Back to Journals » Drug Design, Development and Therapy » Volume 11
Adenosine A2A receptor agonist prevents cardiac remodeling and dysfunction in spontaneously hypertensive male rats after myocardial infarction
Authors da Silva JS ![]() , Gabriel-Costa D, Sudo RT, Wang H, Groban L, Ferraz EB, Nascimento JHM
, Gabriel-Costa D, Sudo RT, Wang H, Groban L, Ferraz EB, Nascimento JHM ![]() , Fraga CAM, Barreiro EJ
, Fraga CAM, Barreiro EJ ![]() , Zapata-Sudo G
, Zapata-Sudo G
Received 21 May 2016
Accepted for publication 30 June 2016
Published 6 March 2017 Volume 2017:11 Pages 553—562
DOI https://doi.org/10.2147/DDDT.S113289
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Jaqueline S da Silva,1 Daniele Gabriel-Costa,1 Roberto T Sudo,1 Hao Wang,2 Leanne Groban,2 Emanuele B Ferraz,3 José Hamilton M Nascimento,3 Carlos Alberto M Fraga,1 Eliezer J Barreiro,1 Gisele Zapata-Sudo1
1Research Program Development of Drugs, Institute of Biomedical Sciences, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil; 2Department of Anesthesiology, Wake Forest School of Medicine, Winston-Salem, NC, USA; 3Institute of Biophysics Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
Background: This work evaluated the hypothesis that 3,4-methylenedioxybenzoyl-2- thienylhydrazone (LASSBio-294), an agonist of adenosine A2A receptor, could be beneficial for preventing cardiac dysfunction due to hypertension associated with myocardial infarction (MI).
Methods: Male spontaneously hypertensive rats (SHR) were randomly divided into four groups (six animals per group): sham-operation (SHR-Sham), and myocardial infarction rats (SHR-MI) were treated orally either with vehicle or LASSBio-294 (10 and 20 mg.kg-1.d-1) for 4 weeks. Echocardiography and in vivo hemodynamic parameters measured left ventricle (LV) structure and function. Exercise tolerance was evaluated using a treadmill test. Cardiac remodeling was accessed by LV collagen deposition and tumor necrosis factor α expression.
Results: Early mitral inflow velocity was significantly reduced in the SHR-MI group, and there was significant recovery in a dose-dependent manner after treatment with LASSBio-294. Exercise intolerance observed in the SHR-MI group was prevented by 10 mg.kg-1.d-1 of LASSBio-294, and exercise tolerance exceeded that of the SHR-Sham group at 20 mg.kg-1.d-1. LV end-diastolic pressure increased after MI, and this was prevented by 10 and 20 mg.kg-1.d-1 of LASSBio-294. Sarcoplasmic reticulum Ca2+ ATPase levels were restored in a dose-dependent manner after treatment with LASSBio-294. Fibrosis and inflammatory processes were also counteracted by LASSBio-294, with reductions in LV collagen deposition and tumor necrosis factor α expression.
Conclusion: In summary, oral administration of LASSBio-294 after MI in a dose-dependent manner prevented the development of cardiac dysfunction, demonstrating this compound’s potential as an alternative treatment for heart failure in the setting of ischemic heart disease with superimposed chronic hypertension.
Keywords: hypertension, myocardial infarction, LASSBio-294 and agonist of adenosine receptor A2A
Introduction
Chronic heart failure (HF) is increasing in incidence and prevalence worldwide. This cardiovascular disease is often preceded by myocardial infarction (MI) and hypertension.1,2 HF presents with a complex constellation of symptoms, including dyspnea and exercise intolerance.3–5 The primary underlying defect in HF is an inability of the left ventricle (LV) to maintain an adequate forward stroke volume to meet the metabolic demands of the body, either because of impaired LV contractile function or insufficient LV filling.6
The dynamic cellular and molecular processes that lead to permanent alterations in the geometry and function of the ventricles after an MI or due to chronic hypertension remain an active area of research. Current interventions used to prevent and treat LV remodeling include beta-blockers, angiotensin-converting enzyme inhibitors, aldosterone antagonists, and cardiac rehabilitation. The overall efficacy of these treatments in terms of cardiovascular morbidity and mortality is modest.7 Therefore, continued exploration of novel pharmacological targets for the treatment of cardiac ischemic and nonischemic HF is reasonable.
Among the potential therapeutic targets are adenosine receptors (ARs), which upon activation by adenosine, appear to ameliorate the deleterious consequences of HF. AR activation inhibits fibroblast proliferation and collagen synthesis,8,9 thus limiting cardiac tissue remodeling and progression to failure. Moreover, activated ARs have favorable regulatory effects on several phenomena associated with ischemic and pressure-overload insults, including cytokine release,10,11 cell death processes,12 and oxidative stress.13,14
Recently, we showed that 3,4-methylenedioxybenzoyl-2-thienylhydrazone (LASSBio-294), an A2AR ligand (IC50 9.5 μM obtained in a binding assay performed by CEREP S.A., Celle-Lévescault, France), prevented the development of diastolic dysfunction15 and attenuated exercise intolerance16 in normotensive rats with MI.
However, the beneficial effect of LASSBio-294 in rats with MI superimposed on preexisting hypertension is still unknown. Indeed, spontaneously hypertensive rat (SHR) strains have provided a useful model of the transition from stable, compensated hypertrophy to decompensated HF in the context of an ischemic insult.17–19 Accordingly, we hypothesized that chronic activation of A2AR by LASSBio-294 after MI would mitigate the progression to HF, as defined by myocardial extracellular matrix remodeling, LV dysfunction, and exercise intolerance, in male SHRs.
Methods
Animals
Male SHRs (12 weeks old) were maintained at 22°C±2°C with a relative humidity of 60%–70%, 12/12-hour light/dark cycles. Food and water were available ad libitum. All procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (National Institute of Health) and the study was approved by the Ethics and Research Committee of the Federal University of Rio de Janeiro (No DFBCICB049A), Rio de Janeiro Brazil.
Coronary artery ligation
To induce MI, under isoflurane anesthesia (3% v/v), rats were subjected to ligation of the anterior descending coronary artery, as previously described by Costa et al.15 The same procedure was employed for a control group (Sham) without suturing the coronary artery. MI was confirmed 4–6 hours after surgery by the presence of anterior wall motion abnormalities and thinning of anterior and posterior LV walls, using M-mode echocardiographic images, obtained in the two-dimensional short-axis view. The SHRs were randomized into the following four groups (six animals per group): without MI treated with vehicle, dimethyl sulfoxide (SHR-Sham), MI treated with dimethyl sulfoxide (SHR-MI), MI treated with 10 mg.kg−1.d−1 LASSBio-294 (SHR-MI + LASSBio-294 [10 mg.kg−1.d−1]), and MI treated with 20 mg.kg−1.d−1 LASSBio-294 (SHR-MI + LASSBio-294 [20 mg.kg−1.d−1]). LASSBio-294 was administered orally by gastric gavage at doses of 10 or 20 mg.kg−1.d−1 for 4 weeks starting on the day of the surgical procedure. LASSBio-294 was synthesized at Laboratório de Avaliação de Substâncias Bioativas of the Federal University of Rio de Janeiro (Rio de Janeiro, RJ, Brazil).
Experimental protocol
The day after the surgical procedure, MI was confirmed by echocardiogram. Only animals that underwent surgery and had absence of motility of the anterior wall of the LV in M-mode were included in the study. After echocardiographic data collection, they were subsequently treated with dimethyl sulfoxide or LASSBio-294 according to their respective groups, which was administered daily for 4 weeks. On the last 2 days, the animals underwent: i) an additional echocardiographic evaluation (26th day) and ii) an exercise fatigue tolerance test (27th day). On the final day, hemodynamic parameters were obtained before euthanasia. After cervical decapitation under anesthesia, the lung and heart were removed for weighing, and the hearts were then prepared for histological evaluation and immunohistochemical analyses.
Exercise fatigue tolerance test
Rats performed a graded treadmill test on a customized rodent treadmill (EP–131, Insight, São Paulo, Brazil). In the fourth week, a treadmill test was conducted as follows: after acclimatization for 3 minutes in the cages, the animals were induced to run for 3 minutes at 8 m.min−1, 3 minutes at 12 m.min−1, and until volitional fatigue at 18 m.min−1. Exercise intolerance was determined by measuring the running distance until volitional fatigue, which was confirmed by loss of the animal’s righting reflex.20
Echocardiographic measures of systolic and diastolic function
LV function was assessed with an echocardiograph equipped with a 10 MHz mechanical transducer (Esaote model, CarisPlus, Firenze, Italy). The animals were anesthetized with 3% (v/v) isoflurane and maintained with 1% (v/v). LV M-mode images were obtained in the two-dimensional short-axis view, close to the papillary muscles. Anterior and posterior wall thickness (AWT and PWT, respectively) and internal diameter during LV end-diastolic and end-systolic dimensions were measured. The LV ejection fraction (EF) and fractional shortening, indices of global systolic function, were calculated as: EF = (LVDV – LVSV)/LVDV, where LVDV is LV end-diastolic volume and LVSV is LV end-systolic volume; and fractional shortening = (LVDD – LVSD)/LVDD) ×100, where LVDD is LV end-diastolic and LVSD end-systolic dimensions. Mitral inflow measurements of early filling velocities (Emax) and late filling velocities (Amax) were used to calculate the E/A ratio to evaluate diastolic function.21 All measurements were obtained according to the American Society of Echocardiography guidelines.
Cardiac mass and hemodynamic measurements
After 4 weeks, the animals were anesthetized with pentobarbital sodium (50 mg.kg−1) and prepared for terminal arterial blood and LV pressure measurements. A catheter (PE-50) was introduced into the right carotid artery and connected to a pressure transducer (MLT884, ADInstruments, Inc.; Colorado Springs, CO, USA) that measured systolic (SBP), diastolic (DBP) and mean (MBP) blood pressures. Subsequently, the catheter was introduced into the LV to record intracavitary pressure. The LV systolic pressure, LV end-diastolic pressure (LVEDP), and LV contraction and relaxation rates were assessed by maximal positive and negative dP/dt, respectively. Data were recorded on a polygraph (Powerlab, ADInstruments, Inc., Sydney, NSW, Australia) using LabChart software (Version 7.0, ADInstruments, Inc., Sydney, NSW, Australia). Then the animals were euthanized, the heart and lung weighed, and the tissues prepared for histological and immunohistochemical analyses.
Cardiac fibrosis
The LV interstitial collagen volume was examined with Picrosirius red staining. The collagen fraction was determined by averaging six fields/heart of the fibrotic areas within the infarct border zone. The amount of collagen in the tissue was determined as a percentage of the total area by optical microscopy.15
Immunohistochemistry
Immunohistochemical analysis of heart sections (4-μm thick) for tumor necrosis factor α (TNF-α) and sarcoplasmic reticulum (SR) Ca2+-ATPase (SERCA2) was performed by standard procedures. Formalin-fixed, paraffin-embedded LV sections were deparaffinized and exposed to 3% (v/v) hydrogen peroxide, then subjected to antigen retrieval via immersion in citric acid (pH 6.0, 0.01 mol/L) at 96°C, followed by slow cooling to 60°C. Subsequently, the sections were incubated with primary anti-TNF-α antibody (AB6671- Abcam, Cambridge, UK) or anti-SERCA2 antibody (1:100) (AB2817, Abcam) overnight at 4°C, rinsed with phosphate-buffered saline, and incubated with biotinylated secondary IgG (Vector Laboratories, Burlingame, CA, USA) at 4°C. Antibody binding was detected by incubation with the peroxidase substrate solution, diaminobenzidine. The tissue sections were counterstained with hematoxylin, dehydrated, mounted, and observed under light microscopy with a 40× objective. The expression was quantified using Image-Pro Plus 6 software (ImageJ), measuring images obtained from the infarct border zone.
Statistical analysis
The data were expressed as the mean ± standard errors of the mean. Differences among groups were considered statistically significant when the P-value was <0.05 using one-way analysis of variance followed by a post hoc Dunnett’s test.
Results
Effects of LASSBio-294 on exercise intolerance in infarcted SHRs
Exercise capacity data, as determined by the graded treadmill test, are shown in Figure 1. The SHR-MI group had a significantly shorter mean running distance (39.01±4.39 m) than the SHR-Sham group (257.9±13.19 m; P<0.05). This exercise intolerance was prevented by treatment with LASSBio-294 at a dose of 10 mg.kg−1.d−1, with the SHR-MI + LASSBio-294 (10 mg.kg−1.d−1) group running a mean distance of 296±26.47 m (P<0.05 vs SHR-MI), which was similar to the distance observed for the noninfarcted group. The mean running distance of SHR-MI + LASSBio-294 (20 mg.kg−1.d−1) animals was 529.8±133.3 m, far exceeding that observed for both the SHR-MI (P<0.01) and SHR-Sham (P<0.05) groups.
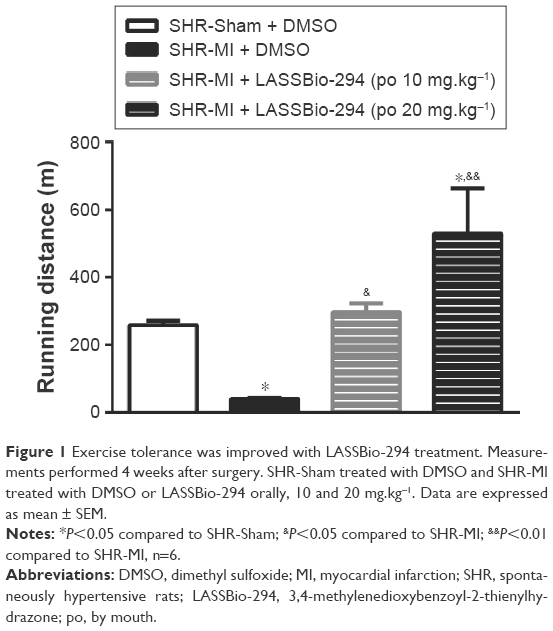 | Figure 1 Exercise tolerance was improved with LASSBio-294 treatment. Measurements performed 4 weeks after surgery. SHR-Sham treated with DMSO and SHR-MI treated with DMSO or LASSBio-294 orally, 10 and 20 mg.kg−1. Data are expressed as mean ± SEM. |
Echocardiographic changes induced in infarcted SHR
Echocardiographic evaluation and assessment of LV structural parameters revealed a significant decrease in diastolic and systolic AWT of the SHR-MI group compared with the SHR-Sham group. Changes in AWT were prevented in the SHR-MI+LASSBio-294 (10 mg.kg−1.d−1) and SHR-MI+LASSBio-294 (20 mg.kg−1.d−1) groups, relative to those observed in the untreated SHR-MI. No significant differences in diastolic and systolic PWT were observed between the groups. Diastolic LV internal chamber diameter was elevated in the SHR-MI group, and treatment with 20 mg.kg−1.d−1 of LASSBio-294 prevented this effect (Table 1). Both doses of LASSBio-294 reversed the MI-induced EF decrease, but only the higher dose (20 mg.kg−1.d−1) prevented the decline in fractional shortening (Table 2).
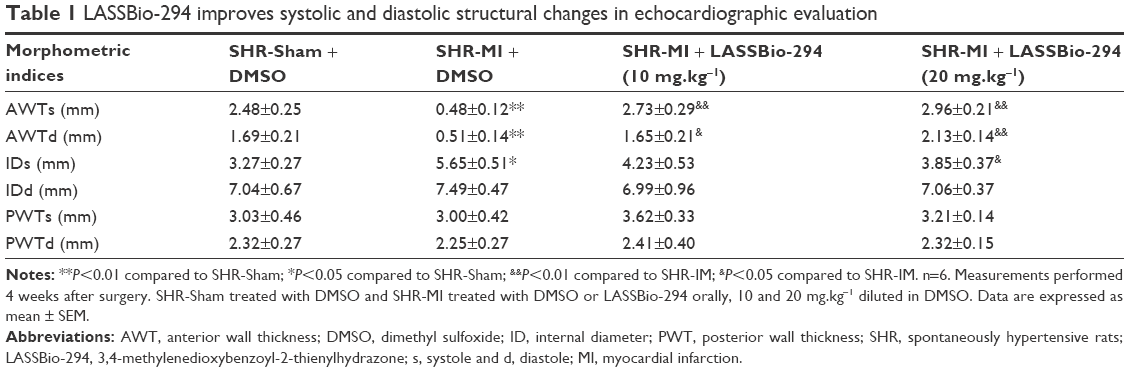 | Table 1 LASSBio-294 improves systolic and diastolic structural changes in echocardiographic evaluation |
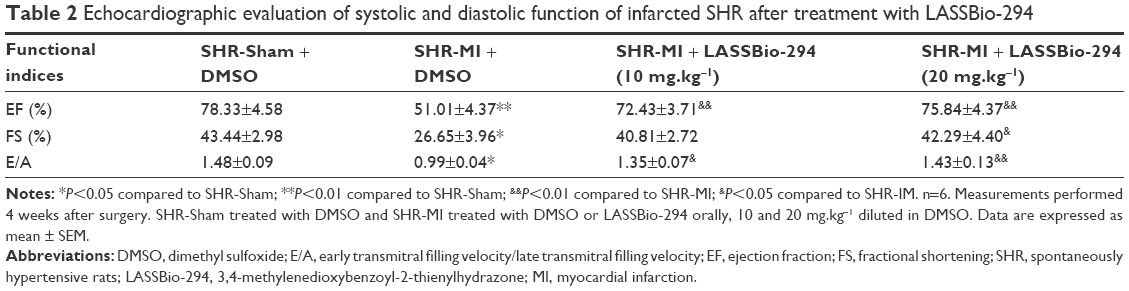 | Table 2 Echocardiographic evaluation of systolic and diastolic function of infarcted SHR after treatment with LASSBio-294 |
Diastolic function obtained through the analyses of early transmitral filling velocity to late transmitral filling velocity ratio (E/A) showed that the SHR-MI group developed a significant impairment of the filling pattern compared with the SHR-Sham group. On the other hand, this index was significantly increased with LASSBio-294 treatment in a dose-dependent manner (Table 2).
LASSBio-294 treatment improves hemodynamic changes in infarcted SHRs
SBP or DBP did not differ significantly between the SHR-Sham and SHR-MI groups. However, as shown in Figure 2A, LASSBio-294 treatment did reduce SBP significantly, at both the 10 mg.kg−1.d−1 (P<0.05 vs SHR-MI and SHR-Sham) and 20 mg.kg−1.d−1 (P<0.01 vs SHR-MI and SHR-Sham) doses. DBP was also reduced in a dose-dependent manner by the treatment (Figure 2B).
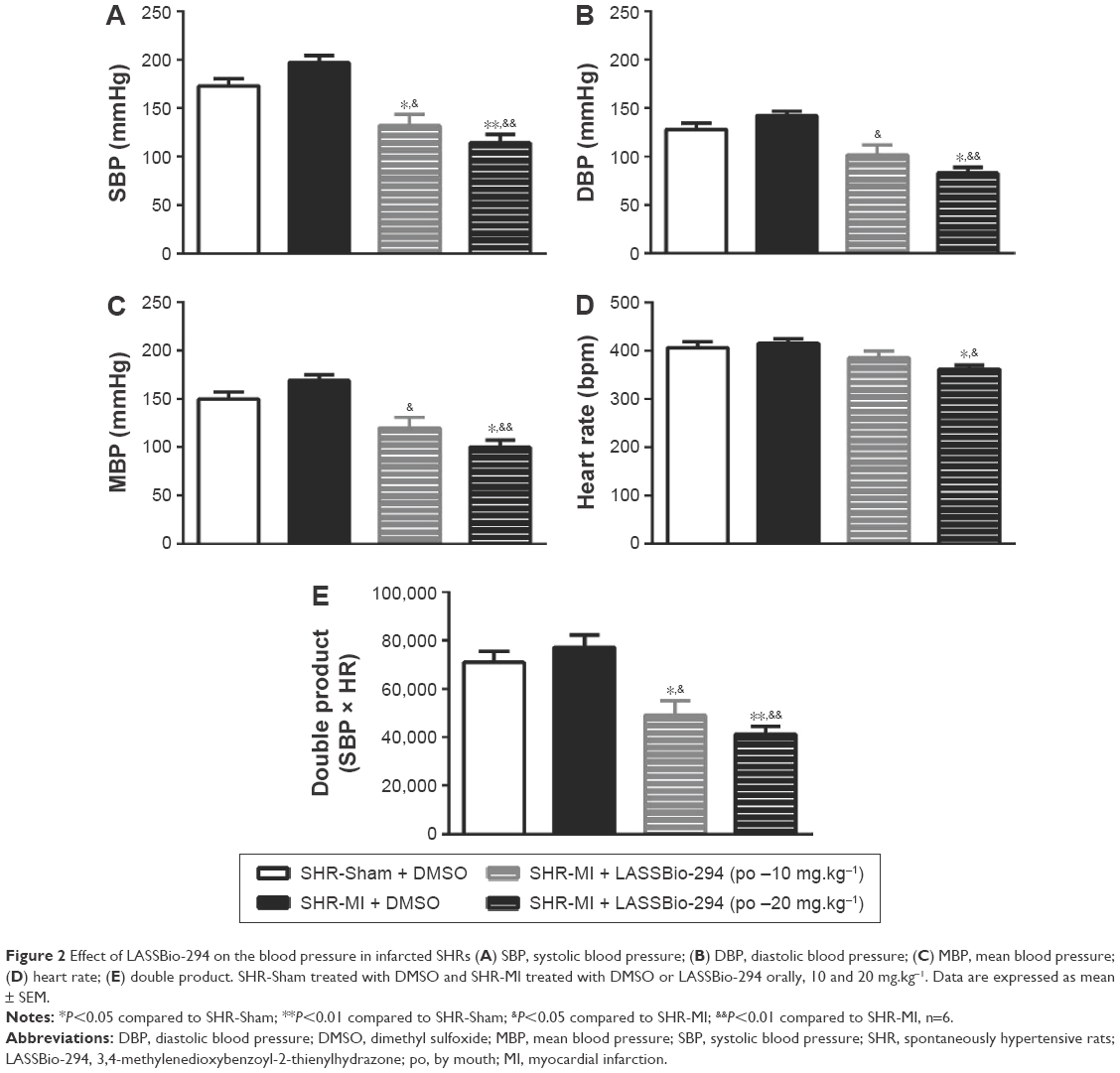 | Figure 2 Effect of LASSBio-294 on the blood pressure in infarcted SHRs (A) SBP, systolic blood pressure; (B) DBP, diastolic blood pressure; (C) MBP, mean blood pressure; (D) heart rate; (E) double product. SHR-Sham treated with DMSO and SHR-MI treated with DMSO or LASSBio-294 orally, 10 and 20 mg.kg−1. Data are expressed as mean ± SEM. |
MBP in the SHR-MI + LASSBio-294 at dose 10 mg.kg−1.d−1 or 20 mg.kg−1.d−1 groups was reduced (P<0.05 and P<0.01, respectively) in a dose-dependent manner compared with the SHR-MI group (Figure 2C). The MBP of the SHR-MI + LASSBio-294 (20 mg.kg−1.d−1) group was even significantly lower than that of the SHR-Sham group (P<0.05).
Only the 20 mg.kg−1.d−1 dose of LASSBio-294 reduced the heart rate significantly in SHR-MI (P<0.05 vs SHR-MI and SHR-Sham) (Figure 2D). Nevertheless, the double rate product was reduced in both the SHR-MI + LASSBio-294 (10 mg.kg−1.d−1) (P<0.05) and SHR-MI + LASSBio-294 (20 mg.kg−1.d−1) (P<0.01) groups when compared with the SHR-MI and SHR-Sham groups (Figure 2E).
LV systolic pressure was reduced in both the SHR-MI + LASSBio-294 (10 mg.kg−1.d−1 and 20 mg.kg−1.d−1) groups compared with that in the SHR-MI group (P<0.05) (Figure 3A). However, there were no significant group differences in +dP/dt (Figure 3B). LVEDP was increased in the SHR-MI group (27.69±3.23 mmHg) compared with the SHR-Sham group (8.98±1.10 mmHg; P<0.01) and reduced in the SHR-MI + LASSBio-294 (10 mg.kg−1.d−1) (7.37±1.05 mmHg) and SHR-MI + LASSBio-294 (20 mg.kg−1.d−1) (4.91±0.76 mmHg) groups, compared with that in the SHR-MI group (both P<0.01) (Figure 3C). The rate of ventricular relaxation, represented by −dP/dt, was reduced in the SHR-MI group compared with that in the SHR-Sham group (P<0.05). Treatment with 10 mg.kg−1.d−1 or 20 mg.kg−1.d−1 of LASSBio-294 resulted in ventricular relaxation rates similar to that observed in the SHR-Sham group (Figure 3D).
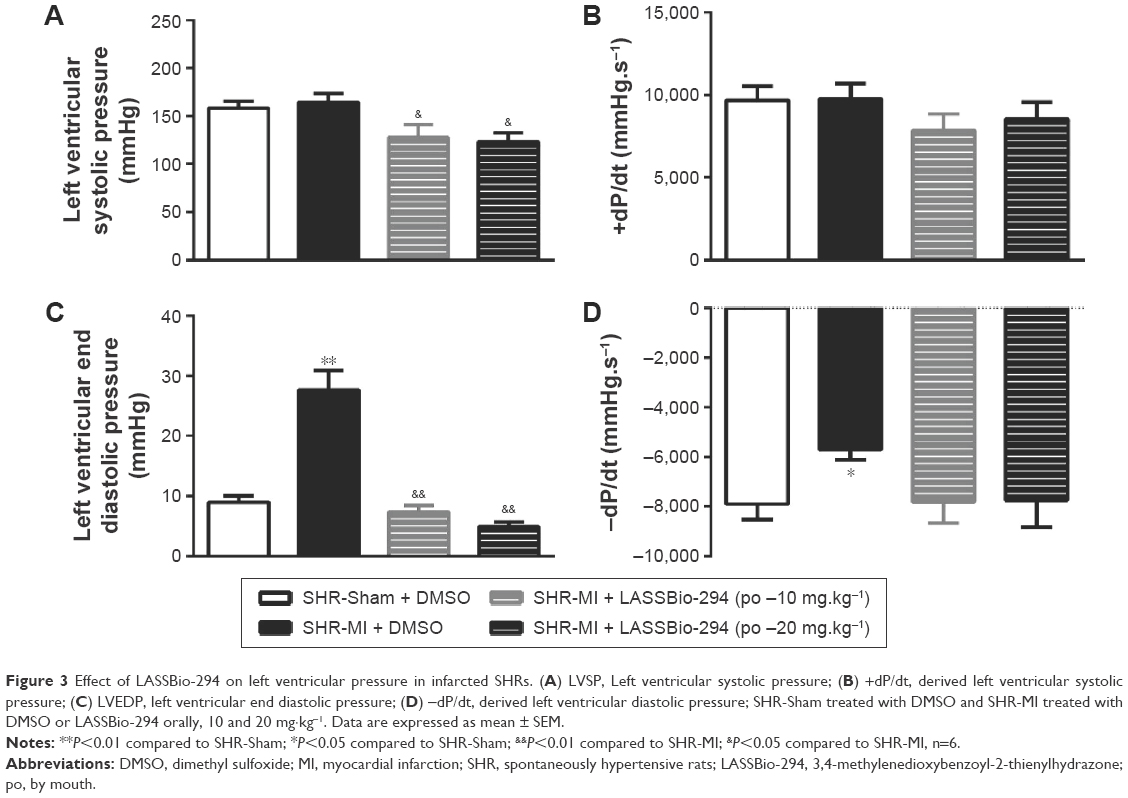 | Figure 3 Effect of LASSBio-294 on left ventricular pressure in infarcted SHRs. (A) LVSP, Left ventricular systolic pressure; (B) +dP/dt, derived left ventricular systolic pressure; (C) LVEDP, left ventricular end diastolic pressure; (D) −dP/dt, derived left ventricular diastolic pressure; SHR-Sham treated with DMSO and SHR-MI treated with DMSO or LASSBio-294 orally, 10 and 20 mg·kg−1. Data are expressed as mean ± SEM. |
Effect of LASSBio-294 on cardiac hypertrophy and pulmonary edema in infarcted SHRs
Absolute heart weight and the heart-to-body weight ratio were significantly higher in the SHR-MI group than in the SHR-Sham group. Treatment with 10 mg.kg−1.d−1 or 20 mg.kg−1.d−1 LASSBio-294 reduced both heart weight and heart-to-body weight ratio, compared with the SHR-MI group. Similar results were observed with lung weight and lung-to-body weight ratio, which were increased in the SHR-MI group compared with the SHR-Sham group, but reduced in SHR-MI rats treated with LASSBio-294 at either dose (Table 3).
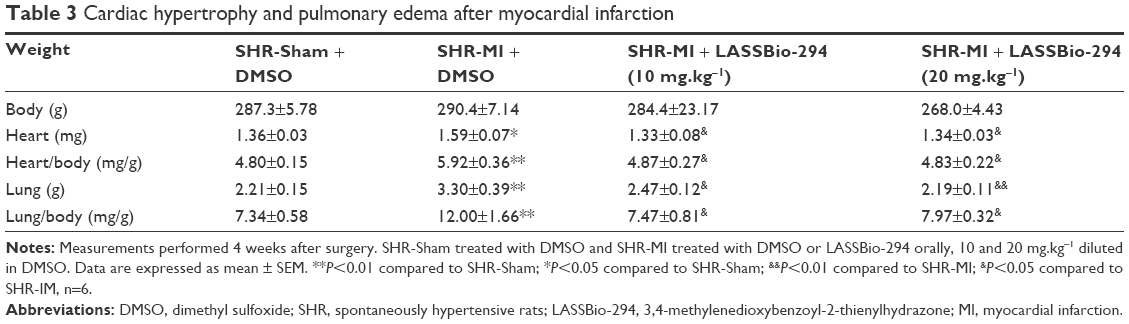 | Table 3 Cardiac hypertrophy and pulmonary edema after myocardial infarction |
Effect of LASSBio-294 on cardiac fibrosis and expression of TNF-α and SERCA2 in cardiac muscle in infarcted SHR
The presence of collagen in the left ventricle tissue was observed in sections stained with picrusirus (Figure 4A). Representative images of TNF-α (Figure 4B) and SERCA (Figure 4C) expression which were analyzed by immunohistochemistry in tissues from different groups.
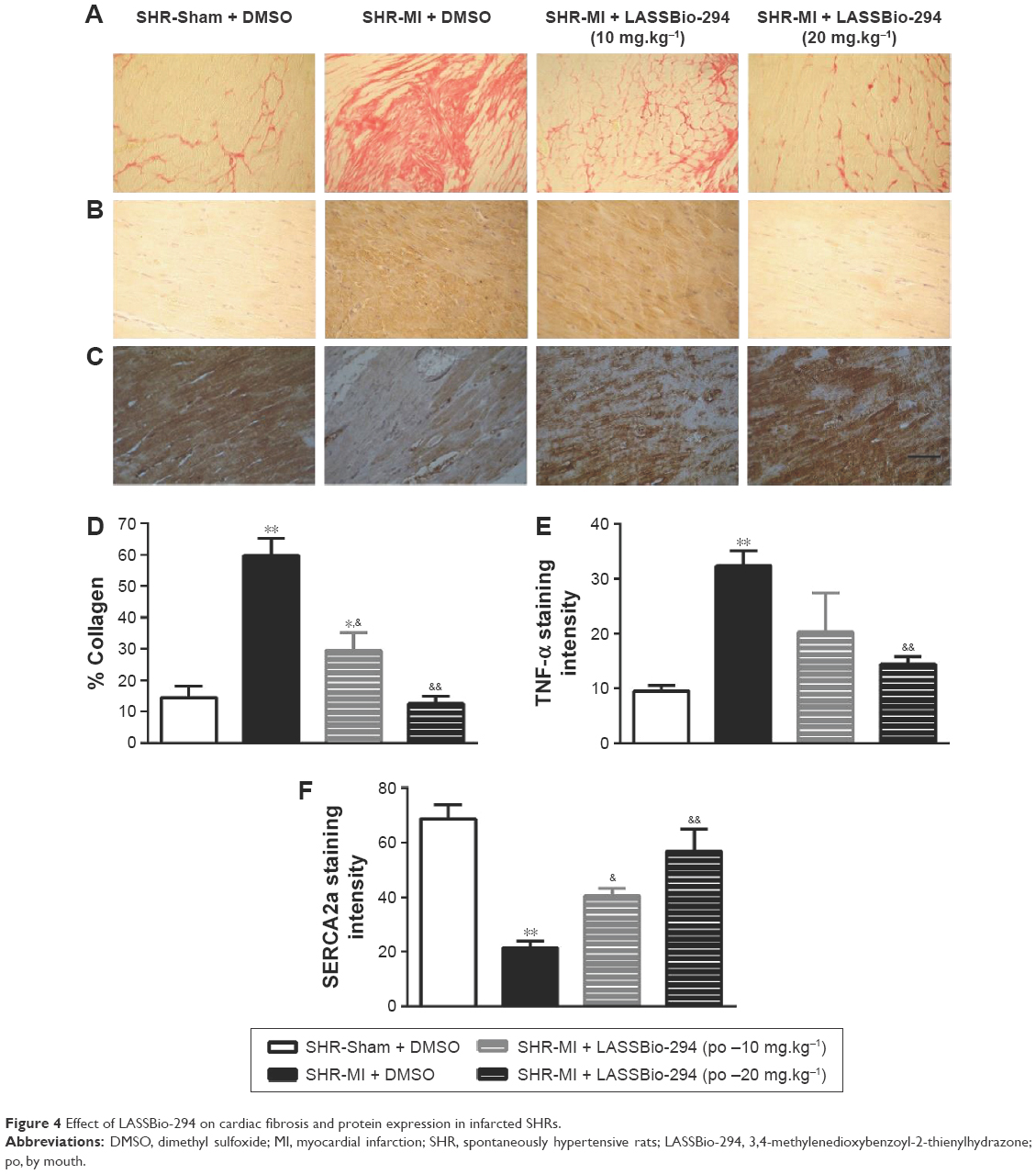 | Figure 4 Effect of LASSBio-294 on cardiac fibrosis and protein expression in infarcted SHRs. |
As expected, the SHR-MI group exhibited an increase in collagen percentage compared with that in the SHR-Sham group (Figure 4D; P<0.01). Treatment of SHR-MI with 10 mg.kg−1.d−1 LASSBio-294 reduced the collagen percentage by nearly half (P<0.05 vs SHR-MI), whereas treatment with 20 mg.kg−1.d−1 LASSBio-294 led to a full recovery, approximating the collagen percentage observed in the SHR-Sham group.
We demonstrated increased TNF-α expression in the SHR-MI group (P<0.01) compared with that observed in the SHR-Sham group (Figure 4E). TNF-α expression was reduced significantly in the SHR-MI + LASSBio-294 (20 mg.kg−1.d−1) group only (P<0.01 vs SHR-MI).
The SERCA2 expression was reduced in the SHR-MI group compared with the SHR-Sham group (21.46%±2.35% vs 68.75%±5.12%; P<0.01). Treatment led to a partial (in SHR-MI + LASSBio-294 [10 mg.kg−1.d−1]) or complete (in SHR-MI + LASSBio-294 [20 mg.kg−1.d−1]) recovery of SERCA2 expression (P<0.05, P<0.01, respectively), relative to the levels observed in animals without MI (Figure 4F).
Discussion
This study showed that chronic treatment with oral LASSBio-294 after MI improved exercise tolerance and attenuated the development of LV dysfunction and remodeling. In a previous work, we had demonstrated that LASSBio-294 prevented the development of HF in normotensive animals when administered intraperitoneally. This is the first time that oral administration of this substance has been shown to be efficacious in preventing the progression of LV dysfunction after MI in a preclinical model of preexisting hypertension.15,16
Exercise intolerance of untreated, infarcted rats was confirmed in our model by the profound reduction in the running distance traveled on the treadmill. Intolerance to physical exercise is one of the main symptoms of chronic diastolic and systolic dysfunction and is a quality of life determinant of patients with HF.22 LASSBio-294 enhanced the running capacity of SHR-MI groups above that of their respective SHR-Sham group. The results of this study suggest that, by improving hemodynamic parameters, LASSBio-294 may contribute to exercise tolerance. However, we cannot exclude direct effects of the substance on skeletal muscle contractility. We have previously demonstrated that LASSBio-294 prevented MI-induced decrease of skeletal muscle function analyzed by the contractile response in vitro and exercise intolerance associated with SERCA2 expression decrease in the soleus muscle.16 Our results are also in agreement with Gonzalez-Serratos et al23 who demonstrated in single fiber frog skeletal muscles that LASSBio-294 reduces development of fatigue and accelerates the recovery of maximal tetanic tension after fatigue is developed. Although we did not directly investigate the effects of LASSBio-294 in SHR-MI rat skeletal muscle, it is reasonable to suggest that LASSBio-294 may act in both cardiac and skeletal muscles to improve exercise tolerance in infarcted SHRs.
LASSBio-294 resulted in maintenance of LV AWT in the injured myocardium and prevented the development of LV systolic and diastolic dysfunction after MI. The normalization of EF in LASSBio-294-treated rats could be attributed to the compound’s positive inotropic activity.15,24 All four AR subtypes, A1, A2A, A2B, and A3, have been detected in the heart,12,13 and stimulation of A2ARs is associated with increased cardiac contractility, SERCA2 expression, and reuptake of Ca2+ by the SR25 effects which would be expected to be beneficial to cardiac function and hemodynamics. Moreover, the improvements in EF, as well as maintenance of SERCA2 expression, could have led to improvements in myocardial relaxation, which were characterized by the increase in E/A ratio, without a concomitant increase in LVEDP after MI. Indeed, either MI or hypertension in isolation can increase LVEDP, particularly when both conditions are present.26 The treatment-associated reductions in LVEDP, increases in E/A, and improvements in systolic function brought the functional profile of the treated SHR-MI groups toward that seen in the SHR-Sham group.
Treatment with LASSBio-294 had an antihypertensive effect. In addition, LASSBio-294 led to a normalized heart rate and double product in SHR-MI in a dose-dependent manner. Indeed, both hypertension and MI promote development of vascular dysfunction. An attenuated vasodilator response to endothelium-dependent vasodilator agents associated with preexisting hypertension and ischemia has been described by Wiemer et al;27 an effect could be related to a marked reduction of NO production. A probable mechanism by which the drug could lead to an antihypertensive effect could be attributed to agonism of A2AR. Stimulation of the A2AR activates the adenylate cyclase system, resulting in increased production of cyclic adenosine monophosphate (cAMP) and protein kinase A, which stimulate K+ channels, leading to adenosine triphosphate-dependent hyperpolarization of vascular smooth muscle cells.28
Furthermore, infusion of the A2AR agonist ATL-146e, which has anti-inflammatory and vasodilatory effects, for 2–5 hours was reported to reduce infarct area,29 associated with a substantial reduction of inflammation and neutrophil infiltration.30 AMP579, an agonist of A1R and A2AR, provided a cardioprotective effect that was inhibited by a selective A2AR antagonist.31 It is possible that LASSBio-294 could have prevented the onset of ventricular hypertrophy by activating A2ARs. In previous studies, we demonstrated that agonists of A2aRs prevented the development of pulmonary vascular remodeling and reduced cardiac hypertrophy in a model of pulmonary hypertension.32,33 Those results were in accordance with the reduction of pulmonary edema in SHR-MI and furthermore, reinforced by the reduction of pulmonary edema induced by stimulation of A2aA/A2B receptors described by Lu et al.34
Other antiremodeling actions of LASSBio-294 in SHR-MI are exemplified by the reduction in cardiac collagen deposition and consequent myocardial fibrosis, which in LASSBio-294-treated rats may be related to the activation of A2ARs.14,31,35,36 This finding suggests that cAMP-dependent signaling by A2AR activation may limit cardiac remodeling and progression to HF by promoting fibroblast-induced inhibition of collagen synthesis.37 According to Cabiati et al,38 MI increased TNF-α gene expression in pigeon’s hearts but did not alter A2aR gene expression. Although A2aR and TNF-α messenger RNA levels apparently have not shown substantial correlation, we consider that LASSBio-294 reduced TNF-α expression by activating A2aR, based on the fact that A2aR activation inhibits cytokine secretion from activated macrophages and prevents TNF-α synthesis.39 Taken together, the cardioprotective effect and reduction of post-myocardial injury induced by LASSBio-294 could be the result of inhibition of pro-inflammatory responses via stimulation of A2AR during ischemia.
Conclusion
Oral administration of the A2AR agonist, LASSBio-294 improved exercise tolerance and reduced hypertension and MI-related increase in blood pressure in a dose-dependent manner. It also prevented the development of diastolic and systolic dysfunction in SHRs after experimentally induced MI. Moreover, A2AR activation with this compound mitigated several consequences in SHR-MI animals, including changes in filling pressure, hypertrophy, fibrosis, and inflammation, apparently at least in part through decreased TNF-α expression. Previously, we observed that LASSBio-294 prevented cardiac remodeling in normotensive myocardial infarcted rats when administered intraperitoneally. The originality of the present work is in the fact that we could demonstrate that the oral administration of a novel A2a agonist rescued cardiovascular system from even more drastic damages induced by hypertension in association with MI, a condition that is often observed in clinics. The present data could be ratified and reinforced by using an A2A antagonist in order to observe if it counteracted the effects induced by LASSBio-294. However, we believe that the present findings have sufficient scientific evidences to support the notion that LASSBio-294 is a promising compound for the treatment of ischemia and hypertension-induced HF.
Acknowledgments
This work was supported by Conselho Nacional de Desenvolvimento Cientifico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Instituto Nacional de Ciência e Tecnologia de Fármacos e Medicamentos (INCT-INOFAR), and Centro Nacional de Biologia Estrutural e Bioimagem (CENABIO).
Disclosure
The authors report no conflicts of interest in this work.
References
Rapsomaniki E, Timmis A, George J, et al. Blood pressure and incidence of twelve cardiovascular diseases: lifetime risks, healthy life-years lost, and age-specific associations in 1.25 million people. Lancet. 2014;383(9932):1899–1911. | ||
Musch TI, Ghaul MR, Tranchitella V, Zelis R. Skeletal muscle glycogen depletion during submaximal exercise in rats with chronic heart failure. Basic Res Cardiol. 1990;85(6):606–618. | ||
Munkvik M, Lunde PK, Aronsen JM, Birkeland JA, Sjaastad I, Sejersted OM. Attenuated fatigue in slow twitch skeletal muscle during isotonic exercise in rats with chronic heart failure. PLoS One. 2011;6(7):e22695. | ||
Ciampi Q, Villari B. Role of echocardiography in diagnosis and risk stratification in heart failure with left ventricular systolic dysfunction. Cardiovasc Ultrasound. 2007;5:34. | ||
Libby P, Bonow RO, Mann DL, Zipes DP. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine, 2-Volume Set. Elsevier Health Sciences; 2007. | ||
Mandinov L, Eberli FR, Seiler C, Hess OM. Diastolic heart failure. Cardiovascular Res. 2000;45(4):813–825. | ||
Tamargo J, Lopez-Sendon J. Novel therapeutic targets for the treatment of heart failure. Nat Rev Drug Discov. 2011;10(7):536–555. | ||
Albrecht-Kupper BE, Leineweber K, Nell PG. Partial adenosine A1 receptor agonists for cardiovascular therapies. Purinergic signal. 2012;8(Suppl 1):91–99. | ||
Cronstein BN, Kramer SB, Rosenstein ED, Korchak HM, Weissmann G, Hirschhorn R. Occupancy of adenosine receptors raises cyclic AMP alone and in synergy with occupancy of chemoattractant receptors and inhibits membrane depolarization. Biochem J. 1988;252(3):709–715. | ||
Ciarka A, van de Borne P, Pathak A. Myocardial infarction, heart failure and sympathetic nervous system activity: new pharmacological approaches that affect neurohumoral activation. Expert Opin Investig Drugs. 2008;17(9):1315–1330. | ||
Jordan JE, Zhao ZQ, Sato H, Taft S, Vinten-Johansen J. Adenosine A2 receptor activation attenuates reperfusion injury by inhibiting neutrophil accumulation, superoxide generation and coronary endothelial adherence. J Pharmacol Exp Ther. 1997;280(1):301–309. | ||
Headrick JP, Peart JN, Reichelt ME, Haseler LJ. Adenosine and its receptors in the heart: regulation, retaliation and adaptation. Biochim Biophys Acta. 2011;1808(5):1413–1428. | ||
Layland J, Carrick D, Lee M, Oldroyd K, Berry C. Adenosine: physiology, pharmacology, and clinical applications. JACC Cardiovasc Interv. 2014;7(6):581–591. | ||
Peart JN, Headrick JP. Adenosinergic cardioprotection: multiple receptors, multiple pathways. Pharmacol Ther. 2007;114(2):208–221. | ||
Costa DG, da Silva JS, Kummerle AE, et al. LASSBio-294, a compound with inotropic and lusitropic activity, decreases cardiac remodeling and improves Ca(2)(+) influx into sarcoplasmic reticulum after myocardial infarction. Am J Hypertens. 2010;23(11):1220–1227. | ||
da Silva JS, Pereira SL, Maia Rdo C, et al. N-acylhydrazone improves exercise intolerance in rats submitted to myocardial infarction by the recovery of calcium homeostasis in skeletal muscle. Life Sci. 2014;94(1):30–36. | ||
Pfeffer J, Pfeffer M, Fletcher P, Braunwald E. Alterations of cardiac performance in rats with established spontaneous hypertension. Am J Cardiol. 1979;44(5):994–998. | ||
Pfeffer JM, Pfeffer MA, Fishbein MC, Frohlich ED. Cardiac function and morphology with aging in the spontaneously hypertensive rat. Am J Physiol. 1979;237(4):H461–H468. | ||
Pfeffer MA, Pfeffer JM, Frohlich ED. Pumping ability of the hypertrophying left ventricle of the spontaneously hypertensive rat. Circ Res. 1976;38(5):423–429. | ||
Liu Q, Anderson C, Broyde A, et al. Glucagon-like peptide-1 and the exenatide analogue AC3174 improve cardiac function, cardiac remodeling, and survival in rats with chronic heart failure. Cardiovasc Diabetol. 2010;9:76. | ||
Lang RM, Bierig M, Devereux RB, et al. Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;18(12):1440–1463. | ||
Kitzman DW, Groban L. Exercise intolerance. Heart Fail Clin. 2008;4(1):99–115. | ||
Gonzalez-Serratos H, Chang R, Pereira EF, et al. A novel thienylhydrazone, (2-thienylidene)3,4-methylenedioxybenzoylhydrazine, increases inotropism and decreases fatigue of skeletal muscle. J Pharmacol Exp Ther. 2001;299(2):558–566. | ||
Sudo RT, Zapata-Sudo G, Barreiro EJ. The new compound, LASSBio 294, increases the contractility of intact and saponin-skinned cardiac muscle from Wistar rats. Br J Pharmacol. 2001;134(3):603–613. | ||
Chan TO, Funakoshi H, Song J, et al. Cardiac-restricted overexpression of the A(2A)-adenosine receptor in FVB mice transiently increases contractile performance and rescues the heart failure phenotype in mice overexpressing the A(1)-adenosine receptor. Clin Transl Sci. 2008;1(2):126–133. | ||
Xia QG, Reinecke A, Dorenkamp M, Daemen MJ, Simon R, Unger T. Effects of endothelin ETA receptor blocker LU 135252 on cardiac remodeling and survival in a hypertensive rat model of chronic heart failure. Acta pharmacol Sin. 2006;27(11):1417–1422. | ||
Wiemer G, Itter G, Malinski T, Linz W. Decreased nitric oxide availability in normotensive and hypertensive rats with failing hearts after myocardial infarction. Hypertension. 2001;38(6):1367–1371. | ||
Gemignani AS, Abbott BG. The emerging role of the selective A2A agonist in pharmacologic stress testing. J Nucl Cardiol. 2010;17(3):494–497. | ||
Glover DK, Riou LM, Ruiz M, et al. Reduction of infarct size and postischemic inflammation from ATL-146e, a highly selective adenosine A2A receptor agonist, in reperfused canine myocardium. Am J Physiol Heart Circ Physiol. 2005;288(4):H1851–H1858. | ||
Patel RA, Glover DK, Broisat A, et al. Reduction in myocardial infarct size at 48 hours after brief intravenous infusion of ATL-146e, a highly selective adenosine A2A receptor agonist. Am J Physiol Heart Circ Physiol. 2009;297(2):H637–H642. | ||
Smits GJ, McVey M, Cox BF, Perrone MH, Clark KL. Cardioprotective effects of the novel adenosine A1/A2 receptor agonist AMP 579 in a porcine model of myocardial infarction. J Pharmacol Exp Ther. 1998;286(2):611–618. | ||
Alencar AK, Pereira SL, da Silva FE, et al. N-acylhydrazone derivative ameliorates monocrotaline-induced pulmonary hypertension through the modulation of adenosine AA2R activity. Int J Cardiol. 2014;173(2):154–162. | ||
Alencar AK, Pereira SL, Montagnoli TL, et al. Beneficial effects of a novel agonist of the adenosine A2A receptor on monocrotaline-induced pulmonary hypertension in rats. Br J Pharmacol. 2013;169(5):953–962. | ||
Lu Q, Harrington EO, Newton J, et al. Adenosine protected against pulmonary edema through transporter- and receptor A2-mediated endothelial barrier enhancement. Am J Physiol Lung Cell Mol Physiol. 2010;298(6):L755–L767. | ||
Geraets D, Kienzle M. Clinical use of adenosine. Iowa Med. 1992;82(1):25–28. | ||
Neubauer S. The failing heart – an engine out of fuel. N Engl J Med. 2007;356(11):1140–1151. | ||
Villarreal F, Epperson SA, Ramirez-Sanchez I, Yamazaki KG, Brunton LL. Regulation of cardiac fibroblast collagen synthesis by adenosine: roles for Epac and PI3K. Am J Physiol Cell Physiol. 2009;296(5):C1178–C1184. | ||
Cabiati M, Martino A, Mattii L, et al. Adenosine receptor expression in an experimental animal model of myocardial infarction with preserved left ventricular ejection fraction. Heart Vessels. 2014;29(4):513–519. | ||
Feng W, Song Y, Chen C, Lu ZZ, Zhang Y. Stimulation of adenosine A(2B) receptors induces interleukin-6 secretion in cardiac fibroblasts via the PKC-delta-P38 signalling pathway. Br J Pharmacol. 2010;159(8):1598–1607. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.