Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Airway gene expression of IL-1 pathway mediators predicts exacerbation risk in obstructive airway disease
Authors Baines KJ ![]() , Fu JJ, McDonald VM
, Fu JJ, McDonald VM ![]() , Gibson PG
, Gibson PG
Received 10 August 2016
Accepted for publication 9 November 2016
Published 8 February 2017 Volume 2017:12 Pages 541—550
DOI https://doi.org/10.2147/COPD.S119443
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Richard Russell
Katherine J Baines,1,2 Juan-juan Fu,3 Vanessa M McDonald,1,2 Peter G Gibson1,2
1Faculty of Health and Medicine, Priority Research Centre for Healthy Lungs, Hunter Medical Research Institute, The University of Newcastle, 2Department of Respiratory and Sleep Medicine, John Hunter Hospital, New Lambton, NSW, Australia; 3Respiratory Group, Department of Integrated Traditional Chinese and West Medicine, West China Hospital, Sichuan University, People’s Republic of China
Background: Exacerbations of asthma and COPD are a major cause of morbidity and mortality and are responsible for significant health care costs. This study further investigates interleukin (IL)-1 pathway activation and its relationship with exacerbations of asthma and COPD.
Methods: In this prospective cohort study, 95 participants with stable asthma (n=35) or COPD (n=60) were recruited and exacerbations recorded over the following 12 months. Gene expressions of IL-1 pathway biomarkers, including the IL-1 receptors (IL1R1, IL1R2, and IL1RN), and signaling molecules (IRAK2, IRAK3, and PELI1), were measured in sputum using real-time quantitative polymerase chain reaction. Mediators were compared between the frequent (≥2 exacerbations in the 12 months) and infrequent exacerbators, and the predictive relationships investigated using receiver operating characteristic curves and area under the curve (AUC) values.
Results: Of the 95 participants, 89 completed the exacerbation follow-up, where 30 participants (n=22 COPD, n=8 asthma) had two or more exacerbations. At the baseline visit, expressions of IRAK2, IRAK3, PELI1, and IL1R1 were elevated in participants with frequent exacerbations of both asthma and COPD combined and separately. In the combined population, sputum gene expression of IRAK3 (AUC=75.4%; P<0.001) was the best predictor of future frequent exacerbations, followed by IL1R1 (AUC=72.8%; P<0.001), PELI1 (AUC=71.2%; P<0.001), and IRAK2 (AUC=68.6; P=0.004). High IL-1 pathway gene expression was associated with frequent prior year exacerbations and correlated with the number and severity of exacerbations.
Conclusion: The upregulation of IL-1 pathway mediators is associated with frequent exacerbations of obstructive airway disease. Further studies should investigate these mediators as both potential diagnostic biomarkers predicting at-risk patients and novel treatment targets.
Keywords: COPD, asthma, exacerbations, interleukin-1
Introduction
Asthma and COPD are common chronic inflammatory airway diseases that are responsible for a large burden of illness worldwide. Exacerbations of asthma and COPD are defined as periods of acute deterioration of symptoms and lung function that can result in hospitalization and increased health care utilization and are a major cause of death.1,2 Exacerbations impose a substantial economic burden and result in a faster decline in lung function3,4 and poorer quality of life.5,6 Some patients experience frequent exacerbations4,7,8 that require more effective management strategies. Although some clinical factors have been identified to be associated with frequent exacerbations of asthma2,9 and COPD,10 the underlying mechanisms, including the role of innate immunity in frequent exacerbations, are not well established. If the activation of certain inflammatory pathways was predictive of exacerbations, this would permit both disease monitoring using biomarkers and novel approaches to treatment.
Inflammation in COPD and asthma has airway and systemic components.11,12 Low-grade systemic inflammation is associated with a rapid decline in lung function,13 increased mortality,14 and a higher exacerbation rate.15,16 We have recently shown that the presence of systemic inflammation, measured by elevated systemic C-reactive protein (CRP) and interleukin (IL)-6, is predictive of future exacerbations in asthma and COPD.17 Further to this, systemic inflammation was associated with elevated IL-1β expression in the airways, and this airway–systemic inflammatory axis was predictive of COPD exacerbations.17
IL-1β is a typical innate immune cytokine involved in the initiation and persistence of inflammation.18,19 IL-1β secretion is increased in stable and exacerbating COPD20,21 and asthma.19,22 We previously reported that airway IL-1β is associated with systemic inflammation in asthma.12,19,22 IL-1β gene expression23 and activation of the IL-1 signaling pathway19 in sputum are associated with airway neutrophilia and inflammasome activation24 in asthma. To extend our findings of the predictive relationship of IL-1β and future exacerbations of airway disease,17 this study assesses selected downstream molecules in the IL-1 signaling pathway. We hypothesized that the activation of the IL-1 pathway is predictive of exacerbations of asthma and COPD, and that these mediators could be used as a biomarker to assess future risk and identify novel therapeutic targets. We, therefore, measured gene expression of selected IL-1 pathway members in our previously conducted prospective cohort study of older people with asthma and COPD17 and conducted a statistical analysis to explore the predictive nature of IL-1 pathway activation and future exacerbations.
Methods
Study population
Patients (n=152) with doctor-diagnosed asthma (n=63) or COPD (n=89) were recruited from research registers, as previously reported.17 Asthma was confirmed by current (past 12 months) episodic respiratory symptoms, doctor’s diagnosis, and prior demonstrated evidence of airway hyperresponsiveness to hypertonic saline and/or increased bronchodilator response, according to American Thoracic Society guidelines.25 COPD was diagnosed according to the Global Initiative for COPD criteria.26 Participants gave written informed consent and the Hunter New England Local Health District Human Research Ethics Committee approved this study. Exclusion criteria included primary respiratory disorders other than asthma and COPD, or current malignancy. An exacerbation in the past month was a cause to delay visits until recovery (4 weeks of stability). Ex-smokers were defined as those having stopped smoking for 6 months or more prior to their participation in this study.
Study design and protocol
This prospective cohort study was conducted between October 2010 and December 2012, and was previously reported.17 After baseline assessment, telephone reviews were conducted with participants to assess medication use and exacerbations every 3 months for 12 months.
Baseline assessment
Participants attended a baseline visit to assess demographics, smoking status, exacerbation history in the preceding year, medical history, medication use, and visual analog scale symptom score. Pre- and post-bronchodilator spirometry, sputum induction,27 and venipuncture with blood collection were performed as described.17
Exacerbation capture
Respiratory hospitalizations, emergency room (ER) visits, unscheduled general practice visits, and medication use including antibiotics and systemic corticosteroids were recorded at each assessment. An exacerbation of COPD was defined as a COPD-related episode with, 1) hospitalization, 2) ER visit, or 3) the need for oral corticosteroid (OCS) and/or antibiotics for at least 3 days.8 An exacerbation of asthma was defined using the severe asthma exacerbation criteria of the American Thoracic Society/European Respiratory Society Task Force,28 or an asthma exacerbation with lower respiratory tract infection requiring antibiotics.29 A frequent exacerbator was defined as a participant with ≥2 exacerbations,8,30 whereas infrequent exacerbators had <2 exacerbations during the follow-up period.
Gene expression analysis
Sputum gene expression was measured on 95 subjects who had sputum stored for RNA analysis using quantitative real-time polymerase chain reaction (qRT-PCR) as previously described.19,23 Briefly, sputum RNA was extracted using the RNeasy Mini Kit (QIAGEN, Hilden, Germany), and 200 ng of RNA was converted to complementary DNA (cDNA) using the High Capacity cDNA RT-PCR kit (Life Technologies) and used to detect relative gene expression using standard Taqman methods (Applied Biosystems, Foster City, CA, USA). IL-1 pathway mediators were selected for measurement (previously identified as differentially regulated),19,23 including the signaling receptor IL1R1, the decoy receptor IL1R2, the antagonist IL1RN, and signaling molecules IRAK2, IRAK3, and PELI1. Statistical analysis was performed on the change in cycle threshold (ΔCt) between the target gene and β-actin (housekeeping gene). Fold-change results were calculated using 2−ΔΔCt relative to both the housekeeping gene (β-actin) and the mean of all samples.
Statistical analysis
Data were analyzed using STATA 11.2 (Stata Corp, College Station, TX, USA) and were reported as mean (SD) or median (interquartile range [25%–75%]) depending on the distribution. Comparisons between two independent groups were performed using Student’s t-test or the two-sample Wilcoxon rank sum test as appropriate. Fisher’s exact test was used to test categorical data. Receiver operating characteristic (ROC) curves were generated and the area under the curve was calculated to determine predictive relationships as previously described.23 Associations were tested using Spearman rank correlation. Significance was accepted if P<0.05.
Results
Clinical characteristics and inflammatory cells
Clinical characteristics and sputum inflammatory cells for the participants are detailed in Table 1. A subset of 95 participants had detailed assessment of clinical characteristics, exacerbation history (prior year) and sputum collected for inflammatory cell counts, and measurement of gene expression of the IL-1 pathway. Of the 95 participants, 89 completed the exacerbation monitoring in the 12 months following their baseline assessment. There were 30 participants who had two or more exacerbations in the year following their baseline assessment, comprising 22 COPD and eight asthma participants. There were 59 individuals who experienced either zero or one exacerbation in the following 12 months, 33 had COPD, while 26 had asthma (Table 1). The distribution of the number of exacerbations recorded over the following 12-month period is shown in Figure 1. Participants with COPD were older, had a greater degree of airflow limitation, and more frequent past exacerbations than those with asthma (Table 1). Asthma and COPD participants were similar in terms of inhaled corticosteroid use and sputum inflammatory cells.
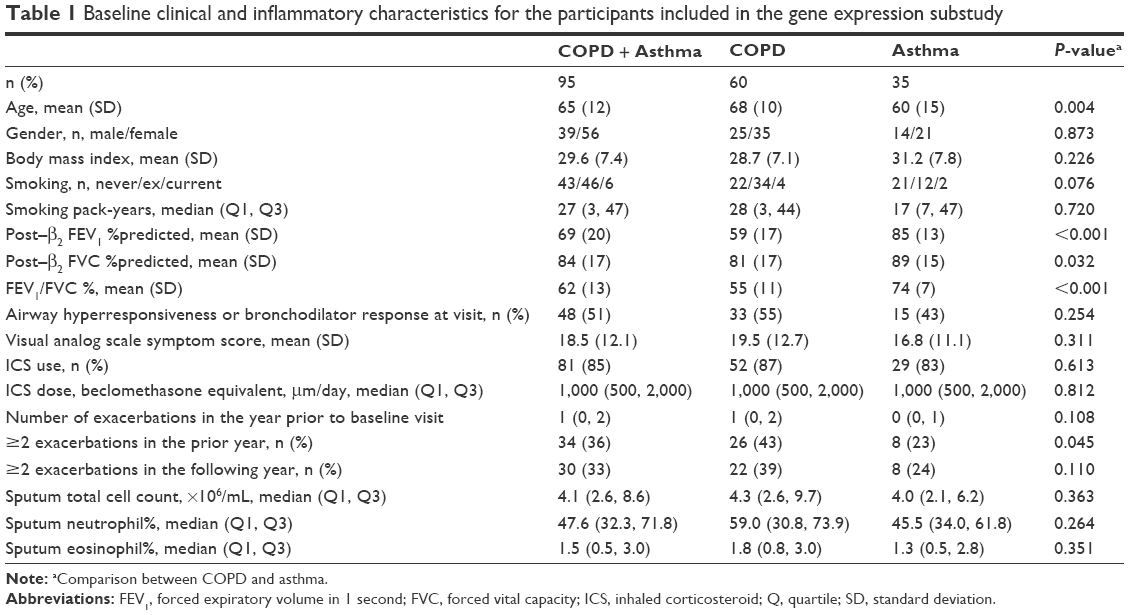 | Table 1 Baseline clinical and inflammatory characteristics for the participants included in the gene expression substudy |
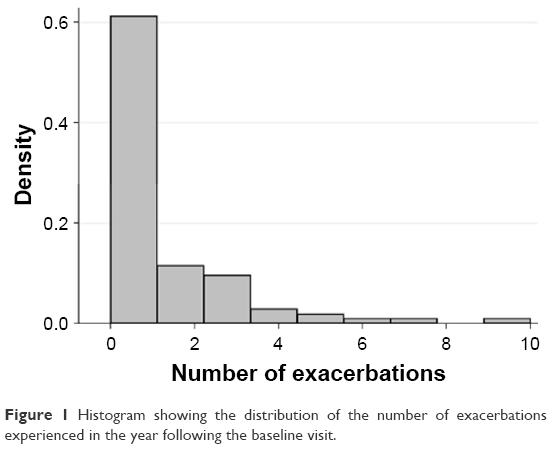 | Figure 1 Histogram showing the distribution of the number of exacerbations experienced in the year following the baseline visit. |
Gene expression of IL-1 pathway mediators predicts future exacerbations
Because of our previously reported associations with IL-1β expression and exacerbation prediction, we further investigated expression of mediators of the IL-1 pathway. At baseline, gene expressions of the IL-1 signaling genes IRAK2, IRAK3, and PELI1 and the signaling receptor IL1R1 were more strongly expressed in the group who had two or more exacerbations (frequent exacerbator group), across the whole combined study population, with a mean fold change of 2.8, 4.6, 2.4, and 3.3, respectively (Table 2; Figure 2). The higher expressions of IRAK2, IRAK3, PELI1, and IL1R1 were also seen in both COPD and asthma when analyzed separately (Table 2; Figure 2). The results were specific for IL-1 signaling because there was no significant difference in the gene expression of the decoy receptor IL1R2 or the receptor antagonist IL1RN (Table 2; Figure 2). IL1A expression was higher in frequent exacerbators with COPD, but not with asthma (Table 2).
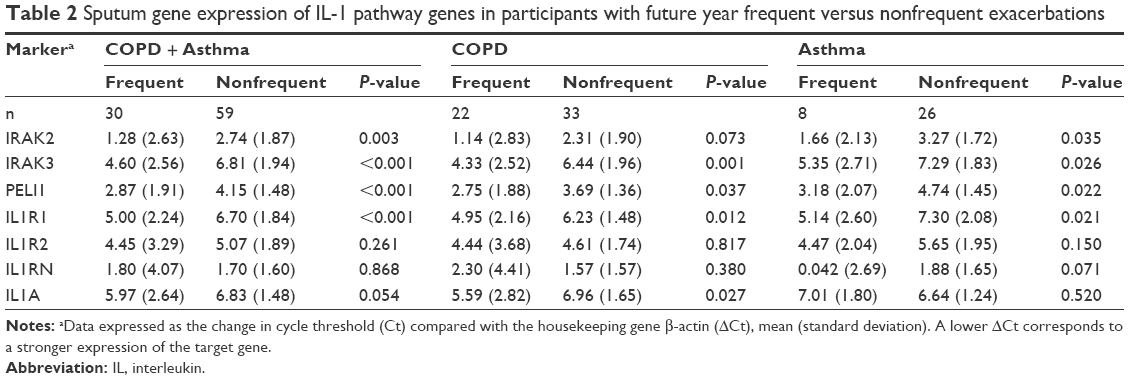 | Table 2 Sputum gene expression of IL-1 pathway genes in participants with future year frequent versus nonfrequent exacerbations |
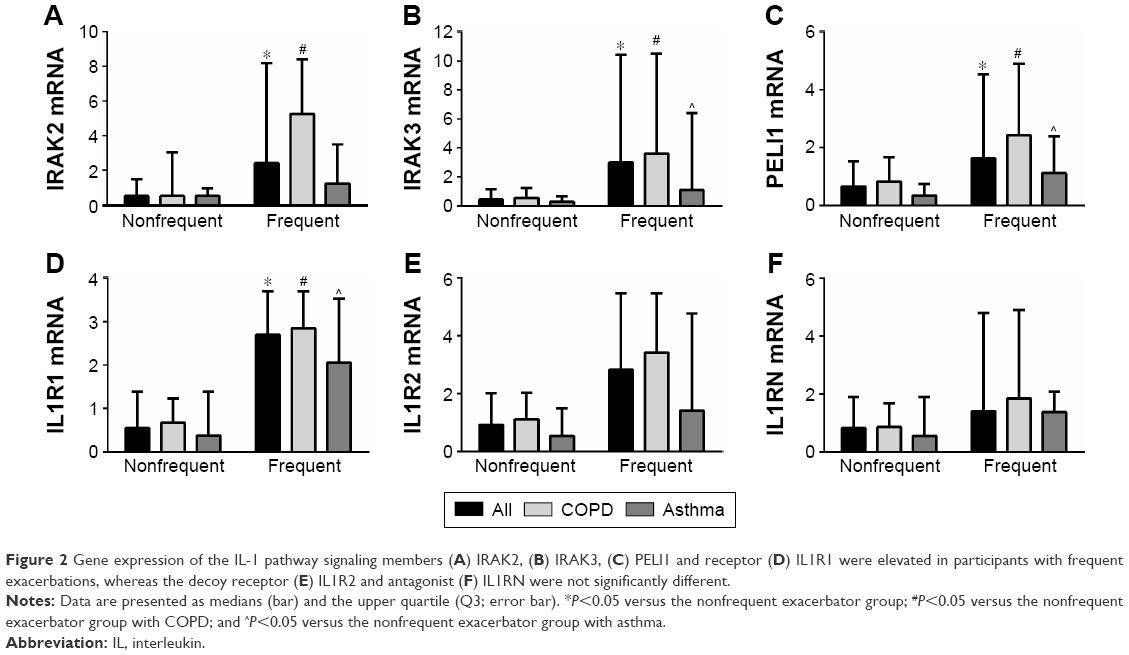 | Figure 2 Gene expression of the IL-1 pathway signaling members (A) IRAK2, (B) IRAK3, (C) PELI1 and receptor (D) IL1R1 were elevated in participants with frequent exacerbations, whereas the decoy receptor (E) IL1R2 and antagonist (F) IL1RN were not significantly different. |
Given the differences in gene expression between groups, the ability of IRAK2, IRAK3, PELI1, and IL1R1 sputum gene expression to predict future frequent exacerbations was investigated using ROC curves. All markers could significantly predict frequent future exacerbations in the combined group, but also in COPD and asthma separately (Table 3; Figure 3). IRAK3 was the best discriminator, followed closely by IL1R1 and PELI1 (Table 3; Figure 3). The level of sputum gene expression of IRAK2, IRAK3, PELI1, and IL1R1 was correlated with the number of exacerbations experienced by the participants (Table 4).
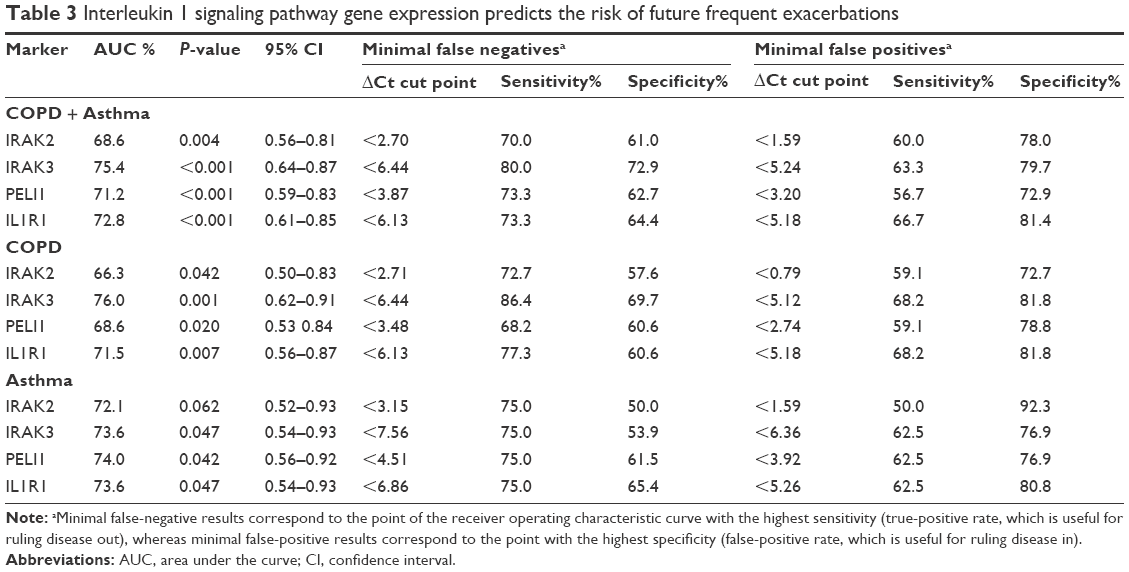 | Table 3 Interleukin 1 signaling pathway gene expression predicts the risk of future frequent exacerbations |
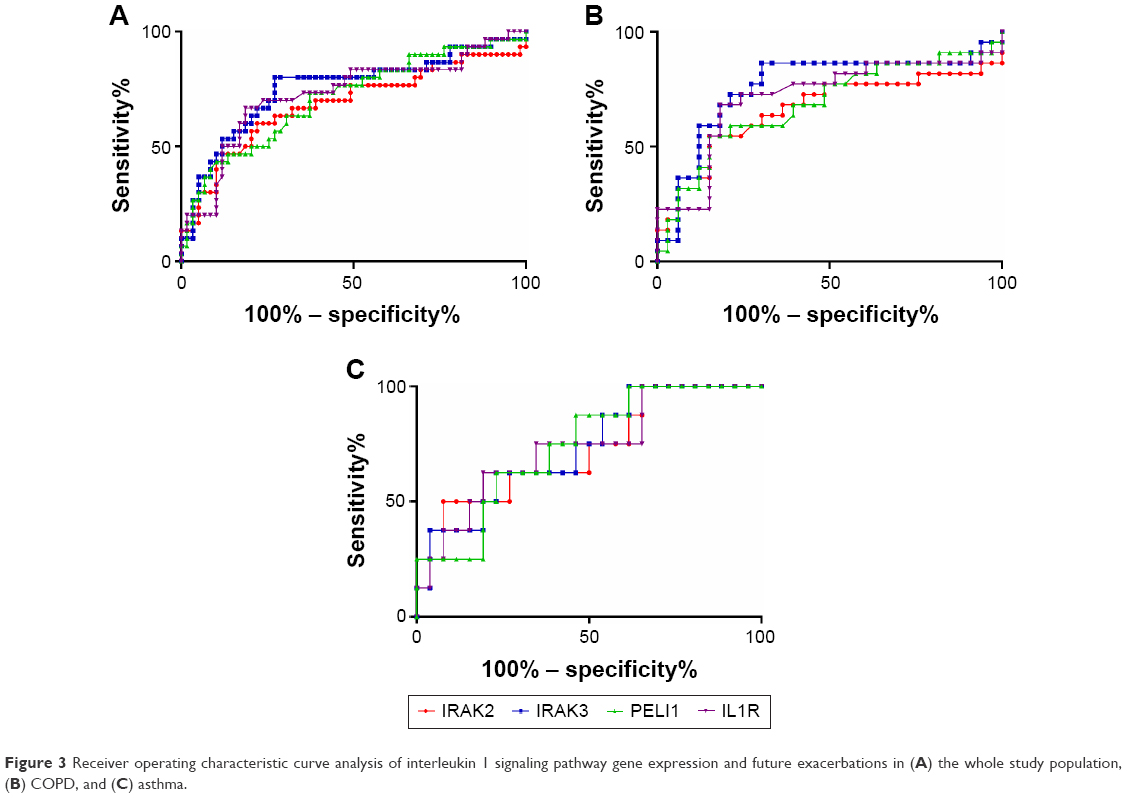 | Figure 3 Receiver operating characteristic curve analysis of interleukin 1 signaling pathway gene expression and future exacerbations in (A) the whole study population, (B) COPD, and (C) asthma. |
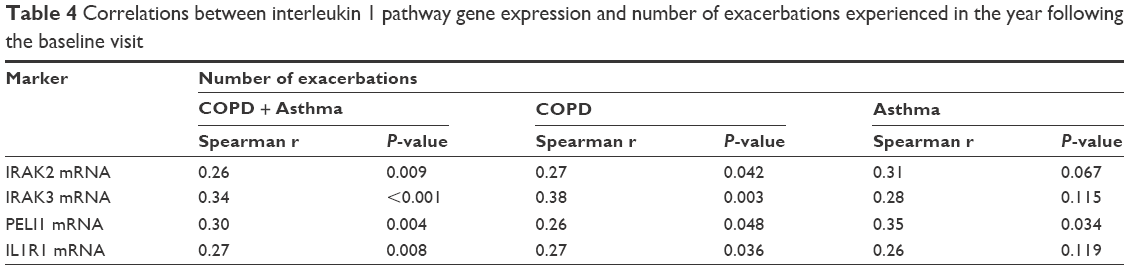 | Table 4 Correlations between interleukin 1 pathway gene expression and number of exacerbations experienced in the year following the baseline visit |
Type and severity of exacerbations
The levels of gene expression of the IL-1 pathway mediators were investigated in relation to the type and severity of the exacerbations experienced, including antibiotic courses (two or more, n=27) and OCS courses (two or more, n=17), general practitioner visits (two or more, n=17), and hospital admissions (n=11). Gene expression of IRAK2 was significantly higher in participants who were admitted to hospital for their exacerbation, and in those who had two or more OCS courses (Figure 4) compared with the nonfrequent exacerbator group. Expressions of IRAK3, PELI1, and IL1R1 were significantly higher in all types of exacerbations compared with the nonfrequent exacerbator group (Figure 4).
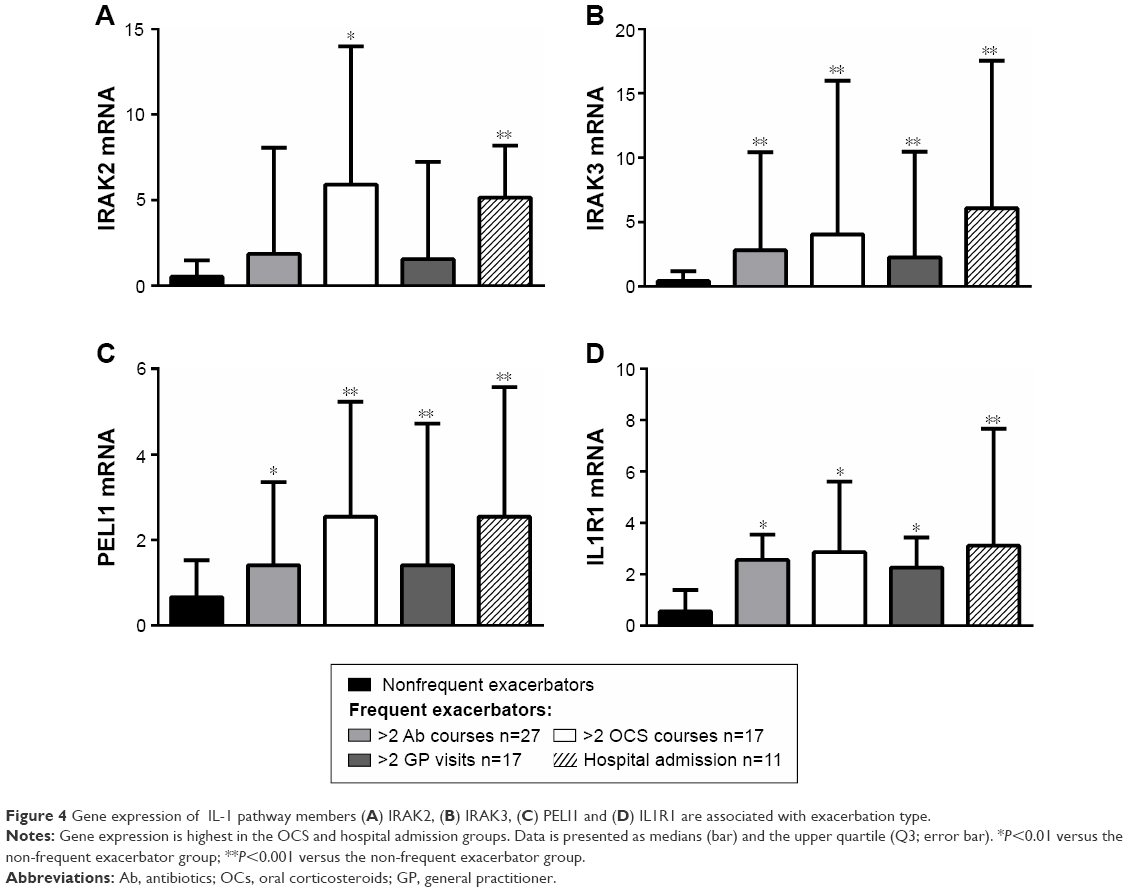 | Figure 4 Gene expression of IL-1 pathway members (A) IRAK2, (B) IRAK3, (C) PELI1 and (D) IL1R1 are associated with exacerbation type. |
Relationship of IL-1 pathway mediators with prior exacerbations
The relationship between the expression levels of the IL-1 pathway genes in the participants with frequent exacerbations was also investigated. Gene expressions of the IL-1 signaling genes IRAK2, IRAK3, and PELI1 and receptor IL1R1 are more strongly expressed in the participants with previous frequent exacerbations, across the study population and in COPD and asthma separately (Table 5; Figure 5). IL1R2 is also more strongly expressed in the frequent exacerbator group across the population and in asthma but not COPD separately, whereas IL1RN was significantly higher in the frequent exacerbators only in asthma (Table 5; Figure 5). IL1A expression was higher in prior year frequent exacerbators with COPD, but not with asthma (Table 5).
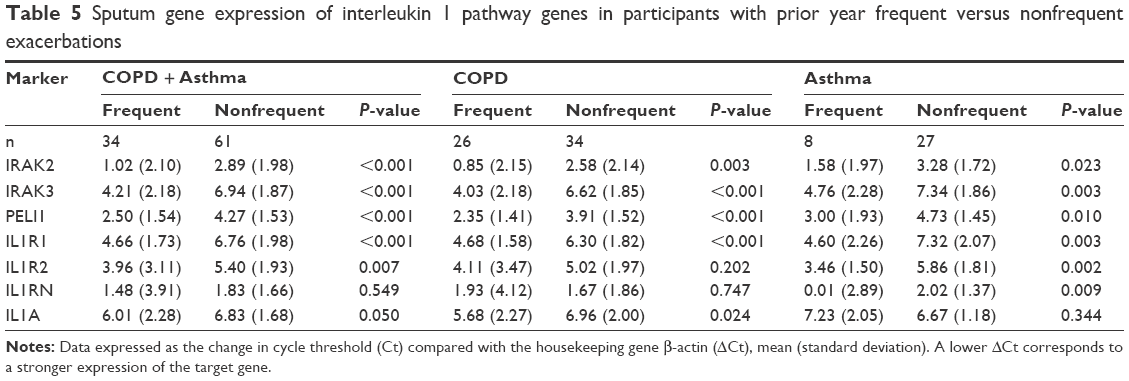 | Table 5 Sputum gene expression of interleukin 1 pathway genes in participants with prior year frequent versus nonfrequent exacerbations |
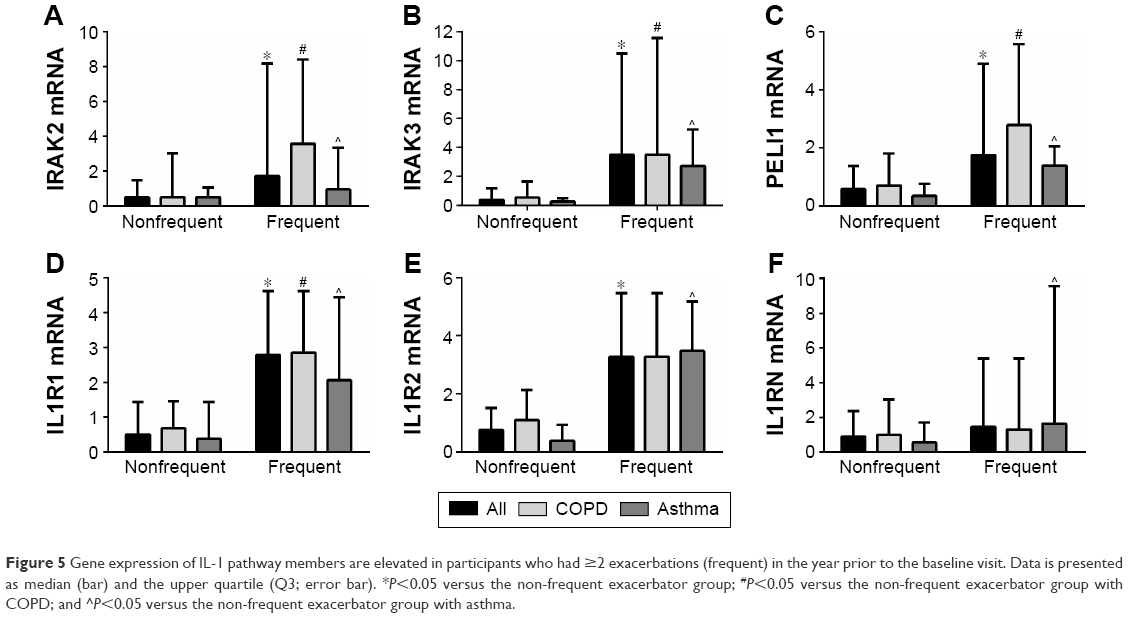 | Figure 5 Gene expression of IL-1 pathway members are elevated in participants who had ≥2 exacerbations (frequent) in the year prior to the baseline visit. Data is presented as median (bar) and the upper quartile (Q3; error bar). *P<0.05 versus the non-frequent exacerbator group; #P<0.05 versus the non-frequent exacerbator group with COPD; and ^P<0.05 versus the non-frequent exacerbator group with asthma. |
Discussion
Severe exacerbations of COPD and asthma lead to rapid health decline and can cause death; preventing severe exacerbations is, therefore, a major treatment goal. The mechanisms underlying exacerbations and their impact are not fully understood, and tools to predict at-risk patients are limited. This study has recorded exacerbations of different types and severities, and linked these to enhanced activation of IL-1 pathway gene expression. This prospective cohort study identifies that gene expressions of airway IL-1 signaling molecules IRAK2, IRAK3, and PELI1 and the signaling receptor IL1R1 are associated with frequent exacerbations and predict exacerbation risk in the obstructive airway diseases COPD and asthma. IL-1 signaling mediators were correlated with the number of exacerbations, and the level of expression was the highest in more severe exacerbations characterized by OCS use or hospital admission. High IL-1 pathway gene expression was also associated with frequent prior year exacerbations. Elevated IL-1 pathway activation is, therefore, potentially both a useful biomarker for exacerbation risk prediction and a novel treatment target for exacerbation reduction in COPD and asthma.
IL-1 family cytokines are major mediators of innate immune responses, controlling proinflammatory reactions to pathogen- or damage-associated molecular patterns.31 Secretion of active IL-1β requires two signals, the first being an upregulation of IL-1β production through toll-like receptor (TLR) signaling, and either cleavage through the NLRP3 inflammasome or neutrophil proteases. IL-1β then acts through its signaling receptor, IL-1R1, to activate a complex signaling cascade involving the IL-1 receptor-associated kinases IRAK1, IRAK2, and IRAK4, resulting in the activation of transcription factors including nuclear factor-κB (NF-κB) and production of inflammatory cytokines. The decoy receptor IL-1R2 and antagonist IL-1Ra (encoded by IL1RN) act to limit this pathway activation through binding IL-1 or IL-1R1, respectively, but not activating a signaling cascade.31 IL-1/TLR pathway signaling is critical in initiating appropriate innate immune responses to infections, and, therefore, malfunction of this pathway is likely to have negative downstream consequences, for example persistence of infection, heightened inflammation.
IRAK-M (encoded by IRAK3) is believed to function as a negative regulator that prevents the dissociation of IRAK-1/2 from the TLR/IL-1 receptor complex, which then inhibits downstream signaling.32 However, these signaling pathways are complex with multiple mechanisms leading to NF-κB activation, but also regulation of mRNA stability. For example, interaction between IRAK2 and IRAK-M can also mediate NF-κB activation through TLR7, while controlling translation of cytokines and chemokines at the mRNA level.33 Expression of IRAK-M is induced by lipopolysaccharide and is expressed in predominantly macrophages. Increased expression of IRAK-M occurs in endotoxin tolerance,34 whereby inflammatory responses to endotoxin are reduced after multiple exposures. Bacterial challenge of IRAK-M-deficient mice produces the elevated levels of cytokines and chemokines in murine lungs.35 However, overexpression of IRAK-M can enhance rhinovirus replication in human airway epithelial cells by enhancing autophagy.36 IRAK-M expression can also be induced by noninfectious triggers, for example adiponectin, a cytokine product of adipose tissue.37 Gene polymorphisms in IRAK-M have been associated with the pathogenesis of early-onset persistent asthma.38
The family of pellino proteins are E3 ubiquitin ligases that play an important role in TLR/IL-1 signaling and immune responses.39 Pellino-1 promotes interactions between the IL-1β and the IRAK4–IRAK1–TRAF6 complex, and mediates NF-κB activation.39 We have previously reported the upregulation of PELI1 gene expression that is associated with airway neutrophilia in asthma19 and recurrent protracted bacterial bronchitis,40 and rhinovirus infection of COPD, but not healthy primary bronchial epithelial cells.41 Pellino-1 knockdown in human primary bronchial epithelial cells reduces the production of IL-1β-induced CXCL8, but does not affect responses to viruses.42 This promisingly suggests that the inhibition of pellino-1 may modulate neutrophilic inflammation without compromising innate immune responses to viral infection.
There are few diagnostic tools presently available that allow clinicians to predict patients who are at high risk of future exacerbations. Airway disease exacerbations are typically associated with neutrophil and sometimes eosinophil influx into the airways;43 however, we did not find a predictive relationship of inflammatory cell counts and future exacerbations,17 so this is likely only relevant at the time of exacerbation. We have previously identified that upregulation of IL-1 pathway gene expression is associated with neutrophilic airway inflammation in asthma,19,23 and so may be related to neutrophil influx. The presence of systemic inflammation, as measured by circulating CRP and IL-6 levels, is also predictive of exacerbations17 and associated with airway neutrophilia.22
Measurement of these mediators would provide a number of advantages over the current marker of future exacerbation, being past history of exacerbation. These advantages include the measurement being objective, with precise and sensitive quantification using qPCR, with the mediators showing clear upregulation that could be used to target treatment directly to underlying mechanisms. Biomarker measurement will not rely on patient recall, and future studies may demonstrate a role in treatment monitoring, based on the levels of IL-1 mediators.
The presence of airway inflammation is often triggered by infection, including bacteria and viruses. Indeed, elevated IL-1β is a feature of a bacteria-predominant acute exacerbation of COPD21 and stable neutrophilic asthma.23 Our study shows that the persistent activation of IL-1 signaling through IL-1R1, IRAK3, IRAK2, and PELI1 influences the future susceptibility of patients to multiple severe exacerbations. Although IL-1 signaling molecules were increased in frequent exacerbators, this was not accompanied by increases in anti-inflammatory actions of IL-1Ra or IL-1R2. The mechanisms underlying these observations need further characterization, as do potential targeted treatments, for example recombinant human IL-1Ra, which inhibits the biological activity of IL-1.44
Although we have found promising insight into the mechanisms underlying exacerbations of COPD and asthma, this study has several limitations. As a single-center study with a relatively small sample size, further studies need to be conducted in a larger cohort, across multiple centers to confirm these relationships. We were unable to assess the heterogeneity of inflammation at the time of exacerbations or test the potential triggers of the exacerbations and their relationship to IL-1 pathway mediators. Further studies are required to assess whether IL-1 pathway activation is associated with a particular infectious trigger of exacerbation, for example, bacteria or viruses, and potential mechanisms for the persistence of this type of inflammation once the exacerbation has resolved, and how this impacts on future susceptibility of the patients. We are also unable to determine the effects of common treatments including antibiotics and OCSs on the expression of IL-1 pathway mediators, of interest in understanding potential strategies to reduce exacerbations. Future studies will need to further characterize any differences between the exacerbations of asthma and COPD on the activation of the IL-1 pathway, such as the increased expression of IL1R2 and IL1RN in frequent prior year exacerbations of asthma but not COPD.
In conclusion, we have shown that the upregulation of IL-1 pathway mediators, including the signaling molecules IRAK2, IRAK3, and PELI1 and the signaling receptor IL1R1, in stable COPD and asthma is associated with frequent exacerbations in the following year, as well as the prior year. Expression of IL-1 pathway mediators correlated with the number and severity of future exacerbations. Further studies are warranted investigating these mediators as both potential diagnostic biomarkers predicting at-risk patients and novel treatment targets for exacerbation reduction.
Acknowledgments
The authors would like to acknowledge the technical assistance of Kelly Steel, Penelope Baines, Rebecca Oldham, Anne-Marie Gibson, Bridgette Ridewood, Michelle Gleeson, Kellie Fakes, Heather Macdonald, and Naomi Fibbens. This study was supported by National Health and Medical Research Council (APP1045371).
Authors’ contributions
Study design and conception: KJB, JF, VMM, and PGG. Data collection and interpretation: KJB, JF, VMM, and PGG. Data analysis: KJB. Writing of the manuscript: KJB. Editing and review of the manuscript: KJB, JF, VMM, and PGG. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
KJB, JF, and PGG have no conflicts of interest to declare in regard to the content of this manuscript. VMM is supported by an NHMRC TRIP fellowship; has participated in educational symposia funded by GlaxoSmithKline, AstraZeneca, Menarini, Boehringer Ingelhiem, and Novartis; and has participated in studies funded by GlaxoSmithKline and advisory boards for GlaxoSmithKline, Novartis, AstraZeneca, and Menarini.
References
Connors AFJ, Dawson NV, Thomas C, et al. Outcomes following acute exacerbation of severe chronic obstructive lung disease. The SUPPORT investigators (Study to Understand Prognoses and Preferences for Outcomes and Risks of Treatments). Am J Respir Crit Care Med. 1996;154(4 Pt 1):959–967. | ||
McDonald VM, Gibson PG. Exacerbations of severe asthma. Clin Exp Allergy. 2012;42(5):670–677. | ||
Bai TR, Vonk JM, Postma DS, Boezen HM. Severe exacerbations predict excess lung function decline in asthma. Eur Resp J. 2007;30(3):452–456. | ||
Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57(10):847–852. | ||
Kupczyk M, ten Brinke A, Sterk PJ, et al. Frequent exacerbators-a distinct phenotype of severe asthma. Clin Exp Allergy. 2014;44(2):212–221. | ||
Spencer S, Calverley PM, Burge PS, Jones PW. Impact of preventing exacerbations on deterioration of health status in COPD. Eur Respir J. 2004;23(5):698–702. | ||
Dougherty RH, Fahy JV. Acute exacerbations of asthma: epidemiology, biology and the exacerbation-prone phenotype. Clin Exp Allergy. 2009;39(2):193–202. | ||
Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363(12):1128–1138. | ||
ten Brinke A, Sterk PJ, Masclee AA, et al. Risk factors of frequent exacerbations in difficult-to-treat asthma. Eur Respir J. 2005;26(5):812–818. | ||
Miravitlles M, Calle M, Soler-Cataluna JJ. Clinical phenotypes of COPD: identification, definition and implications for guidelines. Arch Bronconeumol. 2012;48(3):86–98. | ||
Simpson JL, Grissell TV, Douwes J, Scott RJ, Boyle MJ, Gibson PG. Innate immune activation in neutrophilic asthma and bronchiectasis. Thorax. 2007;62(3):211–218. | ||
Wood LG, Baines KJ, Fu J, Scott HA, Gibson PG. The neutrophilic inflammatory phenotype is associated with systemic inflammation in asthma. Chest. 2012;142(1):86–93. | ||
Donaldson GC, Seemungal TA, Patel IS, et al. Airway and systemic inflammation and decline in lung function in patients with COPD. Chest. 2005;128(4):1995–2004. | ||
Dahl M, Vestbo J, Lange P, Bojesen SE, Tybjaerg-Hansen A, Nordestgaard BG. C-reactive protein as a predictor of prognosis in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(3):250–255. | ||
Agusti A, Edwards LD, Rennard SI, et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One. 2012;7(5):e37483. | ||
Thomsen M, Ingebrigtsen TS, Marott JL, et al. Inflammatory biomarkers and exacerbations in chronic obstructive pulmonary disease. JAMA. 2013;309(22):2353–2361. | ||
Fu J-J, McDonald VM, Baines KJ, Gibson PG. Airway il-1β and systemic inflammation as predictors of future exacerbation risk in asthma and COPD. Chest. 2015;148(3):618–629. | ||
Slack J, McMahan CJ, Waugh S, et al. Independent binding of interleukin-1 alpha and interleukin-1 beta to type I and type II interleukin-1 receptors. J Biol Chem. 1993;268(4):2513–2524. | ||
Baines KJ, Simpson JL, Wood LG, Scott RJ, Gibson PG. Transcriptional phenotypes of asthma defined by gene expression profiling of induced sputum samples. J Allergy Clin Immunol. 2011;127(1):153–160, e1–e9. | ||
Pauwels NS, Bracke KR, Dupont LL, et al. Role of IL-1alpha and the Nlrp3/caspase-1/IL-1beta axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur Respir J. 2011;38(5):1019–1028. | ||
Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med. 2011;184(6):662–671. | ||
Fu JJ, Baines KJ, Wood LG, Gibson PG. Systemic inflammation is associated with differential gene expression and airway neutrophilia in asthma. OMICS. 2013;17(4):187–199. | ||
Baines KJ, Simpson JL, Wood LG, et al. Sputum gene expression signature of 6 biomarkers discriminates asthma inflammatory phenotypes. J Allergy Clin Immunol. 2014;133(4):997–1007. | ||
Simpson JL, Phipps S, Baines KJ, Oreo KM, Gunawardhana L, Gibson PG. Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur Respir J. 2014;43(4):1067–1076. | ||
Global Initiative for Asthma (GINA). Global strategy for asthma management and prevention. [Updated 2012]. Available from: http://www.ginasthma.com | ||
Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the diagnosis, management and prevention of COPD. [Updated 2011]. Available from: www.goldcopd.com. Accessed on June 31st, 2016. | ||
Gibson PG, Wlodarczyk JW, Hensley MJ, et al. Epidemiological association of airway inflammation with asthma symptoms and airway hyperresponsiveness in childhood. Am J Respir Crit Care Med. 1998;158(1):36–41. Accessed on June 31st, 2016. | ||
Reddel HK, Taylor DR, Bateman ED, et al. An official American Thoracic Society/European Respiratory Society statement: asthma control and exacerbations: standardizing endpoints for clinical asthma trials and clinical practice. Am J Respir Crit Care Med. 2009;180(1):59–99. | ||
Brusselle GG, Vanderstichele C, Jordens P, et al. Azithromycin for prevention of exacerbations in severe asthma (AZISAST): a multicentre randomised double-blind placebo-controlled trial. Thorax. 2013;68(4):322–329. | ||
Cote CG, Dordelly LJ, Celli BR. Impact of COPD exacerbations on patient-centered outcomes. Chest. 2007;131(3):696–704. | ||
Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal. 2010;3(105):cm1. | ||
Kobayashi K, Hernandez LD, Galan JE, Janeway CA Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110(2):191–202. | ||
Zhou H, Yu M, Fukuda K, et al. IRAK-M mediates Toll-like receptor/IL-1R-induced NFκB activation and cytokine production. EMBO J. 2013;32(4):583–596. | ||
van’t Veer C, van den Pangaart PS, van Zoelen MAD, et al. Induction of IRAK-M is associated with lipopolysaccharide tolerance in a human endotoxemia model. J Immunol. 2007;179(10):7110–7120. | ||
Deng JC, Cheng G, Newstead MW, et al. Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J Clin Invest. 2006;116(9):2532–2542. | ||
Wu Q, van Dyk LF, Jiang D, et al. Interleukin-1 receptor-associated kinase M (IRAK-M) promotes human rhinovirus infection in lung epithelial cells via the autophagic pathway. Virology. 2013;446(1–2):199–206. | ||
Zacharioudaki V, Androulidaki A, Arranz A, Vrentzos G, Margioris AN, Tsatsanis C. Adiponectin promotes endotoxin tolerance in macrophages by inducing IRAK-M expression. J Immunol. 2009;182(10):6444–6451. | ||
Balaci L, Spada MC, Olla N, et al. IRAK-M is involved in the pathogenesis of early-onset persistent asthma. Am J Hum Genet. 2007;80(6):1103–1114. | ||
Medvedev AE, Murphy M, Zhou H, Li X. E3 ubiquitin ligases Pellinos as regulators of pattern recognition receptor signaling and immune responses. Immunol Rev. 2015;266(1):109–122. | ||
Baines KJ, Upham JW, Yerkovich ST, et al. Mediators of neutrophil function in children with protracted bacterial bronchitis. Chest. 2014;146(4):1013–1020. | ||
Baines K, Hsu A, Tooze M, Gunawardhana LP, Gibson PG, Wark PA. Novel immune genes associated with excessive inflammatory and antiviral responses to rhinovirus in COPD. Respir Res. 2013;14:15. | ||
Bennett JA, Prince LR, Parker LC, et al. Pellino-1 Selectively Regulates Epithelial Cell Responses to Rhinovirus. J Virol. 2012;86(12):6595–6604. | ||
Bathoorn E, Kerstjens H, Postma D, Timens W, MacNee W. Airways inflammation and treatment during acute exacerbations of COPD. Int J Chron Obstruct Pulmon Dis. 2008;3(23):217–229. | ||
Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11(8):633–652. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.