, impairing myocardial function. Studies have shown that inhibition of the Wnt signaling pathway may prevent collagen deposition and improve cardiac function post-MI.11
Aldehyde dehydrogenase-2 (ALDH2) is an NAD+-dependent enzyme that primarily resides in the mitochondria,12 where it participates in aldehyde metabolism in the heart during disease progression.13 For this reason, ALDH2 is thought to play a preventive role in many cardiovascular diseases, including ischemia-reperfusion injury,14 coronary artery disease,15 and cardiac dysfunction.16 The recent evidence indicate that ALDH2 can reduce myocardial fibrosis. Indeed, prolonged ALDH2 activation by Alda-1 affects cardiac fibrosis by decreasing the extent of collagen type I deposition and the collagen type I/III ratio.17 However, the specific mechanisms mediating this anti-fibrotic effect are not clear.
ALDH2 has been shown to antagonize chronic alcohol intake-induced myocardial remodeling through a mechanism that is associated with phosphorylation of glycogen synthase kinase (GSK)-3β (pGSK-3β). Moreover, ALDH2 can reverse alcohol-induced abnormal upregulation of pGSK-3β protein in FVB mice.18 This suggests that ALDH2 may prevent myocardial fibrosis by inhibiting the expression and/or activation of Wnt-related proteins.
In the present study, an MI model was employed to investigate the effects of ALDH2 on myocardial fibrosis by regulating the Wnt/β-catenin signaling pathway. To this end, the levels of collagen types I and III, β-catenin, Wnt-1, GSK-3β, pGSK-3β, WNT1-inducible signaling-pathway protein 1 (WISP-1), and tumor necrosis factor (TNF)-α were examined, and morphometric studies of samples stained with Sirius Red and Masson’s trichrome were carried out in the hearts of rats post-MI, with or without simultaneous administration of ALDH2. The activity of ALDH2 in myocardial tissues was also analyzed. The results provide a theoretical basis for further studies of the activity of ALDH2 in preventing myocardial fibrosis.
Methods
Animals and study design
Male Wistar rats, weighing 200–250 g each, were housed in a specific pathogen-free environment (Certificate no 4402102052). Rats had free access to water and rodent chow and were maintained in rooms at a constant temperature of 20°C–22°C with a 12-hour light–dark cycle. All procedures involving laboratory animals were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of Southern Medical University. Before conducting the experiment, rats were allowed to adapt to the new environment for 1 week.
MI model and experimental design
MI was induced by ligation of the left anterior descending coronary artery, as previously described.19 Rats were anesthetized by isoflurane inhalation, intubated, and placed on a rodent ventilator (SAR-830; IITC, Woodland Hills, CA, USA). Positive pressure artificial respiration was performed, and the body temperature was maintained at 37°C. After performing a left thoracotomy, the left large marginal coronary arteries located approximately 2 mm below the left atrium were ligated with 5-0 Ethicon silk sutures. The development of a pale LV free wall and dilated left atrium indicated successful ligation. Left thoracotomy of equal duration was also performed in the sham group, but the left anterior descending coronary artery ligation was omitted.
In vivo treatment with Alda-1, an activator of ALDH2
Rats were randomly assigned into three groups of 14 animals each: 1) the sham group, in which animals were not treated, 2) the MI model group, and 3) the ALDH2 activator group, in which animals were treated with Alda-1, a small molecule allosteric activator of ALDH2.20,21 Rats in the ALDH2 group were treated with Alda-1 (16 mg/kg, intraperitoneally [ip], once per day) for 10 days. Rats in the sham and MI groups were intraperitoneally injected with the solvent alone (50% polyethylene glycol and 50% dimethyl sulfoxide by volume) every day for 10 days.
Histological analyses
Ten days after MI surgery, all rats were anesthetized with sodium pentobarbital (100 mg/kg ip), and the intact heart was removed and weighed for each rat. The heart weight/body weight (HW/BW) ratio was calculated for each animal. The left ventricle was then dissected and cut perpendicular to the apex-to-base axis to yield two pieces. One piece was frozen in liquid nitrogen and stored at −80°C for subsequent mRNA/protein expression analysis. The other piece was used for histomorphological studies. The hearts were fixed in 4% paraformaldehyde and embedded in paraffin. Serial sections (5 μm) were routinely stained using Masson’s trichrome and Sirius Red staining. Sections were examined under a light microscope (400× magnification) and photographed for morphological analysis.
Transthoracic echocardiography
The morphology and function of the left ventricle were evaluated noninvasively using transthoracic echocardiography, performed in the lateral decubitus position with a commercially available echocardiograph (Philips iE33 Ultrasound System with a 15-MHz transducer; Philips, Bothell, WA, USA). Echocardiography was performed in animals anesthetized with sodium pentobarbital (80 mg/kg, ip; Fluka, Sigma-Aldrich, Inc., St Louis, MO, USA). The procedure was performed in the time-motion mode in a parasternal short-axis view, and perfusion was measured in pulsed Doppler mode. The left ventricular (LV) end-systolic and end-diastolic diameters were also measured. The LV shortening fraction was calculated as follows: ([end-diastolic diameter − end-systolic diameter]/end-diastolic diameter × 100). The end-systolic wall stress was measured as previously described.22 To ensure accurate measurements, intra-animal and intra-observer variabilities were calculated in ten animals before experimentation.
ALDH2 enzymatic activity
ALDH2 activity was assayed using a commercial kit (GenMed Scientifics Inc., Wilmington, DE, USA), according to the manufacturer’s instructions. ALDH2 activity was measured by monitoring the production of NADH from NAD+ at 340 nm in a microplate spectrophotometer (Thermo Fisher Scientific, Vantaa, Finland). The assay mixture (0.25 mL) contained 195 μL GENMED buffer solution, 25 μL GENMED reagent solution, 25 μL GENMED substrate solution, and 200 μg protein. The enzymatic reaction was initiated by gently shaking the samples. ALDH2 activity was expressed as μmol NADH/min/mg protein. The protein concentrations of the lysates were measured using the Bradford method (ProMab Biotechnologies, Inc.; Richmond, CA, USA).
Western blotting analysis
Aliquots of heart tissue (50 mg) were homogenized in liquid nitrogen and extracted in radioimmunoprecipitation assay buffer following the manufacturer’s instructions, and protein concentrations were quantified with a bicinchoninic acid (BCA) protein quantity assay kit. Equal amounts of protein were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) at 4°C, and the protein was then transferred to microporous polyvinylidene fluoride membranes in running buffer with 20% methanol and blocked in 5% nonfat milk in Tris-buffered saline (100 mM NaCl, 50 mM Tris, 0.1% Tween-20, pH 7.5). Membranes were incubated overnight using primary antibodies (anti-ALDH2 [ab166697; Abcam, Cambridge, MA, USA; 1:3,000 dilution], anti-GSK-3β [G6414; Sigma; 1:500 dilution], anti-pGSK-3β [Ser9; G6542; Sigma; 1:1,000 dilution], and anti-β-catenin [ab32572; Abcam; 1:5,000 dilution]). Immuno-blot bands were detected with a Bio-Rad calibrated densitometer. GAPDH was used as the loading control.
Immunohistochemistry
Immunohistochemical staining for WISP-1 and TNF-α was performed using the standard streptavidin peroxidase method with rabbit polyclonal anti-WISP-1 antibodies (sc-25441, H-55; Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:50 dilution) and rabbit polyclonal anti-TNF-α antibodies (Pierce-Endogen, Rockford, IL, USA; 1:200 dilution). Nuclear counterstaining was performed using hematoxylin. Six randomly selected fields from each section were examined at a magnification of 400× and analyzed using NIS-Elements. The positive content (PC) was calculated using the following formula: PC = mean optical density × positive area.23
RNA extraction and real-time polymerase chain reaction
Total RNA was extracted and reverse transcribed from frozen left ventricles as previously described.24 cDNA was diluted in sterile water and used as the template for amplification by polymerase chain reaction (PCR). Primers were synthesized by Sangon Biotech Co. Ltd. (Shanghai, People’s Republic of China). Forward and reverse primers are shown in Table 1. Each specific gene product was amplified by real-time PCR using SYBR Green reactions and an Applied Biosystems 7500 Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA). Cycling conditions were as follows: pre-incubation at 95°C for 15 minutes, denaturation at 95°C for 10 second, and annealing at 58°C for 31 seconds, with 40 cycles. Amplification data were collected by the sequence detector and analyzed with sequence detection software. In order to confirm amplification specificity, the PCR products from each primer pair were subjected to melting curve analysis. The quantitative fold changes in gene expression were calculated using the ΔΔCt method after normalizing to GAPDH.25
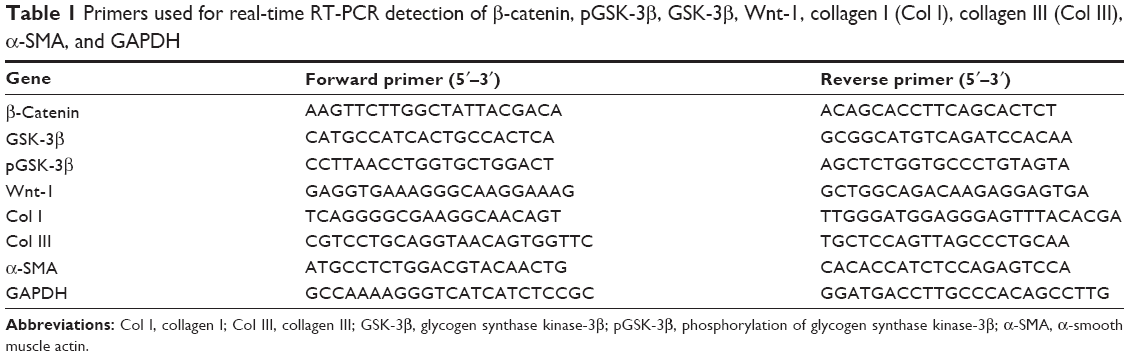 | Table 1 Primers used for real-time RT-PCR detection of β-catenin, pGSK-3β, GSK-3β, Wnt-1, collagen I (Col I), collagen III (Col III), α-SMA, and GAPDH |
Statistical analysis
Each experiment was repeated at least three times. Data are shown as the means ± standard deviations. All data were analyzed using the SPSS statistical package (version 22.0; IBM Corporation, Armonk, NY, USA). Mean values were compared through one-way analysis of variance, and multiple comparisons were performed. Data were also tested through homogeneity tests for variance. If the variances were not homogenous, mean values were compared using Welch’s test. The differences between two groups were analyzed by Games–Howell tests. Differences with P-values of less than 0.05 were considered statistically significant.
Results
ALDH2 activation improved cardiac function and reversed pathological ventricular remodeling after MI in rats
To investigate the protective effects of ALDH2 against ventricular remodeling, we assessed the echocardiographic parameters and HW/BW ratios in rats (Figure 1A, B). Rats in the MI model group exhibited signs of heart failure, as demonstrated by LV dysfunction and pathological cardiac remodeling (Figure 1C–F). These rats also displayed decreased left ventricular ejection fraction (LVEF; P<0.01) and left ventricular fractional shortening (LVFS; P<0.01), as well as increased left ventricular dimension at end diastole (LVEDD; P<0.01) and left ventricular dimension at end systole (LVESD; P<0.01) compared with control animals under basal conditions. As shown in Figure 1C–F, treatment with Alda-1 significantly improved cardiac function compared with that in the MI model group. Moreover, treatment with Alda-1 significantly decreased the HW/BW ratio (P<0.01) compared with that in the MI model group (Figure 1B). This result was consistent with the results of echocardiographic parameters. Figure 1G showed the two-dimensional-guided echocardiographic images of the left ventricle. The results indicated that ALDH2 activation could improve the M-mode echocardiographic images, even similar to the control group.
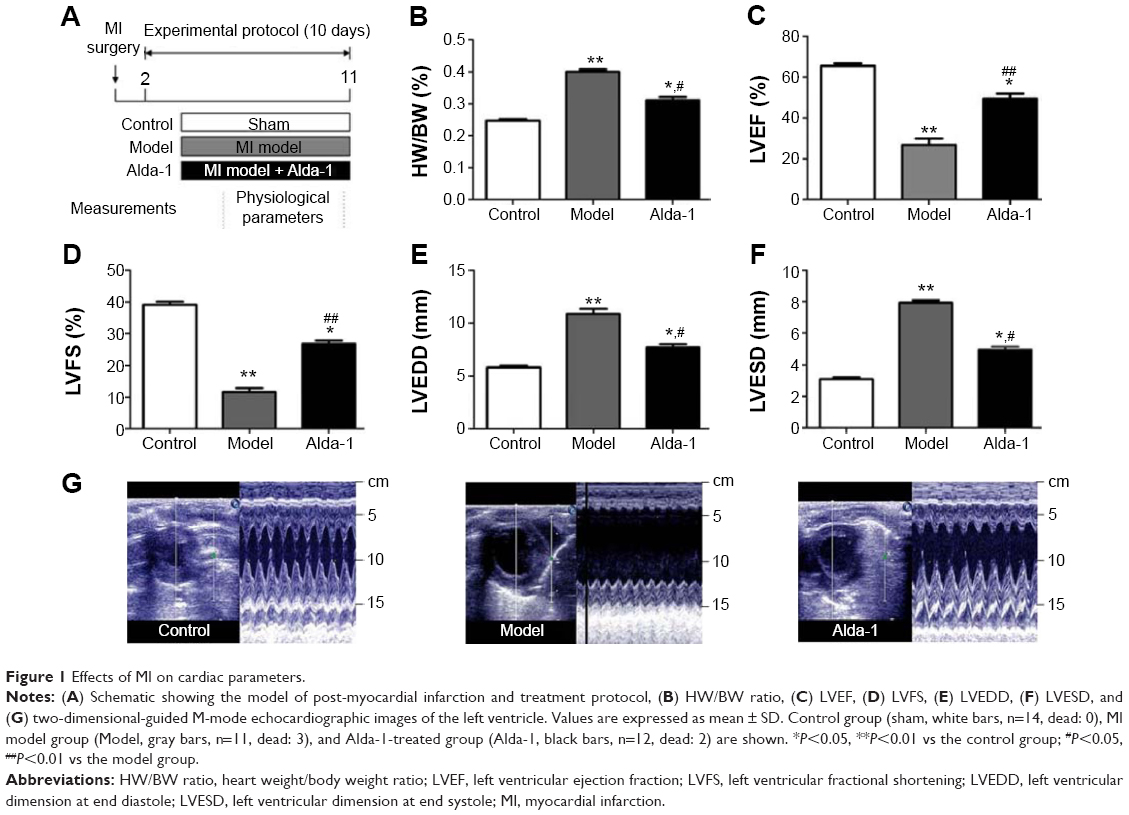 | Figure 1 Effects of MI on cardiac parameters. |
Effects of ALDH2 on cardiac fibrosis
Figure 2 shows representative images of collagen deposition in the heart of one animal from each group of rats. Masson’s trichrome staining revealed an irregular distribution of interstitial cardiac fibrosis (Figure 2B). The extent of fibrosis and the collagen type I/III ratio were evaluated by staining with Sirius Red (Figure 2C). The interstitial space occupied by the extracellular matrix was increased by threefold in the hearts of MI model rats (Figure 2D). However, ALDH2 activation by Alda-1 affected cardiac fibrosis by decreasing the extent of collagen deposition and the collagen type I/III ratio (Figure 2D and E).
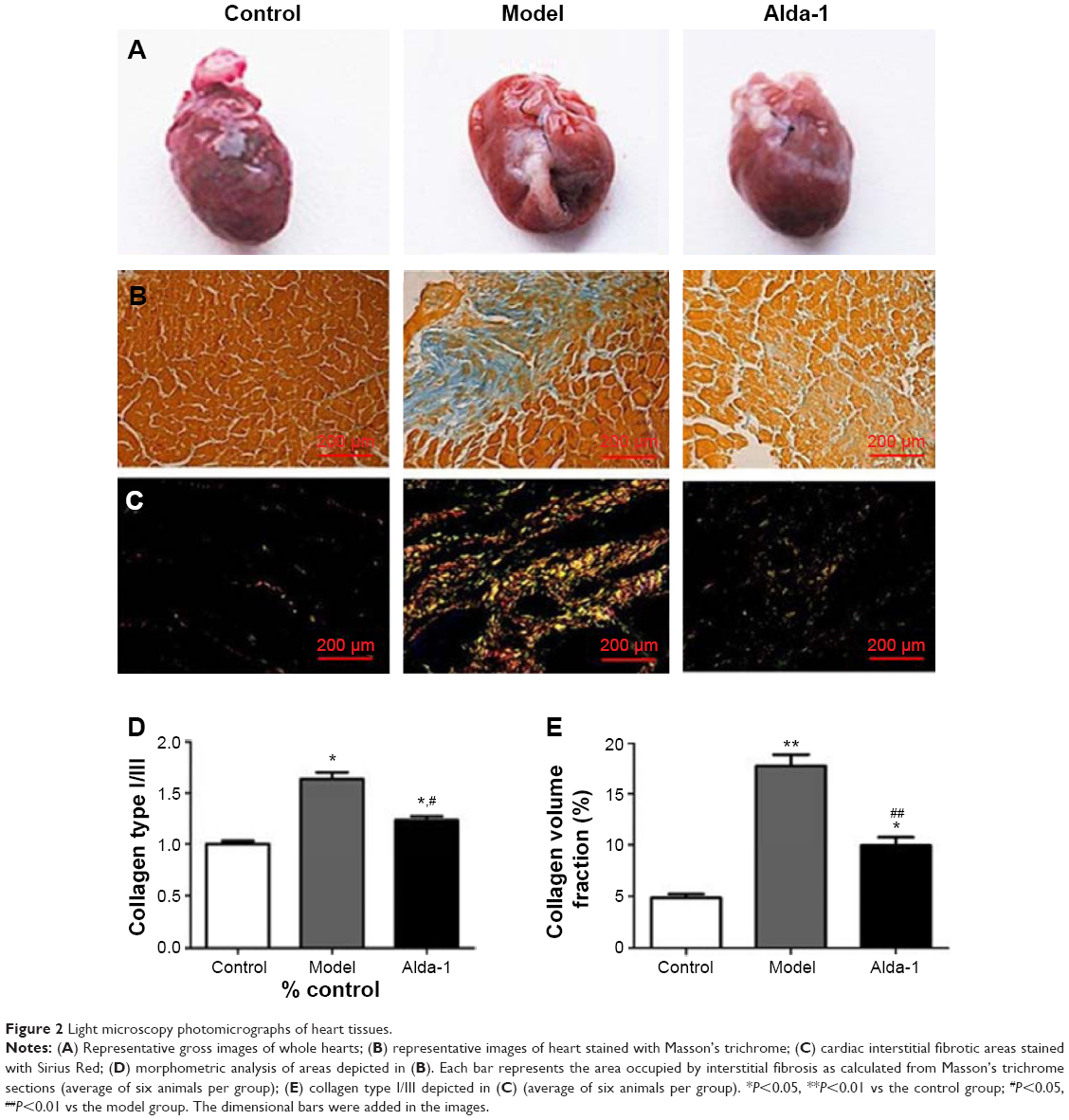 | Figure 2 Light microscopy photomicrographs of heart tissues. |
Activity and expression of ALDH2
As shown in Figure 3A, ALDH2 activity was decreased in the MI model group. After treatment with Alda-1 for 10 days, ALDH2 activity was significantly increased (P<0.01). Western blotting showed that the expression of ALDH2 was significantly decreased in the MI model group (Figure 3B; P<0.01). Moreover, Alda-1 treatment had no effect on ALDH2 expression compared with the MI model group (P>0.05).
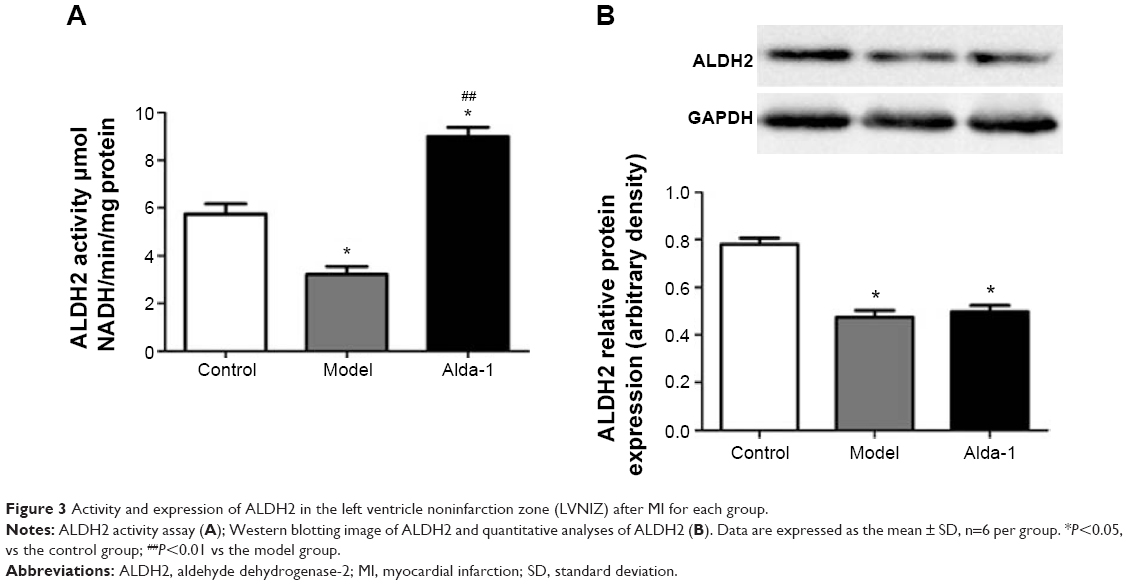 | Figure 3 Activity and expression of ALDH2 in the left ventricle noninfarction zone (LVNIZ) after MI for each group. |
Levels of pGSK-3β, GSK-3β, β-catenin, Wnt-1, collagen types I and III, and α-SMA
To elucidate the potential mechanism(s) involved in ALDH2-dependent cardiac protection against Wnt/β-catenin-regulated cardiac fibrosis, we further examined the levels of β-catenin, Wnt-1, total and phosphorylated GSK-3β by Western blotting and real-time PCR. As shown in Figure 4D–F, mRNA expression levels of β-catenin, phosphorylated GSK-3β, and Wnt-1 were significantly increased in the MI model group (two- to fourfold), compared with that in the sham group (P<0.01). Interestingly, these increases were reversed in rats treated simultaneously with Alda-1. Western blot analysis confirmed that β-catenin and phosphorylated GSK-3β protein levels were increased in the MI model group, and this alteration was partly reversed by Alda-1 treatment (Figure 4A–C).
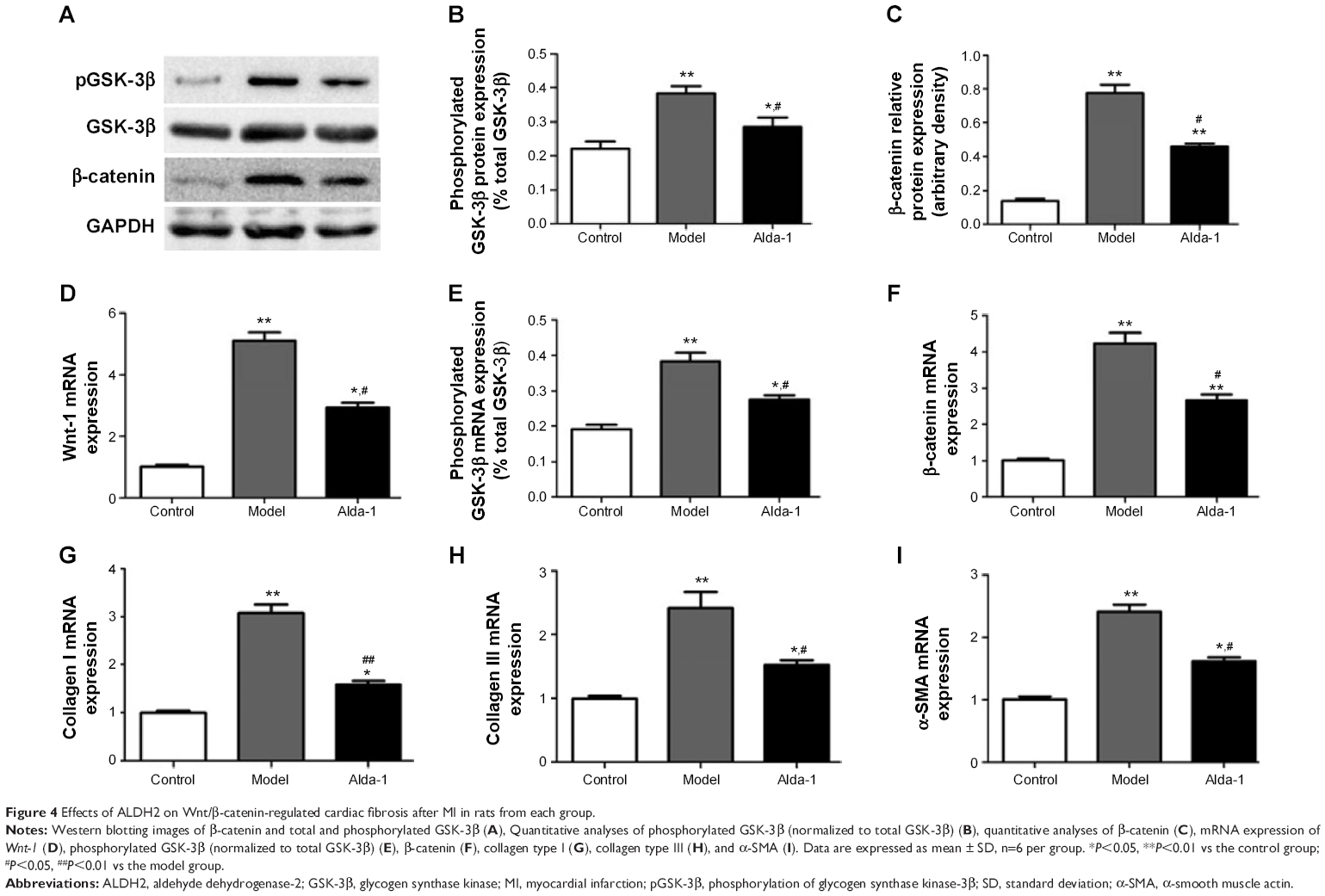 | Figure 4 Effects of ALDH2 on Wnt/β-catenin-regulated cardiac fibrosis after MI in rats from each group. |
As shown in Figure 4E–I, compared with the sham group, expression levels of transcripts encoding collagens types I and III and α-smooth muscle actin (α-SMA) were significantly increased in the MI model group (P<0.01). The levels of these targets were then markedly decreased following treatment with Alda-1 (P<0.01). These results were consistent with those of Masson’s trichrome and Sirius Red staining.
Expression of WISP-1 and TNF-α
Next, we measured the WISP-1 and TNF-α protein levels in the heart (noninfarcted area) at 10 days after MI. Few WISP-1-positive cells were detected in the sham group, whereas numerous WISP-1-positive cells were found in the MI model group (Figure 5A and C); this difference was statistically significant (P<0.01). In the Alda-1 group, the number of 4-hydroxinonenal-positive cells decreased significantly compared with that in the MI model group (P<0.01). Moreover, TNF-α levels were significantly increased in the MI model group (P<0.01) compared with that in the sham group (Figure 5B and D). Treatment with Alda-1 markedly lowered TNF-α levels compared with that in the MI model group (P<0.01). These results were consistent with the results of WISP-1 analysis.
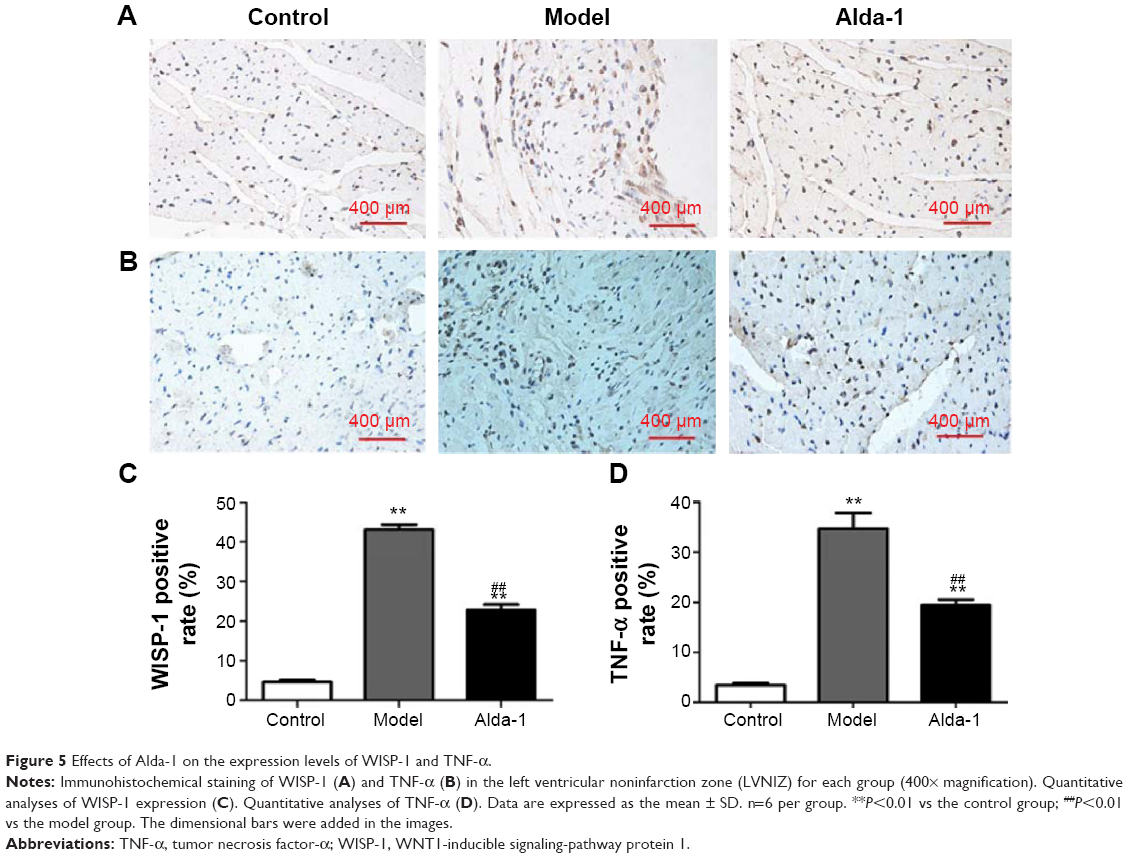 | Figure 5 Effects of Alda-1 on the expression levels of WISP-1 and TNF-α. |
Discussion
MI can lead to myocardial cell necrosis, associated with the loss of necrotic myocardium, myocardial fibroblasts infiltration into the infarction area and noninfarction area, and subsequent transformation of these cells into myofibroblasts.26 This process is part of myocardial remodeling after MI. LV remodeling can occur for weeks or more.27 Remodeling processes are essential for normal healing, but excessive remodeling is associated with abnormal changes in cardiac myocyte size and fibrosis, which will result in further loss of myocardial function. Therefore, decreasing excessive ventricular remodeling should decrease myocardial dysfunction.28
In the present study, our data showed that the HW/BW ratio, LVEDD, and LVESD in the MI model group were greater than those in the control group. In contrast, the LVEF and LVFS were smaller in the MI model and control groups. This basic information describes functional changes in rats at 10 days after MI. Compared with the MI model group, rats in the Alda-1 group exhibited lower HW/BW ratio, LVEDD, and LVESD, but higher LVEF and LVFS. From our results, we found that ALDH2 activation by Alda-1 affected cardiac fibrosis by decreasing the extent of collagen deposition and the collagen type I/III ratio, as shown by Masson’s trichrome and Sirius Red staining. These results are consistent with the results of a previous study.17
Under general physiological conditions, the Wnt/β-catenin signaling pathway is inactive, and β-catenin is phosphorylated by the Axin/APC/CK1/GSK-3β complex; following phosphorylation, β-catenin is identified and degraded by the ubiquitin-mediated proteasomal degradation pathway, maintaining low levels of expression in the cytoplasm. Under stress conditions, the Frizzled receptor binds to LRP5 or LRP6 and forms a coreceptor complex, which actives disheveled proteins, leading to the dissociation of GSK-3β from the degradation complex. Subsequently, GSK-3β is inactivated by phosphorylation of Ser9 by oxidative stress, and β-catenin degradation is prevented. β-Catenin then continuously accumulates in the cytoplasm and nucleus. In the nucleus, β-catenin combination with T-cell factor/lymphoid enhancer factor. Downstream target genes are then activated, including WISP-1.29,30
Laeremans et al reported that the Wnt/β-catenin signaling pathway was involved in the development of heart failure after MI.31 Previous research has shown that the Wnt antagonist secreted Frizzled-related protein 2 has strong antifibrotic effects.32 Moreover, secreted Frizzled-related protein 2 can inhibit collagen deposition after MI and enhance cardiac function.33 In this study, we found that the levels of β-catenin, phosphorylated GSK-3β, and Wnt-1 were significantly increased in the MI model group at 10 days after MI, further verifying that the Wnt/β-catenin signaling pathway was activated post-MI. Interestingly, Alda-1 reversed the elevations in these targets.
WISP-1 is a member of the CCN (CYR61/CTGF/Nov) family of growth factors and a target of the Wnt/β-catenin signaling pathway, showing regulation by β-catenin.34 WISP-1 mediates TNF-α-induced cardiac fibroblast proliferation and collagen synthesis and release. WISP-1 also promotes fibrosis. Fibroblasts are the essential cell type responsible for the formation of fibrous tissue in MI areas.35 A recent study showed that WISP-1 is significantly upregulated in myocardial cells of noninfarcted areas, suggesting that increased WISP-1 signaling may lead to fibroblast proliferation and fibrosis.36 In this study, WISP-1 and TNF-α protein levels were significantly upregulated in the noninfarcted area of the heart at 10 days after MI. Administration of Alda-1 was able to reduce the expression of WISP-1 and TNF-α and to prevent the myocardial fibrosis associated with MI.
ALDH2 plays an important role in regulating cardioprotection against acute ischemic injury37 and pathological ventricular remodeling in animals with heart failure.17 The cardiovascular protective effects of ALDH2 are achieved mainly through its detoxification of mitochondrial reactive aldehydes, such as 4-hydroxinonenal and methane dicarboxylic aldehyde. It is believed that acetaldehyde is highly toxic, mutagenic, and carcinogenic, inducing apoptosis and promoting fibrogenesis through direct cytotoxicity or the release of inflammatory cytokines.38 In this study, treatment of MI rats with Alda-1 decreased the expression of several cardiac genes, including collagen types I and III and α-SMA. In addition, Alda-1 decreased the levels of β-catenin, phosphorylated GSK-3β, and Wnt-1 in MI rats. These results indicated that the reduction in cardiac fibrosis achieved with Alda-1 in MI rats may be, at least in part, associated with downregulation of Wnt/β-catenin signaling.
Though the present study provided some sufficient data for the conclusion, there were also some limitations. Except for the indexes examined above, the evaluation of cardiac microcirculation and angiogenesis is also the important feature of cardiac pathophysiology.39 Therefore, in the following study, we would examine the microcirculation and angiogenesis in the MI model for the cardiac pathophysiology study. Also, the effects of ALDH2 on the other heart diseases have not been investigated. To apply the ALDH2 in the drug design or clinical therapy, the ALDH2 should be studied with extensive cardiac diseases.
In this study, Alda-1 exhibited cardiovascular protective effects. These protective effects may involve different mechanisms, including reduction of aldehyde load, elevation of mitochondrial respiratory control ratios, decreasing the mitochondrial Ca2+-induced permeability transition and cytochrome c release,17 and differential regulation of autophagy through AMP-activated protein kinase and Akt/mammalian target of rapamycin signaling.40 Although additional studies are required to determine the specific relationships between Wnt/β-catenin signaling and Alda-1, identification of these relationships will facilitate elucidation of the mechanisms involved in these processes.
In clinical, the cardiac fibrosis is the common symptom for the MI. Therefore, inhibiting the cardiac fibrosis in MI may play an important role in the therapy. The present study discovered the specific pathway of the ALDH2-blocked MI-related cardiac fibrosis, which could provide the clues for the drug design and development for MI. Also, the findings of our study may provide important insights into the pharmacological basis of clinical application of Alda-1 in preventing myocardial fibrosis after MI.
Conclusion
The reduction of cardiac fibrosis and the down-regulation of β-catenin, phosphorylated GSK-3β, Wnt-1, and WISP-1 may be mediated by increased ALDH2 activity, leading to reduction of MI-related cardiac fibrosis.
Acknowledgments
The present study was granted by National Natural Science Foundation of People’s Republic of China (Nos 81302892, 81173459, 81373575, and 81202841), Guangdong Natural Science Foundation (Nos S2013040016226 and S2013010014777), Specialized Research Fund for the Doctoral Program of Higher Education (No 20124433110019), and People’s Republic of China Postdoctoral Science Foundation (No 2013M530363).
Disclosure
The authors report no conflicts of interest in this work.
References
Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics – 2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. | ||
WHO Regional Office for Europe. The European Health Report 2012: Charting the Way to Well-Being. Copenhagen, Denmark: WHO Regional Office for Europe; 2012. | ||
Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death of 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. | ||
World Health Organization. Cardiovascular Diseases (CVDS). Geneva: World Health Organization. Available from: http://www.who.int/mediacentre/factsheets/fs317/en/. Accessed August 7, 2015. | ||
MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;1:9–26. | ||
Gurley KA, Rink JC, Sanchez AA. Beta-catenin defines head versus tail identity during planarian regeneration and homeostasis. Science. 2008;5861:323–327. | ||
Surendran K, McCaul SP, Simon TC. A role for Wnt-4 in renal fibrosis. Am J Physiol Renal Physiol. 2002;3:F431–F441. | ||
Tzouvelekis A, Bonella F, Spagnolo P. Update on therapeutic management of idiopathic pulmonary fibrosis. Ther Clin Risk Manag. 2015;11:359–370. | ||
Duan J, Xu H, Ma S, et al. Cre-mediated targeted gene activation in the middle silk glands of transgenic silkworms (Bombyx mori). Transgenic Res. 2013;3:607–619. | ||
Hahn JY, Cho HJ, Bae JW, et al. Beta-catenin overexpression reduces myocardial infarct size through differential effects on cardiomyocytes and cardiac fibroblasts. J Biol Chem. 2006;41:30979–30989. | ||
Jones SE, Jomary C. Secreted Frizzled-related proteins: searching for relationships and patterns. Bioessays. 2002;9:811–820. | ||
Endo J, Sano M, Katayama T, Hishiki T, Shinmura K, Morizane S. Metabolic remodeling induced by mitochondrial aldehyde stress stimulates tolerance to oxidative stress in the heart. Circ Res. 2009;11:1118–1127. | ||
Ma H, Guo R, Yu L, Zhang Y, Ren J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur Heart J. 2011;8:1025–1038. | ||
Chen CH, Budas GR, Churchill EN, Disatnkik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;5895:1493–1495. | ||
Guo YJ, Chen L, Bai YP, et al. The ALDH2 Glu504Lys polymorphism is associated with coronary artery disease in Han Chinese: relation with endothelial ADMA levels. Atherosclerosis. 2010;2:545–550. | ||
Liao J, Sun A, Xie Y, et al. Aldehyde dehydrogenase-2 deficiency aggravates cardiac dysfunction elicited by endoplasmic reticulum stress induction. Mol Med. 2012;18:785–793. | ||
Gomes KM, Campos JC, Bechara LR, et al. Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodeling. Cardiovasc Res. 2014;4:498–508. | ||
Doser TA, Turdi S, Thomas DP, Epstein PN, Li SY, Ren J. Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation. 2009;14:1941–1949. | ||
Johns TN, Olson BJ. Experimental myocardial infarction. I. A method of coronary occlusion in small animals. Ann Surg. 1954;5:675–682. | ||
Perez-Miller S, Younus H, Vanam R, Chen CH, Mochly-Rosen D, Hurley TD. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat Struct Mol Biol. 2010;2:159–164. | ||
Sun L, Ferreira JC, Mochly-Rosen D. ALDH2 activator inhibits increased myocardial infarction injury by nitroglycerin tolerance. Sci Transl Med. 2011;107:107r–111r. | ||
Boissiere J, Eder V, Machet MC, Courteix D, Bonnet P. Moderate exercise training does not worsen left ventricle remodeling and function in untreated severe hypertensive rats. J Appl Physiol. 1985;104:321–327. | ||
Cai HB, Sun XG, Liu ZF, et al. Effects of dahuangzhechong pills on cytokines and mitogen activated protein kinase activation in rats with hepatic fibrosis. J Ethnopharmacol. 2010;1:157–164. | ||
Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal Biochem. 1987;1:156–159. | ||
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods. 2001;4:402–408. | ||
Benito B, Gay-Jordi G, Serrano-Mollar A, et al. Cardiac arrhythmogenic remodeling in a rat model of long-term intensive exercise training. Circulation. 2011;1:13–22. | ||
Daskalopoulos EP, Janssen BJ, Blankesteijn WM. Myofibroblasts in the infarct area: concepts and challenges. Microsc Microanal. 2012;1:35–49. | ||
Cohn JN. Structural basis for heart failure. Ventricular remodeling and its pharmacological inhibition. Circulation. 1995;10:2504–2507. | ||
Kuhl M, Sheldahl LC, Malbon CC, Moon RT. Ca(2+)/calmodulin-dependent protein kinase II is stimulated by Wnt and frizzled homologs and promotes ventral cell fates in Xenopus. J Biol Chem. 2000;17:12701–12711. | ||
MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;1:9–26. | ||
Laeremans H, Hackeng TM, van Zandvoort MA, et al. Blocking of frizzled signaling with a homologous peptide fragment of wnt3a/wnt5a reduces infarct expansion and prevents the development of heart failure after myocardial infarction. Circulation. 2011;15:1626–1635. | ||
Jones SE, Jomary C. Secreted frizzled-related proteins: searching for relationships and patterns. Bioessays. 2002;9:811–820. | ||
He W, Zhang L, Ni A, et al. Exogenously administered secreted frizzled related protein 2 (Sfrp2) reduces fibrosis and improves cardiac function in a rat model of myocardial infarction. Proc Natl Acad Sci U S A. 2010;49:21110–21115. | ||
Shi-Wen X, Leask A, Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008;2:133–144. | ||
Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol. 1999;1:C1–C9. | ||
Colston JT, de la Rosa SD, Koehler M, et al. Wnt-induced secreted protein-1 is a prohypertrophic and profibrotic growth factor. Am J Physiol Heart Circ Physiol. 2007;293:H1839–H1846. | ||
Chen CH, Sun L, Mochly-Rosen D. Mitochondrial aldehyde dehydrogenase and cardiac diseases. Cardiovasc Res. 2010;1:51–57. | ||
Mello T, Ceni E, Surrenti C, Galli A. Alcohol induced hepatic fibrosis: role of acetaldehyde. Mol Aspects Med. 2008;1–2:17–21. | ||
Santulli G, Cipolletta E, Sorriento D, et al. CaMK4 gene deletion induces hypertension. J Am Heart Assoc. 2012;1:e001081. | ||
Ma H, Guo R, Yu L, Zhang Y, Ren J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur Heart J. 2011;8:1025–1038. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.