Back to Archived Journals » Oncolytic Virotherapy » Volume 4
Advances in the design and development of oncolytic measles viruses
Authors Hutzen B, Raffel C, Studebaker A ![]()
Received 28 May 2015
Accepted for publication 14 July 2015
Published 27 August 2015 Volume 2015:4 Pages 109—118
DOI https://doi.org/10.2147/OV.S66078
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Brian Hutzen,1 Corey Raffel,2 Adam W Studebaker1
1Center for Childhood Cancer and Blood Diseases, The Research Institute at Nationwide Children’s Hospital, Columbus, OH, USA; 2Department of Neurological Surgery and Pediatrics, University of California, San Francisco, San Francisco, CA, USA
Abstract: A successful oncolytic virus is one that selectively propagates and destroys cancerous tissue without causing excessive damage to the normal surrounding tissue. Oncolytic measles virus (MV) is one such virus that exhibits this characteristic and thus has rapidly emerged as a potentially useful anticancer modality. Derivatives of the Edmonston MV vaccine strain possess a remarkable safety record in humans. Promising results in preclinical animal models and evidence of biological activity in early phase trials contribute to the enthusiasm. Genetic modifications have enabled MV to evolve from a vaccine agent to a potential anticancer therapy. Specifically, alterations of the MV genome have led to improved tumor selectivity and delivery, therapeutic potency, and immune system modulation. In this article, we will review the advancements that have been made in the design and development of MV that have led to its use as a cancer therapy. In addition, we will discuss the evidence supporting its use, as well as the challenges associated with MV as a potential cancer therapeutic.
Keywords: virotherapy, measles virus, oncolytic therapy
Introduction
To minimize risk to the patient and general population, an ideal oncolytic virus should selectively kill tumor cells while being nonpathogenic to noncancerous tissue. The Edmonston strain of measles virus (MV-Edm) and its various derivatives meet these criteria. MV is a member of the genus Morbillivirus in the Paramyxoviridae family. MV is a spherical, enveloped virus that has a nonsegmented, single-stranded, negative-sense RNA genome that comprises approximately 16,000 nucleotides, encoding six genes that are translated into eight viral proteins.1,2
The vast majority of MV oncolytic therapy studies utilize derivatives of the MV-Edm strain. This strain was isolated in 1954 by John Enders and Thomas Peebles from a throat culture of a young boy named David Edmonston.3 Serial passaging of MV-Edm in human and monkey kidney cells resulted in the loss of the virus’s pathogenicity, allowing for the creation of the first live, attenuated MV vaccine in 1963.4 The safety of using MV-Edm clinically has been demonstrated over the last 50 years with over a billion human recipients worldwide.5 Furthermore, there has been no documentation of the reversion of MV-Edm back to pathogenic MV.
Three receptors that permit MV entry into human cells have been identified: signaling lymphocyte activation molecule, membrane cofactor protein (CD46), and nectin-4.6–8 CD46, a regulator of complement activation, is the preferred receptor for all laboratory strains of MV-Edm. This tropism was acquired following a single amino acid substitution at position 481, changing an asparagine to a tyrosine.9–11 Tumor selectivity is conferred by MV-Edm’s acquired tropism for CD46.9 Overexpression of CD46 is frequently seen in human cancer cells, where it most likely serves as a survival mechanism to protect the transformed cells from complement-mediated lysis.12–14 CD46 overexpression has been documented in numerous cancers including brain, breast, cervical, colorectal, endometrial, gastrointestinal, hepatocellular, lung, renal and ovarian carcinomas, and has also been reported in hematopoietic malignancies such as leukemia and multiple myeloma.15–25 Although CD46 is ubiquitously expressed on every nucleated cell in the human body, MV-Edm requires a minimum threshold of CD46 expression on the cell surface to initiate infection and fusion.26 The low CD46 densities associated with normal cells usually preclude MV-Edm infection and any subsequent intercellular fusion.14 Conversely, tumor cells express high levels of CD46, thus making them susceptible to MV-Edm infection, which leads to extensive intercellular fusion (syncytia) and subsequent cell death.26 The dependence on receptor density to generate a productive virus infection allows oncolytic viruses derived from MV-Edm to functionally discriminate between normal and transformed cells.
In addition to its predilection for infecting tumor cells and its overall safety when administered clinically, the genome of MV-Edm is amenable to genetic manipulation. In 1996, Radecke et al developed a reverse genetic system for MV rescue that allowed recombinant MV to be generated from cDNA ushering in a new era of measles-based virotherapies.14,27 Genetic manipulation of MV cDNA has made it possible to build upon the already considerable strengths of MV-Edm mentioned above by creating novel MV with enhanced attributes and functions. In the last 20 years, investigations have centered around creating recombinant MV that produces detectable markers that monitor viral infection, express transgenes that confer enhanced oncolytic or immune-modulatory activity, and contain modifications that increase their selectivity for neoplastic tissue.14 In this article, we will review the advancements that have been made in the design and development of the original MV-Edm vaccine strain that have ultimately led to its use as a cancer therapy.
“Monitoring” oncolytic MVs
A critical component in evaluating oncolytic virus efficacy is the ability to monitor infection and spread. To facilitate its detection, recombinant MV-GFP and MV-CEA, two MV-Edm derivatives that encode green fluorescent protein (GFP) and carcinoembryonic antigen (CEA), respectively, were developed.28,29 Sequences coding for GFP and CEA were inserted into the 3′ end of MV genome before the N gene (Figure 1). Placement of the sequences here ensures maximal transcription and protein expression, thereby increasing the sensitivity of virus detection.30,31 MV-GFP is routinely used in in vitro evaluation when fluorescence microscopy techniques can be used to detect MV infection, whereas MV-CEA is used as a biomarker of in vivo infection when direct observation is not possible.28,31 Following MV-CEA infection of tumor cells, CEA, a nonimmunogenic soluble peptide with no biological function, is released into the blood stream.29 As CEA has a constant circulation half-life, measurement of CEA levels in the patient’s serum can therefore provide useful information on the kinetics of MV infection. Important parameters including virus bioavailability, immune system involvement, and any potential dose-limiting toxicity can all be evaluated, making it possible to provide individualized medicine.29 The therapeutic potential of MV-CEA has already been demonstrated in multiple preclinical tumor models and is currently being tested in Phase I clinical trials for the treatment of recurrent ovarian cancer and glioblastoma multiforme (Table 1).32,33
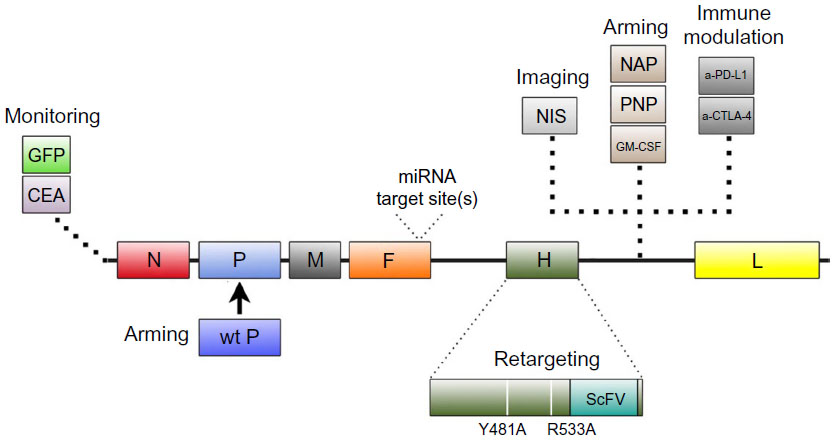 | Figure 1 Summary of modifications introduced into MV-Edm through genetic engineering. |
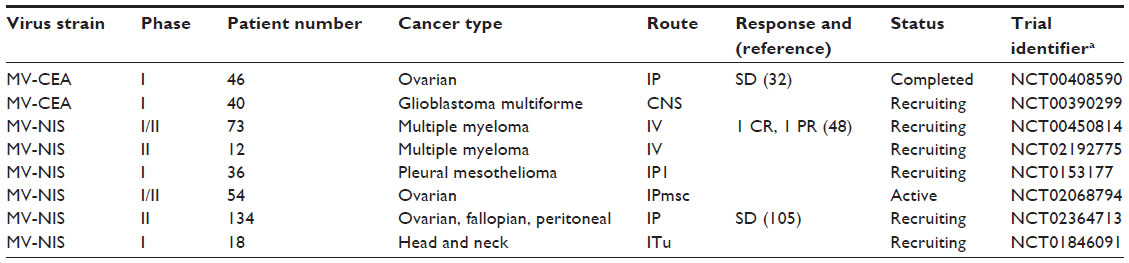 | Table 1 Summary of completed, ongoing, and actively recruiting clinical trials using measles virus |
To demonstrate tumor-specific infection in vivo, MV-NIS, a recombinant MV expressing the thyroidal sodium iodide symporter (NIS) gene, was constructed (Figure 1). NIS is a transmembrane ion channel that facilitates iodide transport into thyroid follicular cells.34,35 The administration of radioactive iodine and subsequent active uptake of iodide by cells can be used for imaging or ablation of the thyroid.36 Tumors infected with MV-NIS similarly acquire the ability to concentrate radioiodine, allowing anatomical mapping of the tumor and infectivity status to be monitored with single photon emission computed tomography or positron emission tomography imaging techniques using 123I and 124I, respectively, as tracers.34,37,38 Furthermore, MV-NIS therapy can also be combined with the β−-emitting radioiodine isotope 131I to enhance the therapeutic potency of the virus (see “Arming”).14 The efficacy of MV-NIS treatment has been evaluated in preclinical models of multiple myeloma, ovarian cancer, pancreatic cancer, mesothelioma, prostate cancer, malignant gliomas, cancers of the head and neck, osteosarcoma, and medulloblastoma.34,39–46 Phase I/II clinical trials investigating MV-NIS in recurrent or refractory multiple myeloma, malignant pleural mesothelioma, ovarian epithelial cancer, and recurrent or metastatic squamous cell carcinoma of the head and neck are presently underway (Table 1).47–52
“Re-targeting” oncolytic MVs
Successful oncolytic viruses selectively kill cancer cells without causing excessive toxicity to non-neoplastic cells. To further assist in this outcome, recombinant MV has been engineered to target specific receptors overexpressed on tumor cells. As sequence determinants in the H gene are responsible for virus specificity, manipulation of the H gene to allow for targeted infection does not negatively impact subsequent virus fusion and syncytia formation mediated by the F protein.14 Mutations introduced in the H gene that ablates virus entry through the natural receptors CD46 and signaling lymphocyte activation molecule, combined with display of a tumor-specific ligand or single-chain antibody in the H gene, allow for the creation of retargeted MV (Figure 1).53–55 To ensure rescue of retargeted MV that no longer recognize their normal receptors, a pseudoreceptor STAR system was developed.56 This system utilizes Vero cells stably expressing a single-chain antibody against 6-His, which then allows the rescue of retargeted viruses that have been modified to express a 6-histidine tag at the N-terminus of the retargeted H protein.
Recombinant MV has successfully been developed to target multiple tumor-associated markers including the epidermal growth factor receptor, epidermal growth factor receptor-vIII, and interleukin-13 receptor α2 found on gliomas, myeloma markers CD38 and CD138, folate receptor α receptor expressed in ovarian cancers, prostate stem cell antigen expressed by prostatic and pancreatic cancers, CD20-positive non-Hodgkin’s lymphoma, and Her-2/neu.56–65 In each case, retargeted viruses retained their antitumor potency while restricting viral infection and replication to cells expressing the receptor. More importantly, retargeted strains, unlike their natural receptor-utilizing counterparts, displayed no off-target pathology following administration in a CD46 transgenic mouse model of MV toxicity.66
Although the ability to redirect MV tropism to generate more tumor-selective viruses is appealing, the utility of this approach remains clinically unproven. The impetus to reengineer MV tropism was based on the assumption that the ubiquitous expression of CD46 would result in MV infection and killing of normal cells.14 However, detailed studies determined that MV requires a minimum density of CD46 receptor density to induce cytopathic effect, and CD46 expression levels in normal cells are below this minimum threshold.26 Furthermore, no dose limiting toxicities have been reported in the Phase I clinical trials with the CD46-tropic MV-CEA and MV-NIS viruses that would warrant using an MV with altered tropism (Table 1).67 In the future, use of retargeted MV may be beneficial when the cellular CD46 expression level is insufficient to support virus-induced cytopathic effect.
An alternative approach to restrict viral tropism to tumor cells is the incorporation of microRNA target sites (miRTSs) into the viral genome.68,69 miRTS, which can inhibit translation, has been cloned into the 3′-untranslated region of both the viral fusion and hemagglutinin genes (Figure 1).68,69 Virus replication is controlled by differential microRNA expression documented in cancer cells. As numerous microRNAs are downregulated in cancer cells, virus replication is allowed to proceed, whereas the endogenous microRNA expression in normal cells prevents virus replication. Recombinant strains of MV have been engineered to contain target sites for brain-specific microRNA-7, liver-specific microRNA-122, and gastrointestinal-specific microRNA-148a.68,69 In vitro and in vivo studies demonstrated that normal cells and tissues restricted MV replication. In comparison, malignant cells and tissues were permissive for virus replication. In addition, miRTS specific for microRNA-7 protected mice susceptible to MV infection following an intracerebral challenge.68
“Arming” oncolytic MVs
Advances in genetic engineering have allowed investigators to create “armed” viruses that have increased antitumor efficacy. Armed viruses combine the lytic potential of the virus with the therapeutic capacity of a transgene cloned into the viral genome. The most widely studied therapeutic transgene utilized to arm recombinant MV is NIS. Apart from providing a noninvasive means of imaging tumors, MV-NIS (described in “Monitoring”) enhances the efficacy of MV against radiosensitive malignancies by concentrating radioiodine in virus-infected cells.34 MV-NIS in combination with the β− particle emitting radioiodine isotope 131I significantly improved survival in preclinical models of multiple myeloma and prostate cancer.34,43 More recently, the combination of MV-NIS and 131I was found to have significant antitumor activity in orthotopic models of glioblastoma multiforme and medulloblastoma, radiosensitive brain tumors of adulthood and childhood, respectively.44,46
Investigators have also evaluated MV-PNP, an armed MV encoding the Escherichia coli purine nucleoside phosphorylase (PNP) gene (Figure 1).70 PNP is a prodrug convertase that catalyzes the prodrugs 6-methylpurine-2′-deoxyriboside (MeP-dR) and fludarabine (9-beta-D-arabinofuranosyl-2-fluoroadenine 5’-monophosphate) into the highly cytotoxic 6-methylpurine and 2-fluoroadenine, respectively.71 These highly diffusible products are metabolized to toxic adenosine triphosphate analogs, which can subsequently arrest DNA, RNA, and protein synthesis.71 Co-administration of MV-PNP and MeP-dR significantly prolonged survival in a subcutaneous syngeneic model of murine colon adenocarcinoma.70 Complete tumor regression was also observed in nine out of ten animals when MV-PNP/MeP-dR was co-administered with the immunosuppressive agent cyclophosphamide.70 In separate studies, fludarabine increased the oncolytic efficacy of MV-PNP in xenograft models of Burkitt’s lymphoma and pancreatic cancer.62,64 Clinical trials with MV-PNP have yet to be formally proposed.
Another novel approach to increase the efficacy of MV-Edm is to replace the defective P gene associated with the vaccine strain with a wild-type P gene (Figure 1).72 During vaccine development, mutations in the MV-Edm P gene resulted in defects in the P, C, and V proteins transcribed from the P gene, thus rendering these proteins incapable of suppressing the type I interferon (IFN) response.8,73 As a result, tumor cells infected with MV-Edm produce substantially more IFNs than those infected with a wild-type MV, which can compromise viral gene expression.72 In a study by Haralambieva et al, the antitumor activity of a chimeric MV-GFP virus armed with the wild-type P gene was evaluated in vitro and in vivo.72 The chimeric virus induced significantly lower levels of type I IFN than unmodified MV-GFP and displayed greater oncolytic potency against human multiple myeloma xenografts. Despite the improved efficacy, clinical testing of this chimeric MV-Edm has not been pursued due to concerns in the potential pathogenicity associated with the wild-type P gene.14
“Immune-modulating” oncolytic MVs
Another strategy for improving the oncolytic potential of MV-Edm is to construct recombinant MV that expresses a transgene that stimulates the native antitumor immune response or alters the tumor microenvironment.74–76 MV-Edm derivatives have been constructed to express the immunostimulatory transgenes granulocyte macrophage colony-stimulating factor (GM-CSF) and IFN-β (Figure 1).42,77,78 GM-CSF potentiates many neutrophil functions including stimulation of phagocytosis, lysozyme release, oxidative metabolism, and recruitment of complement.79 IFN-β is involved in antibody production, natural killer and T-cell activation, and macrophage function.80,81 Treatment of a mouse xenograft model of Burkitt’s lymphoma with a recombinant MV expressing the murine GM-CSF (MV-mGM-CSF) induced infiltration of activated neutrophils and an antitumor response.78 MV strains expressing the murine IFN-β (MV-mIFN-B) induced CD68-positive immune cell filtration, decreased CD31-positive vascular endothelial cells, and a significant antitumor response in xenograft models of human mesothelioma.42
MV-NAP, encoding a secreted form of the Helicobacter pylori neutrophil-activating protein (NAP), was also developed to modulate the immune system (Figure 1). NAP is a virulence factor involved in the pathogenesis of H. pylori infection and a potent modulator of proinflammatory cytokines.82 Treatment of xenograft models of lung and intrapleural metastatic breast cancer with MV-NAP significantly prolonged survival.83 Increased survival was mediated in part by the induction of a nonspecific inflammatory reaction in the tumor microenvironment.83
MV derivatives were recently generated expressing antibodies against the immune checkpoint modulators cytotoxic T-lymphocyte antigen 4 (MV-aCTLA-4) and programmed death-1 ligand 1 (MV-aPD-L1) (Figure 1).84 CTLA-4 and PD-L1 are T-cell inhibitory factors that play critical roles in T-cell activation.85,86 Tumor cells co-opt these checkpoint modulators to escape cellular immunity, particularly against T-cells specific for tumor antigens. Results from clinical trials evaluating antibodies targeting CTLA-4 and PD-L1 have been encouraging, with antibodies blocking CTLA-4 being the first in the class of immune checkpoints to achieve US Food and Drug Administration approval. Recombinant MV expressing immune checkpoint modulators were constructed to restrict the toxicity associated with systemic antibody treatment to the tumor bed, as well as stimulate antitumor immunity. To evaluate the immunotherapeutic effects of oncolytic MV in vivo, a syngeneic model of malignant melanoma was established.84 MV-aCTLA-4 and MV-aPD-L1 treatment delayed tumor progression, while animals treated with MV-aPD-L1 had a significantly prolonged median overall survival. Both viruses were associated with a significant increase in CD3+ T-cells in the tumor and a decrease in FoxP3+ regulatory T-cells. Treatment with MV-aPD-L1 was associated with increased levels of CD8+ cytotoxic T-cells and activated IFN-γ expressing CD8+ cells, as well as an increased CD8/T-regulatory cell ratio. In vivo oncolytic efficacy of MV-aCTLA-4 and MV-aPD-L1 was evaluated in human melanoma xenografts.84 Tumor regression was observed in all treated mice, with complete remission achieved in 80% of the animals. Coupling the oncolytic potential of MV-Edm with immunotherapeutics may serve as novel treatment strategy.
“Immune-evading” oncolytic MVs
Due to previous vaccination or natural infection most candidates for measles virotherapy will have prior immunity to the virus, which may significantly impact the therapeutic efficacy.14,87,88 Circulating anti-MV antibodies and T-lymphocytes can rapidly neutralize an oncolytic MV. Furthermore, antibody titers progressively increase following each successive exposure, thus making re-administration of MV very difficult.89 Multiple approaches to circumvent or modulate anti-measles immunity are being evaluated. One possible strategy to modulate the immune response is to combine MV therapy with immunosuppressive agents such as cyclophosphamide.14 Previous studies with oncolytic strains of herpes virus demonstrated a decrease in the innate immune response, enhanced oncolytic activity, and prolonged viral gene expression in tumors following cyclophosphamide treatment.90–92 A preclinical toxicology study with MV-NIS performed in immunocompetent squirrel monkeys (Saimiri sciureus) reported similar findings.93 Cyclophosphamide treatment prior to intravenous administration of MV-NIS resulted in a decreased humoral immune response to the virus and a prolongation of viral gene expression.93 Importantly, no significant toxicity was reported in these animals.93 These preclinical observations have led to the inclusion of cyclophosphamide in a Phase I clinical trial evaluating MV-NIS in patients with recurrent or refractory multiple myeloma.47,48
A second novel strategy to circumvent the anti-measles immune response and improve viral delivery is to use infected cell carriers. In this approach, MV is delivered to the tumor in pre-infected cells such as monocytoid cell lines or mesenchymal stem cells.94,95 Since no naked virions are present, antibodies cannot neutralize the virus. Ideally, cell carriers are permissive to MV infection, display some capacity to traffic and deliver MV to tumor sites, and protect the virus from antibody neutralization.67 Cell carriers would also reduce sequestration of the virus by lung, liver, and spleen macrophages following systemic administration to treat disseminated and hematopoietic malignancies.96,97 In a preclinical study of ovarian cancer in passively immunized athymic mice, mesenchymal stem cells were shown to be susceptible to MV infection, migrate to the ovarian tumor xenograft, and provide a therapeutic benefit.95 Similar findings were reported in an orthotopic model of hepatocellular carcinoma in passively immunized SCID mice.98 These encouraging preclinical results have led to the creation of a Phase I clinical trial evaluating patient derived mesenchymal stem cells as carriers of MV-NIS in recurrent ovarian cancer (Table 1).50
Clinical considerations
Before initiating clinical testing, the safety of MV-CEA and MV-NIS strains was extensively evaluated in mouse and primate models. Studies performed in a transgenic mouse lacking the IFN-α/β receptor and expressing the human CD46 receptor in a tissue-specific pattern similar to humans (Ifnarko CD46 GE), demonstrated no toxicity following intraperitoneal, intravenous, or CNS delivery of the virus.66,99,100 Subsequent toxicology studies in measles-susceptible primates also demonstrated MV-NIS safety following CNS delivery in rhesus macaques (Macaca mulatta)101 and intravenous delivery in cynomolgus monkeys (Saimiri sciureus).93
The safety and maximum tolerated dose of MV-CEA and MV-NIS are currently being evaluated in numerous Phase I clinical trials (Table 1).33,47–52 While data from many of these trials are still forthcoming, there have been no reports of dose-limiting toxicity following administration of intraperitoneal doses up to 109 TCID50, intravenous doses up to 1011 TCID50, and CNS delivery of doses up to 107 TCID50. Results from the completed dose escalation trial involving intraperitoneal delivery of MV-CEA (103–109 TCID50) demonstrated no dose-limiting toxicity or virus induced immunosuppression.32 Serum CEA levels, a marker of virus replication, were observed in patients receiving the highest dose (109 TCID50). Significant decreases in cancer antigen-125 levels were observed in five patients, and median survival of patients in the trial (12.15 months) was double the historical expected median survival in this patient population (6 months).102 Based upon these results, a Phase I/II trial evaluating intraperitoneal administration of MV-NIS in treatment-resistant ovarian cancer was performed (Table 1).103 No dose-limiting toxicity was observed with MV-NIS doses up to 109 TCID50. 125I uptake was detected in the tumors of three patients indicating virus infection, and the overall median survival of 26.5 months compared favorably to studies evaluating novel therapeutics in this patient population (6–12 months).103 Interestingly, post-treatment evaluation showed an increase in IGFBP2 and Frα-specific effector T-cells, indicating a Th1 response against the ovarian cancer cells.103
Finally, a recent report from a clinical trial investigating MV-NIS in recurrent drug-refractory multiple myeloma builds upon the encouraging results observed in ovarian cancer (Table 1).48 In the trial, two patients with multiple plasmacytomas responded to therapy following intravenous delivery of MV-NIS at a dose of 1011 TCID50, with one patient experiencing durable complete remission at all disease sites.48 As MV-NIS infected cells express NIS and therefore concentrate iodine, single photon emission computed tomography was able to confirm tumor-specific infection following 123I administration. In the future, MV-NIS could be combined with high-energy beta-emitting 131I to increase the bystander effect surrounding infected cells. It should be noted that two factors may have contributed to the favorable response to therapy observed in these two patients. First, both patients had low pretreatment serum titers of anti-measles antibodies. Second, a very high dose of virus was administered. Previous experience in the ovarian cancer trials also suggested a dose-dependent response to therapy.32,103
Advantages and disadvantages of oncolytic MVs
There is an ever-growing list of oncolytic viruses in preclinical and clinical testing. Although there are reviews describing the advantages and disadvantages associated with these viruses, there is virtually no information comparing their head-to-head efficacy. MVs offer numerous advantages when deciding to include oncolytic viruses as part of the therapeutic approach. As discussed in previous sections, the excellent safety profile associated with MV-Edm strains makes it an attractive candidate for oncolytic virotherapy compared with other oncolytic viruses not used as vaccine agents.5 In contrast to the oncolytic DNA genome containing adenoviruses (Ads) and herpes simplex viruses (HSVs), MV with its RNA genome replicates in the cytoplasm of infected cells thus eliminating the possibility of insertional DNA mutagenesis. Similar to polioviruses and vaccinia viruses (VVs), MV has been genetically manipulated to select for preferential replication in cancer cells. In contrast, Ad, HSV, and vesicular stomatitis viruses (VSVs) have been genetically engineered with mutations or deletions in genes required for replication in normal but not cancer cells. There are multiple mechanisms by which oncolytic viruses lead to the death of tumor cells. While many oncolytic viruses cause tumor death via direct cell lyses, including MV, Ad, and HSV, the ability of MV to form syncytia provides an additional mechanism of killing that many other oncolytic viruses do not possess.104 Expression of viral hemagglutinin and fusion proteins in MV infected cells interacts with CD46 expressed by noninfected neighboring cells thus creating a bystander effect.14 Recently multiple oncolytic viruses (MV, Ad, VV, and HSV) have been demonstrated to induce antitumor immunity.105 In this process, local infection induces inflammation leading to immune stimulation and recruitment of immune cells. Cellular debris generated by oncolysis is taken up by antigen-presenting cells. Tumor antigens can then trigger cellular or antibody-mediated immune responses.105
While MV offer many advantages compared with other oncolytic viruses when deciding to conduct oncolytic virotherapy, there are disadvantages associated with MV. Although immunization has demonstrated the safety of MV-Edm administration and provides a safety barrier for subsequent exposure, serum neutralizing antibodies can potentially compromise oncolytic MV efficacy. This is extremely important when attempting to treat metastatic tumors where intravenous delivery of oncolytic viruses is necessary. In contrast, VV and VSV are two oncolytic viruses that have the potential to exhibit efficacy when delivered intravenously.106,107
Conclusion and future directions
MV-Edm derivatives are a promising experimental approach to the treatment of cancer as they have demonstrated significant antitumor activity in multiple preclinical models. Furthermore, results from completed clinical trials demonstrate their safety and show early evidence of biologic activity in humans. Numerous genetic advancements have been made in the design and development of MV-Edm derivatives. These enhancements have attempted to increase their safety, potency, and ability to be monitored. Recombinant strains targeting tumor-specific markers, or containing microRNA recognition sites, were designed to restrict virus replication to tumor cells, therefore leaving normal cells unharmed. MV-Edm derivatives that express the E. coli PNP gene, contain the P gene from the wild-type virus, or express NIS have been constructed to increase virus CPE. Furthermore, CEA and NIS reporter genes have made real-time in vivo monitoring possible. There has been a recent impetus to construct MV-Edm derivatives that either evade the systemic immune response via infected cell carriers or illicit an antitumor immune response. Results regarding the safety and efficacy of MV therapy from ongoing clinical trials, coupled with continual evolution of MV-Edm derivatives, will help guide future development strategies, leading to a new generation of safer and more effective oncolytic MV.
Disclosure
The authors report no conflicts of interest in this work.
References
Castaneda SJ, Wong TC. Leader sequence distinguishes between translatable and encapsidated measles virus RNAs. J Virol. 1990;64(1):222–230. | |
Billeter MA, Naim HY, Udem SA. Reverse genetics of measles virus and resulting multivalent recombinant vaccines: applications of recombinant measles viruses. Curr Top Microbiol Immunol. 2009;329:129–162. | |
Enders JF, Peebles TC. Propagation in tissue cultures of cytopathogenic agents from patients with measles. Proc Soc Exp Biol Med. 1954;86(2):277–286. | |
Griffin DE, Pan CH. Measles: old vaccines, new vaccines. Curr Top Microbiol Immunol. 2009;330:191–212. | |
Moss WJ, Griffin DE. Measles. Lancet. 2012;379(9811):153–164. | |
Dhiman N, Jacobson RM, Poland GA. Measles virus receptors: SLAM and CD46. Rev Med Virol. 2004;14(4):217–229. | |
Muhlebach MD, Mateo M, Sinn PL, et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature. 2011; 480(7378):530–533. | |
Yanagi Y, Takeda M, Ohno S. Measles virus: cellular receptors, tropism and pathogenesis. J Gen Virol. 2006;87(Pt 10):2767–2779. | |
Naniche D, Varior-Krishnan G, Cervoni F, et al. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J Virol. 1993;67(10):6025–6032. | |
Dorig RE, Marcil A, Chopra A, Richardson CD. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell. 1993; 75(2):295–305. | |
Nielsen L, Blixenkrone-Moller M, Thylstrup M, Hansen NJ, Bolt G. Adaptation of wild-type measles virus to CD46 receptor usage. Arch Virol. 2001;146(2):197–208. | |
Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol Immunol. 2003;40(2–4):109–123. | |
Hara T, Suzuki Y, Semba T, Hatanaka M, Matsumoto M, Seya T. High expression of membrane cofactor protein of complement (CD46) in human leukaemia cell lines: implication of an alternatively spliced form containing the STA domain in CD46 up-regulation. Scand J Immunol. 1995;42(6):581–590. | |
Russell SJ, Peng KW. Measles virus for cancer therapy. Curr Top Microbiol Immunol. 2009;330:213–241. | |
Ulasov IV, Tyler MA, Zheng S, Han Y, Lesniak MS. CD46 represents a target for adenoviral gene therapy of malignant glioma. Hum Gene Ther. 2006;17(5):556–564. | |
Thorsteinsson L, O’Dowd GM, Harrington PM, Johnson PM. The complement regulatory proteins CD46 and CD59, but not CD55, are highly expressed by glandular epithelium of human breast and colorectal tumour tissues. APMIS. 1998;106(9):869–878. | |
Simpson KL, Jones A, Norman S, Holmes CH. Expression of the complement regulatory proteins decay accelerating factor (DAF, CD55), membrane cofactor protein (MCP, CD46) and CD59 in the normal human uterine cervix and in premalignant and malignant cervical disease. Am J Pathol. 1997;151(5):1455–1467. | |
Murray KP, Mathure S, Kaul R, et al. Expression of complement regulatory proteins-CD 35, CD 46, CD 55, and CD 59-in benign and malignant endometrial tissue. Gynecol Oncol. 2000;76(2):176–182. | |
Juhl H, Helmig F, Baltzer K, Kalthoff H, Henne-Bruns D, Kremer B. Frequent expression of complement resistance factors CD46, CD55, and CD59 on gastrointestinal cancer cells limits the therapeutic potential of monoclonal antibody 17-1A. J Surg Oncol. 1997;64(3):222–230. | |
Kinugasa N, Higashi T, Nouso K, et al. Expression of membrane cofactor protein (MCP, CD46) in human liver diseases. Br J Cancer. 1999;80(11):1820–1825. | |
Varsano S, Rashkovsky L, Shapiro H, Ophir D, Mark-Bentankur T. Human lung cancer cell lines express cell membrane complement inhibitory proteins and are extremely resistant to complement-mediated lysis; a comparison with normal human respiratory epithelium in vitro, and an insight into mechanism(s) of resistance. Clin Exp Immunol. 1998;113(2):173–182. | |
Blok VT, Daha MR, Tijsma OM, Weissglas MG, van den Broek LJ, Gorter A. A possible role of CD46 for the protection in vivo of human renal tumor cells from complement-mediated damage. Lab Invest. 2000; 80(3):335–344. | |
Seya T, Hara T, Matsumoto M, Akedo H. Quantitative analysis of membrane cofactor protein (MCP) of complement. High expression of MCP on human leukemia cell lines, which is down-regulated during cell differentiation. J Immunol. 1990;145(1):238–245. | |
Bjorge L, Hakulinen J, Wahlstrom T, Matre R, Meri S. Complement-regulatory proteins in ovarian malignancies. Int J Cancer. 1997;70(1):14–25. | |
Ong HT, Timm MM, Greipp PR, et al. Oncolytic measles virus targets high CD46 expression on multiple myeloma cells. Exp Hematol. 2006;34(6):713–720. | |
Anderson BD, Nakamura T, Russell SJ, Peng KW. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004;64(14):4919–4926. | |
Radecke F, Spielhofer P, Schneider H, et al. Rescue of measles viruses from cloned DNA. EMBO J. 1995;14(23):5773–5784. | |
Duprex WP, McQuaid S, Hangartner L, Billeter MA, Rima BK. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. J Virol. 1999;73(11):9568–9575. | |
Peng KW, Facteau S, Wegman T, O’Kane D, Russell SJ. Non-invasive in vivo monitoring of trackable viruses expressing soluble marker peptides. Nat Med. 2002;8(5):527–531. | |
Rima BK, Duprex WP. The measles virus replication cycle. Curr Top Microbiol Immunol. 2009;329:77–102. | |
Duprex WP, Rima BK. Using green fluorescent protein to monitor measles virus cell-to-cell spread by time-lapse confocal microscopy. Method Mol Biol. 2002;183:297–307. | |
Galanis E, Hartmann LC, Cliby WA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70(3):875–882. | |
Mayo Clinic. Viral therapy in treating patients with recurrent glioblastoma multiforme. Available from: www.clinicaltrials.gov/ct2/show/NCT00390299. ClinicalTrials.gov identifier: NCT00390299. Accessed May 22, 2015. | |
Dingli D, Peng KW, Harvey ME, et al. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004;103(5):1641–1646. | |
Dohan O, De la Vieja A, Paroder V, et al. The sodium/iodide Symporter (NIS): characterization, regulation, and medical significance. Endocr Rev. 2003;24(1):48–77. | |
Riesco-Eizaguirre G, Santisteban P. A perspective view of sodium iodide symporter research and its clinical implications. Eur J Endocrinol. 2006;155(4):495–512. | |
Dingli D, Kemp BJ, O’Connor MK, Morris JC, Russell SJ, Lowe VJ. Combined I-124 positron emission tomography/computed tomography imaging of NIS gene expression in animal models of stably transfected and intravenously transfected tumor. Mol Imaging Biol. 2006;8(1):16–23. | |
Carlson SK, Classic KL, Hadac EM, et al. In vivo quantitation of intratumoral radioisotope uptake using micro-single photon emission computed tomography/computed tomography. Mol Imaging Biol. 2006;8(6):324–332. | |
Hutzen B, Pierson CR, Russell SJ, Galanis E, Raffel C, Studebaker AW. Treatment of medulloblastoma using an oncolytic measles virus encoding the thyroidal sodium iodide symporter shows enhanced efficacy with radioiodine. BMC Cancer. 2012;12:508. | |
Hasegawa K, Pham L, O’Connor MK, Federspiel MJ, Russell SJ, Peng KW. Dual therapy of ovarian cancer using measles viruses expressing carcinoembryonic antigen and sodium iodide symporter. Clin Cancer Res. 2006;12(6):1868–1875. | |
Carlson SK, Classic KL, Hadac EM, et al. Quantitative molecular imaging of viral therapy for pancreatic cancer using an engineered measles virus expressing the sodium-iodide symporter reporter gene. AJR Am J Roentgenol. 2009;192(1):279–287. | |
Li H, Peng KW, Dingli D, Kratzke RA, Russell SJ. Oncolytic measles viruses encoding interferon beta and the thyroidal sodium iodide symporter gene for mesothelioma virotherapy. Cancer Gene Ther. 2010;17(8):550–558. | |
Msaouel P, Iankov ID, Allen C, et al. Noninvasive imaging and radiovirotherapy of prostate cancer using an oncolytic measles virus expressing the sodium iodide symporter. Mol Ther. 2009;17(12):2041–2048. | |
Opyrchal M, Allen C, Iankov I, et al. Effective radiovirotherapy for malignant gliomas by using oncolytic measles virus strains encoding the sodium iodide symporter (MV-NIS). Hum Gene Ther. 2012;23(4):419–427. | |
Li H, Peng KW, Russell SJ. Oncolytic measles virus encoding thyroidal sodium iodide symporter for squamous cell cancer of the head and neck radiovirotherapy. Hum Gene Ther. 2012;23(3):295–301. | |
Domingo-Musibay E, Allen C, Kurokawa C, et al. Measles Edmonston vaccine strain derivatives have potent oncolytic activity against osteosarcoma. Cancer Gene Ther. 2014;21(11):483–490. | |
Mayo Clinic. Vaccine therapy with or without cyclophosphamide in treating patients with recurrent or refractory multiple myeloma. Available from: http://www.clinicaltrials.gov/ct2/show/NCT00450814. ClinicalTrials.gov identifier: NCT00450814. Accessed May 22, 2015. | |
Russell SJ, Federspiel MJ, Peng KW, et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc. 2014;89(7):926–933. | |
Mayo Clinic. Intrapleural measles virus therapy in patients with malignant pleural mesothelioma. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01503177. ClinicalTrials.gov identifier: NCT01503177. Accessed May 22, 2015. | |
Mayo Clinic. MV-NIS infected mesenchymal stem cells in treating patients with recurrent ovarian cancer. Available from: http://www.clinicaltrials.gov/ct2/show/NCT02068794. ClinicalTrials.gov identifier: NCT02068794. Accessed May 22, 2015. | |
Mayo Clinic. MV-NIS or investigator’s choice chemotherapy in treating patients with ovarian, fallopian, or peritoneal cancer. Available from: http://www.clinicaltrials.gov/ct2/show/NCT02364713. ClinicalTrials.gov identifier: NCT02364713. Accessed May 22, 2015. | |
Mayo Clinic. Viral therapy in treating patients with recurrent or metastatic squamous cell carcinoma of the head and neck cancer. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01846091. ClinicalTrials.gov identifier: NCT01846091. Accessed May 22, 2015. | |
Hashiguchi T, Kajikawa M, Maita N, et al. Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc Natl Acad Sci U S A. 2007;104(49):19535–19540. | |
Xie MF, Tanaka K, Ono N, Minagawa H, Yanagi Y. Amino acid substitutions at position 481 differently affect the ability of the measles virus hemagglutinin to induce cell fusion in monkey and marmoset cells co-expressing the fusion protein. Arch Virol. 1999;144(9):1689–1699. | |
Vongpunsawad S, Oezgun N, Braun W, Cattaneo R. Selectively receptor-blind measles viruses: Identification of residues necessary for SLAM- or CD46-induced fusion and their localization on a new hemagglutinin structural model. J Virol. 2004;78(1):302–313. | |
Nakamura T, Peng KW, Harvey M, et al. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat Biotechnol. 2005;23(2):209–214. | |
Paraskevakou G, Allen C, Nakamura T, et al. Epidermal growth factor receptor (EGFR)-retargeted measles virus strains effectively target EGFR- or EGFRvIII expressing gliomas. Mol Ther. 2007;15(4):677–686. | |
Allen C, Vongpunsawad S, Nakamura T, et al. Retargeted oncolytic measles strains entering via the EGFRvIII receptor maintain significant antitumor activity against gliomas with increased tumor specificity. Cancer Res. 2006;66(24):11840–11850. | |
Peng KW, Donovan KA, Schneider U, Cattaneo R, Lust JA, Russell SJ. Oncolytic measles viruses displaying a single-chain antibody against CD38, a myeloma cell marker. Blood. 2003;101(7):2557–2562. | |
Hummel HD, Kuntz G, Russell SJ, et al. Genetically engineered attenuated measles virus specifically Infects and kills primary multiple myeloma cells. J Gen Virol. 2009;90:693–701. | |
Hasegawa K, Nakamura T, Harvey M, et al. The use of a tropism-modified measles virus in folate receptor-targeted virotherapy of ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6170–6178. | |
Bossow S, Grossardt C, Temme A, et al. Armed and targeted measles virus for chemovirotherapy of pancreatic cancer. Cancer Gene Ther. 2011;18(8):598–608. | |
Liu C, Hasegawa K, Russell SJ, Sadelain M, Peng KW. Prostate-specific membrane antigen retargeted measles virotherapy for the treatment of prostate cancer. Prostate. 2009;69(10):1128–1141. | |
Ungerechts G, Springfeld C, Frenzke ME, et al. Lymphoma chemovirotherapy: CD20-targeted and convertase-armed measles virus can synergize with fludarabine. Cancer Res. 2007;67(22):10939–10947. | |
Hasegawa K, Hu C, Nakamura T, Marks JD, Russell SJ, Peng KW. Affinity thresholds for membrane fusion triggering by viral glycoproteins. J Virol. 2007;81(23):13149–13157. | |
Mrkic B, Pavlovic J, Rulicke T, et al. Measles virus spread and pathogenesis in genetically modified mice. J Virol. 1998;72(9):7420–7427. | |
Msaouel P, Iankov ID, Dispenzieri A, Galanis E. Attenuated oncolytic measles virus strains as cancer therapeutics. Curr Pharm Biotechnol. 2012;13(9):1732–1741. | |
Leber MF, Bossow S, Leonard VH, et al. MicroRNA-sensitive oncolytic measles viruses for cancer-specific vector tropism. Mol Ther. 2011;19(6):1097–1106. | |
Baertsch MA, Leber MF, Bossow S, et al. MicroRNA-mediated multi-tissue detargeting of oncolytic measles virus. Cancer Gene Ther. 2014;21(9):373–380. | |
Ungerechts G, Springfeld C, Frenzke ME, et al. An immunocompetent murine model for oncolysis with an armed and targeted measles virus. Mol Ther. 2007;15(11):1991–1997. | |
Parker WB, Allan PW, Shaddix SC, et al. Metabolism and metabolic actions of 6-methylpurine and 2-fluoroadenine in human cells. Biochem Pharmacol. 1998;55(10):1673–1681. | |
Haralambieva I, Iankov I, Hasegawa K, Harvey M, Russell SJ, Peng KW. Engineering oncolytic measles virus to circumvent the intracellular innate immune response. Mol Ther. 2007;15(3):588–597. | |
Ohno S, Ono N, Takeda M, Takeuchi K, Yanagi Y. Dissection of measles virus V protein in relation to its ability to block alpha/beta interferon signal transduction. J Gen Virol. 2004;85(Pt 10):2991–2999. | |
Boisgerault N, Tangy F, Gregoire M. New perspectives in cancer virotherapy: bringing the immune system into play. Immunotherapy. 2010;2(2):185–199. | |
Wojton J, Kaur B. Impact of tumor microenvironment on oncolytic viral therapy. Cytokine Growth Factor Rev. 2010;21(2–3):127–134. | |
De Silva N, Atkins H, Kirn DH, Bell JC, Breitbach CJ. Double trouble for tumours: exploiting the tumour microenvironment to enhance anticancer effect of oncolytic viruses. Cytokine Growth Factor Rev. 2010; 21(2–3):135–141. | |
Grossardt C, Engeland CE, Bossow S, et al. Granulocyte-macrophage colony-stimulating factor-armed oncolytic measles virus is an effective therapeutic cancer vaccine. Human Gene Ther. 2013;24(7):644–654. | |
Grote D, Cattaneo R, Fielding AK. Neutrophils contribute to the measles virus-induced antitumor effect: enhancement by granulocyte macrophage colony-stimulating factor expression. Cancer Res. 2003;63(19):6463–6468. | |
Quesniaux V, Jones T. Granulocyte-macrophage colony stimulating factor. In: Thomson A, editor. The Cytokine Handbook. Burlington, MA: Academic Press Ltd; 1998:637–670. | |
Chan CW, Crafton E, Fan HN, et al. Interferon-producing killer dendritic cells provide a link between innate and adaptive immunity. Nat Med. 2006;12(2):207–213. | |
Le Bon A, Tough DF. Links between innate and adaptive immunity via type I interferon. Curr Opin Immunol. 2002;14(4):432–436. | |
D’Elios MM, Amedei A, Cappon A, Del Prete G, de Bernard M. The neutrophil-activating protein of Helicobacter pylori (HP-NAP) as an immune modulating agent. FEMS Immunol Med Bicrobiol. 2007;50(2):157–164. | |
Iankov ID, Allen C, Federspiel MJ, et al. Expression of immunomodulatory neutrophil-activating protein of Helicobacter pylori enhances the antitumor activity of oncolytic measles virus. Mol Ther. 2012;20(6):1139–1147. | |
Engeland CE, Grossardt C, Veinalde R, et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Ther. 2014;22(11):1949–1959. | |
Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182(2):459–465. | |
Bour-Jordan H, Esensten JH, Martinez-Llordella M, Penaranda C, Stumpf M, Bluestone JA. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/ B7 family. Immunol Rev. 2011;241(1):180–205. | |
McQuillan GM, Kruszon-Moran D, Hyde TB, Forghani B, Bellini W, Dayan GH. Seroprevalence of measles antibody in the US population, 1999–2004. J Infect Dis. 2007;196(10):1459–1464. | |
Audet S, Virata-Theimer ML, Beeler JA, et al. Measles-virus-neutralizing antibodies in intravenous immunoglobulins. J Infect Dis. 2006;194(6):781–789. | |
Hangartner L, Zinkernagel RM, Hengartner H. Antiviral antibody responses: the two extremes of a wide spectrum. Nature Rev Immunol. 2006;6(3):231–243. | |
Fulci G, Breymann L, Gianni D, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci U S A. 2006;103(34):12873–12878. | |
Kambara H, Saeki Y, Chiocca EA. Cyclophosphamide allows for in vivo dose reduction of a potent oncolytic virus. Cancer Res. 2005;65(24):11255–11258. | |
Ikeda K, Ichikawa T, Wakimoto H, et al. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat Med. 1999;5(8):881–887. | |
Myers RM, Greiner SM, Harvey ME, et al. Preclinical pharmacology and toxicology of intravenous MV-NIS, an oncolytic measles virus administered with or without cyclophosphamide. Clin Pharmacol Ther. 2007;82(6):700–710. | |
Iankov ID, Blechacz B, Liu C, et al. Infected cell carriers: a new strategy for systemic delivery of oncolytic measles viruses in cancer virotherapy. Mol Ther. 2007;15(1):114–122. | |
Mader EK, Maeyama Y, Lin Y, et al. Mesenchymal stem cell carriers protect oncolytic measles viruses from antibody neutralization in an orthotopic ovarian cancer therapy model. Clin Cancer Res. 2009;15(23):7246–7255. | |
Fisher K. Striking out at disseminated metastases: the systemic delivery of oncolytic viruses. Curr Opin Mol Ther. 2006;8(4):301–313. | |
Wang Y, Yuan F. Delivery of viral vectors to tumor cells: extracellular transport, systemic distribution, and strategies for improvement. Ann Biomed Eng. 2006;34(1):114–127. | |
Ong HT, Federspiel MJ, Guo CM, et al. Systemically delivered measles virus-infected mesenchymal stem cells can evade host immunity to inhibit liver cancer growth. J Hepatol. 2013;59(5):999–1006. | |
Allen C, Paraskevakou G, Liu C, et al. Oncolytic measles virus strains in the treatment of gliomas. Expert Opin Biol Ther. 2008;8(2):213–220. | |
Peng KW, Frenzke M, Myers R, et al. Biodistribution of oncolytic measles virus after intraperitoneal administration into Ifnar-CD46Ge transgenic mice. Hum Gene Ther. 2003;14(16):1565–1577. | |
Myers R, Harvey M, Kaufmann TJ, et al. Toxicology study of repeat intracerebral administration of a measles virus derivative producing carcinoembryonic antigen in rhesus macaques in support of a phase I/II clinical trial for patients with recurrent gliomas. Hum Gene Ther. 2008;19(7):690–698. | |
Markman M, Webster K, Zanotti K, Peterson G, Kulp B, Belinson J. Survival following the documentation of platinum and taxane resistance in ovarian cancer: a single institution experience involving multiple phase 2 clinical trials. Gynecol Oncol. 2004;93(3):699–701. | |
Galanis E, Atherton PJ, Maurer MJ, et al. Oncolytic measles virus expressing the sodium iodide symporter to treat drug-resistant ovarian cancer. Cancer Res. 2015;75(1):22–30. | |
Atherton MJ, Lichty BD. Evolution of oncolytic viruses: novel strategies for cancer treatment. Immunotherapy. 2013;5(11):1191–1206. | |
Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14(8):559–567. | |
Kirn DH, Wang Y, Liang W, Contag CH, Thorne SH. Enhancing poxvirus oncolytic effects through increased spread and immune evasion. Cancer Res. 2008;68(7):2071–2075. | |
Liu YP, Steele MB, Suksanpaisan L, et al. Oncolytic measles and vesicular stomatitis virotherapy for endometrial cancer. Gynecol Oncol. 2014;132(1):194–202. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.