Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 5
Advances in the management of relapsing–remitting multiple sclerosis: role of oral dimethyl fumarate (BG-12)
Authors Nielsen AS
Received 1 March 2015
Accepted for publication 21 April 2015
Published 21 May 2015 Volume 2015:5 Pages 51—61
DOI https://doi.org/10.2147/DNND.S68723
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Müller
A Scott Nielsen1,2
1Virginia Mason Multiple Sclerosis Center, Seattle, WA, USA; 2Department of Neurology, University of Washington, Seattle, WA, USA
Abstract: Multiple sclerosis is a complex and chronic inflammatory disease of the central nervous system which affects an estimated 2.3 million individuals worldwide. Genetic research has uncovered over 100 immune-related genes associated with the disease and has provided a multitude of potential therapeutic targets. To date, 13 US Food and Drug Administration-approved disease-modifying therapies designed to influence the aberrant immune system are available for the indication of relapsing forms of the disease. BG-12 is a novel oral multiple sclerosis therapeutic with a unique putative mechanism of action that activates the Nrf2 anti-oxidant pathway. Despite the enthusiasm for multiple therapeutic options, including oral options, the practitioner is faced with the difficult task of providing guidance for patients regarding optimal sequencing of therapeutics without sensitive clinical biomarkers to match a particular therapy’s putative mechanism of action to the patient’s specific pathophysiology. Moreover, while BG-12 has a preferred route of administration, there is limited safety data with which to guide counseling in the clinic. Dimethyl fumarate (DMF or BG-12) is one of three available oral therapies which will be discussed in this review in terms of its pharmacokinetic profile, putative mechanism of action, clinical effectiveness, safety, tolerance, and patient-reported experience. BG-12’s potential as a first-line therapy and as a sequencing therapeutic to aid in transition off natalizumab will be discussed.
Keywords: BG-12, disease-modifying therapy, Tecfidera
Introduction: natural history, epidemiology, and therapeutic landscape of multiple sclerosis
Multiple sclerosis (MS) is classically described as a chronic inflammatory condition of the central nervous system (CNS) resulting in demyelination and axonal injury.1 The timing of these inflammatory insults are unpredictable and may result in transient bouts of neurologic dysfunction that is followed by full or partial recovery.2 While approximately 85% of individuals with MS initially experience a “relapsing–remitting” course, 15% experience a progressive course of unrelenting accumulation of disability – with or without superimposed relapses – from the outset. The advent of magnetic resonance imaging (MRI) has provided in vivo inflammatory markers of the disease that have been parlayed into successful proof-of-concept experiments, Phase II and pivotal clinical trial data, ultimately hastening the regulatory approval of our current disease-modifying therapies (DMTs).3,4 At the time of this publication, there exist 13 US Food and Drug Administration (FDA)-approved therapies for the indication of relapsing–remitting MS, with several more in early- and late-phase development (Table 1).5
 | Table 1 Landscape of current and emerging DMTs for multiple sclerosis |
MS usually presents between the ages of 20 and 50, affects nearly all ethnic groups with a predilection for northern European ancestry, and disproportionately affects women more than men.2 The National MS Society estimates that 2.3 million individuals worldwide are living with MS. Epidemiologic studies have typically demonstrated a latitudinal gradient of MS incidence and prevalence where the farther away from the equator a population is sampled, the greater the point estimates (prevalence estimates in Europe are reported as high as 1 in 500).6,7 This observation, in addition to immigration studies demonstrating that an individual’s risk of MS is tied to their place of residence established prior to adolescence, has supported the hypothesis that environmental factors play a key role in early MS pathogenesis – perhaps decades before the first clinical manifestations.8 A recent meta-analysis identified 44 environmental risk factors associated with MS, and three were identified as the most methodologically and statistically credible: anti-EBNA IgG seropositivity, infectious mononucleosis, and smoking.9 Recently, the hypothesis that vitamin D confers a favorable effect on MS development and clinical course through modulation of the immune system has entered more definitive clinical trial testing.10 Moreover, MS genome-wide association studies have uncovered over 100 susceptibility genes largely implicating an aberrant immune system.11 While population research has shed considerable light on the pathophysiology of MS and has suggested a complex genetic/epidemiologic model, the etiologic touchstone of the disease remains elusive.
Regardless, much is known about the natural history of the disease considering the relatively recent introduction of the first DMTs in the early 1990s. Irreversible disability in terms of ambulatory capacity from time of disease onset to early walking impairment, the need to use a unilateral ambulatory device, or the need to use a wheelchair is expected within a median time of 8, 20, and 30 years, respectively.12 Favorable early clinical predictors implicating a longer interval to experiencing disability include female sex; relapsing MS phenotype; complete recovery after the first clinical attack; a longer inter-attack interval between first and second relapses; and the location of the first clinical attack affecting sensory rather than motor tracts.13–16 Younger age at disease onset has been associated with a longer time interval between first clinical manifestations and ambulatory difficulties. However, age is a double-edged sword as later onset MS cases typically reach the same physical disability milestones at a later age, albeit, at a faster rate once the diagnosis is realized.12
A contemporary epidemiologic analysis of the French Rennes MS database has further confirmed the earlier natural history findings among the relapsing-onset subgroup in a cohort of 2,054 patients dated from 1976 to 2004, many of whom received some form of therapy (approximately 17.2% of reported patient-years were exposed to interferons, mitoxantrone, glatiramer acetate, or off-label use of various cytotoxic agents).17 The authors conducted their study under the working hypothesis that MS represents a two-stage progression paradigm: Phase 1, defined as the time from disease onset to first signs of irreversible disability; and Phase 2, defined as the epoch between the end of Phase 1 and the need to use a cane to ambulate 100 meters. Results indicate that the rate of Phase 1 progression was highly variable and appeared to be influenced by age, sex, history of relapses (particularly during the first 2 years of disease onset), and MS phenotype (defined as relapsing or progressive onset). However, the rate of Phase 2 progression was largely independent of these factors and was invariable. The authors concluded that focal inflammation occurring early in the disease course likely influences the rate of Phase 1 progression, and therefore, a narrow window of opportunity may exist to intervene therapeutically for the benefit of the patient. This study did not evaluate the timing of therapeutic exposure and its effect of disability progression.17 Although ambulatory dysfunction is a well-known manifestation of the disease, MS disability and quality of life can be greatly affected by interrelated symptoms of cognitive impairment, neuropathic pain, visual obscuration, bowel and bladder dysfunction, sleep disorders, and disabling fatigue, to name a few.18
MS poses significant challenges to the patient, physician, and society as a whole. While MS typically manifests during early adulthood at a time of educational, familial, and professional aspirations, the mean survival after disease onset may extend 45 years, resulting in decades of uncertainty, psychological stress, and physical limitations for patients and caregivers.2,19,20 Currently approved DMTs have demonstrated the ability to reduce: 1) the frequency of relapses; 2) CNS inflammatory disease activity manifest on MRI; and 3) accumulation of disability over the short-term. Published 21-year follow-up data from the pivotal interferon-β-1b clinical trial suggests a long-term mortality benefit for early exposure.21 Over the course of the past two decades, the MS clinician now find themselves in the enviable position of having access to numerous proven therapies that significantly change the MS disease course for the better. However, the choice, timing, and sequencing of DMTs can be complicated in their pragmatic application when considering variable tolerance and safety profiles and differences in putative mechanism of action. The recent advent of oral therapies for MS has generally been well received by patients who seem to prefer oral over traditional injectable DMTs.22 Faced with this patient preference, clinicians must weigh the reported benefit of these newer therapies in the context of an absence of long-term risk and safety data. This review focuses on the oral DMT dimethyl fumarate (BG-12) in context of the other approved DMTs, its potential role as a first-line agent, and consideration of BG-12 as a sequencing agent from natalizumab.
Pharmacology, mode of action, and pharmacokinetics of dimethyl fumarate (BG-12)
Over 60 years ago, the German chemist Schweckendiek, who suffered from psoriasis, hypothesized that fumaric acid esters (FAEs) may help correct a metabolic disorder of the citric acid cycle which he presumed resulted in his immunologic skin condition. After initial success with topically administered FAEs, he developed an oral version that eventually was tested and approved in 1994 in Germany for this indication, and marketed as Fumaderm® (Fumapharm AG, Lucerne, Switzerland): a mixture of dimethyl and ethylhydrogenfumarate.23,24 In 2003, Biogen purchased licensing rights to develop a second generation fumarate, dimethyl fumarate (DMF or BG-12), for psoriasis and MS.25
DMF is a lipophilic ester of fumaric acid (a component of the citric acid cycle) and is immediately cleaved by intestinal esterases into its active metabolite monomethyl fumarate (MMF).26 Among psoriasis patients, a single 120 mg dose of DMF yields a median maximal plasma MMF concentration of 0.84 mg/L while demonstrating a half-life of 44 minutes.27 MMF is absorbed into the pre-systemic circulation where it likely translocates into blood cells due to its lipophilic properties and interacts with intracellular electrophilic molecules such as glutathione, and has the capacity to pass through the blood–brain barrier.26,28,29 Elimination of MMF is primarily through the lungs via exhalation, although lesser contributions are excreted through the feces and urine (as mercapturic acids).26,27,30
While the exact mechanism of action by which BG-12 confers its benefit for MS is unknown, several observations from preclinical studies suggest pleiotropic putative mechanisms. BG-12 appears to induce an anti-inflammatory Th2 cytokine shift through reduced interferon-gamma and increased IL-4 and -5 production.31 Moreover, an anti-inflammatory and antioxidative effect is likely facilitated through BG-12’s interaction with nuclear factor-kappa B (NF-κB) and nuclear (erythroid-derived2)-related factor (Nrf2) protein complexes, respectively. BG-12 initially depletes intracellular glutathione which is followed by a rebound increase in concentration that subsequently inhibits the movement of NF-κB to the nucleus, thereby suppressing transcription of inflammatory cytokines and adhesion molecules.27,32–34
Moreover, BG-12 appears to inhibit transcription of neuroinflammatory mediators – iNOS, TNF-α, IL-1β, and IL-6 – inside activated microglia and astrocytes.35 MMF also has been observed to impair monocyte-derived dendritic cell differentiation.36 At high doses, DMF (but not MMF) has demonstrated T-cell apoptosis in vitro.37 However, it is unclear if T-cell apoptosis occurs by this mechanism in vivo as DMF is rapidly hydrolyzed into MMF rendering DMF plasma levels undetectable.28 An alternative explanation for undetectable DMF is a rapid intracellular coupling of DMF with glutathione.26 Regardless, DMF and MMF have demonstrated reduced CNS inflammatory infiltrates and improved disease course in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitic (MOG-EAE) mice.38
Alternatively, BG-12 encourages activation of a putative antioxidative stress pathway.34 At homeostasis, kelch-like ECH-associated protein (Keap-1) is bound to Nrf2 which remains in the cytosol.39 In the presence of BG-12, DMF or MMF can irreversibly bind to Keap-1 which leads to a conformational change and uncoupling of Keap-1 from Nrf2.40 Nrf2 is then free to translocate inside the nucleus where it binds to the antioxidant responsive element.41 This interaction results in the induction of antioxidative genes – which may explain the observation of prolonged survival of neurons and glial cells in vitro – and appears to mitigate the disease course of MOG-EAE mice.40–42
Comparative efficacy, safety, and tolerability of oral dimethyl fumarate (BG-12)
Efficacy
Over a decade after Fumaderm was approved in Germany, ten MS subjects were administered compounded fumarate in a pilot study that demonstrated a favorable effect on gadolinium-enhancing (Gd+) MRI brain lesions.43 A Phase II study of 257 randomized relapsing–remitting MS subjects, dosed with BG-12 at 240 mg three times a day (TID), demonstrated a reduction in new Gd+ brain lesions by 69% compared with placebo over a 12-week interval.44 A post hoc subgroup analysis of this data revealed a 34% reduction in the evolution of these enhancing lesions into T1-hypointense lesions – a putative marker of permanent axonal damage associated with irreversible disability – among subjects exposed to BG-12 compared to placebo.45
Two pivotal clinical trials of BG-12 followed: 1) Determination of the Efficacy and Safety of Oral Fumarate in RRMS (DEFINE); and 2) Comparator and an Oral Fumarate in RRMS (CONFIRM). Both trials were similar in their targeted population. Both required subjects between the ages of 18 and 55, diagnosed with relapsing–remitting MS by the McDonald criteria with at least one relapse in the previous year or 1 Gd+ lesion within 6 weeks prior to randomization, and a physical disability level defined by the Expanded Disability Status Scale (EDSS) at ≤5.0.46–49 The EDSS is an ordinal disability rating scale based on the neurologic examination that ranges from 0 (no disability) to 10 (death).48 Moreover, both studies were randomized, double-blinded, and placebo-controlled to determine the efficacy and safety of BG-12 over a 2-year period in a multinational setting. While DEFINE and CONFIRM shared equal randomization to placebo and BG-12 240 mg dosed at twice a day (BID) and TID, CONFIRM also provided a rater-blinded active comparator with glatiramer acetate 20 mg subcutaneous daily injection (superiority analysis between glatiramer and BG-12 was not performed as the study size was not designed to sufficient statistical power).46,47
Standard metrics for MS clinical trials were captured; however, the primary endpoint differed between the pivotal studies. The primary endpoint in DEFINE was the proportion of patients with a relapse while CONFIRM reported the annualized relapse rate (ARR) at 2 years. It is the author’s opinion that these two differing primary endpoints provide unique and complementary information about the study drug’s effectiveness. While ARR is commonly reported in MS pivotal trials, this metric can be skewed by a few highly inflammatory MS subjects with aggressive disease which is not representative of the entire treatment group, thereby making it difficult to extrapolate the data into routine clinical care. Alternatively, the proportion of patients with a relapse is a better estimate of the effect of BG-12 on the treatment population. Both primary endpoints for the pivotal trials were statistically significant, providing complementary proof of BG-12’s clinical effectiveness in relapsing–remitting MS.
Secondary endpoints in both studies included confirmed disability at 12 weeks and MRI metrics (new or enlarging T2-weighted lesions, Gd+ enhancing lesions) while CONFIRM had the additional MRI endpoint of new T1-hypointense lesions. Confirmed disability was defined as a 1.0 increase (or 1.5 increase if baseline EDSS was 0) in the EDSS that was documented at two separate evaluations at least 12 weeks apart. Statistically significant results based on the eventual FDA-approved dosing of 240 mg BID compared to placebo were similar between studies with a reduction in the following metrics for DEFINE and CONFIRM, respectively: ARR (47%, 44%); proportion of patients relapsing (49%, 46%); confirmed disability progression (38%, DEFINE only); new or enlarging T2 lesions (85%, 71%); and Gd+ lesions (90%, 74% reduced odds). T1-hypointense MRI lesions were reduced by 57% in the BG-12 BID group compared to placebo. The DEFINE cohort also demonstrated a 21% attenuated rate of whole brain atrophy compared to baseline (P=0.02), which may be explained by increased myelin density suggested by the observation of increased brain magnetization transfer ratio measurements.50,51 All study endpoints comparing BG-12 at both doses to placebo were statistically significant in favor of BG-12, with the exception of confirmed disability progression in the CONFIRM study (Table 2).46,47 The placebo group of CONFIRM demonstrated a low incidence of confirmed disability progression during the trial, rendering an insignificant reduction among the BG-12-treated groups.
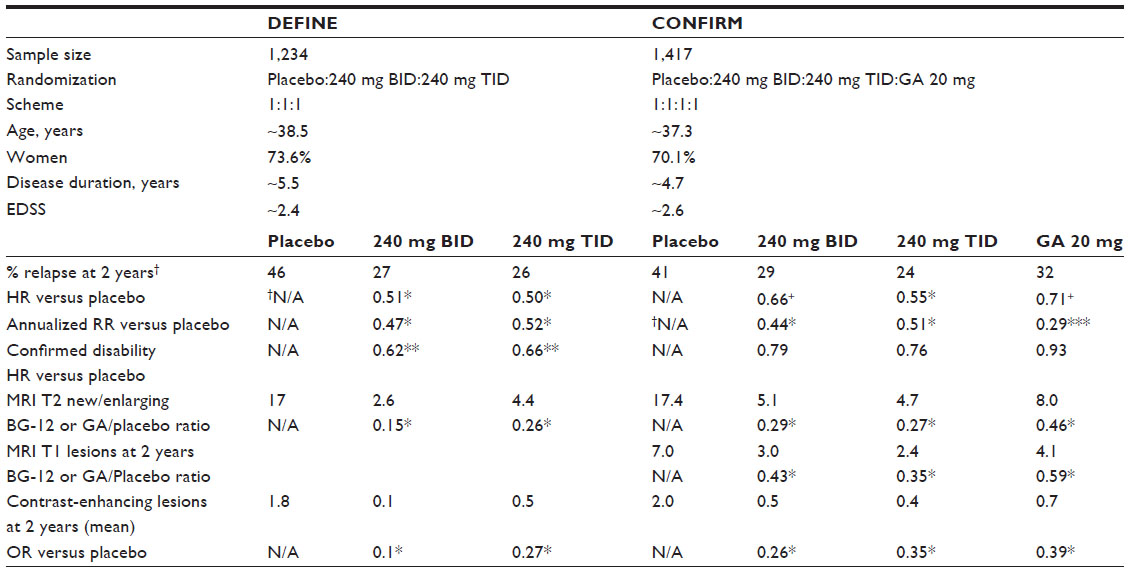 | Table 2 Pivotal clinical trials’ demographic and efficacy summary |
Tolerability and safety
BG-12 was well tolerated during the pivotal trials, with a reported 12%–16% cohort discontinuation of study drug (BID or TID dosing) due to an adverse event.46,47 In the CONFIRM trial, the discontinuation rate of BG-12 (12%) was comparable to the rate among subjects randomized to glatiramer acetate (10%). The principle side effects experienced include gastrointestinal (10%–17% incidence) and flushing reaction (28%–34% incidence) which were graded as mild or moderate severity and were typically limited to the 1st month of therapy (Table 3). Flushing may be prostaglandin-mediated and this reaction may be mitigated by pretreatment with aspirin administered 30 minutes before BG-12 dosing.52 Appropriate dose and frequency of administration of aspirin for this purpose is currently unknown, and is under clinical investigation.53 Gastrointestinal distress may be related to the observation of transient eosinophilia of the gastric mucosa in patients exposed to BG-12. Early research suggests that an initial daily dose of montelukast 10 mg may help relieve gastrointestinal side effects during the 1st month of therapy.54 Moreover, consuming a high-fat and -protein meal at time of BG-12 administration may reduce gastrointestinal and flushing side effects by effectively delaying absorption.55 Also reported was a slightly increased risk of infection including gastroenteritis as well as upper respiratory and urinary tract infection.
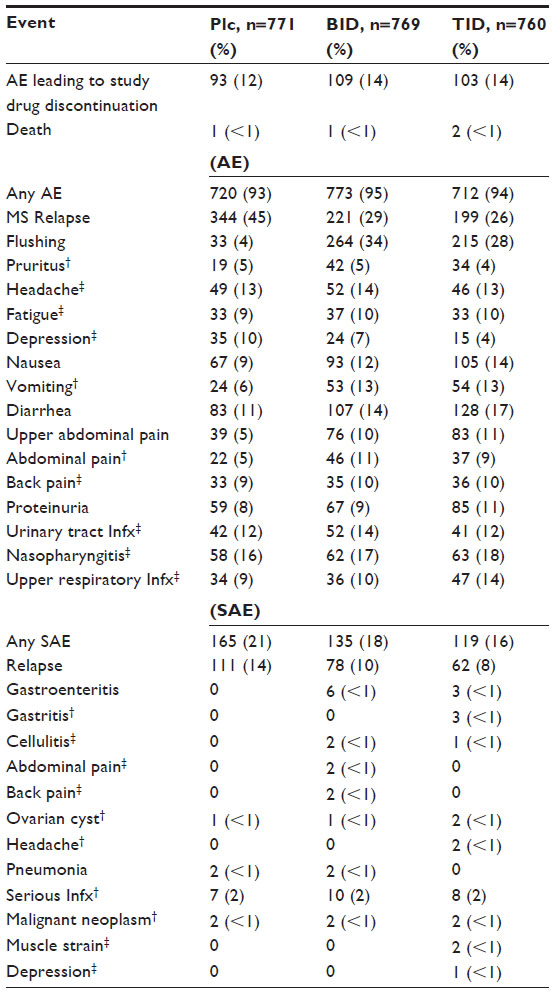 | Table 3 Combined pivotal clinical trials’ AE |
Labeled a pregnancy category C therapeutic, BG-12 has demonstrated embryolethality, impaired growth, and birth defects in rats and rabbits.56 Indeed, BG-12 is a small molecule and has the ability to cross the placenta into the fetal circulation. Limited human pregnancy registry data have not identified deleterious effects to children brought to term or an increased frequency of spontaneous abortions.56 Regardless, the potential for harmful effects of BG-12 to a developing fetus remains, prompting the recommendation for patients and partners to use effective birth control while exposed to BG-12. Metabolism of BG-12 does not utilize the CYP450 hepatic system, and therefore there is low concern for drug–drug interactions between BG-12 and oral contraceptives.57
Mixed results of preclinical animal research of DMF suggested the possibility of deleterious renal tissue changes while other publications reported a possible mitigating effect of nephrotoxicity by DMF.58–60 Clinical renal indices were followed during the pivotal trials which reported a comparable incidence of proteinuria among study groups including placebo (8% placebo patients and 9% combined incidence between pivotal trials of BG-12 BID-dosed patients). There were no reported cases of renal failure during the pivotal trials and proteinuria was largely found to be mild and reversible.46 Postmarketing Phase IV clinical monitoring of BG-12, which is prospectively following renal indices, is currently ongoing.61
Of major clinical concern is the role BG-12 may play in the pathogenesis of progressive multifocal leukoencephalopathy (PML): a rare infection of oligodendrocytes by the John Cunningham virus. PML tends to begin with an insidious onset and is experienced as a relentless progression of neurologic disability and possible death. Indeed, a case of PML occurred in a patient being treated with efalizumab for psoriasis who had a history of fumaric acid exposure 7 years prior to PML confirmation.62 Since FDA approval of BG-12, five additional cases of PML were reported with the contemporary use of Fumaderm or compounded FAE for the indication of psoriasis at the time of PML diagnosis.63–67 In all but the Nieuwkamp et al case,67 the patients experienced severe lymphopenia, and in most cases, persistent lymphopenia (200 lymphocytes/mm3) for 2–5 years prior to PML onset. The manufacturer’s response at the time of these case reports emphasized the differences in ingredients, metabolites, and formulations between BG-12 and Fumaderm/FAEs, and the fact that PML had not been associated with BG-12.68 The theoretical risk for PML on BG-12 stems from immunosuppression, and data from the pivotal trials inform that there is a reduction of mean lymphocyte count by 30% of baseline among those exposed to BG-12.46,47 Subsequent experience with BG-12 indicates that approximately 6% of patients on BG-12 experience a grade 3 (<500 lymphocytes/mm3) lymphopenia which has clinical implications for cell-mediated (antiviral) immunity.69 In November 2014, the FDA published a safety warning after a patient who was initially enrolled in the DEFINE study and assigned to the placebo arm (and continued in the open-label extension study ENDORSE) subsequently died of PML.70,71 To date, this is the only case of PML in a patient exposed to BG-12. In this particular case, the patient had persistently low (grade 3) lymphopenia for over 3 years prior to PML onset. The manufacturer now recommends that lymphocyte counts be monitored every 6–12 months and BG-12 be held if lymphocytes remain at <500 cells/μL for >6 months.72 There is a divergence of opinion among MS specialists regarding the frequency of lab monitoring, and it is the author’s practice to monitor every 3 months for the 1st year while monitoring every 6 months thereafter, assuming lymphocytes stabilized within normal limits. This practice is borne out of the observation that lymphocyte counts tend to plateau between the 9- and 12-month mark on BG-12. In addition, it is advisable to monitor CD8+ lymphocyte counts (involved in antiviral immunity) which appear to be disproportionately suppressed compared to CD4+ counts among patients exposed to BG-12.72
Patient-reported outcome measures, satisfaction/acceptability, adherence, and uptake
Patient-derived health-related quality of life (HR-QoL) metrics were prospectively collected during the pivotal trials and analyzed.73,74 A preplanned tertiary analysis of HR-QoL outcomes was performed on the combined data from DECIDE and CONFIRM.75 At baseline and approximately every 3–6 months, patients were administered surveys including the SF-36 questionnaire consisting of eight multi-item domains which were used to measure two composite scores: the Physical and Mental Component Summaries (PCS and MCS, respectively). Both composite scores were derived from four subscales. PCS comprised physical functioning, role-physical, bodily pain, and general health subscales, while MCS included vitality, social functioning, role-emotional, and mental health. Over 2,300 patients’ data were analyzed which demonstrated a mean increase in PCS and MCS scores (higher scores translate to better HR-QoL) at 2 years for patients exposed to DMF, while those randomized to placebo demonstrated reduced mean scores over the same time period. These observations were seen as early as 6 months from first dose of the study drug. PCS scores at 2 years increased by 0.47 and 0.43 from baseline (DMF BID and TID, respectively) while placebo mean PCS scores dropped by 1.05 (P<0.0001 DMF versus placebo). MCS score change from baseline observed by +0.31, +0.63, and –0.60 (DMF BID, DMF TID, and placebo, respectively; P<0.025).
The authors found that subjects who experienced clinical relapses had a mean PCS score that was lower at 2 years compared to those without a relapse (P<0.0001). While not statistically significant, the relapsing cohort trended for a lower mean MCS score compared with those who did not relapse (P=0.0622). Moreover, confirmed disability progression was associated with reduced mean PCS and MCS scores at 2 years (P<0.0001 and P=0.0076, respectively). Logistic regression modeling identified baseline clinical predictors of improved PCS and MCS at 2 years: exposure to DMF treatment, less physical disability at baseline (lower EDSS), and lower PCS and MCS scores at baseline. Predictive modeling also identified an age ≤40 years as a predictor of improved PCS at 2 years (but not MCS). The estimated increased odds for PCS score improvement over 2 years with DMF exposure compared to placebo was 42%. The authors concluded that their exploratory analyses suggest a parallel improvement in patient-reported HR-QoL in association with DMF exposure that appears to be related to improved clinical trial metrics of reduced clinical relapses and mitigation of sustained disability.75
Social media research supports the perception that patients prefer oral therapies in the treatment of MS, and the comparable study drug discontinuation rate from the pivotal trials of BG-12 compared with traditional platform therapies further support acceptable adherence and general acceptance among patients for DMF.22 Furthermore, patient-reported outcomes collected during the pivotal trials clarifies the benefits of BG-12 treatment from a patient’s perspective and further validates the efficacy of BG-12 in MS clinical care.76
Conclusion and place in therapy
A topic of great interest in the MS community is the appropriate DMT sequencing options for patients on natalizumab with multiple risk factors for PML. Established risk factors for PML in the setting of natalizumab administration include: 1) John Cunningham virus antibody seropositivity (particularly an antibody index titer >1.5); 2) any exposure to cytotoxic therapy; and 3) duration of natalizumab exposure >2 years. The concept of a “drug holiday” – of variable time off natalizumab – has been postulated as a way to theoretically reduce the risk of PML in the high-risk population. The RESTORE study, an exploratory investigation of MS disease response to a 24-week interruption of natalizumab with a double-blinded placebo or partially-blinded active comparator DMT versus natalizumab continuation, revealed a substantial risk of MS disease recurrence in the form of a clinical relapse or MRI inflammatory activity.77 While no MRI activity was seen in the natalizumab continuation group, 46% of placebo patients demonstrated MRI activity that was generally observed between the 3rd and 6th month after randomization. Although it is not clear whether a drug holiday confers a more favorable PML risk stratification, published data have identified cases of PML occurring 6 months after the last natalizumab infusion.78
In this context, the clinician and patient must weigh the PML risk of continued natalizumab exposure to the more likely risk of a significant recrudescence of MS disease activity after discontinuing natalizumab. To date, there is a paucity of data to aid the clinician. An MSBase Registry study reported on 89 patients who were transitioned from natalizumab to fingolimod with varying washout periods.79 ARRs were slightly increased during the fingolimod treatment period (0.38) compared to the natalizumab treatment epoch (0.26, P=0.002); however, ARR prior to natalizumab and fingolimod was reported in the range of 1.5, suggesting that there was not a significant rebound in disease activity after transitioning from natalizumab to fingolimod. Moreover, stratification of the cohort based on washout-period duration suggests that a period of <2 months is more favorable than 2–4 months (adjusted hazard ratio: 1.54, P=0.10 versus adjusted hazard ratio: 2.12, P=0.04, respectively).79 Indeed, a small clinical and MRI study of 22 patients transitioning from natalizumab to fingolimod found that a 3-month washout period prior to fingolimod initiation was insufficient to prevent reactivation of MS inflammatory disease activity in half the cohort.80 Early data suggests that extended washout periods from natalizumab are hazardous and sequencing to fingolimod may be optimal with short washouts, or none at all.81
The application of BG-12 as a sequencing agent off of natalizumab is variable among clinicians in the absence of guidelines. An academic MS center retrospectively reviewed the course of transition from natalizumab to BG-12 in 40 patients with or without the use of pulsed-monthly methylprednisolone infusions.82 Prescriber habits demonstrated the use of steroids in approximately half of the cohort (n=21), particularly in patients experiencing relapses in the washout period which ranged from 2 to 82 weeks (average 18.2-week washout period). Relapses occurred in 12.5% of the cohort in the washout period, while 15% experienced a relapse after BG-12 initiation. No stratification by duration of washout period was analyzed. The authors concluded that lengthy washout periods with pulsed-monthly steroids may not improve the transition process, and therefore, larger population studies are needed. A separate academic MS center reported a cohort of 51 patients who switched to BG-12 after a 4-week washout from natalizumab. Among this group, 25% relapsed (85% women) over a 6-month observational period.83 In an additional exploratory study, patients at high risk for PML underwent a natalizumab to BG-12 transition (n=29) after a 3-week washout period. Approximately 38% of the cohort experienced an MS relapse while only 21% of patients demonstrated new, enlarging, or Gd+ lesions on MRI. There was no observed increase in disability (measured by the EDSS) during the 6-month follow-up period. Among this cohort, 76% of patients successfully transitioned to BG-12.84
Actionable data to inform DMT sequencing guidelines is clearly lacking. At present, a Phase IV observational study (STRATEGY) is underway to evaluate clinical relapse activity during the natalizumab to BG-12 transition in a real-world setting.85 Despite the lack of available data, some MS experts suggest that a washout period of 2 months or less is advisable when sequencing to BG-12.86 It is the author’s opinion that in cases of historically aggressive MS disease activity, consideration for no washout period when transitioning to BG-12 should be taken with screening MRI of the brain with gadolinium at 3 months after the last natalizumab infusion. Demargination of leukocytes in the systemic circulation due to natalizumab typically results in higher than normal levels of functional circulating lymphocytes. Therefore, introduction of BG-12 immediately after stopping natalizumab may not result in severe lymphopenia (and subsequent increased risk for opportunistic infection) immediately during the critical period of transition where there remains the heightened risk of recurrent MS disease activity. Recommendations for shorter washout periods are further supported by the observation that lymphocyte reduction due to BG-12 is first noticeable at 4–6 weeks after initiation and begins to plateau between 24 and 48 weeks.47,69 This is the critical timeframe for expected recurrent disease activity after discontinuing natalizumab, and relatively low risk for PML. Currently, there is data demonstrating the successful use of BG-12 in sequencing patients off of natalizumab. Further data is needed to clarify the optimal washout period and sequencing agents for future guidelines regarding natalizumab discontinuation.
BG-12 offers a unique putative mechanism of action with potential neuroprotective and anti-inflammatory benefit; it has a demonstrated record of effectiveness in ameliorating the deleterious course of relapsing MS; and is generally well tolerated. The safety profile is also considered favorable, particularly when practitioners vigilantly monitor leukocyte and lymphocyte counts, and T-cell subsets in order to identify the expected 6% of patients that may reach significant levels of immunosuppression and risk for opportunistic infection such as PML. While there is a divergence of opinion among MS experts in regards to lab monitoring, the author’s practice is to monitor a complete blood count (with differential to include absolute lymphocyte count) and CD4/CD8 T-cell subsets every 3 months during the 1st year of BG-12 therapy, and if counts are acceptable, every 6 months thereafter. Absolute lymphocyte counts typically reach a nadir around the 6-month mark, although this can be delayed out to 1 year.58 If grade 3 lymphopenia (<500 cells/mm3) is observed for 6 or more months, it is recommended to discontinue BG-12. Clinical data indicate that lymphocyte counts are observed to begin approaching baseline levels within 1 month of BG-12 discontinuation.58
Combined analysis of CONFIRM and DEFINE allowed subgroup analysis of early MS patients (diagnosed within 1 year of study entry).87 A total of 688 early MS patients that were approximately evenly distributed between placebo and BID or TID dosing of BG-12. ARR was reduced by 56% (BID) and 60% (TID) compared to placebo (P<0.0001). Risk of confirmed 12-week disability progression was reduced by 71% (BID) and 47% (TID) when compared to placebo (P<0.009). These post hoc analyses demonstrate that early treatment of MS is efficacious and the beneficial effects may be more robust when compared to the combined CONFIRM and DEFINE cohort as a whole. In the setting of interferon-β-1b treatment, early exposure to DMT also demonstrated favorable long-term outcomes when compared to delayed exposure.21 These data support the narrative that a narrow window of opportunity exists to intervene for optimal short- and long-term benefit – ostensibly before considerable CNS inflammatory damage and the degenerative changes of MS ensue. In this setting, BG-12 240 mg dosed BID may be considered a valid first-line treatment of relapsing forms of MS.88–90
Disclosure
The author has participated in the speaker’s bureau for Biogen Idec and Teva.
References
Peterson JW, Bö L, Mörk S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50:389–400. | |
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343(13):938–952. | |
Li DK, Paty DW. Magnetic resonance imaging results of the PRISMS trial: a randomized, double-blind, placebo-controlled study of interferon-beta1a in relapsing-remitting multiple sclerosis. Prevention of Relapses and Disability by Interferon-beta1a Subcutaneously in Multiple Sclerosis. Ann Neurol. 1999;46(2):197–206. | |
Sormani MP, Bonzano L, Roccatagliata L, Cutter GR, Mancardi GL, Bruzzi P. Magnetic resonance imaging as a potential surrogate for relapses in multiple sclerosis: a meta-analytic approach. Ann Neurol. 2009;65(3):268–275. | |
Loleit V, Biberacher V, Hemmer B. Current and future therapies targeting the immune system in multiple sclerosis. Curr Pharm Biotechnol. 2014;15:276–296. | |
Trojano M, Lucchese G, Graziano G, et al. Geographical variations in sex ratio trends over time in multiple sclerosis. PloS One. 2012;7(10):e48078. | |
Salmen A, Gold R. Mode of action and clinical studies with fumarates in multiple sclerosis. Exp Neurol. 2014;262 Pt A:52–56. | |
Wolfson C, Wolfson DB. The latent period of multiple sclerosis: a critical review. Epidemiology. 1993;4(5):464–470. | |
Belbasis L, Bellou V, Evangelou E, Ioannidis JP, Tzoulaki I. Environmental risk factors and multiple sclerosis: an umbrella review of systematic and meta-analyses. Lancet Neurol. 2015;14:263–273. | |
Dörr J, Döring A, Paul F. Can we prevent or treat multiple sclerosis by individualised vitamin D supply? EPMA J. 2013;4(1):4. | |
International Multiple Sclerosis Genetics Consortium (IMSGC), Beecham AH, Patsopoulos NA, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013;45(11):1353–1360. | |
Confavreux C, Vukusic S, Adeleine P. Early clinical predictors and progression of irreversible disability in multiple sclerosis: an amnesic process. Brain. 2003;126:770–782. | |
Kurtzke JF, Beebe GW, Nagler B, Kurland LT, Auth TL. Studies on the natural history of multiple sclerosis – 8. Early prognostic features of the later course of the illness. J Chronic Dis. 1977;30(12):819–830. | |
Poser S, Poser W, Schlaf G, et al. Prognostic indicators in multiple sclerosis. Acta Neurol Scand. 1986;74(5):387–392. | |
Runmarker B, Andersen O. Prognostic factors in a multiple sclerosis incidence cohort with twenty-five years of follow-up. Brain. 1993; 116(Pt 1):117–134. | |
Weinshenker BG, Rice GP, Noseworthy JH, Carriere W, Baskerville J, Ebers GC. The natural history of multiple sclerosis: a geographically based study. 3. Multivariate analysis of predictive factors and models of outcome. Brain. 1991;114(Pt2):1045–1056. | |
Leray E, Yaouang J, Le Page E, et al. Evidence for a two-stage disability progression in multiple sclerosis. Brain. 2010;133:1900–1913. | |
Veauthier C, Paul F. Sleep disorders in multiple sclerosis and their relationship to fatigue. Sleep Med. 2014;15:5–14. | |
Hillman L. Caregiving in multiple sclerosis. Phys Med Rehabil Clin N Am. 2013;24(4):619–627. | |
Ragonese P, Aridon P, Salemi G, D’Amelio M, Savettieri G. Mortality in multiple sclerosis: a review. Eur J Neurol. 2008;15:123–127. | |
Goodin DS, Reder AT, Ebers GC, et al. Survival in MS: a radomized cohort study 21 years after the start of the pivotal IFNβ-1b trial. Neurology. 2012;78(17):1315–1322. | |
Ramagopalan S, Wasiak R, Cox AP. Using Twitter to investigate opinions about multiple sclerosis treatments: a descriptive, exploratory study. F1000Research. 2014;3:216. | |
Schweckendiek W. [Treatment of psoriasis vulgaris]. Med Monatsschr. 1959;13(2):103–104. German. | |
Mrowietz U, Christophers E, Altmeyer P. Treatment of severe psoriasis with fumaric acid esters: scientific background and guidelines for therapeutic use. The German Fumaric Acid Ester Consensus Conference. Br J Dermatol. 1999;141(3):424–249. | |
No authors listed. BG 12: BG 00012, BG 12/Oral Fumarate, FAG-201, second-generation fumarate derivative – Fumapharm/Biogen Idec. Drugs R D. 2005;6(4):229–230. | |
Kees F. Dimethyl fumarate: a Janus-faced substance? Expert Opin Pharmacother. 2013;14(11):1559–1567. | |
Litjens NH, van Strijen E, van Gulpen C, et al. In vitro pharmacokinetics of anti-psoriatic fumaric acid esters. BMC Pharmacol. 2004;4:22. | |
Rostami-Yazdi M, Clement B, Mrowietz U. Pharmacokinetics of anti-psoriatic fumaric acid esters in psoriasis patients. Arch Dermatol Res. 2010;302(7):531–538. | |
Werdenberg D, Joshi R, Wolffram S, Merkle HP, Langguth P. Presystemic Metabolism and intestinal absorption of antipsoriatic fumaric acid esters. Biopharm Drug Dispos. 2003;24:259–273. | |
Rostami-Yazdi M, Clement B, Schmidt TJ, Schinor D, Mrowietz U. Detection of metabolites of fumaric acid esters in human urine: implications for their mode of action. J Invest Dermatol. 2009;129:231–234. | |
Zoghi S, Amirghofran Z, Nikseresht A, Ashjazadeh N, Kamali-Sarvestani E, Rezaei N. Cytokine secretion pattern in treatment of lymphocytes of multiple sclerosis patients with fumaric acid esters. Immunol Invest. 2011;40:581–596. | |
Rubant SA, Ludwig RJ, Diehl S, et al. Dimethylfumarate reduces leukocyte rolling in vivo through modulation of adhesion molecule expression. J Invest Dermatol. 2008;128:326–331. | |
Vandermeeren M, Janssens S, Wouters H, et al. Dimethylfumarate is an inhibitor of cytokine-induced nuclear translocation of NF-kappa B1, but not ReIA in normal human dermal fibroblast cells. J Invest Dermatol. 2001;116(1):124–130. | |
Albrecht P, Bouchachia I, Goebels N, et al. Effects of dimethyl fumarate on neuroprotection and immunomodulation. J Neuroinflammation. 2012;9:163. | |
Wilms H, Sievers J, Rickert U, Rostami-Yazdi M, Mrowietz U, Lucius R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J Neuroinflammation. 2010;7:30. | |
Zhu K, Mrowietz U. Inhibition of dendritic cell differentiation by fumaric acid esters. J Invest Dermatol. 2001;116(2):203–208. | |
Treumer F, Zhu K, Gläser R, Mrowietz U. Dimethylfumarate is a potent inducer of apoptosis in human T cells. J Invest Dermatol. 2003;121(6):1338–1388. | |
Schilling S, Goelz S, Linker R, Luehder F, Gold R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophase infiltration. Clin Exp Immunol. 2006;145:101–107. | |
Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13(1):76–86. | |
Linker RA, Lee DH, Ryan S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134(Pt 3):678–692. | |
Foresti R, Bains SK, Pitchumony TS, et al. Small molecule activators of the Nrf2-HO-1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol Res. 2013;76:132–148. | |
Scannevin RH, Chollate S, Jung MY, et al. Fumarates promote cytoprotection of central nervious system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther. 2012;341(1):274–284. | |
Schimrigk S, Brune N, Hellwig K, et al. Oral fumaric acid esters for the treatment of active multiple sclerosis: an open-label, baseline-controlled pilot study. Eur J Neurol. 2006;13:604–610. | |
Kappos L, Gold R, Miller DH, et al. Effect of BG-12 on contrast-enhanced lesions in patients with relapsing – remitting multiple sclerosis: subgroup analyses from the phase 2b study. Mult Scler. 2012;18(3):314–321. | |
MacManus DG, Miller DH, Kappos L, et al. BG-12 reduces evolution of new enhancing lesions to T1-hypointense lesions in patients with multiple sclerosis. J Neurol. 2011;258:449–456. | |
Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–1107. | |
Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012; 367(12):1087–1097. | |
Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33(11):1444–1452. | |
Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol. 2005;58(6):840–846. | |
Arnold DL, Gold R, Kappos L, et al. Effects of delayed-release dimethyl fumarate on MRI measures in the Phase 3 DEFINE study. J Neurol. 2014;261:1794–1802. | |
Arnold DL, Gold R, Kappos L, et al. Magnetization transfer ratio in the delayed-release dimethyl fumarate DEFINE study. J Neurol. 2014;261:2429–2437. | |
Sheikh SI, Nestorov I, Russell H, et al. Tolerability and pharmacokinetics of delayed-release dimethyl fumarate administered with and without aspirin in healthy volunteers. Clin Ther. 2013;35(10):1582–1594. | |
Biogen Idec. Phase 4 Study of Effect of Aspirin on Flushing in Dimethyl Fumarate (DMF)-Treated Participants With Relapsing-Remitting Multiple Sclerosis (RRMS) (ASSURE). Available from: https://clinicaltrials.gov/ct2/show/NCT02090413. NLM identifier: NCT02090413. Accessed April 18, 2015. | |
Tornatore C, Amjad F. Attenuation of dimethyl fumarate-related gastrointestinal symptoms with montelukast (P7.251). Neurology. 2014;82(10 Suppl P7):251. | |
US Food and Drug Administration. FDA Approved Labeling (Tecfidera, NDA 204063). Silver Spring: US Food and Drug Administration; 2013. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/204063lbl.pdf. Accessed February 26, 2015. | |
Lu E, Wang BW, Alwan S, et al. A Review of safety-related pregnancy data surrounding the oral disease-modifying drugs for multiple sclerosis. CNS Drugs. 2014;28:89–94. | |
Burness CB, Deeks ED. Dimethyl fumarate: a review of its use in patients with relapsing-remitting multiple sclerosis. CNS Drugs. 2014;28(4):373–387. | |
Center for Drug Evaluation and Research. Medical Review(s). Application Number: 204063Orig1s000. Silver Spring: US Food and Drug Administration; 2013. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204063Orig1s000MedR.pdf. Accessed February 26, 2015. | |
Oh CJ, Kim JY, Choi YK, et al. Dimethylfumarate Attenuates renal fibrosis via NF-E2-related factor 2-mediated inhibition of transforming growth factor-β/Smad signaling. PloS One. 2012;7(10):e45870. | |
Munday R, Smith BL, Munday CM. Effect of inducers of DT-diaphorase on the haemolytic activity and nephrotoxicity of 2-amino-1,4-naphthoquinone in rats. Chem Biol Interact. 2005;155:140–147. | |
Biogen Idec. Dimethyl Fumarate (DMF) Observational Study (ESTEEM). Available from: https://clinicaltrials.gov/ct2/show/NCT02047097?term=MS+and+ESTEEM&rank=1. NLM identifier: NCT02047097. Accessed February 26, 2015. | |
Kothary N, Diak IL, Brinker A, Bezabeh S, Avigan M, Dal Pan G. Progressive multifocal leukoencephalopathy associated with efalizumab use in psoriasis patients. J Am Acad Dermatol. 2011;65(3):546–551. | |
van Oosten BW, Killestein J, Barkhof F, Polman CH, Wattjes MP. PML in a patient treated with dimethyl fumarate from a compounding pharmacy. N Engl J Med. 2013;368(17):1658–1659. | |
Ermis U, Weis J, Schulz J. PML in a patient treated with fumaric acid. N Engl J Med. 2013;368:1657–1658. | |
Stoppe M, Thomä E, Liebert UG, et al. Cerebellar manifestation of PML under fumarate and after efalizumab treatment of psoriasis. J Neurol. 2014;261:1021–1024. | |
Buttmann M, Stoll G. Case reports of PML in patients treated for psoriasis. N Engl J Med. 2013;369(11):1081. | |
Nieuwkamp DJ, Murk JL, van Oosten BW, et al. PML in a patient without severe lymphocytopenia receiving dimethyl fumarate. N Engl J Med. 2015;372(15):1474–1476. | |
Sweetser MT, Dawson KT, Bozic C. Manufacturer’s response to case reports of PML. N Engl J Med. 2013;368(17):1659–1661. | |
Fox R, Gold R, Phillips JT, et al. Lymphocyte count reductions in relapsing-remitting multiple sclerosis (RRMS) patients treated with delayed-release dimethyl fumarate: an integrated analysis of the placebo-controlled studies (P3.179). Neurology. 2014; 82(10 Suppl P3):179. | |
US Food and Drug Administration. FDA Drug Safety Communication: FDA warns about case of rare brain infection PML with MS drug Tecfidera (dimethyl fumarate). Silver Spring: US Food and Drug Administration; 2014. Available from: http://www.fda.gov/Drugs/DrugSafety/ucm424625.htm. Accessed February 26, 2015. | |
Rosenkranz T, Novas M, Terborg C. PML in a patient with lymphocytopenia treated with dimethyl fumarate. N Engl J Med. 2015;372(15):1476–1478. | |
Spencer CM, Crabtree-Hartman EC, Lehmann-Horn K, Cree BA, Zamvil SS. Reduction of CD8+ T lymphocytes in multiple sclerosis patients treated with dimethyl fumarate. Neurol Neuroimmunol Neuroinflamm. 2015;2(3):e76. | |
Kappos L, Gold R, Arnold DL, et al. Quality of life outcomes with BG-12 (dimethyl fumarate) in patients with relapsing-remitting multiple sclerosis: the DEFINE study. Mult Scler. 2014;20(2):243–252. | |
Kita M, Fox RJ, Phillips JT, et al. Effects of BG-12 (dimethyl fumarate) on health-related quality of life in patients with relapsing-remitting multiple sclerosis: findings from the CONFIRM study. Mult Scler. 2014;20(2):253–257. | |
Kita M, Fox RJ, Gold R, et al. Effects of delayed-release dimethyl fumarate (DMF) on health-related quality of life in patients with relapsing-remitting multiple sclerosis: an integrated analysis of the phase 3 DEFINE and CONFIRM studies. Clin Ther. 2014;36(12):1958–1971. | |
Heesen C, Cohen JA. Does the patient know best? Quality of Life assessment in multiple sclerosis trials. Mult Scler. 2014;20(2):131–132. | |
Fox RJ, Cree BA, De Sèze J, et al. MS disease activity in RESTORE: a randomized 24-week natalizumab treatment interruption study. Neurology. 2014;82:1491–1498. | |
Fine AJ, Sorbello A, Kortepeter C, Scarazzini L. Progressive multifocal leukoencephalopathy after natalizumab discontinuation. Ann Neurol. 2014;75:108–115. | |
Jokubaitis VG, Li V, Kalincik T, et al. Fingolimod after natalizumab and the risk of short-term relapse. Neurology. 2014;82:1204–1211. | |
Rinaldi F, Seppi D, Calabrese M, Perini P, Gallo P. Switching therapy from natalizumab to fingolimod in relapsing-remitting multiple sclerosis: clinical and magnetic resonance imaging findings. Mult Scler. 2012;18(11):1640–1643. | |
Giovannoni G, Naismith RT. Natalizumab to fingolimod washout in patients at risk of PML: when good intentions yield bad outcomes. Neurology. 2014;82:1196–1197. | |
Vo KH, Barra M, Drago N, et al. Clinical Management of Patients with Relapsing-Remitting Multiple Sclerosis Transitioning from Natalizumab to Dimethyl Fumarate. Presented at 2014 Joint Congress of European Neurology; May 31 – June 3; Istanbul, Turkey. | |
Hillman L, Gallaro D. Incidence of relapse during the first 6 months in a cohort of patients switched to dimethyl fumarate. 2014; Presented at: 2014 CMSC ACTRIMS Cooperative Meeting; May 28–31; 2014; Dallas, TX. | |
Foley J, Hoyt T, Christensen A, Blatter A. A pilot study to assess disease state stability, efficacy, and tolerability in a natalizumab to dimethyl fumarate crossover design. Available from: http://onlinelibrary.ectrims-congress.eu/ectrims/2014/ACTRIMS-ECTRIMS2014/64578/john.foley.a.pilot.study.to.assess.disease.state.stability.efficacy.and.html. Accessed February 26, 2014. | |
Biogen Idec. Real-world Outcomes on Tecfidera® (BG00012, Dimethyl Fumarate) Post-Tysabri® (BG00002, Natalizumab) (STRATEGY). Available from: https://clinicaltrials.gov/ct2/show/NCT02159573. NLM identifier: NCT02159573. Accessed April 1, 2015. | |
Mellen Center. Mellen Center Approaches: Use of DMF in MS. Cleveland: Cleveland Clinic. Available from: http://my.clevelandclinic.org/ccf/media/files/Neurological-Institute/mellen-center/13-neu-718-dimethyl-fumarate-fact-sheet.pdf. Accessed April 1, 2015. | |
Gold R, Giovannoni G, Phillips JT, et al. Efficacy and safety of delayed-release dimethyl fumarate in patients newly diagnosed with relapsing-remitting multiple sclerosis (RRMS). Mult Scler. 2015;21(1):57–66. | |
Nicholas JA, Boster AL, Imitola J, O’Connell C, Racke MK. Design of oral agents for the management of multiple sclerosis: benefit and risk assessment for dimethyl fumarate. Drug Des Devel Ther. 2014;8:897–908. | |
Bomprezzi R. Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: an overview. Ther Adv Neurol Disord. 2015;8(1):20–30. | |
Fox RJ. In the coming year we should abandon interferons and glatiramer acetate as first-line therapy for MS: yes. Mult Scler. 2013;19(1):24–25. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.