Back to Journals » Clinical Interventions in Aging » Volume 9
Add-on prolonged-release melatonin for cognitive function and sleep in mild to moderate Alzheimer’s disease: a 6-month, randomized, placebo-controlled, multicenter trial
Authors G Wade A, Farmer M, Harari G, Fund N, Laudon M, Nir T, Frydman-Marom A, Zisapel N
Received 6 April 2014
Accepted for publication 2 May 2014
Published 18 June 2014 Volume 2014:9 Pages 947—961
DOI https://doi.org/10.2147/CIA.S65625
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Alan G Wade,1 Mildred Farmer,2 Gil Harari,3 Naama Fund,3 Moshe Laudon,4 Tali Nir,4 Anat Frydman-Marom,4 Nava Zisapel4,5
1CPS Research, Glasgow, UK; 2Meridien Research Inc., St Petersburg, FL, USA; 3Medistat, Ltd, 4Neurim Pharmaceuticals Ltd, Tel Aviv, Israel; 5Department of Neurobiology, Faculty of Life Sciences, Tel Aviv University, Tel Aviv, Israel
Purpose: A link between poor sleep quality and Alzheimer’s disease (AD) has recently been suggested. Since endogenous melatonin levels are already reduced at preclinical AD stages, it is important to ask whether replenishing the missing hormone would be beneficial in AD and whether any such effects would be related to the presence of sleep disorder in patients.
Patients and methods: The effects of add-on prolonged-release melatonin (PRM) (2 mg) to standard therapy on cognitive functioning and sleep were investigated in 80 patients (men [50.7%], women [49.3%], average age 75.3 years [range, 52–85 years]) diagnosed with mild to moderate AD, with and without insomnia comorbidity, and receiving standard therapy (acetylcholinesterase inhibitors with or without memantine). In this randomized, double-blind, parallel-group study, patients were treated for 2 weeks with placebo and then randomized (1:1) to receive 2 mg of PRM or placebo nightly for 24 weeks, followed by 2 weeks placebo. The AD Assessment Scale–Cognition (ADAS-Cog), Instrumental Activities of Daily Living (IADL), Mini–Mental State Examination (MMSE), sleep, as assessed by the Pittsburgh Sleep Quality Index (PSQI) and a daily sleep diary, and safety parameters were measured.
Results: Patients treated with PRM (24 weeks) had significantly better cognitive performance than those treated with placebo, as measured by the IADL (P=0.004) and MMSE (P=0.044). Mean ADAS-Cog did not differ between the groups. Sleep efficiency, as measured by the PSQI, component 4, was also better with PRM (P=0.017). In the comorbid insomnia (PSQI ≥6) subgroup, PRM treatment resulted in significant and clinically meaningful effects versus the placebo, in mean IADL (P=0.032), MMSE score (+1.5 versus -3 points) (P=0.0177), and sleep efficiency (P=0.04). Median ADAS-Cog values (–3.5 versus +3 points) (P=0.045) were significantly better with PRM. Differences were more significant at longer treatment duration. PRM was well tolerated, with an adverse event profile similar to that of placebo.
Conclusion: Add-on PRM has positive effects on cognitive functioning and sleep maintenance in AD patients compared with placebo, particularly in those with insomnia comorbidity. The results suggest a possible causal link between poor sleep and cognitive decline.
Keywords: acetylcholinesterase inhibitors, memantine, insomnia
Introduction
Alzheimer’s disease (AD), a degenerative brain disorder, is the leading cause of dementia in the elderly. The classic hallmarks of AD are cognitive dysfunction and psychiatric and behavioral disturbances, which lead to progressive deterioration of memory, language, and intellect.1 The degenerative process often produces neurobehavioral symptoms, including sleep disturbances, mainly characterized by nighttime awakenings.2 Sleep has an important role in memory consolidation.3,4 Emerging evidence links poor sleep to increased AD risk and memory loss.5–8 However, to prove causality, it is important to show that improvement in sleep can ameliorate the disease.
The loss of cholinergic function is believed to contribute significantly to memory loss and cognitive dysfunction in AD. This deficiency can be partially alleviated by treatment with cholinergic agents, such as acetylcholinesterase inhibitors.9 Acetylcholinesterase inhibitors, alone or together with memantine, an N-methyl-D-aspartate (NMDA) receptor antagonist aimed at neuroprotection against glutamate neurotoxicity,10 are the first-line drugs for AD today. However, the sleep problem is not addressed by these medications, and most current hypnotics are not useful because they further impair cognitive functioning11–14 and may themselves be associated with an increased risk of dementia.15,16
Melatonin is the major hormone produced and secreted at night by the pineal gland into the cerebrospinal fluid (CSF) and circulation. It has a major role in regulation of the biological clock, particularly the sleep–wake cycle and the induction of physiological sleep.17 Early neuropathological changes in AD are accompanied by decreased CSF melatonin levels.18 The reduced melatonin levels are already found in the preclinical stages and correlate significantly with the severity of mental and sleep impairments in demented patients.19
Several studies, mostly open label, have reported on the beneficial effects of melatonin on cognitive decline and sleep in AD and in patients with mild cognitive impairment.20–25 Of these, six were randomized, double-blind, placebo-controlled trials, with a total of 310 AD patients, mostly with advanced disease. These studies differed considerably in design, patient inclusion/exclusion criteria, melatonin preparations and doses used, end points, and treatment duration. Therefore, the questions, whether melatonin has beneficial effects on cognitive functions in AD, whether its effects are beyond those provided by the standard AD therapy, whether they are sustained over time, and to what extent the effects are driven by improvement in sleep, remain unanswered. These questions were addressed in the current study.
A prolonged-release melatonin (PRM) formulation (Circadin® 2 mg; Neurim Pharmaceuticals Ltd, Tel Aviv, Israel) was developed in order to circumvent the fast clearance of melatonin in the body (half-life [T1/2] =0.54–0.67 hours) and has been licensed since 2007 in Europe, Australia, and other countries, for insomnia in patients aged 55 and older.17 In the target population, PRM provides significant and clinically meaningful improvements in sleep quality, sleep onset latency, and quality of life and importantly, morning alertness and psychomotor performance.26–28 In particular, it is not associated with negative effects on anterograde memory or cognitive functioning that are impaired in AD.29 Because good sleep quality is imperative for cognitive functioning, particularly at older age,5,6,30 the improvement of nighttime sleep and daytime alertness with PRM in AD patients may potentially also alleviate the sleep-related deficits in cognitive functioning. In addition, there is a growing body of evidence suggesting a beneficial effect of melatonin on behavioral deficits associated with cholinergic dysfunction.31
The aim of this randomized, placebo-controlled, 6-month study was to evaluate the effects of add-on PRM versus placebo on cognition and sleep, in patients with mild to moderate AD who are treated with standard AD therapy (acetylcholinesterase inhibitors, with and without memantine). For optimal treatment, patients were also instructed to have outdoor light exposure for at least 2 hours per day.32
Methods
Study design and participants
The study was performed in five centers, one in the UK (N=35 patients) and four in the USA (N=45 patients). The study protocol, informed consents, and amendments were approved in writing by the appropriate local site Independent Ethics Committee (IEC)/Institutional Review Boards (IRB) (National Health Service [NHS]-Scotland) (IEC, Helsinki Committee/IRB).
All potential candidates for the study were given a current copy of the Informed Consent Form (ICF) to read. Appropriate translations were provided in the native language of the subject. The investigator, study physician, or authorized investigative staff member explained all aspects of the study in lay language and answered all of the candidates’ questions regarding the study. The candidates wishing to participate in the study were asked to sign the ICF. No study procedure was performed prior to signing the ICF. Subjects who refused to participate or who withdrew from the study were treated without prejudice. All subjects were given a copy of the signed ICF. Altogether, three ICFs were obtained for each participant outlining: the patient consent to participate, the caregiver consent for patient participation, and the caregiver consent to participate.
The patients were recruited outpatients. A total of 80 male and female outpatients (ages 50–85) diagnosed with mild to moderate AD and Mini–Mental State Examination (MMSE) score of ≥15 were recruited to the study. Patients had to have no evidence of focal disease to account for dementia, as established by computed tomography (CT), positron emission tomography (PET), or magnetic resonance imaging (MRI) scans. Following diagnosis, all patients underwent a 2-week single-blind, placebo run-in period, followed by double-blind randomization to treatment with PRM (Circadin® 2 mg) or placebo for 24 weeks and a 2-week placebo run-out period (Figure 1). To prevent bias, matching placebo tablets, which were identical in appearance, taste, and odor, were used. The treatment was double-blinded, with two parallel treatment groups. Selection for a treatment group was determined by a computer-generated randomization list, in a 1:1 ratio (PRM 2 mg:placebo), using the randomized permuted blocks method. The patients were not synchronized in their living habits, except that study medication was administered orally, one tablet/day, 1–2 hours before bedtime, preferably at 9 pm, after dinner. In addition, patients were instructed to spend 2 hours a day in outdoor daylight. Patients had to have been on stable doses of acetylcholinesterase inhibitor (with or without memantine) for 2 months prior to recruitment. Patients who were using melatonin during the preceding 2 weeks or benzodiazepines or other hypnotics during the preceding 4 weeks and throughout the run-in period, were excluded.
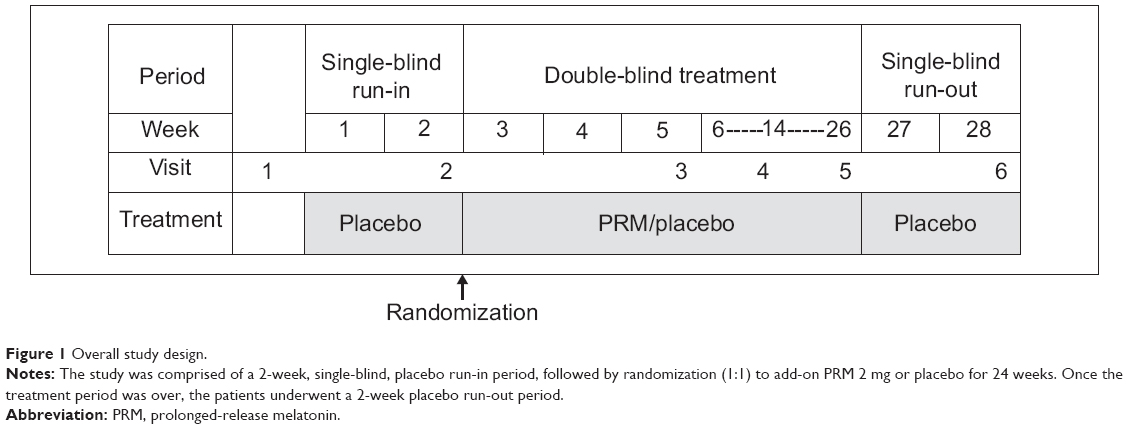 | Figure 1 Overall study design. |
Assessments
The efficacy measures included the change, from baseline to 12 and 24 weeks of the double-blind treatment period, on cognitive parameters assessed by the AD Assessment Scale–Cognition (ADAS-Cog),33 Instrumental Activities of Daily Living (IADL),34 and MMSE score35 (at 24 weeks). Sleep parameters were assessed after 3, 12, and 24 weeks, by the Pittsburgh Sleep Quality Index (PSQI)36 global score and individual components and by a sleep diary that documented number and duration of midsleep awakenings. The PSQI was completed by the investigator, or investigator designee, with the spouse or caregiver, and patient. In case of contradiction between the caregiver and patient response, the response of the caregiver was chosen. Overall clinical status was assessed by the Clinical Global Impression (CGI) scale,37 behavioral signs and symptoms by the Neuropsychiatric Inventory (NPI) scale,38 and patients’ well-being by the World Health Organization (WHO)-5 Well-Being Index.39 Caregiver’s sleep was assessed by the Sleep Disorders Inventory (SDI).40 The safety parameters were assessed at each visit and included spontaneously reported adverse events (AEs) or serious AEs (SAEs), and vital signs (heart rate and blood pressure), physical examination, and laboratory tests.
Statistical analysis
Based on extrapolation of the effect sizes and standard deviations reported by Asayama et al21 a difference from baseline in the cognitive ADAS-Cog parameter of −3.29 for PRM and 0.5 for placebo after 24 weeks of treatment, a residual standard deviation of 4.5, and the use of a 1:1 randomization ratio, it was assumed that 140 completed patients (70 PRM, 70 placebo) would be sufficient to achieve 95% power at the 5% two-sided significance level for the amended primary objective (ADAS-Cog). The study was thus planned to look at the effects of add-on PRM on cognition and sleep in 140 mild to moderate AD patients, with and without insomnia comorbidity, treated with standard AD therapy.
Due to severe difficulties in recruitment of patients, the study was stopped after about half of the intended patients (80) were recruited. The database was locked, and all data were analyzed on an exploratory basis, according to the preplanned statistical analysis plan.
Two data sets and a subpopulation were thus analyzed: a) the safety set, which included all patients who were randomized to treatment and who took at least one dose of the study medication; b) the full analysis set (FAS), which included all patients in the safety population who had efficacy data for the ADAS-Cog recorded for baseline and at least one postbaseline-period assessment; and c) the insomnia comorbidity subpopulation, which included all patients in the FAS who had insomnia comorbidity at baseline (PSQI ≥6). Differences between groups were tested at a 5% two-sided significance level to achieve 80% power. The data were analyzed using SAS® version 9.1 (SAS Institute, Cary, NC, USA). We used an observed case approach with regards to missing data.41 Given the exploratory nature of the study, P-values were reported without any adjustment for multiplicity.
Results
A total of 80 patients were enrolled into the study, seven were excluded before randomization, due to noncompliance, and 73 patients were randomized (39 patients into the PRM and 34 into the placebo cohort). A total of 60 patients (82.2%) completed the study (31 [79.5%] in the PRM and 29 [85.3%] in the placebo cohorts). A total of 13 patients were in the insomnia comorbid subpopulation (seven in the PRM and six in the placebo cohorts). Subject disposition is presented in Figure 2.
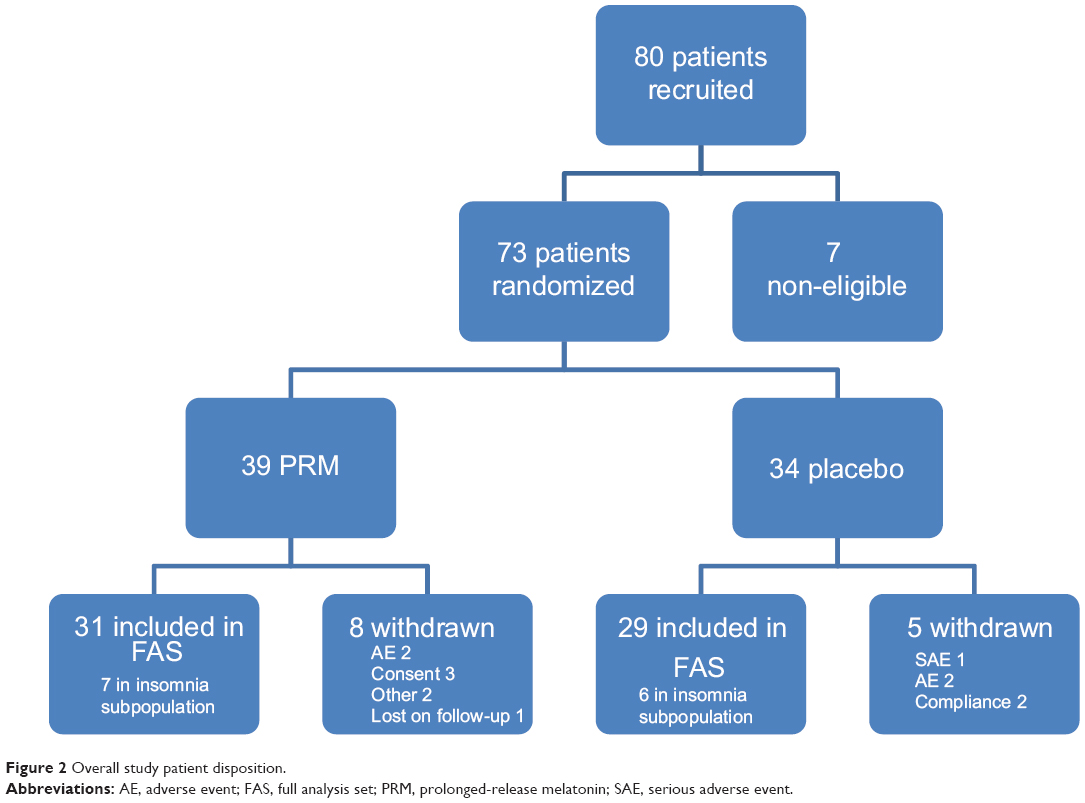 | Figure 2 Overall study patient disposition. |
There were no statistically significant differences in demography or baseline characteristics between the PRM and placebo cohorts (Table S1). In the PRM group, there was a higher percentage of males (59.0% versus 41.2%) (not significant) and a lower proportion of patients who took memantine (29.3% versus 51.6%) (P=0.068) compared with the placebo cohort. Treatment compliance remained above 90% in both treatment groups throughout the study.
Cognition
Table 1 depicts the effects of 24 weeks of PRM and placebo treatment on cognitive skills in the FAS population and the comorbid insomnia subpopulation. By the end of the 24-week treatment period, there was no difference in mean ADAS-Cog score between the treatment groups of the FAS. Median ADAS-Cog score levels improved by 2 points in the PRM-treated group and deteriorated by 0.5 points in the placebo-treated group, but the difference was not statistically significant (Table 1, Figure 3A). In the subpopulation of patients suffering from insomnia comorbidity, there was an improvement (decrease) of −3.5 points in the median ADAS-Cog score in the PRM and a deterioration (increase) of +3 points in the placebo group, and the difference in treatment effect between the groups was significant (P=0.045) (Table 1, Figure 3A). After the shorter period (12 treatment weeks), the median ADAS-Cog score improved −2.0 points in the PRM and deteriorated +1 point in the placebo group, and the difference between groups was not statistically significant for this time (data not shown).
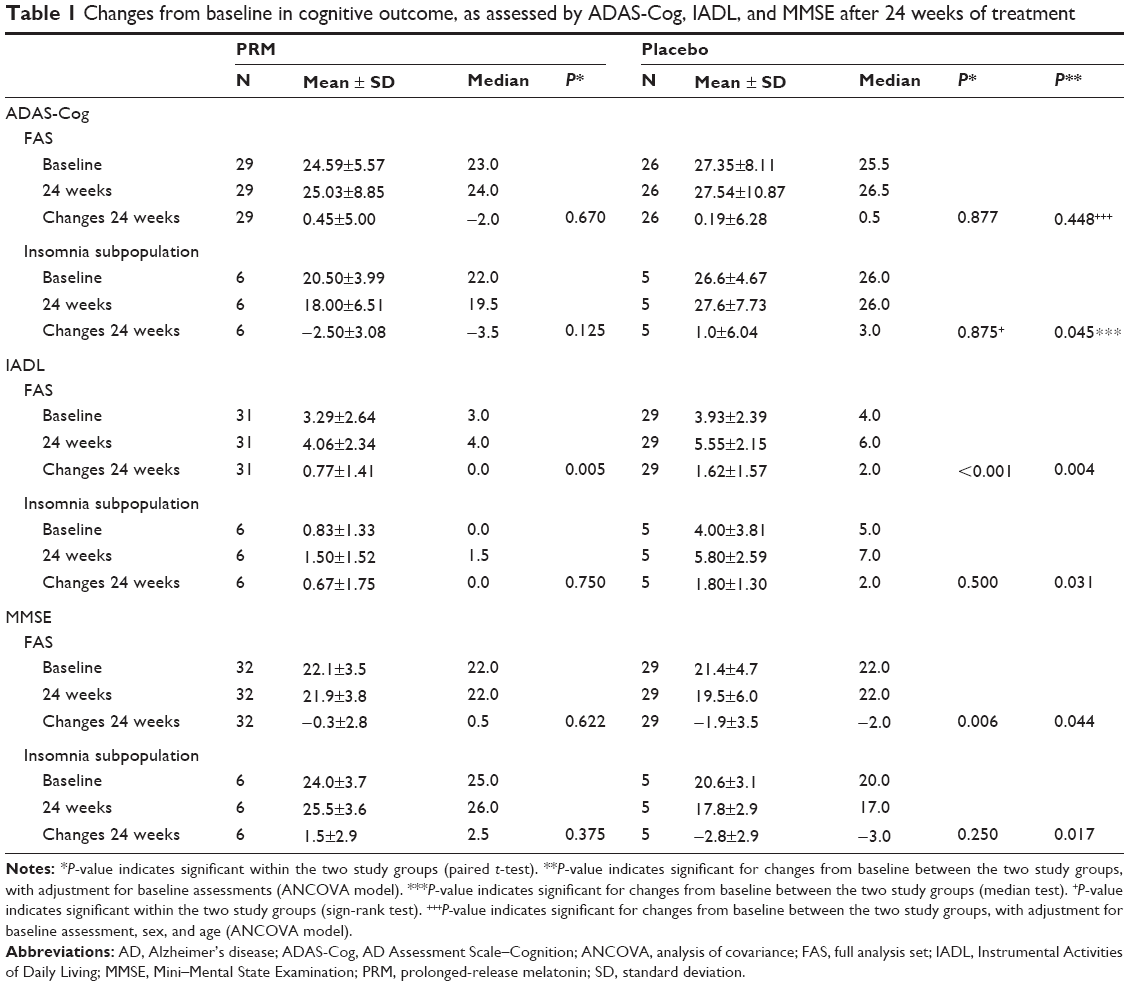 | Table 1 Changes from baseline in cognitive outcome, as assessed by ADAS-Cog, IADL, and MMSE after 24 weeks of treatment |
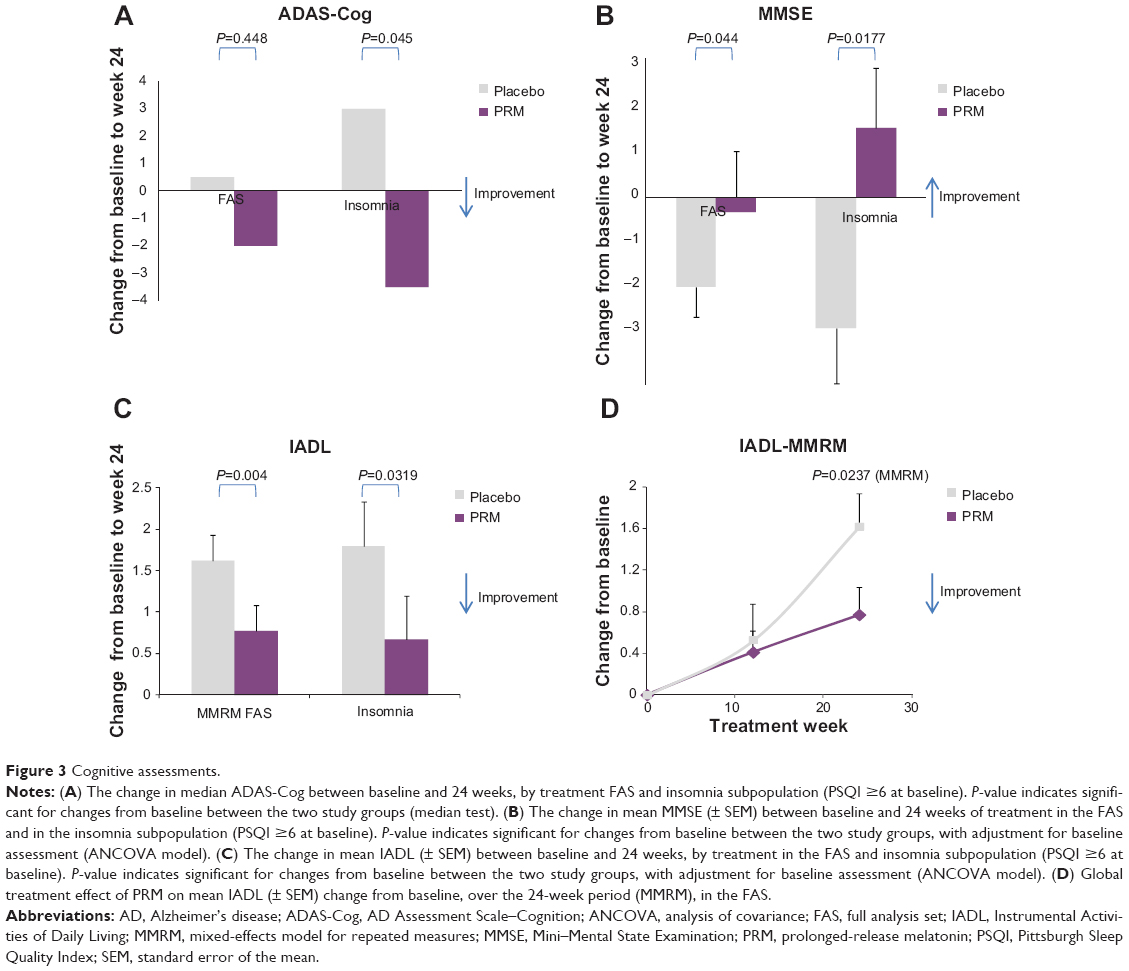 | Figure 3 Cognitive assessments. |
By the end of the 24-week treatment period, the decline in MMSE in the FAS population was significantly less in the PRM compared with the placebo group (P=0.044, baseline adjusted analysis of covariance [ANCOVA]) (Table 1, Figure 3B). The mean decline in MMSE score in the FAS population deteriorated significantly over the 24-week period in the placebo group (P=0.006), while it did not change in the PRM group. In the subpopulation of patients suffering from comorbid insomnia, MMSE scores significantly improved with PRM over placebo, showing an increase in MMSE after 24 weeks of 1.5 points, while the placebo group deteriorated by 2.8 points, and the difference in treatment effect between the groups was significant (P=0.0177, baseline adjusted ANCOVA) (Table 1, Figure 3B).
A significant effect of PRM compared with placebo was demonstrated in self-care and activities of daily living, assessed by the IADL after 24 weeks of double-blind treatment (P=0.004) (Table 1, Figure 3C). These differences remained significant after adjusting for sex and age (P=0.005), AD severity, insomnia severity, and memantine intake (P=0.019). The effect on IADL was more pronounced at longer treatment duration, and the global treatment effect of PRM over the 24-week period (mixed-effects model for repeated measures [MMRM]) was also significantly greater with PRM compared with placebo (P=0.0237) (Figure 3D).
Likewise, the mean IADL score in the subpopulation of patients suffering from comorbid insomnia was significantly better with 24 weeks of PRM as compared with placebo (P=0.0319, baseline adjusted ANCOVA model) (Table 1, Figure 3C).
Sleep
Most patients (>70%) in the study did not have insomnia. Following 24 weeks of treatment, there were no significant differences in treatment effects in the PSQI global score between the groups. The PSQI global score significantly decreased (improved) compared with baseline in the PRM- (−1.62±2.74) (P=0.004) but not in the placebo-treated group (−0.74±2.52) (P=0.139) (Table 2).
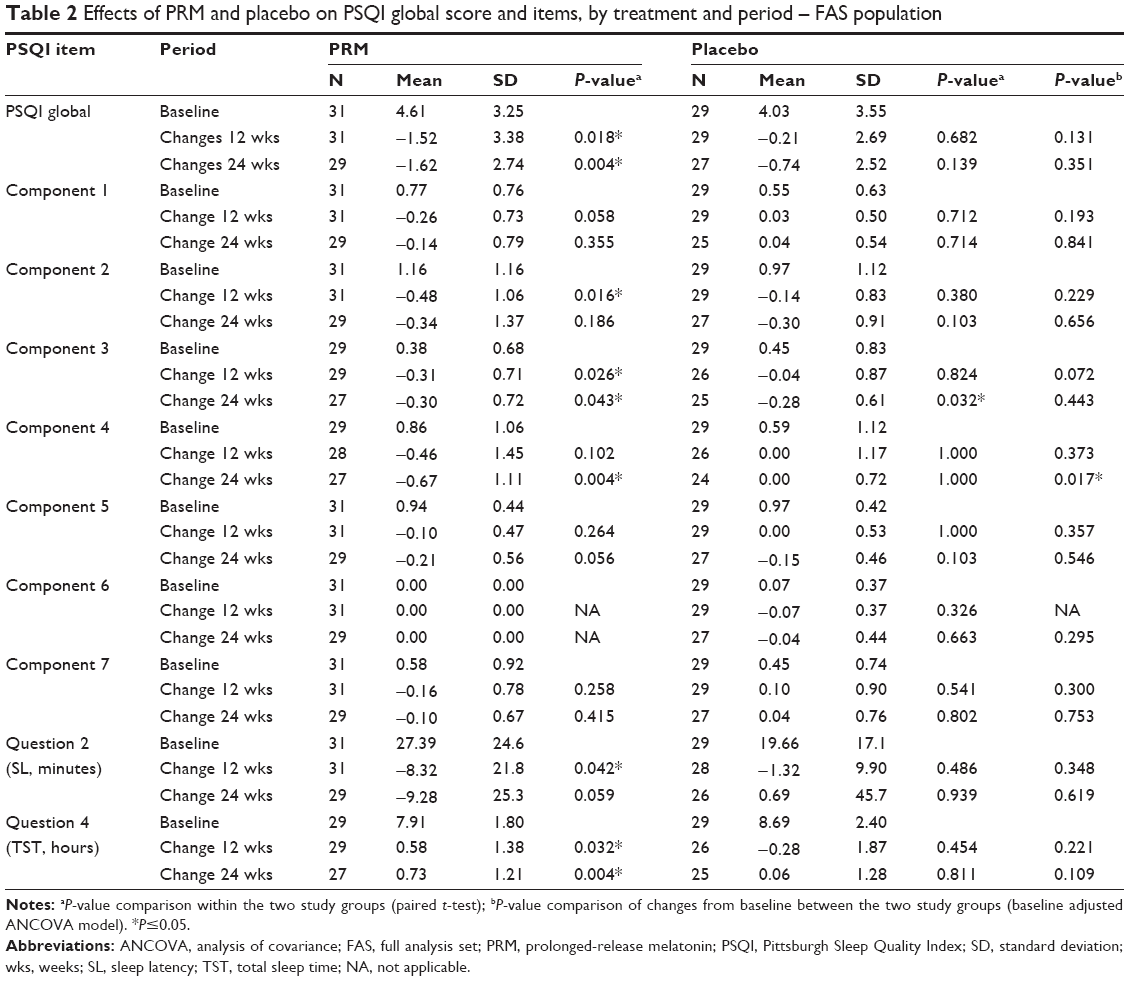 | Table 2 Effects of PRM and placebo on PSQI global score and items, by treatment and period – FAS population |
Despite the absence of insomnia comorbidity in the FAS population, PSQI component 4 scores, measuring sleep efficiency, improved significantly in the PRM group over placebo after 24 weeks of treatment (P=0.017, baseline adjusted ANCOVA) (Table 2) and over the entire 24-week, double-blind treatment period (MMRM, P=0.0312) (Figure 4A). In the comorbid insomnia subpopulation, despite the small sample size, sleep efficiency improved significantly in the PRM group over placebo after 24 weeks of treatment (P=0.04) (Table 3, Figure 4B).
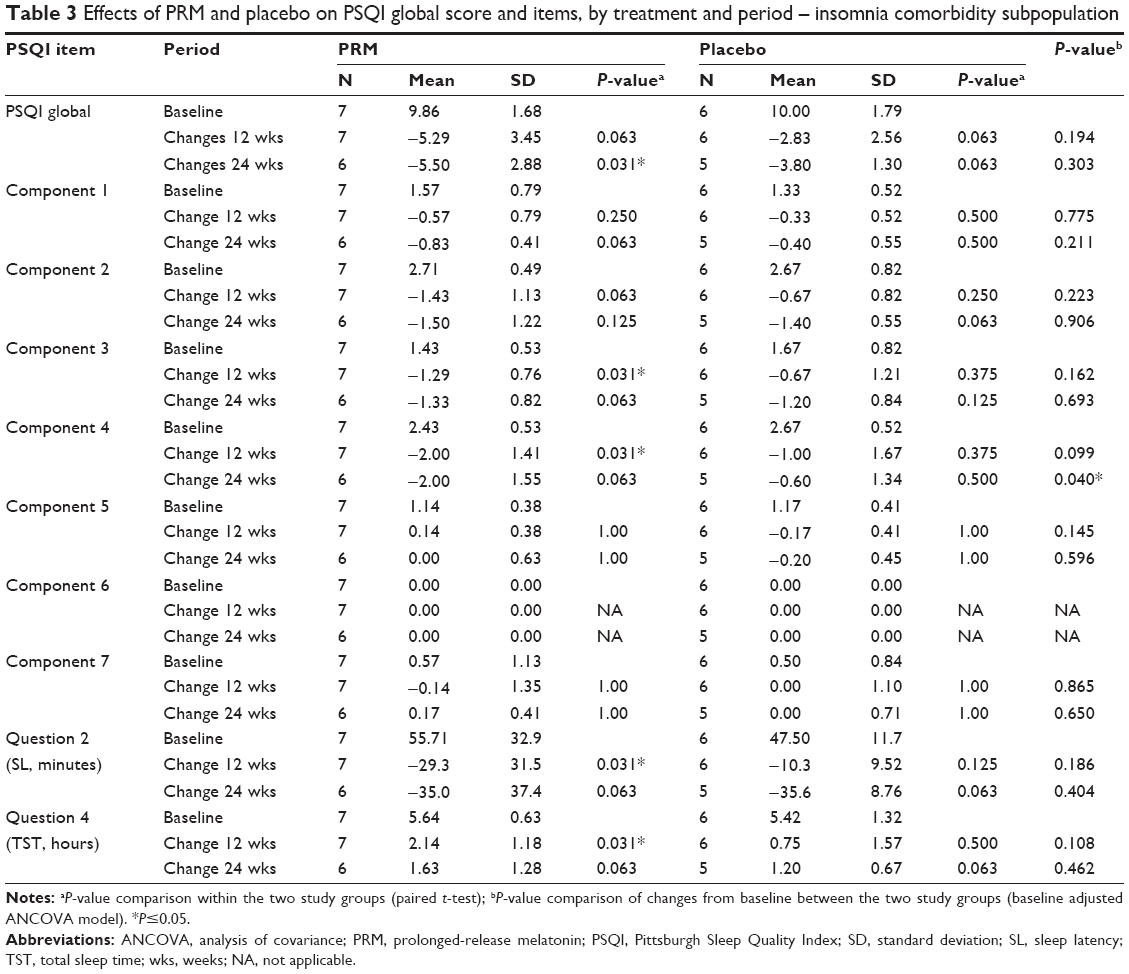 | Table 3 Effects of PRM and placebo on PSQI global score and items, by treatment and period – insomnia comorbidity subpopulation |
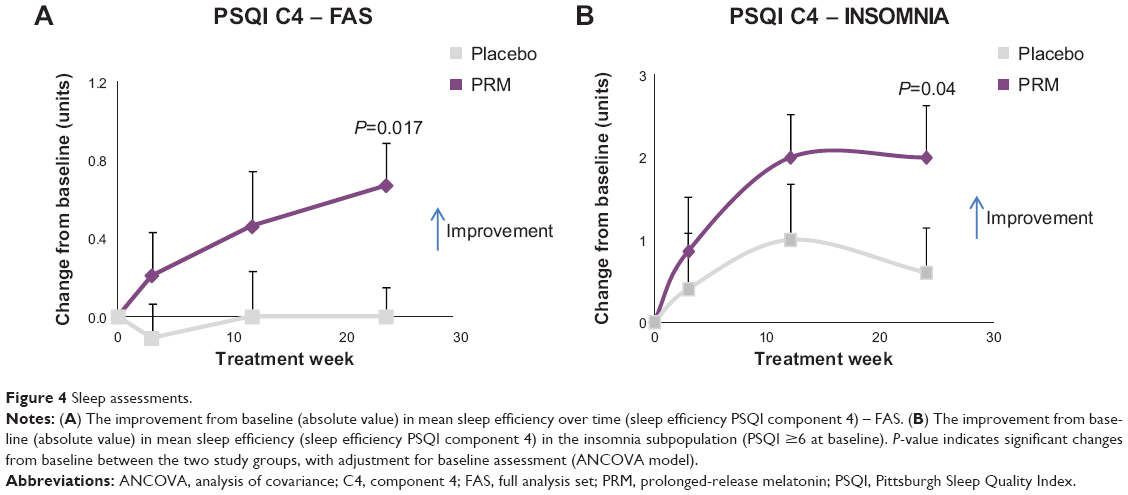 | Figure 4 Sleep assessments. |
An improvement (decrease) of −5.5 (P=0.031) points in the PRM compared with −3.8 in the placebo group was observed after 24 weeks (Table 3) in PSQI global scores, but the difference between groups did not reach the predefined statistical significance level (P=0.303).
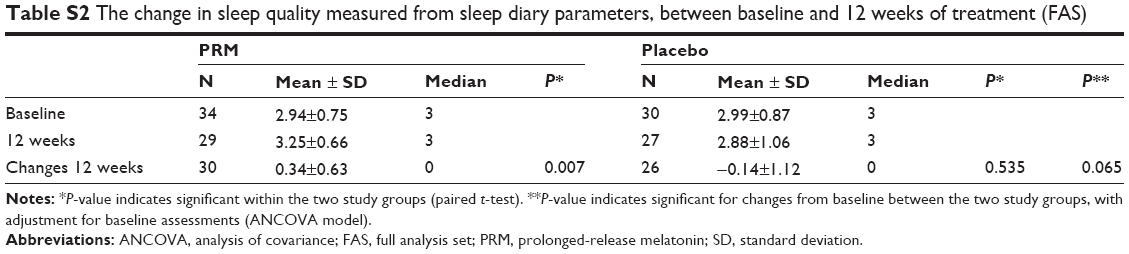
The quality of sleep at week 12 (sleep diary) was significantly increased in the PRM group (P=0.007) and did not change significantly in the placebo group (Table S2). A trend for a statistically significant difference was observed between the treatment groups at week 12 (P=0.065), and a statistically significant difference between groups was measured over the 12-week period (MMRM, P=0.0295). In the subpopulation of patients suffering from comorbid insomnia, a trend for a statistically significant difference in quality of sleep was observed at week 12 (P=0.091, by median test). The difference in mean change in quality of sleep assessed in the diary after 12 weeks of treatment was 0.48 units (Table S2).
Other parameters
No difference in NPI severity score was observed for the change from baseline to either week 12 or week 24 between groups. A statistically significant difference in NPI distress score was observed between the groups at week 24 (P=0.026). NPI distress scores increased significantly in the PRM group between baseline and week 24 (3.1±7.59) (P=0.033) and decreased nonsignificantly in the placebo group (−0.24±4.17) (P=0.758). However, the increase in the NPI distress score of the PRM group was not considered to be clinically relevant.42 No differences in NPI severity score or in NPI distress score were observed for the comorbid insomnia subpopulation (data not shown).
Sleep, measured by the SDI, improved in the PRM group as compared with the placebo after 24 weeks of treatment, demonstrating trends of significance (P=0.09) (data not shown). Caregiver distress, measured by the SDI, decreased in both treatment groups, in the FAS and in the insomnia subpopulation. No other significant differences between groups were demonstrated. No significant interactions between treatment effects and concomitant memantine intake or disease severity were found.
Safety
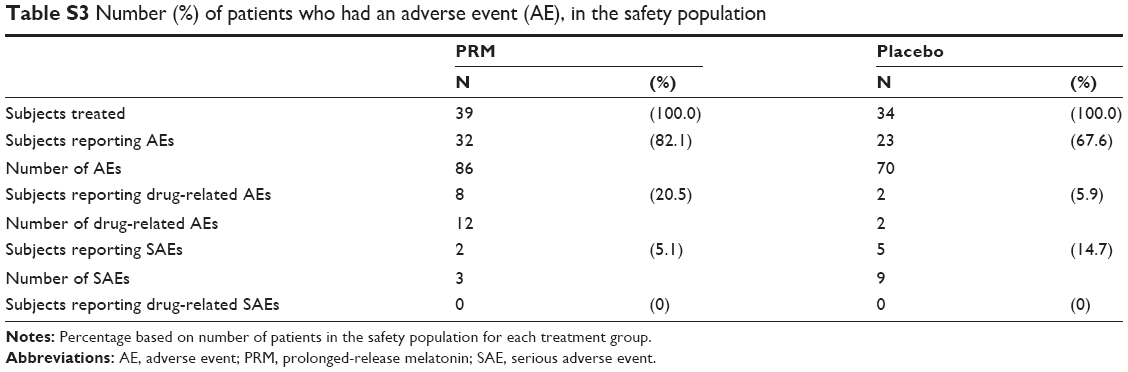
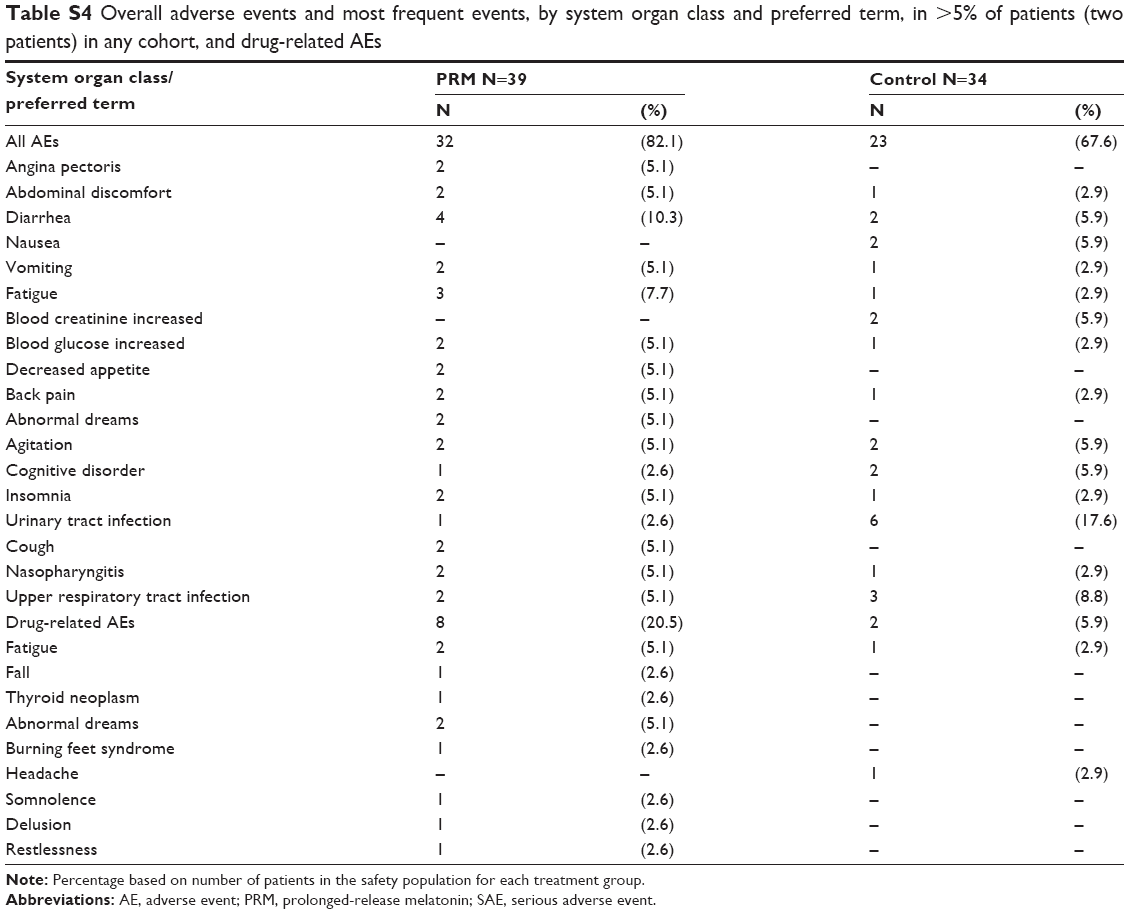
The incidence of AEs occurring in two or more patients (>5% of patients) is displayed in Table S3 and of drug-related AEs in Table S4. Most AEs reported during the study were transient, mild or moderate in severity, and not considered to be related to the study drug. The most common AEs were urinary tract infection (17.6% of patients in the placebo compared with 2.6% in the PRM cohort), diarrhea (10.3% of patients in the PRM compared with 5.9% in the placebo cohort), and upper respiratory tract infection (8.8% of patients in the placebo compared with 5.1% in the PRM cohort). Drug-related AEs were reported by eight patients (20.5%) in the PRM and by two (5.9%) in the placebo cohort, with no statistical difference between the two groups (post hoc analysis) (chi-squared Fisher’s exact test, P=0.0933). Three patients from each cohort discontinued due to AEs.
No deaths occurred during the study. Three SAEs were reported by two patients (5.1%) in the PRM and nine by five patients (14.7%) in the placebo cohort, with no statistical difference between the two groups (post hoc analysis) (chi-squared Fisher’s exact test, P=0.2396); none were related to the study drug. Two patients (5.9%) in the placebo cohort withdrew from the study due to SAEs – one patient due to severe myocardial infarction and one due to esophageal stenosis.
No clinically significant changes in physical examination, vital signs, or lab parameters were observed during the study in either of the study groups.
Discussion
The current study assessed the efficacy and safety of once daily add-on PRM compared with placebo over a 24-week period, in patients diagnosed with mild to moderate AD who were on stable doses of acetylcholinesterase inhibitors, with or without memantine. Assessments were made of changes in cognitive functioning and sleep since insomnia itself might negatively impact cognitive functioning. All end points were analyzed, both for the total FAS population (N=65) and for the subpopulation of patients suffering from comorbid insomnia (PSQI ≥6 at baseline [N=13]). The results show evidence of efficacy and safety of long-term add-on PRM in mild and moderate AD and particularly, in patients with comorbid insomnia.
Effects on cognition
In agreement with published data on the change in the ADAS-Cog parameter in AD patients treated with acetylcholinesterase inhibitors, with and without memantine,9 there was no significant deterioration in mean group cognitive state, as measured by the ADAS-Cog, for both treatment groups, after 24 weeks of the double-blind period. The median ADAS-Cog deteriorated somewhat in the placebo and improved in the PRM group, but the difference between groups was not statistically significant. Compatible with this notion, longer treatment periods and a larger sample size than those used in our study are needed to demonstrate significant decline in the ADAS-Cog score in the placebo group and demonstrate PRM treatment effects with this instrument. The other two instruments (MMSE and IADL) were evidently more sensitive to change within a 24-week period. Thus, cognition, as measured by the MMSE, deteriorated significantly in the placebo group over the 24-week period but did not change in the PRM group. The difference between the PRM and placebo effects was significant (P=0.044). Furthermore, after the 24-week, double-blind treatment period, PRM demonstrated clinically meaningful and statistically significant amelioration over placebo for self-care and activities of daily living, as measured by the IADL (P=0.004).
The benefit was even more pronounced in the subpopulation suffering from insomnia comorbidity. There was an improvement (decrease) of −3.5 points in median ADAS-Cog scores in the PRM and deterioration of +3 points in the placebo group after 24 weeks of double-blind treatment, and the difference in treatment effect between groups was significant (P=0.045). The difference in treatment effect between the groups (6.5 points on the median ADAS-Cog) was considered clinically meaningful.43,44
Moreover, PRM improved MMSE scores over placebo, showing an increase in MMSE scores after 24 weeks of 1.5 points, while the placebo group scores deteriorated by 2.8 points. The difference in treatment effects in MMSE (4.5 points) with PRM over placebo in the insomnia subpopulation was statistically significant (P=0.0177) and was considered clinically meaningful.45 Furthermore, there was a significantly lesser deterioration in mean IADL score with PRM compared with placebo (P=0.0319).
The effect on IADL was more pronounced after 24 weeks than after 12 weeks of treatment. This might suggest amelioration of disease progression with PRM compared with placebo. However, it is quite clear that the decline in IADL in the placebo arm at 12 weeks was low, if a linear decline was to be expected. This can be possibly attributed to time-dependent changes in the placebo effect previously shown to mostly affect the first assessments of cognitive functioning in AD trials.46,47 Further long-term trials will be needed to examine whether the greater effect of PRM with longer treatment duration represents attenuation of cognitive decline.
Effects on sleep
Most patients in the FAS population did not have insomnia, and thus, as expected, the treatment effect of PRM compared with that of placebo on the global PSQI score did not reach statistical significance.
Despite the absence of insomnia comorbidity in the FAS population, PSQI component 4, measuring sleep efficiency, improved significantly in the PRM group over placebo after 24 weeks of treatment (P=0.017) and over the entire 24-week, double-blind treatment period (MMRM, P=0.0312). In the comorbid insomnia subpopulation, despite the small sample size, sleep efficiency improved significantly in the PRM group over placebo after 24 weeks of treatment (P=0.04).
Altogether, the results obtained for the sleep variables are suggestive of beneficial effects of add-on PRM therapy on sleep maintenance in the AD patients. Notably, the sleep parameter mostly affected by PRM in AD patients is sleep efficiency, which is also the main parameter affected by the disease.2,48 Such effects, according to studies in children with neurodevelopmental disorders, may be more pronounced with PRM formulations because they are able to sustain active melatonin levels throughout the night.49–51 Beside sleep, the ability of melatonin to affect hippocampal networks and subsequently cognition may rely on presence of the hormone at high levels during the night, when this area is sensitive to the hormone.52,53
Several studies, including six randomized, double-blind, placebo-controlled trials, with a total of 310 AD patients, have previously reported on the effects of melatonin on sleep in AD patients, mostly with advanced disease. In three of these studies (N=83), there were no significant effects of melatonin (1.5–6 mg sustained-release formulations, 3–8.5 mg immediate-release formulations, 10 days −7 weeks) compared with placebo on sleep or behavioral aspects.54–56 In one study (N=50), light treatment plus melatonin (5 mg, 10 weeks) increased daytime wake, activity levels, and strengthened the rest–activity cycle,57 and in another study (N=20 unmedicated patients), there were significant improvements in ADAS-Cog and ADAS-non-Cog scores with melatonin (3 mg, 4 weeks) compared with placebo but no significant differences in MMSE scores and sleep, although it is unknown whether such effects were sustained at longer treatment duration.21 It is interesting to note that most studies reporting negative effects of melatonin on sleep in AD used actigraphy to assess sleep. In another study of 2.5 mg sustained-release melatonin for 2 months in AD patients (N=157), both actigraphy and caregiver ratings of sleep were used. Whereas caregiver ratings of sleep quality showed improvement with melatonin compared with placebo treatment, the actigraphy data failed to show significant differences between treatment groups, and no other benefit was reported.58 The use of actigraphy as a substitute for sleep polysomnography in AD patients was recently debated.59,60 On the other hand, sleep quality can be negatively affected by emotional distress and caregiver burden. It is thus possible that actigraphy and subjective assessments of sleep will differ in reported sleep parameters and treatment effects. These methodological differences, in addition to differences in study design, duration, melatonin formulation, and dose, may account for the inconsistencies between results obtained in the previous studies.
A pertinent question is whether the effects of add-on PRM treatment on sleep were similar in AD patients with insomnia comorbidity to those previously seen in nondemented patients with insomnia aged 55 years and older. The results of this study show that as for AD patients, the improvement in sleep develops progressively, reaching stable plateau levels at 12 weeks (Figure 4A and B). A similar evolution of response with time of treatment was reported previously in nondemented patients with insomnia aged 55 and older61 and was considered to represent consolidation of the circadian system.
The greater effects of PRM on cognition in the insomnia subpopulation support the notion that sleep is critical for memory performance. Recent studies indicate that sleep duration and sleep quality may play a role in cognitive performance in healthy older adults.5 A recent study on sleep quality and preclinical AD concluded that beta-amyloid deposition in the preclinical stage of AD is associated with worse sleep quality but not with changes in sleep quantity.62 PRM restores the body’s normal sleep–wake cycle, improves sleep quality and daytime functioning, maintains the physiological sleep structure in insomnia patients, and as found in the present study, improves sleep maintenance in AD patients.26,63 This may explain the improvement in ADAS-Cog, IADL, and MMSE observed in AD patients with insomnia comorbidity after PRM treatment. Importantly, treatment of the sleep complaints with traditional hypnotics may not be beneficial and may even be deleterious in AD patients because of the impairments in cognitive and memory functioning associated with these drugs.11–13 Furthermore, long-term use of benzodiazepine hypnotics is associated with an increased risk for dementia.15,16 Because PRM improves sleep without the risk of memory or cognitive impairment, the improvement of sleep can further contribute to the overall efficacy of PRM in AD, as found in the present study.13,29,64 The efficacy results obtained in AD patients with insomnia comorbidity are most interesting, in particular with respect to the interaction between insomnia and AD, and larger studies of PRM in this population are warranted.
An important question is whether the add-on PRM treatment was attenuating disease progression. Evaluation of the change in the cognitive parameters over time indicates that while the placebo group deteriorated progressively during the 24 weeks (as measured by the IADL and MMSE), a significantly lesser decline in cognitive functioning was seen in the PRM-treated cohort after 12 and 24 weeks of treatment, suggesting that PRM attenuated the cognitive decline. Further studies will have to be undertaken to understand whether this trend could represent disease modification.
There is now compelling evidence for the association between shorter sleep duration and poor sleep quality to greater beta amyloid burden and higher risk for AD.7,8,62,65,66 Based on the aforementioned evidence and the results obtained from our study, a plausible mechanism for PRM effects in AD could be that the improved sleep efficiency leads to lower risk of accumulation of beta amyloid deposition62 and/or increase in beta amyloid clearance from the brain,65 which can ultimately result in the attenuation of AD progression. If so, patients with good sleep quality can potentially also benefit from the neuroprotective effects of the hormone.67,68
Overall, PRM was well tolerated as most AEs were of mild or moderate severity and resolved without sequelae. No deaths occurred during the study, and none of the SAEs reported were considered related to the study drug.
Conclusion
We conclude that the add on of PRM to acetylcholinesterase inhibitor, with or without memantine therapy, might be an effective and safe means for improvement in cognitive functioning and sleep maintenance in mild to moderate AD patients. The benefit may be greater for those with insomnia comorbidity, probably because good sleep is critical for proper cognition and memory processing. Keeping in mind that larger sample size and longer study duration are required in order to validate these results, the results are compatible with the accumulating evidence on a causal relationship between poor sleep and cognitive decline in AD.
Acknowledgments
The authors would like to acknowledge and thank the general practitioners Dr SS Shrivastava and partners, Dr M Farmer and partners, Dr J Andersen, and partners who all managed the patients so admirably. They are also indebted to the research nurses and management staff of Criterium, Inc., without whom the study would not have taken place.
Disclosure
AGW, MF, GH, and NF have acted as paid consultants to Neurim Pharmaceuticals. AGW is the director of CPS research. NZ, TN, ML, and AFM are employees of Neurim Pharmaceuticals. The study was funded by Neurim Pharmaceuticals. The authors report no other conflicts of interest.
References
Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010; 362(4):329–344. | ||
Pollak CP, Perlick D. Sleep problems and institutionalization of the elderly. J Geriatr Psychiatry Neurol. 1991;4(4):204–210. | ||
Maquet P. The role of sleep in learning and memory. Science. 2001; 294(5544):1048–1052. | ||
Backhaus J, Junghanns K, Born J, Hohaus K, Faasch F, Hohagen F. Impaired declarative memory consolidation during sleep in patients with primary insomnia: Influence of sleep architecture and nocturnal cortisol release. Biol Psychiatry. 2006;60(12):1324–1330. | ||
Miyata S, Noda A, Iwamoto K, Kawano N, Okuda M, Ozaki N. Poor sleep quality impairs cognitive performance in older adults. J Sleep Res. 2013;22(5):535–541. | ||
Murre JM, Kristo G, Janssen SM. The effect of self-reported habitual sleep quality and sleep length on autobiographical memory. Memory. 2014;22(6):633–645. | ||
Lim AS, Yu L, Kowgier M, Schneider JA, Buchman AS, Bennett DA. Modification of the relationship of the apolipoprotein E ε4 allele to the risk of Alzheimer disease and neurofibrillary tangle density by sleep. JAMA Neurol. 2013;70(12):1544–1551. | ||
Ju YE, Lucey BP, Holtzman DM. Sleep and Alzheimer disease pathology – a bidirectional relationship. Nat Rev Neurol. 2014;10(2):115–119. | ||
Hansen RA, Gartlehner G, Webb AP, Morgan LC, Moore CG, Jonas DE. Efficacy and safety of donepezil, galantamine, and rivastigmine for the treatment of Alzheimer’s disease: a systematic review and meta-analysis. Clin Interv Aging. 2008;3(2):211–225. | ||
Rive B, Gauthier S, Costello S, Marre C, François C. Synthesis and comparison of the meta-analyses evaluating the efficacy of memantine in moderate to severe stages of Alzheimer’s disease. CNS Drugs. 2013; 27(7):573–582. | ||
Chavant F, Favrelière S, Lafay-Chebassier C, Plazanet C, Pérault-Pochat MC. Memory disorders associated with consumption of drugs: updating through a case/noncase study in the French PharmacoVigilance Database. Br J Clin Pharmacol. 2011;72(6):898–904. | ||
Leufkens TR, Lund JS, Vermeeren A. Highway driving performance and cognitive functioning the morning after bedtime and middle-of-the-night use of gaboxadol, zopiclone and zolpidem. J Sleep Res. 2009; 18(4):387–396. | ||
Otmani S, Demazières A, Staner C, et al. Effects of prolonged-release melatonin, zolpidem, and their combination on psychomotor functions, memory recall, and driving skills in healthy middle aged and elderly volunteers. Hum Psychopharmacol. 2008;23(8):693–705. | ||
Vermeeren A, Coenen AM. Effects of the use of hypnotics on cognition. Prog Brain Res. 2011;190:89–103. | ||
Billioti de Gage S, Bégaud B, Bazin F, et al. Benzodiazepine use and risk of dementia: prospective population based study. BMJ. 2012; 345:e6231. | ||
Wu CS, Wang SC, Chang IS, Lin KM. The association between dementia and long-term use of benzodiazepine in the elderly: nested case-control study using claims data. Am J Geriatr Psychiatry. 2009;17(7):614–620. | ||
Zisapel N. Melatonin and sleep. Open Neuroendocrinol J. 2010; 3:85–95. | ||
Tohgi H, Abe T, Takahashi S, Kimura M, Takahashi J, Kikuchi T. Concentrations of serotonin and its related substances in the cerebrospinal fluid in patients with Alzheimer type dementia. Neurosci Lett. 1992;141(1):9–12. | ||
Mishima K, Tozawa T, Satoh K, Matsumoto Y, Hishikawa Y, Okawa M. Melatonin secretion rhythm disorders in patients with senile dementia of Alzheimer’s type with disturbed sleep-waking. Biol Psychiatry. 1999; 45(4):417–421. | ||
Srinivasan V, Kaur C, Pandi-Perumal S, Brown GM, Cardinali DP. Melatonin and its agonist ramelteon in Alzheimer’s disease: possible therapeutic value. Int J Alzheimers Dis. 2010;(2011):1–15. | ||
Asayama K, Yamadera H, Ito T, Suzuki H, Kudo Y, Endo S. Double blind study of melatonin effects on the sleep-wake rhythm, cognitive and non-cognitive functions in Alzheimer type dementia. J Nippon Med Sch. 2003;70(4):334–341. | ||
Brusco LI, Márquez M, Cardinali DP. Monozygotic twins with Alzheimer’s disease treated with melatonin: Case report. J Pineal Res. 1998;25(4):260–263. | ||
Brusco LI, Márquez M, Cardinali DP. Melatonin treatment stabilizes chronobiologic and cognitive symptoms in Alzheimer’s disease. Neuro Endocrinol Lett. 2000;21(1):39–42. | ||
Cardinali DP, Vigo DE, Olivar N, Vidal MF, Furio AM, Brusco LI. Therapeutic application of melatonin in mild cognitive impairment. Am J Neurodegener Dis. 2012;1(3):280–291. | ||
Rondanelli M, Opizzi A, Faliva M, et al. Effects of a diet integration with an oily emulsion of DHA-phospholipids containing melatonin and tryptophan in elderly patients suffering from mild cognitive impairment. Nutr Neurosci. 2012;15(2):46–54. | ||
Lemoine P, Zisapel N. Prolonged-release formulation of melatonin (Circadin) for the treatment of insomnia. Expert Opin Pharmacother. 2012;13(6):895–905. | ||
Wade A, Downie S. Prolonged-release melatonin for the treatment of insomnia in patients over 55 years. Expert Opin Investig Drugs. 2008;17(10):1567–1572. | ||
Garfinkel D, Laudon M, Nof D, Zisapel N. Improvement of sleep quality in elderly people by controlled-release melatonin. Lancet. 1995; 346(8974):541–544. | ||
Otmani S, Metzger D, Guichard N, et al. Effects of prolonged-release melatonin and zolpidem on postural stability in older adults. Hum Psychopharmacol. 2012;27(3):270–276. | ||
Altena E, Ramautar JR, Van Der Werf YD, Van Someren EJ. Do sleep complaints contribute to age-related cognitive decline? Prog Brain Res. 2010;185:181–205. | ||
Markus RP, Silva CL, Franco DG, Barbosa EM, Ferreira ZS. Is modulation of nicotinic acetylcholine receptors by melatonin relevant for therapy with cholinergic drugs? Pharmacol Ther. 2010;126(3): 251–262. | ||
Riemersma-van der Lek RF, Swaab DF, Twisk J, Hol EM, Hoogendijk WJ, Van Someren EJ. Effect of bright light and melatonin on cognitive and noncognitive function in elderly residents of group care facilities: a randomized controlled trial. JAMA. 2008;299(22):2642–2655. | ||
Mohs RC, Knopman D, Petersen RC, et al. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer’s Disease Assessment Scale that broaden its scope. The Alzheimer’s Disease Cooperative Study. Alzheimer Dis Assoc Disord. 1997;11 Suppl 2:S13–S21. | ||
Lawton MP, Brody EM. Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist. 1969;9(3): 179–186. | ||
Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198. | ||
Buysse DJ, Reynolds CF, Monk TH, Berman SR, Kupfer DJ. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res. 1989;28(2):193–213. | ||
Guy W. ECDEU Assessment Manual for Psychopharmacology. Rockville, MD: US Department of Health, Education, and Welfare; 1976. | ||
Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44(12): 2308–2314. | ||
Bech P, Olsen LR, Kjoller M, Rasmussen NK. Measuring well-being rather than the absence of distress symptoms: a comparison of the SF-36 Mental Health subscale and the WHO-Five Well-Being Scale. Int J Methods Psychiatr Res. 2003;12(2):85–91. | ||
Tractenberg RE, Singer CM, Cummings JL, Thal LJ. The Sleep Disorders Inventory: an instrument for studies of sleep disturbance in persons with Alzheimer’s disease. J Sleep Res. 2003;12(4):331–337. | ||
Prakash A, Risser RC, Mallinckrodt CH. The impact of analytic method on interpretation of outcomes in longitudinal clinical trials. Int J Clin Pract. 2008;62(8):1147–1158. | ||
Howard R, Phillips P, Johnson T, et al. Determining the minimum clinically important differences for outcomes in the DOMINO trial. Int J Geriatr Psychiatry. 2011;26(8):812–817. | ||
Schrag A, Schott JM; Alzheimer’s Disease Neuroimaging Initiative. What is the clinically relevant change on the ADAS-Cog? J Neurol Neurosurg Psychiatry. 2012;83(2):171–173. | ||
Rockwood K, Fay S, Gorman M. The ADAS-cog and clinically meaningful change in the VISTA clinical trial of galantamine for Alzheimer’s disease. Int J Geriatr Psychiatry. 2010;25(2):191–201. | ||
Andrew MK, Rockwood K. A five-point change in Modified Mini-Mental State Examination was clinically meaningful in community-dwelling elderly people. J Clin Epidemiol. 2008;61(8):827–831. | ||
Rogers SL, Farlow MR, Doody RS, Mohrs R, Friedhoff LT; Donepezil Study Group*. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Neurology. 1998;50(1): 136–145. | ||
Knopman DS. Clinical trial design issues in mild to moderate Alzheimer disease. Cogn Behav Neurol. 2008;21(4):197–201. | ||
Yu JM, Tseng IJ, Yuan RY, Sheu JJ, Liu HC, Hu CJ. Low sleep efficiency in patients with cognitive impairment. Acta Neurol Taiwan. 2009;18(2):91–97. | ||
Jan JE, Hamilton D, Seward N, Fast DK, Freeman RD, Laudon M. Clinical trials of controlled-release melatonin in children with sleep-wake cycle disorders. J Pineal Res. 2000;29(1):34–39. | ||
Gringras P, Gamble C, Jones AP, et al; MENDS Study Group. Melatonin for sleep problems in children with neurodevelopmental disorders: randomised double masked placebo controlled trial. BMJ. 2012; 345:e6664. | ||
De Leersnyder H, Zisapel N, Laudon M. Prolonged-release melatonin for children with neurodevelopmental disorders. Pediatr Neurol. 2011; 45(1):23–26. | ||
Gorfine T, Zisapel N. Late evening brain activation patterns and their relation to the internal biological time, melatonin, and homeostatic sleep debt. Hum Brain Mapp. 2009;30(2):541–552. | ||
Gorfine T, Zisapel N. Melatonin and the human hippocampus, a time dependent interplay. J Pineal Res. 2007;43(1):80–86. | ||
Serfaty M, Kennell-Webb S, Warner J, Blizard R, Raven P. Double blind randomised placebo controlled trial of low dose melatonin for sleep disorders in dementia. Int J Geriatr Psychiatry. 2002;17(12): 1120–1127. | ||
Mahlberg R, Walther S. Actigraphy in agitated patients with dementia. Monitoring treatment outcomes. Z Gerontol Geriatr. 2007;40(3): 178–184. | ||
Gehrman PR, Connor DJ, Martin JL, Shochat T, Corey-Bloom J, Ancoli-Israel S. Melatonin fails to improve sleep or agitation in double-blind randomized placebo-controlled trial of institutionalized patients with Alzheimer disease. Am J Geriatr Psychiatry. 2009;17(2): 166–169. | ||
Dowling GA, Burr RL, Van Someren EJ, et al. Melatonin and bright-light treatment for rest-activity disruption in institutionalized patients with Alzheimer’s disease. J Am Geriatr Soc. 2008;56(2):239–246. | ||
Singer C, Tractenberg RE, Kaye J, et al; Alzheimer’s Disease Cooperative Study. A multicenter, placebo-controlled trial of melatonin for sleep disturbance in Alzheimer’s disease. Sleep. 2003;26(7):893–901. | ||
Kawada T. Sleep evaluation by actigraphy for patients with Alzheimer disease. JAMA Neurol. 2013;70(8):1074. | ||
Ju YE, Holtzman DM. Sleep evaluation by actigraphy for patients with Alzheimer disease – reply. JAMA Neurol. 2013;70(8):1074–1075. | ||
Wade AG, Crawford G, Ford I, et al. Prolonged release melatonin in the treatment of primary insomnia: evaluation of the age cut-off for short- and long-term response. Curr Med Res Opin. 2011;27(1):87–98. | ||
Ju YE, McLeland JS, Toedebusch CD, et al. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013;70(5):587–593. | ||
Wade AG, Ford I, Crawford G, et al. Efficacy of prolonged release melatonin in insomnia patients aged 55–80 years: quality of sleep and next-day alertness outcomes. Curr Med Res Opin. 2007;23(10):2597–2605. | ||
Luthringer R, Muzet M, Zisapel N, Staner L. The effect of prolonged-release melatonin on sleep measures and psychomotor performance in elderly patients with insomnia. Int Clin Psychopharmacol. 2009;24(5): 239–249. | ||
Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373–377. | ||
Spira AP, Gamaldo AA, An Y, et al. Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurol. 2013; 70(12):1537–1543. | ||
Lipartiti M, Franceschini D, Zanoni R, et al. Neuroprotective effects of melatonin. Adv Exp Med Biol. 1996;398:315–321. | ||
García-Mesa Y, Giménez-Llort L, López LC, et al. Melatonin plus physical exercise are highly neuroprotective in the 3xTg-AD mouse. Neurobiol Aging. 2012;33(6):1124.e13–e29. |
Supplementary materials
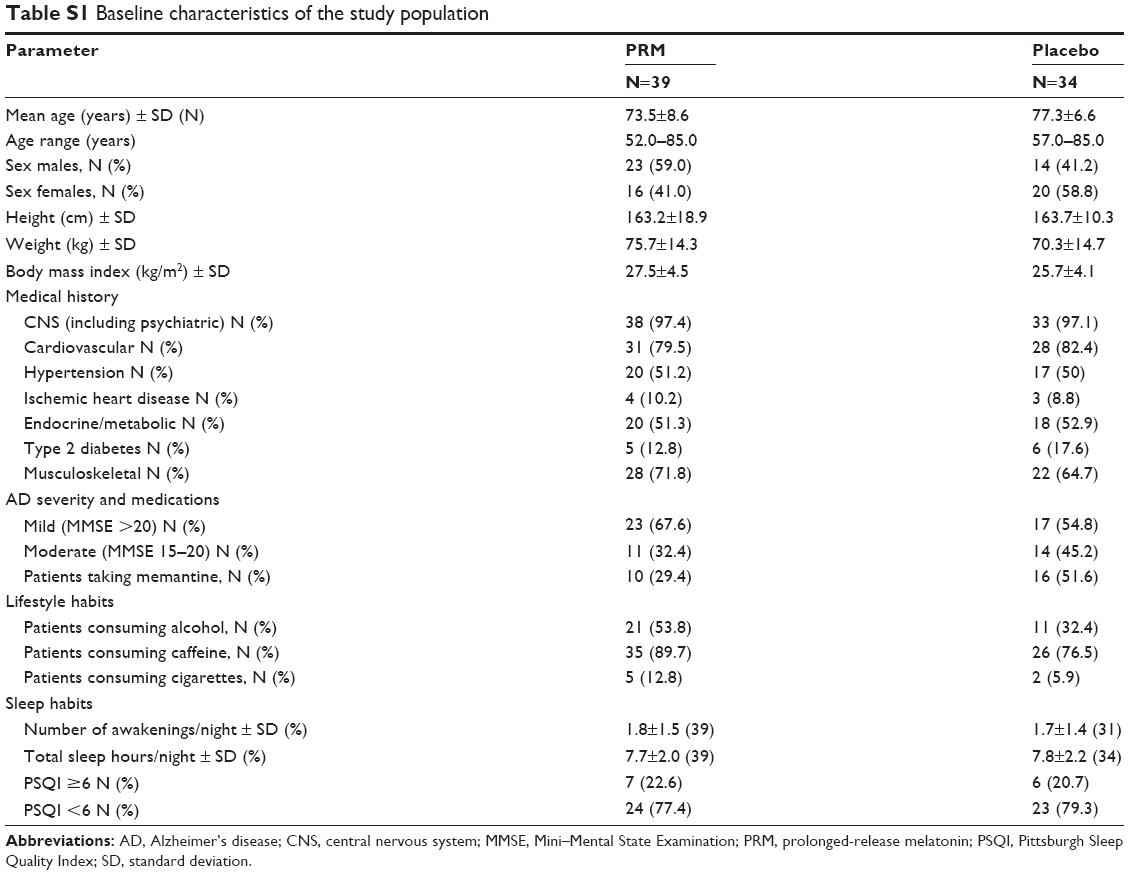 | Table S1 Baseline characteristics of the study population |
 | Table S2 The change in sleep quality measured from sleep diary parameters, between baseline and 12 weeks of treatment (FAS) |
 | Table S3 Number (%) of patients who had an adverse event (AE), in the safety population |
 | Table S4 Overall adverse events and most frequent events, by system organ class and preferred term, in >5% of patients (two patients) in any cohort, and drug-related AEs |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.