Back to Archived Journals » Orphan Drugs: Research and Reviews » Volume 6
Adverse events reported for hereditary angioedema medications: a retrospective study of spontaneous reports submitted to the EudraVigilance database, 2007-2013
Received 19 August 2015
Accepted for publication 29 January 2016
Published 3 May 2016 Volume 2016:6 Pages 1—8
DOI https://doi.org/10.2147/ODRR.S94790
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Abdel Abdel-Rahman
Lise Aagaard,1 Anette Bygum,2
1Section for Clinical Pharmacology, Institute of Public Health, Faculty of Health Sciences, University of Southern Denmark, 2Department of Dermatology and Allergy Centre, Odense University Hospital, Odense, Denmark
Abstract: Information about long-term safety issues from use of orphan drugs in treatment of hereditary angioedema (HAE) is limited and must be studied further. As clinical trials in patients with rare diseases are limited, prescribers and patients have to rely on spontaneous adverse drug reaction (ADR) reports for obtaining major information about the serious, rarely occurring, and unknown ADRs. In this study, we aimed to characterize ADRs reported for HAE medications in Europe from 2007 to 2013. ADR reports submitted for C1-inibitors and bradykinin receptor antagonists to the European ADR database, EudraVigilance (EV), were included in this study. The ADR reports were categorized with respect to age and sex of the patients, category of the reporter, type and seriousness of the reported ADRs, and medications. The unit of analysis was one adverse event (AE). Totally, 187 AEs were located in EV, and of these, 138 AEs were reported for Cinryze® (C1-inhibitor) (73% of the total) and 49 AEs for Firazyr® (icatibant) (26% of the total AEs). Approximately 60% of all AEs were serious, including three fatal cases. Less than 5% of AEs were reported in children. In total, 62% of AEs were reported for women and 38% for men. For both Cinryze® and Firazyr®, the majority of reported AEs were of the type “general disorders and administration site conditions”. For Cinryze®, a large number of AEs of the type “HAE” and “drug ineffective” was reported, but only few of these were serious. For Firazyr®, several nonserious reports on injection site reactions were reported. In conclusion, this study showed that in EV, several ADR reports from use of HAE medications were identified, and a large number of these were serious, including fatal cases.
Keywords: orphan drugs, hereditary angioedema, adverse drug reactions, adverse events, C1-inhibitor, Cinryze®, icatibant, Firazyr®, EudraVigilance
Introduction
Hereditary angioedema (HAE) is an autosomal dominant and rare disease which is caused by either a diminished level (HAE type 1) or dysfunction (HAE type 2) of complement C1-inhibitor.1–3 Complement C1-inhibitor deficiency causes an uncontrolled activation of the contact system with excessive bradykinin formation as the main outcome.1–3 Symptoms of HAE are recurrent angioedema attacks without urticarial signs, typically characterized by swelling of the skin and/or mucosa. Angioedema episodes most frequently affect the extremities, abdomen, head/neck area, and the upper respiratory airways.1–3 HAE patients do not respond to traditional angioedema treatments, for example, antihistamines, glucocorticoids, and epinephrine, and for many years, HAE patients have been treated off-label with androgens and/or tranexamic acid. Hence, the need for development of more effective and specific HAE treatments has been prevailing for both patients and health care professionals.4–6 Traditionally, the pharmaceutical industry did not find it profitable to develop medicines for rare diseases, but with the worldwide implementation of national Orphan Diseases Acts, it has became more economically profitable to develop specific medications for treatment of HAE, the so-called orphan drugs.7,8 In the European Union (EU), a rare disease is defined as any disease occurring in less than 5 in 10,000 people, and consequently, orphan drugs are being licensed under favorable conditions with respect to demonstration of efficacy and safety.8 The EU Orphan Disease Act has led to the development of several new HAE-specific medical treatments, for example, C1-inhibitors,9–11 bradykinin receptor antagonists (icatibant),12,13 and the recombinant plasma kallikrein inhibitor ecallantide (Kalbitor®; Dyax Corp. Burlington, Vermont, USA).14–16 Lack of sufficient knowledge of long-term safety aspects from use of orphan drugs in treatment of HAE patients makes spontaneous ADR reporting systems an important source of information about medicine safety.17 As premarketing clinical trials within the area of rare diseases are scarce, clinicians and health authorities have to rely on spontaneous reports as the major source of new information on ADRs, particularly data on serious and rarely occurring ADRs.17 Since 2012, researchers have been allowed to access the EU ADR database, EudraVigilance (EV), and this has opened for cross-national analysis based on a standardized reporting format.18 By February 2016, no studies have systematically analyzed postmarketing safety data reported for orphan drugs licensed for treatment of HAE and submitted to national ADR databases in Europe.19 The aim of this study was to characterize the adverse events (AEs) reported for orphan drugs licensed for treatment of HAE submitted to the EV database from 2007 to 2013.
Materials and methods
Study design
We retrospectively reviewed ADR reports submitted to the EV database for orphan drugs licensed for treatment of HAE in Europe. The reported AEs were analyzed with respect to their type and seriousness, age and sex of the patient, suspected medicines, and type of the reporter. The unit of analysis was one ADR. The age groups of the patients were categorized into two: children (0–17 year olds) and adults (≥18 year olds). AEs reported for HAE medications licensed through the central licensing procedure in the EU by the European Medicines Agency (EMA) were included in this study. The included HAE medications were Berinert® (CLS Behring, Danderyd, Sweden) Cinryze® (Shire, Danderyd, Sweden), and Ruconest® (Shire, Danderyd, Sweden) (C1-inhibitors – ATC code B06AC01) and Firazyr® (Shire, Danderyd, Sweden) (icatibant) (bradykinin receptor antagonist – ATC code B06AC02). Table 1 displays the characteristics of the included HAE medications, their approved indications and age groups, time of licensing in the EU, name of the marketing authorization holder, and occurrence and type of reported adverse drug reactions listed in the latest version of the official product information. Information about the orphan drugs was retrieved through the website of the EMA. The C1-inhibitor Berinert®, which is also licensed for acute and prophylactic treatment of HAE, was excluded from this study. Berinert® is not centrally authorized in the EU, and therefore, we could not get access to the ADR reports reported for Berinert® through EMA.
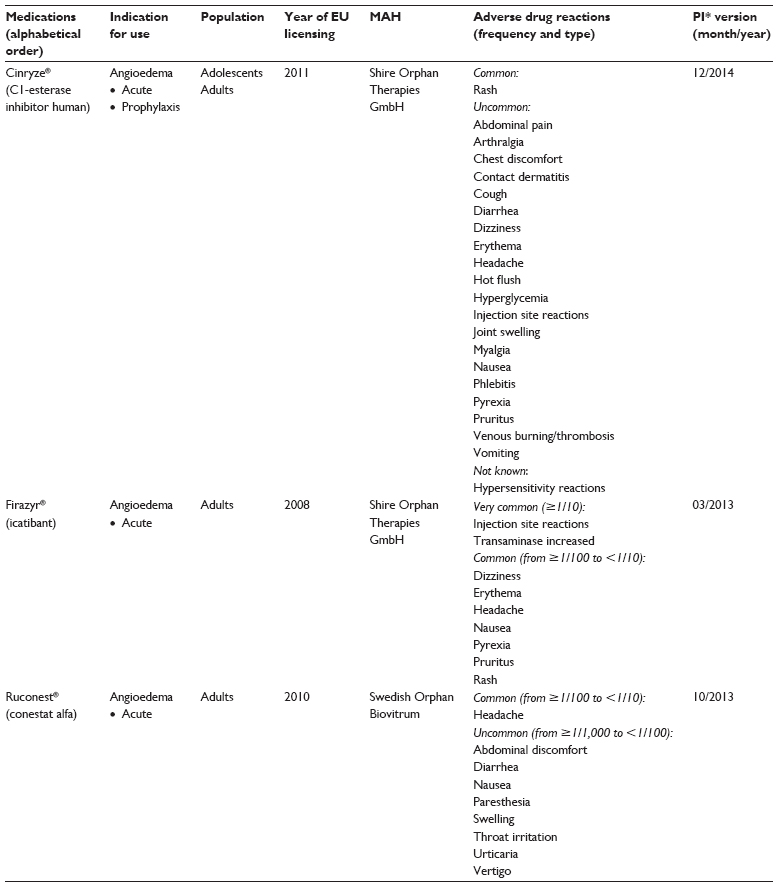 | Table 1 Characteristics of orphan drugs licensed for treatment of hereditary angioedema in Europe and information about adverse drug reactions (frequency and type) available in the official product information |
Material
The EV database contains information about all suspected spontaneous ADR reports and the ADRs reported in clinical trials and postmarketing for all medicinal products authorized in the EU.20 ADR data should be transmitted electronically to the database in accordance with the International council on harmonisation of technical requirements for registration of Pharmaceuticals for human use (ICH E2B) (R2) standard.21 The EV database is not publicly accessible, and authorization for data access was given by the EMA.22 ADR information was provided for this study in anonymous form with encrypted personal identification.23 Data extraction and data analyses of the raw material were comprehensive and time consuming. ADR information was extracted from the EV database with respect to the type of reporter and the criteria of seriousness and medications for which the AEs were reported. Criteria of seriousness were evaluated and made by the reporters and the evaluations were confirmed by the national regulatory agencies before forwarding the ADR data to the EV database. The reported AEs were coded according to their type and seriousness using the Council for International Organizations of Medical Sciences criteria by the academic staff in the national regulatory agencies.24 ADR data were extracted from the EV database in Microsoft Excel files using the following criteria: patient’s sex and age, medicines (active substance), adverse drug reaction, and severity.22 The material comprised all ADR reports submitted to the EV database from 2007 to 2013. Data were extracted from the EV database and delivered to us as several large Excel files. In STATA® statistical software package (StataCorp LP, College Station, TX, USA), the Excel files were merged into one major file and the ADR reports were searched for duplicates. Data analysis including coding of ADR reports was conducted in an Access database. Each ADR report may refer to one or more suspected ADR(s) as well as to one or more medicinal products. In this study, we only included the AEs reported for medications, which were listed as suspect drug by the reporter, meaning that a causal relationship between the reported ADR and the drug was expected.
Classification of AEs by type and seriousness
The different types of reported AEs were classified according to the Medical Dictionary for Regulatory Activities System Organ Class (SOC).24 Serious AEs were defined as: fatal, life-threatening, requiring hospitalization or prolongation of existing hospitalization, and resulting in persistent or significant disability/incapacity in the reporter’s assessment, in a congenital anomaly/birth defect, and other medically important conditions. All other AEs were classified as nonserious.24 The scientific staffs working in the different national regulatory agencies made the seriousness evaluations, and these evaluations were forwarded to us together with the copies of the ADR reports.
Ethical approval
According to Danish law, no ethical approval was required for this study.
Results
From 2007 to 2013, a total of 79 individual ADR reports containing information about 187 AEs were found in EV for HAE medications licensed by the EMA (Table 2). For Cinryze®, a total of 60 ADR reports covering information about 138 AEs were reported, and for Firazyr®, the number was 19 reports and 49 AEs. No ADR reports were found for Ruconest® in the EV database. Health care professionals reported all ADR cases. In total, 62% of AEs were reported for women and 38% for men. Approximately 5% of AEs were reported in 0–17 year olds. Approximately 10% of ADR reports were found without information about patient age in EV.
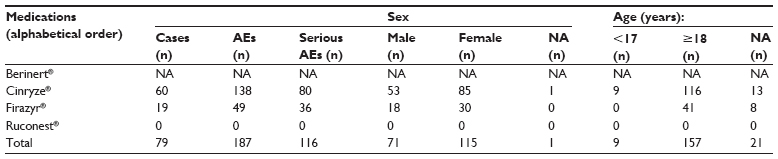 | Table 2 Distribution of cases and adverse drug reactions (AEs) for orphan drugs licensed for treatment of HAE by medication, seriousness, age, and sex, found in the EudraVigilance database, 2007–2013 |
AEs by seriousness
In total, 60% (n=116) of all reported AEs (n=187) were classified as serious, and among these, three fatal cases were reported (Table 2). For Cinryze®, the share of serious AEs was 57% (n=80) and for Firazyr®, it was 70% (n=36). The characteristics of the three fatal cases are displayed in Table 3. For Cinryze®, one fatal case of sudden death presumably due to “basilar artery thrombosis” was reported in a 52-year-old male, and one fatal case due to the AEs “cytomegalovirus infection” and “cytomegalovirus test positive” in a male infant was also found in EV for Cinryze®. For Firazyr®, one fatal case of sudden death presumably due to “myocardial infarction” was reported in a 67-year-old male.
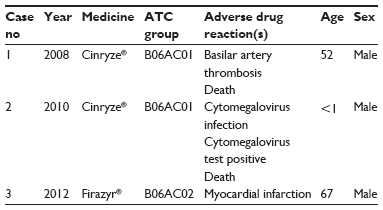 | Table 3 Fatal adverse drug reaction cases reported for Cinryze® and Firazyr® in Europe, 2007–2013 |
AEs by type and seriousness
Table 4 shows the distribution of reported AEs by SOC, seriousness, and medicine. For Cinryze®, the largest shares of AEs were reported for the SOCs “general disorders and administration site conditions” (25% of the total AEs) and “skin and subcutaneous disorders” (25% of the total AEs). For Cinryze®, the majority of serious AEs were also found within the SOC “general disorders and administration site conditions” (60% of the reported AEs). For Firazyr®, majority of the reported AEs were of the type “general disorders and administration site conditions” (49% of the total AEs), and approximately 45% of these AEs were serious.
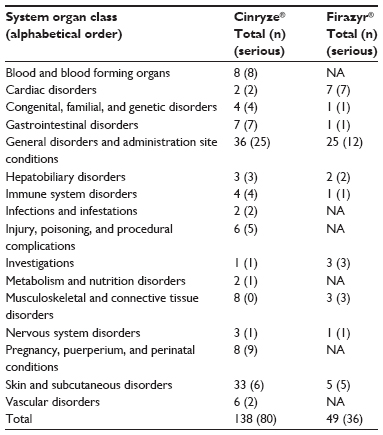 | Table 4 Number of adverse drug reactions reported for HAE medications distributed by system organ class, seriousness (in parentheses), and medicine, 2007–2013 |
AEs by medication
Table 5 displays the number of reported AEs by medicines and seriousness. For Cinryze®, the largest number of reported AEs was “HAE” (n=25) and “drug ineffective” (n=10); however, only a few of these ADR cases were serious. For Firazyr®, a large number of nonserious ADR reports on injection site reactions were found in EV. For both Cinryze® and Firazyr®, only 10% of the reported AEs were already mentioned in the official product information.
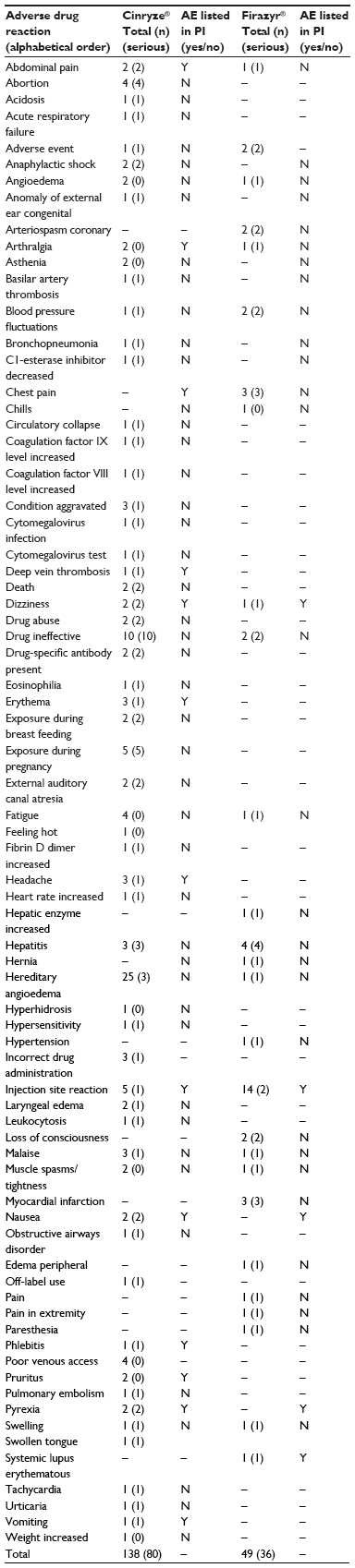 | Table 5 Number of reported adverse events by type (preferred term), seriousness (in parentheses), and medicine reported in Europe from 2007 to 2013 |
Discussion
This is the first study conducted to analyze postmarketing ADR data reported for orphan drugs licensed for treatment of HAE in Europe and submitted to the EV ADR database. Only few ADR reports, all for the medications Cinryze® and Firazyr®, were found in EV, and a large number of these were serious. More than one half of AEs were reported for women, and physicians reported all the AEs. A large number of AEs reported for Cinryze® and Firazyr® were of the type “general disorders and administration site conditions”. The majority of AEs reported for both Cinryze® and Firazyr® were not mentioned in the official product information.17
Reported AEs by type and seriousness
The majority of AEs reported for Cinryze® and Firazyr® were of the type “general disorders and administration site conditions”, for example, injection site reactions, and this ADR pattern was expected due to their route of administration parenteral. Additionally, for Cinryze®, a large number of AEs of the type “skin and subcutaneous disorders” were also reported, and these reactions were all listed in the product information. The findings are in line with the ADR patterns reported in another study monitoring the safety of use of C1-inbibitors in different clinical settings in the US and in Europe.25 Thromboembolic events due to administration of C1-inhibitors have been of concern and discussed in the literature.26,27 In this study, only few cases were reported for Cinryze® (“basilar artery thrombosis” “increased level of coagulation factors IX and VIII”, “deep vein thrombosis”, “pulmonary embolism”, and “tachycardia”). Analyses of postmarketing data submitted to the US Food and Drug Administration ADR database showed ten cases of thromboembolic events from use of Cinryze®.28,29 More than one half of the AEs found in EV were serious; however, this reporting pattern was not surprising, since physicians – from a clinical perspective – may find it more relevant to report serious reactions than nonserious reactions.30 Due to the increasing number of patients diagnosed with HAE, more patients will get access to treatment with orphan drugs, despite the problems with patients’ access due to high prices and national restrictions in patients’ access,31 and therefore, a larger number of ADR reports occurring from use of these medications is expected to be reported in the future.31–33
Reported AEs by therapeutic groups and medications
The largest number of AEs was reported for the medication Cinryze®, which was expected since Cinryze® is frequently recommended and used in HAE patients.4–6 As expected, no ADR reports were found for Ruconest®, because this medicine is not prescribed very often for treatment of HAE. For Cinryze®, a large number of AEs of the type “HAE” were reported, indicating that the medicine may increase the severity of the disease or that the patients/physicians have difficulties in distinguishing between disease symptoms and possible AEs from medication use. The use of orphan drugs in treatment of HAE is considered being low due to the small disease prevalence in EU, the price of the treatments,28 and limited access to these treatments in some member states.31 Therefore, we also expected a low number of ADR reports present in EV, and our assumption was confirmed. However, the many serious cases may also indicate that health care professionals and patients report only unknown and serious events, which is also mandatory for the health care professionals operating in the individual EU member states. However, it is also well-known that a high rate of underreporting is biasing the spontaneous reporting systems and probably a much higher number of AEs have occurred in real life.34 Therefore, we encourage the health care professionals and consumers/patients to continue reporting of suspected AEs in order to increase knowledge about ADRs from use of orphan drugs.
Study limitations
The strength of this study is that it comprised all AEs reported for HAE medications in Europe, which were forwarded to the EV database during a 7-year period, hence a limitation to this study is that we do not know to which extent the causality of the reported AEs can be confirmed, and this has implications for the interpretation of the findings. The respective pharmaceutical companies make causality assessment of ADR cases as a part of their legal obligations to monitor the safety of their marketed drugs. In this study, we did not evaluate the validity of the ADR reports since we only had access to the data entered into the EV database and not the ADR original reports; however, we expect the data to be valid, as they were predominantly entered into EV by the regulatory agencies. Spontaneous reporting systems suffer from various barriers, such as incomplete recognition of AEs, administrative barriers to reporting, and low data quality, all of which may result in underreporting of important serious and rarely occurring AEs.
Hence, AEs that are classified as being nonserious or already known may be underreported; however, this study provides information on the reported AEs, which contributes to broadening the knowledge on medicine safety. Hence, it is not possible to generalize from data reported to the EV database to the other EU member states, as the prescribing practice and disease diagnostic procedures may be very different. Although ADR reporting is mandatory in the EU, it is unknown whether the analyzed ADR data represent all information available in each country, and therefore, the actual ADR prevalence could not be estimated. Spontaneous reports are an important source of information about new and previously unrecognized AEs, and the value of spontaneous reporting schemes lies in their ability to act as hypothesis-generating procedures.35 Therefore, EMA and the national regulatory agencies should continue to systematically survey and analyze AEs reported by health care professionals/consumers and patients in order to signal previously unknown and rarely occurring AEs presumably related to use of orphan drugs.
Conclusion
Only a few AEs from use of HAE medications were identified in the EV database, but a large majority of these were serious, including fatal cases. There is a need of more research into the prescribing of these medicines to patients with HAE, as well as tighter reporting of AEs for HAE medicines prescribed for this population, particularly from use in children and adolescents.
Acknowledgments
The authors would like to thank the European Medicines Agency for providing data.
Author contributions
LA and AB designed the study, analyzed data, and wrote the first version of the manuscript. LA carried out the sampling. Both authors saw and approved the final version of the manuscript. No sources of funding were used to assist in the preparation of this study.
Disclosure
The authors report no conflicts of interest in relation to the content of this manuscript.
References
Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. 2006;119(3):267–274. | |
Rasmussen ER, Bindslev-Jensen C, Bygum A. Angioedema – assessment and treatment. Tidsskr Nor Legeforen. 2012(21); 2391–2395. | |
Bowen T, Cicardi M, Farkas H, et al. 2010 International consensus algorithm for the diagnosis, therapy and management of hereditary angioedema. Allergy Asthma Clin Immunol. 2010;6(1):24. | |
Cicardi M, Bork K, Caballero T, et al. Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy. 2012;67(2):147–157. | |
Bork K. Current drugs in early development for hereditary angioedema: potential for effective treatment. Expert Opin Investig Drugs. 2014;23(7):887–891. | |
Bygum A. Hereditary angioedema—consequences of a new treatment paradigm in Denmark. Acta Derm Venereol. 2014;94(4):436–441. | |
Wilson DA, Bork K, Shea EP, Rentz AM, Blaustein MB, Pullman WE. Economic costs associated with acute attacks and long-term management of hereditary angioedema. Ann Allergy Asthma Immunol. 2010; 104(4):314–320. | |
Eur-Lex.europa.eu [website on the Internet]. Regulation (EC) no. 141/2000 of the European Parliament and of the council of 16 December 1999 on orphan medicinal products; 1999. Available from: http://ec.europa.eu/health/files/eudralex/vol-1/reg_2000_141/reg_2000_141_en.pdf. Accessed December 7, 2015. | |
Craig TJ, Levy RJ, Wasserman RL, et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J Allergy Clin Immunol. 2009;124(4):801–808. | |
Zuraw B, Cicardi M, Levy RJ, et al. Recombinant human C1-inhibitor for the treatment of acute angioedema attacks in patients with hereditary angioedema. J Allergy Clin Immunol. 2010;126(4):821–827. | |
Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363(6):513–522. | |
Cicardi M, Banerji A, Bracho F, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med. 2010; 363(5):532–541. | |
Lumry WR, Li HH, Levy RJ, et al. Randomized placebo-controlled trial of the bradykinin B2 receptor antagonist icatibant for the treatment of acute attacks of hereditary angioedema: the FAST-3 trial. Ann Allergy Asthma Immunol. 2011;107(6):529–537. | |
Cicardi M, Levy RJ, McNeil DL, et al. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010; 363(6):523–531. | |
Levy RJ, Lumry WR, McNeil DL, et al. EDEMA4: a phase 3, double-blind study of subcutaneous ecallantide treatment for acute attacks of hereditary angioedema. Ann Allergy Asthma Immunol. 2010;104(6):523–529. | |
Schneider L, Lumry W, Vegh A, Williams AH, Schmalbach T. Critical role of kallikrein in hereditary angioedema pathogenesis: a clinical trial of ecallantide, a novel kallikrein inhibitor. J Allergy Clin Immunol. 2007;120(2):416–422. | |
Aagaard L, Hansen EH. Information about ADRs explored by pharmacovigilance approaches: a qualitative review of studies on antibiotics, SSRIs and NSAIDs. BMC Clin Pharmacol. 2009;9:4. | |
EU directive of the European Parliament and of the council amending Directive 2001/83/EC as regards information to the general public on medicinal products subject to medical prescription. COM (2012) 48 final. Brussels; 2012. Available from: http://eur-lex.europa.eu/index.html. Accessed December 7, 2015. | |
Sabharwal G, Craig T. Recombinant human C1 esterase inhibitor for the treatment of hereditary angioedema due to C1 inhibitor deficiency C1-INH-HAE. Expert Rev Clin Immunol. 2015;11(3):319–327. | |
European Medicines Agency: Note for guidance – EudraVigilance Human –Processing of safety messages and individual case safety reports (ICSRs). EMA/H/20665/04/Final Rev 2. Available from: https://eudravigilance.ema.europa.eu/human/docs/guid-P-Technical%20 Documentation-EMEA-H-20665-04-en-Final_Revision_2.pdf. Accessed December 7, 2015. | |
European Medicines Agency: Volume 9. Pharmacovigilance: medicinal products for human use and veterinary products. Available from: http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev9.html. Accessed December 7, 2015. | |
Office Journal of the European Commission. Regulation (EC) No 1049/2001 of the European Parliament and of the Council of 30 May 2001 regarding public access to European Parliament, Council and Commission documents. L 145/43. Available from: www.europarl.europa.eu/register/pdf/r1049_en.pdf. Accessed December 7, 2015. | |
Health Action International Europe: Patient reporting of adverse drug reactions – seminar report; 2005. Available from: http://haieurope.org/wp-content/uploads/2015/09/Direct-Patient-Reporting-in-the-EU.pdf. Accessed December 7, 2015. | |
MedDRA. Available from: http://www.meddra.org/. Accessed December 7, 2015. | |
Busse P, Bygum A, Edelman J, et al. Safety of c1-esterase inhibitor in acute and prophylactic therapy of hereditary angioedema: findings from the ongoing international Berinert patient registry. J Allergy Clin Immunol Pract. 2015;3(2):213–219. | |
Kalaria S, Craig T. Assessment of hereditary angioedema treatment risks. Allergy Asthma Proc. 2013;34(6):519–522. | |
Zuraw LB, Kalfus I. Safety and efficacy of prophylactic nanofiltered C1-inhibitor in hereditary angioedema. Am J Med. 2012;125(9):938.e1–e7. | |
Crowther M, Bauer K, Kaplan AP. The thrombogenicity of C1 esterase inhibitor (human): Review of the evidence. Allergy Asthma Proc. 2014;35(6):444–453. | |
Ghandi PK, Gentry WM, Bottorf MB. Thrombotic events associated with C1 esterase inhibitor products in patients with hereditary angioedema: investigations from the United States Food and Drug Administration Adverse Event Reporting System database. Pharmacotherapy. 2012(10);32:902–909. | |
Herdeiro MT, Figueiras A, Polónia J, Gestal-Otero JJ. Physicians’ attitudes and adverse drug reaction reporting: a case-control study in Portugal. Drug Saf. 2005;28(9):825–833. | |
Aagaard L, Kristensen K. Access to cross-border health care services for patients with rare diseases in the European Union. Orphan Drugs: Res Rev. 2014:4;39–45. | |
Westermark K, Holm BB, Söderholm M, et al. European regulation on orphan medicinal products: 10 years of experience and future perspectives. Nat Rev Drug Discov. 2011;10(5):341–349. | |
Riedl MA, Lumry WR, Busse P, et al. Prevalence of hereditary angioedema in untested first-degree blood relatives of known subjects with hereditary angioedema. Allergy Asthma Proc. 2015;36(3):206–212. | |
Pérez García M, Figueras A. The lack of knowledge about the voluntary reporting system of adverse drug reactions as a major cause of underreporting: direct survey among health professionals. Pharmacoepidemiol Drug Saf. 2011;20(12):1295–1302. | |
Kesselheim AS, Gagne JJ. Strategies for postmarketing surveillance of drugs for rare diseases. Clin Pharmacol Ther. 2014;95(3):265–268. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.