Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
Aerobic Exercise Alleviates Inflammation, Oxidative Stress, and Apoptosis in Mice with Chronic Obstructive Pulmonary Disease
Authors Wang X ![]() , Wang Z, Tang D
, Wang Z, Tang D
Received 3 March 2021
Accepted for publication 19 April 2021
Published 17 May 2021 Volume 2021:16 Pages 1369—1379
DOI https://doi.org/10.2147/COPD.S309041
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Xishuai Wang, 1, 2 Zhiqing Wang, 1 Donghui Tang 1
1Department of College of P.E and Sport, Beijing Normal University, Beijing, People’s Republic of China; 2Department of Animal Genetic Resources, Institute of Animal Science, Chinese Academy of Agricultural Sciences, Beijing, People’s Republic of China
Correspondence: Donghui Tang; Xishuai Wang
Department of College of P.E and Sport, Beijing Normal University, No. 19, Xinjiekouwai St, Haidian District, Beijing, 100875, People’s Republic of China
Email [email protected]; [email protected]
Background: Chronic inflammation, oxidative stress, and apoptosis play critical roles in chronic obstructive pulmonary disease (COPD) pathogenesis. Here, we attempted to determine whether aerobic exercise (AE) could improve COPD by counteracting the COPD-associated inflammatory response, oxidative stress, and apoptosis in mice.
Methods: Thirty male ICR mice were assigned into one of three groups: control (Con), COPD, and COPD + AE. COPD was simulated by intratracheal injection of lipopolysaccharide (LPS) for 4 weeks. Low-intensity AE was performed for 4 weeks. Bronchoalveolar lavage fluid (BALF) cell counts and the levels of inflammatory cytokine in BALF and serum were detected. Hematoxylin and eosin (HE), Masson trichrome, and Sirius Red staining as well as terminal deoxynucleotidyl transferase dUTP nick end labeling were performed to identify the degree of pulmonary emphysema, bronchial mucus cell hyperplasia, pulmonary fibrosis, and cell apoptosis. Oxidative stress parameters were measured. Furthermore, gene expression levels for the CXCL1, IL-1β, IL-10, IL-17, matrix metalloproteinase (MMP)9, TGF-β, TNF-α, and silent information regulator (sirt)1 were detected in mice lung tissues.
Results: AE improved LPS-induced emphysema, pulmonary fibrosis, bronchial mucus cell hyperplasia, bronchoconstriction, and cell apoptosis. AE prevented an LPS-induced increase in the total cell, neutrophil, and macrophage counts. AE decreased malondialdehyde (MDA) and myeloperoxidase (MPO) levels but increased glutathione (GSH) and superoxide dismutase (SOD) levels. AE decreased BALF levels of IL-1β, TNF-α, and TGF-β but increased BALF IL-10 levels. AE suppressed the gene expression levels of pro-inflammatory factors CXCL1, IL-1β, IL-17, and TNF-α and profibrotic factors MMP-9 and TGF-β but activated those of anti-inflammatory factor IL-10 and lung-protective factor sirt1.
Conclusion: AE is a potential therapeutic approach for COPD. AE improved emphysema, bronchial mucus cell hyperplasia, and pulmonary fibrosis in mice with COPD by alleviating the inflammatory response, oxidative stress injury, and cell apoptosis as well as activating sirt1.
Keywords: exercise, COPD, pulmonary fibrosis, emphysema, pulmonary inflammation, oxidative stress injury
Corrigendum for this paper has been published
Introduction
Chronic obstructive pulmonary disease (COPD), which is characterized by lung emphysema, airway remodeling, and not fully reversible airflow limitation, involves alterations in inflammatory parameters and oxidative status.1,2 Although significant progress has been made, morbidity and mortality in COPD patients are still very high worldwide.3 The pathogenesis of COPD is complex. It is well documented that inflammation and oxidative stress play central roles in the pathogenesis of COPD.4,5 Moreover, cell apoptosis also contributes to COPD.6 We attempted to identify the physiological functions of these factors in response to regular aerobic exercise (AE) in mice with COPD.
COPD is characterized by a chronic inflammatory response involving inflammatory mediators, including interleukin (IL)-1β and 1L-10, and effector cells, in which neutrophils play an integral role.7,24 Deleterious accumulation of neutrophils is a critical mechanism underlying COPD in part because neutrophils release multiple factors, including transforming growth factor beta (TGF-β) and matrix metalloproteinase 9 (MMP-9), which are important in the pathogenesis of COPD.8,9 Previous studies have demonstrated that exercise improved immune functions by altering the number of immune cells, including neutrophils.10 Inflammatory mediators such as IL-1β and 1L-10, and effector cells, including neutrophils, have been established as crucial mechanisms of COPD, but their physiological functions in response to regular AE in mice with COPD remain unclear.
Pulmonary fibrosis, which is characterized by progressive dyspnea and hypoxemia; is a serious condition with few available treatments.11 Neutrophils release multiple factors, including TGF-β and MMP-9, which are essential in the pathogenesis of pulmonary fibrosis.8,9 MMP-9 is associated with emphysema, pulmonary inflammation, and lung remodeling in patients with COPD.10 Pro-fibrosis factors, such as IL-17, MMP-9, and TGF-β, also contribute to the pathogenesis of pulmonary fibrosis;12–14 however, their physiological functions in response to regular AE in mice with COPD remain unclear.
Pulmonary fibrosis, which is characterized by progressive dyspnea and hypoxemia; leads to expiratory dyspnea; and is associated with an unacceptably high incidence, is a seriously endangered condition with few available treatments.11 Neutrophils release multiple factors, including TGF-β and MMP-9, which play prominent roles in the pathogenesis of pulmonary fibrosis.8,9 MMP-9 is associated with emphysema, pulmonary inflammation, and lung remodeling in patients with COPD.10 TGF-β plays vital roles in the pathogenesis of pulmonary fibrosis.11 Pro-fibrosis factors, such as IL-17, MMP-9, and TGF-β play prominent roles in the pathogenesis of pulmonary fibrosis.12–14 However, the physiological role of pro-fibrosis factors in response to regular AE in mice with COPD remains unclear.
Silent information regulator (sirt1) plays a vital role in the development of COPD.15 Compared to the normal population, the levels of sirt1 in COPD patients were significantly reduced.16–18 Activation of sirt1 by agents, such as resveratrol, protects against COPD because sirt1 modulates the inflammatory/anti-inflammatory and oxidation/antioxidation balances.19 Previous studies have found that exercise can activate sirt1 in muscle tissue, increasing the level of cellular NAD+ and the NAD/NADH ratio.20 In the present study, we determined whether AE could increase sirt1 and modulate the inflammatory/anti-inflammatory and oxidation/antioxidation balances in COPD mice.
Materials and Methods
Animals and Experimental Design
All protocols used in this study were approved by the Animal Experimental Welfare of the Institute of Animal Science, Chinese Academy of Agricultural Sciences (Beijing, China). All experiments were performed in accordance with the Animal Experimental Welfare of the Institute of Animal Science, Chinese Academy of Agricultural Sciences, and the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. The authors complied with the ARRIVE guidelines and every effort was made to minimize suffering. Thirty male ICR mice (6 weeks old, 18–21 g) were provided by Beijing HFK Biotechnology Co., Ltd. (Beijing, China). The conditions of the husbandry room were temperature at 22 ± 5 °C, relative humidity at 50% ± 10%, and the light cycle was 12 h/day. The body weight and health status of the mice were recorded. The mice were anesthetized via intraperitoneal injection of pentobarbital (50 mg/kg).
The COPD model was produced by intratracheal injection of 0.2 mg/kg LPS (L2880) twice a week for 4 weeks as previously described.21–25 The mice were divided into three groups with eight individuals each: (1) control (Con), in which mice received the same volume of saline via intratracheal injection; (2) COPD, in which mice received 0.2 mg/kg LPS via intratracheal injection; or (3) COPD + AE, in which mice received 0.2 mg/kg LPS via intratracheal injection and were subjected to AE for 4 weeks, simultaneously. LPS was dissolved in saline solution. Saline or LPS was injected twice a week (days 1, 4, 8, 11, 15, 18, 22, and 25); thus, the mice received a total of eight injections of LPS or saline. All mice were anesthetized via intraperitoneal injection of pentobarbital (50 mg/kg). All mice were euthanized on day 28.
Exercise Protocol
The mice were trained using a six-lane treadmill. Adaptive training of the mice was performed for 3 days (20 min, 25% inclination, 3.3 m/min). After the adaptive training, the maximal exercise capacity test of each mouse was performed. The maximal aerobic capacity was determined based on the maximal speed. The mean maximal speed reached by each animal was 23.6 m/s. Hence, the maximal aerobic capacity (100%) was 23.6 m/s. Mice from AE and COPD + AE groups were trained in low-intensity exercise (50% maximal speed, 11.8 m/s). The exercise intervention lasted for 4 weeks, 5 days/week, 1 h/day.
Collection of Tissue Samples
Blood samples were collected and placed in a blood collection vessel containing an anticoagulant, followed by centrifugation at 3000 rpm for 15 min. The serum was transferred to Ep tubes and immediately stored at −20 °C. We collected bronchoalveolar lavage fluid (BALF) using 2 mL of ice-cold 0.9% NaCl. The whole lung was flushed four times, and the returned fluid was collected. The supernatant was stored at −80 °C until subsequent analysis. After sampling, the lung tissue samples were immediately placed in an incubator filled with liquid nitrogen. The tissue was then transferred to a liquid nitrogen tank for long-term preservation. For histological analysis, lung tissue samples were fixed in 4% paraformaldehyde (PFA).
Histopathology
For histological analysis, 4% PFA-fixed lung tissue samples were embedded in paraffin. Using the paraffin section method, 5-µm-thick sections were stained with HE, Masson, Sirius Red, and TUNEL. Photographs taken using an inverted microscope (Olympus Corp., Japan) were used to identify the degree of pulmonary emphysema, pulmonary fibrosis, and cell apoptosis.
Detection of Pulmonary and Systemic Inflammation
The levels of the pro-inflammatory factors CXCL1, IL-17, TGF-β, IL-1β, and TNF-α as well as that of the anti-inflammatory factor IL-10 in the BALF were detected using mouse enzyme-linked immunosorbent assay (ELISA) kits (NeoBioscience Technology Company, Shenzhen, China). Similarly, the levels of the TNF-α and IL-10 in serum were detected using mouse ELISA kits (NeoBioscience Technology Company, Shenzhen, China).
Determination of the Gene Expression Levels in Lung Tissue
RNA from lung tissues was extracted using TRIzol (Promega, Madison, WI), their concentration was measured to detect the quality of total RNA, and cDNA was synthesized using an Applied Biosystems 2720 Thermal Cycler (Applied Biosystems, Singapore). GAPDH was selected as the internal reference gene, and the 2−ΔΔCT method was used for quantitative analysis. All primers were designed and synthesized by the Shanghai Sangon Biotech Company (Shanghai, China). Table 1 lists the murine PCR primer sequences.
 |
Table 1 PCR Primer Sequence Information of Mouse |
Statistical Analysis
SPSS 21.0 software was used for data processing. The experimental results are indicated by the mean ± standard deviation (x ± SD). Statistical differences among groups were assessed using one-way analysis of variance (ANOVA). The significance level was set at P < 0.05. GraphPad Prism 6 software was used to draw figures.
Results
Protective Effects of AE Against Pulmonary Inflammation
After LPS administration for 6 h, the number of total cells (P < 0.001), neutrophils (P < 0.001), and macrophages (P < 0.001) were significantly increased. AE played an essential role in preventing the increase in the number of total cells (P = 0.0374), neutrophils (P = 0.0203), and macrophages (P = 0.0236) compared to the COPD group. Treatment with LPS and AE did not influence the number of lymphocytes (P > 0.05) or eosinophils (P > 0.05) in the BALF (Table 2).
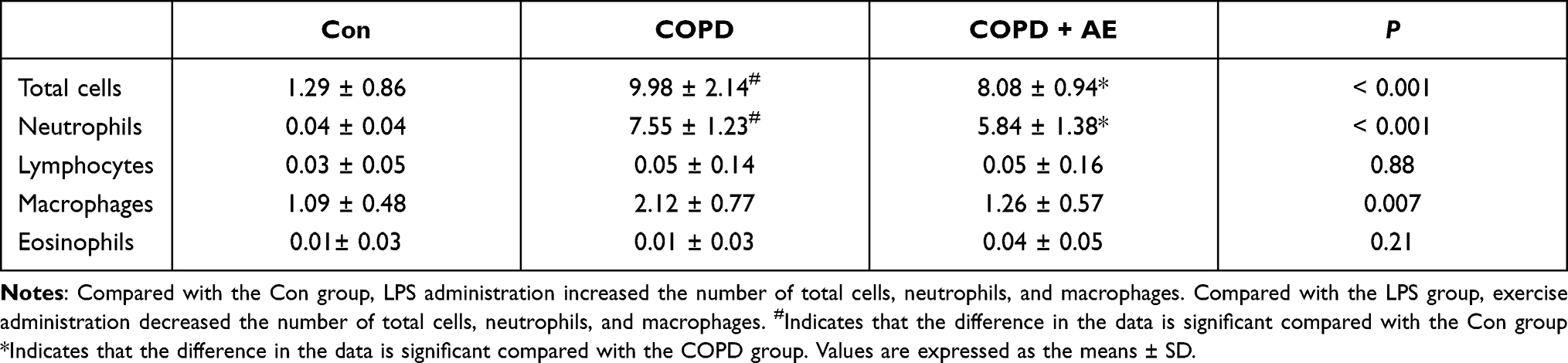 |
Table 2 Total and Differential Cell Counts in BALF (Cells/ml) |
LPS administration markedly upregulated BALF levels of CXCL1 (P < 0.001), IL-1β (P < 0.001), IL-10 (P < 0.001), IL-17 (P < 0.001), TNF-α (P < 0.001) and TGF-β (P < 0.001) compared to the Con group. AE upregulated BALF levels of IL-10 (P = 0.0014) and CXCL1 (P = 0.0036); downregulated BALF levels of TGF-β (P = 0.0285), IL-1β (P = 0.0378), and TNF-α (P = 0.0284); and did not influence BALF levels of IL-17 (P = 0.96) compared to the COPD group. These results demonstrate that the 4-week AE regimen relieved pulmonary inflammation in mice with COPD (Table 3).
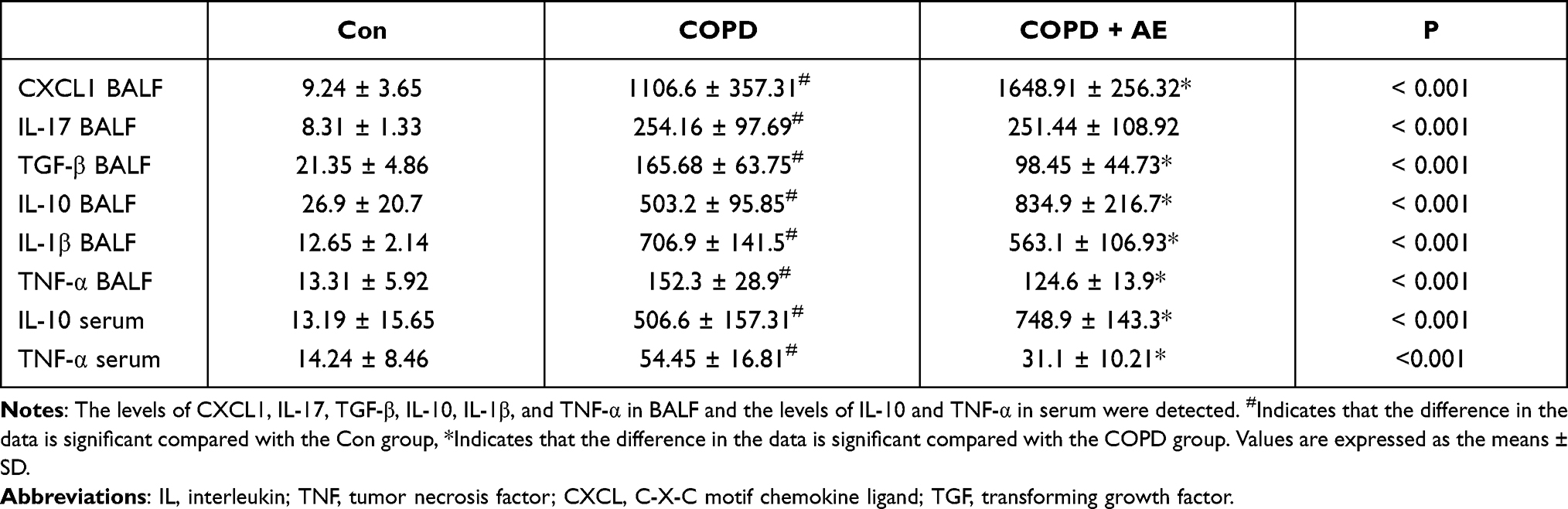 |
Table 3 The Levels of Inflammatory Factors in BALF and Serum (pg/ml) |
Protective Effects of AE Against Systemic Inflammation
The serum levels of IL-10 (P < 0.001) and TNF-α (P = 0.012) were significantly increased after compared to the Con group. AE upregulated serum levels of IL-10 (P = 0.0062) and downregulated serum levels of TNF-α (P = 0.0249) in mice with COPD (Table 3). These results demonstrate that the 4-week AE relieved systemic inflammation in mice with COPD.
Protective Effects of AE Against Lung Emphysema and Airway Remodeling
HE staining revealed the degree of emphysema. Compared to the Con group (Figure 1A), LPS administration increased the mean linear intercept (MLI; Figure 1B). 4 weeks of AE significantly decreased the MLI (Figure 1C) compared to the COPD group. Similarly, the MLI increased after LPS administration (P < 0.001), whereas exercise intervention decreased the MLI (P = 0.0005) (Figure 1D). The difference in the MLI between the Con and COPD + AE groups was significant (P = 0.0218).
 |
Figure 1 Detection of emphysema. Abbreviation: MLI, mean linear intercept. Notes: (A) Con group. (B) COPD group. (C) COPD+AE group. Hematoxylin-eosin staining revealed evidence of the degree of emphysema. (D) Detection of MLI. LPS administration increased the MLI, while 4 weeks of exercise intervention significantly decreased the MLI. The difference in MLI was significant between the Con and COPD + AE groups. #P<0.05 compared with the Con group. *P<0.05 compared with the COPD group (magnification, x100). |
Protective Effects of AE Against Pulmonary Fibrosis
Masson staining revealed the degree of pulmonary fibrosis. Compared to that in the Con group (Figure 2A), the degree of pulmonary fibrosis in the airway wall increased after LPS administration (Figure 2B). Compared to the COPD group, exercise intervention decreased the degree of pulmonary fibrosis in the airway wall (Figure 2C). Similarly, the fibrotic score increased after LPS administration (P < 0.001), whereas exercise intervention decreased the fibrotic score (P < 0.001; Figure 2D).
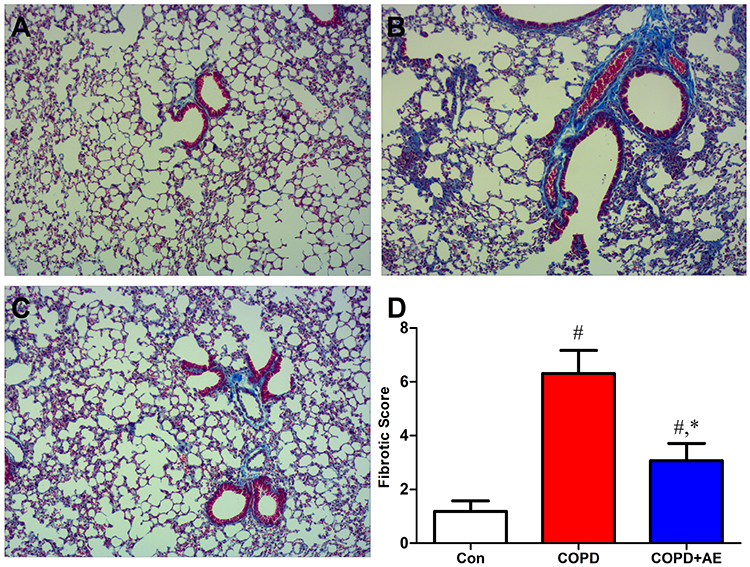 |
Figure 2 Detection of pulmonary fibrosis. Notes: (A) Con group. (B) COPD group. (C) COPD+AE group. (D) Detection of the fibrotic score. Mason staining revealed evidence of the degree of pulmonary fibrosis. The fibrotic score increased after LPS administration, while exercise administration decreased fibrotic score. #P<0.05 compared with the Con group. *P<0.05 compared with the COPD group (magnification, x200). |
Sirius Red staining revealed evidence of collagen fiber deposition. Compared to the Con group (Figure 3A), LPS administration increased collagen fiber deposition in the airway wall (Figure 3B). Compared to the COPD group, 4 weeks of exercise intervention decreased collagen fiber deposition in the airway wall (Figure 3C). Similarly, airway collagen increased after LPS administration (P < 0.001), whereas exercise intervention decreased the fibrotic score (P < 0.001; Figure 3D).
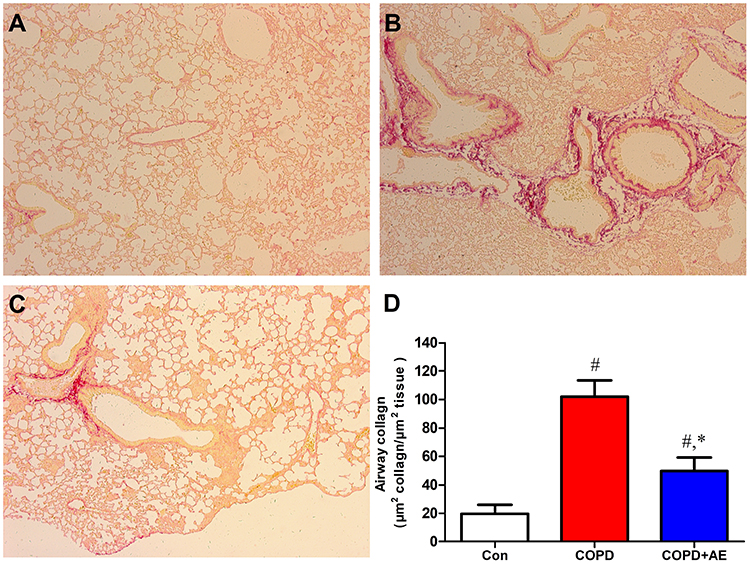 |
Figure 3 Detection of airway fibrosis. Notes: (A) Con group. (B) COPD group. (C) COPD+AE group. (D) Detection of collagen fiber deposition in the airway wall. The airway collagen increased after LPS administration, while exercise administration decreased fibrotic score. The results were expressed as μm2 of collagen fibers per μm2 of tissue area. #P<0.05 compared with the Con group. *P<0.05 compared with the COPD group (magnification, x40). |
LPS administration increased the levels of pro-fibrotic factor TGF-β in the BALF compared to the Con group (P < 0.001; Table 3). Compared to the COPD group, AE decreased the levels of TGF-β (P = 0.0285) in the BALF. LPS administration increased the gene expression levels of profibrotic factors IL-17 (P < 0.001; Figure 4C), TGF-β (P < 0.001; Figure 4F), and MMP-9 (P < 0.001; Figure 4G) compared to the Con group. 4-week AE regimen decreased gene expression levels of IL-17 (P < 0.001), TGF-β (P < 0.001), and MMP-9 (P < 0.001) compared to the COPD group.
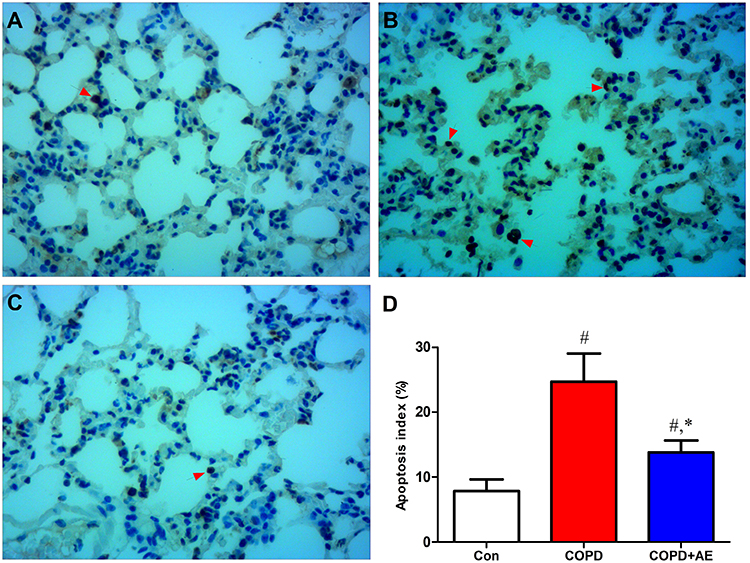 |
Figure 4 Detection of gene expression levels in lung tissue. Notes: The gene expression levels of pro-inflammatory factors IL-1β (A), IL-17 (C), TNF-α (D), and CXCL1 (E) and profibrotic factors TGF-β (F) and MMP-9 (G) and decreased anti-inflammatory factor IL-10 (B) and lung-protective factor sirt1 (H) were detected. #P<0.05 compared with the Con group. *P<0.05 compared with the COPD group. |
Protective Effects of AE Against Cell Apoptosis
TUNEL staining revealed evidence of cell apoptosis in the lung tissue. In the control group, a small number of cells were apoptotic (Figure 5A), and many apoptotic cells were detected after LPS administration (Figure 5B). AE of COPD mice inhibited the number of apoptotic cells (Figure 5C). Similarly, the apoptosis index increased after LPS administration (P < 0.001), while exercise intervention decreased it in mice with COPD (P < 0.001; Figure 5D).
 |
Figure 5 Detection of cell apoptosis. Notes: (A) Con group. (B) COPD group. (C) COPD+AE group. (D) Detection of apoptosis index. TUNEL staining revealed evidence of cell apoptosis in the lung. LPS administration increased the number of apoptotic cells (red arrow) and apoptosis index. 4 weeks of exercise intervention decreased the number of apoptotic cells and apoptosis index in COPD mice. #P<0.05 compared with the Con group. *P<0.05 compared with the COPD group (magnification, x200). |
AE Attenuates Oxidative Stress Injury in Lung Tissue
The levels of MDA (Figure 6A), MPO (Figure 6B), GSH (Figure 6C), and SOD (Figure 6D) were detected as indicators of oxidative stress injury. After LPS administration, the levels of MDA (P < 0.001) and MPO (P < 0.001) increased, while the levels of SOD (P < 0.001) and GSH (P < 0.001) decreased. Compared to the COPD group, 4 weeks of exercise intervention upregulated the levels of SOD (P < 0.001) and GSH (P < 0.001) but downregulated the levels of MDA (P < 0.001) and MPO (P < 0.001).
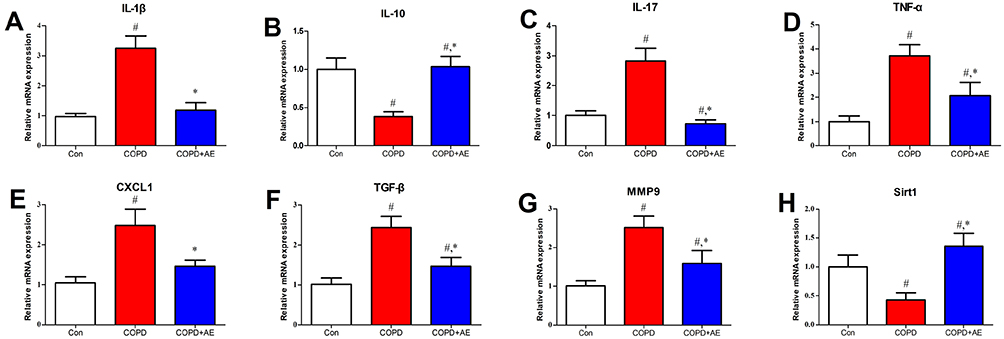 |
Figure 6 Detection of oxidative stress injury. Notes: The levels of MDA (A), MPO (B), GSH (C), and SOD (D) as indicators of oxidative stress injury were detected. LPS administration increased the levels of MDA and MPO and decreased the levels of SOD and GSH. 4 weeks of exercise intervention decreased the levels of MDA and MPO and increased the levels of SOD and GSH in mice with COPD. #P<0.05 compared with the Con group. *P<0.05 compared with the COPD group. |
Effects of LPS Injection and AE on Gene Expression Levels
LPS injection upregulated the gene expression levels of pro-inflammatory factors IL-1β (P < 0.001; Figure 4A), IL-17 (P < 0.001; Figure 4C), TNF-α (P < 0.001; Figure 4D), and CXCL1 (P < 0.001; Figure 4E) as well as those of profibrotic factors TGF-β (P < 0.001; Figure 4F) and MMP-9 (P < 0.001; Figure 4G) while downregulating the levels of anti-inflammatory factor IL-10 (P < 0.001; Figure 4B) and lung protective factor sirt1 (P < 0.001; Figure 4H). AE of COPD mice activated the gene expression levels of IL-10 (P < 0.001) and sirt1 (P < 0.001) but inhibited gene expression levels of CXCL1 (P < 0.001), IL-1β (P < 0.001), IL-17 (P < 0.001), MMP-9 (P < 0.001), TGF-β (P < 0.001), and TNF-α (P < 0.001).
Detection of Bronchial Mucus Cell Hyperplasia and Bronchoconstriction
Compared to the Con group (Figure 7A), LPS administration led to severe bronchial mucus cell hyperplasia and bronchoconstriction (Figure 7B). Compared to the COPD group, 4 weeks of exercise intervention improved bronchial mucus cell hyperplasia and bronchoconstriction (Figure 7C).
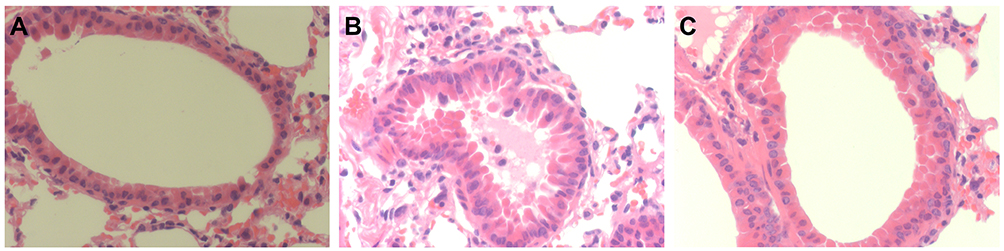 |
Figure 7 Detection of bronchial mucus cell hyperplasia and bronchoconstriction. Notes: (A) Con group. (B) COPD group. (C) COPD+AE group. LPS administration led to severe bronchial mucus cell hyperplasia and bronchoconstriction, while 4 weeks of AE improved bronchial mucus cell hyperplasia and bronchoconstriction (magnification, x400). |
Discussion
The present study aimed to investigate whether AE could protect against LPS-induced inflammation, oxidative stress, and apoptosis in mice with COPD and explore the related mechanisms.
Previous studies have demonstrated that LPS administration induces an inflammatory response, neutrophil recruitment, emphysema, bronchial mucus cell hyperplasia, and bronchoconstriction, which are the main features of COPD.21 The experimental results were consistent with previous conclusions.21–25 The results of Masson and Sirius Red staining demonstrated that 4 weeks of AE prevented the upregulation of the degree of pulmonary fibrosis, suggesting that AE had an anti-fibrotic effect. We found that LPS injection increased the BALF levels of pro-fibrogenic factors (IL-17 and TGF-β) and increased the gene expression levels of IL-17, MMP-9, and TGF-β in the lung tissue. In contrast, AE inhibited the levels of TGF-β in the BALF and decreased the gene expression levels of IL-17, MMP-9, and TGF-β, suggesting that AE exhibited an anti-fibrogenic effect in mice with COPD.
Previous studies have found that TNF-α is chemotactic for neutrophils, and anti-TNF-α antiserum administration reduces neutrophil influx into bronchoalveolar spaces.21 In the present study, we found that AE reduced the levels of TNF-α in serum and the BALF as well as the gene expression levels of TNF-α in the lung tissue. We also found that LPS administration led to a deleterious accumulation of neutrophils in the lung tissue, while 4 weeks of AE inhibited neutrophil infiltration in mice with COPD. These results demonstrate that AE inhibits neutrophil infiltration in part by decreasing TNF-α levels.
Neutrophils release a variety of cytokines, including MMP-9 and TGF-β, which play prominent roles in the development of COPD.12–14 A 4-week AE regimen prevented the upregulation of TGF-β and MMP-9 levels in mice with COPD. These results demonstrated that AE improved COPD in part by decreasing the levels of MMP-9 and TGF-β. Chronic inflammation characterized by increasing pro-inflammatory factor levels and decreasing anti-inflammatory factor levels is closely related to COPD and that this association is causal.26 In the present study, AE decreased the gene expression levels of CXCL1, IL-1β, IL-17, and TNF-α while increasing the gene expression levels of IL-10. Similarly, AE decreased BALF levels of IL-1β, TNF-α, and TGF-β, while AE increased the IL-10 levels. These results demonstrate that regular AE in COPD mice is essential in modulating the inflammatory/anti-inflammatory balance.
Previous studies have found that the oxidant/antioxidant balance plays a prominent role in the pathogenesis of COPD, and exercise improves COPD by exerting protective effects against oxidative stress through the exercise-irisin-nuclear factor erythroid 2-related factor 2 axis in mice.5,27 In the present study, we identified the antioxidant effects of exercise. We demonstrated that AE in COPD mice downregulated the levels of MDA and MPO while upregulating the levels of SOD and GSH. Hence, AE attenuated oxidative stress injury and modulated the oxidative/antioxidative balance in mice with COPD. AE is a preventive tool for COPD, and one of the mechanisms involved in this process is linked to the increase in antioxidant capacity. Moreover, sirt1 has a therapeutic effect in a COPD model, which is related to the inhibition of oxidative stress.18 These results demonstrated that AE attenuated oxidative stress in part by activating sirt1 expression in mice with COPD.
Physical exercise has been frequently used as a therapeutic tool for COPD. One of the mechanisms involved in this process is associated with increases in antioxidant and anti-inflammatory properties. Cell apoptosis is also responsible for the pathogenesis of COPD. However, the effect of AE on cell apoptosis in mice with COPD has been overlooked.6 In the present study, we found that 4 weeks of AE inhibited apoptosis in mice with COPD. Hence, AE improved COPD in part by inhibiting cell apoptosis. To the best of our knowledge, this is the first study to demonstrate that AE exerts a significant protective effect against apoptosis in mice with COPD.
Sirt1 is an important lung-protective factor, but its physiological roles in the response to AE in COPD mice are unknown. We identified that LPS administration inhibited the gene expression levels of sirt1, while a 4-week AE regimen activated these levels. By activating sirt1 expression, AE improved COPD.
Limitations
AE reduced apoptosis in mice with COPD. However, the underlying mechanism by which exercise reduces apoptosis remains unclear. Even though AE could improve exercise capacity, we did not account for the effects of exercise on the mice. Furthermore, we also did not account for the effects of exercise on the complications of COPD, such as cardiovascular comorbidities, pulmonary arterial hypertension, and muscle wasting.
Conclusions
AE is a potential therapeutic approach for treating COPD. AE improved COPD by counteracting the COPD-associated inflammatory response, oxidative stress injury, and apoptosis. In addition, AE exhibited an anti-fibrogenic effect by inhibiting the levels of the pro-fibrogenic factors IL-17, MMP-9, and TGF-β. Sirt1, which can be activated by regular AE, and played a protective role in mice with COPD.
Abbreviations
AE, aerobic exercise; COPD, chronic obstructive pulmonary disease; BALF, bronchoalveolar lavage fluid; HE, Hematoxylin and eosin; MDA, malondialdehyde; MPO, myeloperoxidase; GSH, glutathione; SOD, superoxide dismutase; IL, interleukin; TNF, tumor necrosis factor; CXCL, C-X-C motif chemokine ligand; TGF, transforming growth factor; MMP, matrix metalloproteinase; MLI, mean linear intercept; LPS, lipopolysaccharide; PFA, paraformaldehyde; sirt, silent information regulator; dUTP, terminal deoxynucleotidyl transferase; TUNEL, nick end labeling.
Data Sharing Statement
The datasets used or analyzed during the current study are available from the corresponding author Donghui Tang on reasonable request.
Acknowledgment
This study was supported by the National Natural Science Foundation of China (NSFC) (Grant No. 71874017, 81472992), the Agricultural Science and Technology Innovation Program (ASTIP) (cxgc-ias-01), and the BNU Interdisciplinary Research Foundation for the First-Year Doctoral Candidates (Grant No. BNUXKJC1814).
Disclosure
The authors report no conflicts of interest in this work, and the manuscript has been approved by all authors for publication.
References
1. Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet. 2017;389(10082):1931–1940. doi:10.1016/S0140-6736(17)31222-9
2. Labaki WW, Rosenberg SR. Chronic obstructive pulmonary disease. Ann Intern Med. 2020;173(3):ITC17–ITC32. doi:10.7326/AITC202008040
3. Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet. 2004;364(9434):613–620. doi:10.1016/S0140-6736(04)16855-4
4. Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8(3):183–192. doi:10.1038/nri2254
5. Fischer BM, Voynow JA, Ghio AJ. COPD: balancing oxidants and antioxidants. Int J Chron Obstruct Pulmon Dis. 2015;2(10):261–276. doi:10.2147/COPD.S42414
6. Sun X, Feng X, Zheng D, et al. Ergosterol attenuates cigarette smoke extract-induced COPD by modulating inflammation, oxidative stress and apoptosis in vitro and in vivo. Clin Sci. 2019;133(13):1523–1536. doi:10.1042/CS20190331
7. Stockley RA. Neutrophils and the pathogenesis of COPD. Chest. 2002;121(5):151–155. doi:10.1378/chest.121.5_suppl.151s
8. Ong J, Faiz A, Timens W, et al. Marked TGF-beta-regulated miRNA expression changes in both COPD and control lung fibroblasts. Sci Rep. 2019;9(1):18214. doi:10.1038/s41598-019-54728-4
9. Matin S, Nemati A, Ghobadi H, et al. The effect of conjugated linoleic acid on oxidative stress and matrix metalloproteinases 2 and 9 in patients with COPD. Int J Chron Obstruct Pulmon Dis. 2018;3(13):1449–1454. doi:10.2147/COPD.S155985
10. Simpson RJ, Kunz H, Agha N, et al. Exercise and the regulation of immune functions. Prog Mol Biol Transl Sci. 2015;135(355–380). doi:10.1016/bs.pmbts.2015.08.001
11. Thannickal VJ, Toews GB, White ES, et al. Mechanisms of pulmonary fibrosis. Annu Rev Med. 2004;55(1):395–417. doi:10.1146/annurev.med.55.091902.103810
12. Bellaye PS, Yanagihara T, Granton E, et al. Macitentan reduces progression of TGF-beta1-induced pulmonary fibrosis and pulmonary hypertension. Eur Respir J. 2018;52(2):1701857. doi:10.1183/13993003.01857-2017
13. Zhang J, Wang D, Wang L, et al. Profibrotic effect of IL-17A and elevated IL-17RA in idiopathic pulmonary fibrosis and rheumatoid arthritis-associated lung disease support a direct role for IL-17A/IL-17RA in human fibrotic interstitial lung disease. Am J Physiol Lung Cell Mol Physiol. 2019;316(3):L487–L497. doi:10.1152/ajplung.00301.2018
14. Wu L, Luo Z, Zheng J, et al. IL-33 can promote the process of pulmonary fibrosis by inducing the imbalance between MMP-9 and TIMP-1. Inflammation. 2018;41(3):878–885. doi:10.1007/s10753-018-0742-6
15. Rahman I, Kinnula VL, Gorbunova V, Yao H. SIRT1 as a therapeutic target in inflammaging of the pulmonary disease. Prev Med. 2012;54:20–28. doi:10.1016/j.ypmed.2011.11.014
16. Rajendrasozhan S, Yang S-R, Kinnula VL, et al. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177(8):861–870. doi:10.1164/rccm.200708-1269OC
17. Yao H, Chung S, Hwang JW, et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J Clin Invest. 2012;122(6):2032–2045. doi:10.1172/JCI60132
18. Wang X-L, Li T, Li J-H, et al. The effects of resveratrol on inflammation and oxidative stress in a rat model of chronic obstructive pulmonary disease. Molecules. 2017;22(9):1529. doi:10.3390/molecules22091529
19. Yao H, Sundar K, Ahmad T, et al. SIRT1 protects against cigarette smoke-induced lung oxidative stress via a FOXO3-dependent mechanism. Am J Physiol Lung Cell Mol Physiol. 2014;306(9):816–828. doi:10.1152/ajplung.00323.2013
20. Cantó C
21. Stolk J, Rudolphus A, Davies P, et al. Induction of emphysema and bronchial mucus cell hyperplasia by intratracheal instillation of lipopolysaccharide in the hamster. J Pathol. 1992;167(3):349–356. doi:10.1002/path.1711670314
22. Stolk J, Rossie W, Dijkman JH. Apocynin improves the efficacy of secretory leukocyte protease inhibitor in experimental emphysema. Am J Respir Crit Care Med. 1994;150:1628–1631. doi:10.1164/ajrccm.150.6.7952625
23. Wang X, Wang Y, Zhao X, et al. Potential effects of peroxisome proliferator-activated receptor activator on LPS-induced lung injury in rats. Pulm Pharmacol Ther. 2009;22(4):318–325. doi:10.1016/j.pupt.2009.01.004
24. Jones B, Donovan C, Liu G, et al. Animal models of COPD: what do they tell us? Respirology. 2017;22(1):21–32. doi:10.1111/resp.12908
25. Ghorani V, Boskabady MH, Khazdair MR, et al. Experimental animal models for COPD: a methodological review. Tob Induc Dis. 2017;2:15–25. doi:10.1186/s12971-017-0130-2
26. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
27. Kubo H, Asai K, Kojima K, et al. Exercise ameliorates emphysema of cigarette smoke-induced COPD in mice through the exercise-irisin-Nrf2 axis. Int J Chron Obstruct Pulmon Dis. 2019;14:2507–2516. doi:10.2147/COPD.S226623
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.