Back to Archived Journals » Metalloproteinases In Medicine » Volume 7
ADAMDEC1 and Its Role in Inflammatory Disease and Cancer
Authors Kumagai T ![]() , Fan S
, Fan S ![]() , Smith AM
, Smith AM ![]()
Received 21 May 2020
Accepted for publication 21 July 2020
Published 17 August 2020 Volume 2020:7 Pages 15—28
DOI https://doi.org/10.2147/MNM.S263813
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yoshifumi Itoh
Tomoko Kumagai,1 Shuangqi Fan,2 Andrew Mark Smith1
1UCL Eastman Dental Institute, University College London (UCL), Eastman Dental Institute, Microbial Diseases, Royal Free Campus, London NW3 2PF, UK; 2College of Veterinary Medicine, South China Agricultural University, Guangzhou, People’s Republic of China
Correspondence: Andrew Mark Smith
UCL Eastman Dental Institute, University College London (UCL), Eastman Dental Institute, Microbial Diseases, Royal Free Campus, Rowland Hill Street, London NW3 2PF, UK
Tel +44 (0)20 8016 7756
Email [email protected]
Abstract: A disintegrin and metalloprotease like decysin (ADAMDEC1) is a highly conserved secreted metalloprotease that belongs to a family of A disintegrin and metalloprotease (ADAMs). It is expressed exclusively in the gastrointestinal tract of animals and is known to possess a very rare zinc-binding motif (HEXXHXXGXXD) within the metalloprotease domain. The biological function of ADAMDEC1 as well as its true biological substrates remains unknown although its characteristic features reported to date suggest it plays a fundamental role in the physiology of mammals. Historically its expression, in healthy state, was believed to be limited to the monocyte-derived macrophages (MDMs) and dendritic cells within the gastrointestinal tract; however, the recent development of single-cell sequencing has provided evidence supporting its expression in a wider range of cell types. There is an increasing body of evidence linking the alterations in ADAMDEC1 expression and various inflammatory diseases and cancers. Although a detailed mechanistic role of ADAMDEC1 in these conditions remains elusive. In this review, we aim to summarise the characteristic features of this unique metalloprotease, discuss the associations with various human diseases and define the potential mechanistic role of ADAMDEC1 in mammalian physiology.
Keywords: A disintegrin and metalloproteases, metalloprotease, epithelial defense against cancer, mucosal inflammation, inflammatory bowel disease, colorectal cancer
Introduction
A disintegrin and metalloprotease like decysin 1 (ADAMDEC1) is a unique metazincin metalloprotease belonging to the A disintegrin and metalloproteases (ADAMs) family. To date there have been very few studies conducted on the biological function of ADAMDEC1, nevertheless, the information currently available points to a number of fundamental roles for this intriguing metalloprotease in animal physiology and disease. In this review, we aim to summarise the characteristic features of ADAMDEC1, associations with various diseases and the potential mechanistic role of this unusual ADAMs family member.
Molecular and Genetic Characteristics of ADAMDEC1
ADAMDEC1 and ADAM Proteins
ADAMDEC1 was first discovered through sequencing of mRNA extracted from the dendritic cells of human tonsils in 1997.1 Following its discovery, it was categorised to a family of ADAMs due to its high sequence homology with ADAM28. ADAMs are highly conserved secreted or transmembrane metalloproteases that share a common structural composition, consisting of an N-terminal signalling peptide, prodomain, metalloprotease, disintegrin-like domain, cysteine-rich region, as well as a transmembrane domain and cytoplasmic tail (Figure 1). ADAMDEC1 is considered to be a unique member of the ADAMs family for a number of reasons. First of all, ADAMDEC1 is missing the cytoplasmic tail, transmembrane, cysteine-rich domains thus consisting of only a signalling peptide, prodomain, metalloprotease and disintegrin-like domains.1 Additionally, its disintegrin-like domain is truncated resulting in a protein structure comprised 470 amino acids, which is almost half the size of the typical ADAMs (Figure 1).1 It is believed to be cleaved at the furin-recognition site releasing the signalling peptide intracellularly and secreted as a mature active form as indicated by the absence of a transmembrane domain. Furin has been, in fact, shown to process and cleave off the signalling peptide of ADAMDEC1 in vitro.2
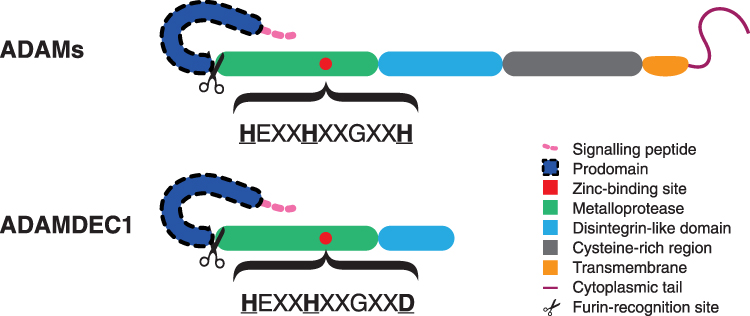 |
Figure 1 Schematic diagram illustrating the structural differences between the typical ADAMs and ADAMDEC1. The structural composition of ADAMDEC1 consists of a signaling peptide, prodomain, metalloprotease and disintegrin-like domain. The third histidine (H) of the characteristic histidine repeat motif in the zinc-binding site is replaced by aspartate (D) within the metalloprotease domain of ADAMDEC1. |
The catalytically active ADAMs possess a highly characteristic histidine (H) repeat motif (HEXXHXXGXXH) within the zinc-binding site of the metalloprotease domain.3 Within the zinc-binding site of ADAMDEC1, however, the third histidine is replaced by aspartate (D) resulting in the motif sequence of HEXXHXXGXXD.1 The HEXXHXXGXXD motif within the zinc-binding site is extremely rare and has only previously been found in two other proteins, immune inhibitor A and extracellular small neutral protease, which are both bacterial proteases. These bacterial proteases are known to be catalytically active thus ADAMDEC1 is expected to also possess a metalloprotease function despite the alteration in the zinc-binding site.4,5 The replacement of the third histidine to aspartate, however, has been shown to confer a reduction in the catalytical activity and diminish the substrate specificity compared to the typical ADAMs.6,7 Furthermore, the alteration in the zinc-binding site also seems to confer a resistance to the usual regulation by tissue inhibitors of metalloproteases (TIMPs), which are naturally occurring inhibitors of ADAMs.6 These findings suggest that ADAMDEC1 may have a physiological role that differs from the normal metalloproteases and is under an alternative regulatory system.
Conservation of ADAMDEC1 Through Evolution
To date, ADAMDEC1 has been identified in 130 mammalian species and no other class of animals. ADAMDEC1 resembles ADAM7 and ADAM28 at both the nucleotide and amino acid level with a sequential amino acid homology of 36% and 47%, respectively.8 In humans, the genes of ADAMDEC1, ADAM7 and ADAM28 are all found clustered together on the short arm of Chromosome 8 (8p). The clustering of these three ADAMs is similarly found in other mammalian species including marsupials and monotremes.9 ADAM7 and ADAMDEC1 are absent with no existing orthologs in the genomes of the non-mammalian vertebrates such as fishes, amphibians, reptiles and birds.9,10 Instead, however, in these animals the ADAM28 locus is present in this region. It is, therefore, suggested that ADAMDEC1 is likely to have arisen by a partial gene duplication from ADAM28 at this locus and acquired a novel function after the divergence of mammals from reptiles. Of the 130 species of the mammals that possess ADAMDEC1, the zinc-binding site amino acid sequence of HELGHXLGMXD within the metalloprotease domain has been either detected or predicted in 126 species so far, with variations of amino acid seen at only two positions (Figure 2). This high level of conservation through evolution suggests that ADAMDEC1 is likely to play a fundamental role in mammalian physiology.
 |
Figure 2 The zinc-binding motif in the metalloprotease catalytic site of ADAMDEC1 is highly conserved in mammals. The bold letters represent the histidine repeat motif in which the third histidine is replaced by aspartate in the zinc-binding site of ADAMDEC1. Only two and nine variations of amino acid exist at the two amino acid positions, shaded blue and red respectively, within the zinc-binding motif among the 126 species known to possess ADAMDEC1. |
Proteolytic Target of ADAMDEC1
Very little is known about the catalytic activity of ADAMDEC1 or its true biological targets. The mature active recombinant human ADAMDEC1 has demonstrated a catalytic ability to process α2-macroglobulin, casein and carboxymethylated transferrin in vitro.6,7 The ability of ADAMDEC1 to cleave α2-macroglobulin and casein was lost when the glutamate following the first histidine in the zinc-binding motif was replaced by alanine (HAXXHXXGXXD) to form the catalytically inactive mutant ADAMDEC1E353A, confirming that the catalytic function depends on the zinc-binding motif in the metalloprotease domain.6 Interestingly, however, the mature active form of recombinant mouse ADAMDEC1 does not demonstrate any catalytic activity against casein or carboxymethylated transferrin. Furthermore, the cleavage of α2-macroglobulin by ADAMDEC1 in vitro required ~40 hours of incubation, which is much slower than expected if α2-macroglobulin was a true physiological substrate for ADAMDEC1.6 These findings suggest that these compounds are unlikely to be the true natural biological substrates for ADAMDEC1.
It seems likely that ADAMDEC1 possesses sheddase activity in accordance with the majority of the ADAMs family. Chen et al showed that the mature active form of recombinant human ADAMDEC1 was able to cleave the membrane-bound pro-epidermal growth factor (EGF) on the surface of thrombin-activated platelets.11 Chymostatin, a naturally occurring protease inhibitor, which is known to inhibit the hydrolysis reaction of several ADAMs, was also able to inhibit the cleavage of pro-EGF by ADAMDEC1. The authors, therefore, suggested that the shedding of pro-EGF off the activated platelets by ADAMDEC1 might be physiologically relevant. The extremely high expression of ADAMDEC1 and low abundance of platelets in the lamina propria of intestine under healthy status, however, would suggest pro-EGF on the activated platelets is unlikely to be the only or main biological target. Another study by Jimenez-Pascual et al demonstrated the ability of ADAMDEC1 to cleave the membrane-bound fibroblast growth factor-2 (FGF2) on glioblastoma (GBM) cancer stem cells.12 FGF2 was detected in the media minutes after the GBM cancer stem cells were incubated with recombinant ADAMDEC1. Although FGF2 is known to be expressed in the lamina propria of intestine and has been suggested to play roles in preserving intestinal stem cells, further studies are needed to provide the relevance of ADAMDEC1 on FGF2 regulation.13
Overall, studies to date have provided evidence to support a potential catalytic ability of ADAMDEC1 that depends on the metalloprotease domain. However, it is anticipated that the true physiological substrates of ADAMDEC1 are yet to be fully elucidated.
Potential Function of Disintegrin-Like Domain of ADAMDEC1
It had been speculated that the disintegrin-like domain of ADAMDEC1 was unlikely to function as an integrin-recognition adhesion molecule based on the fact that it is truncated. However, a study by Yako et al reported that the apical extrusion of oncogene, RasV12, expressing Madin-Darby Canine Kidney (MDCK) cells was suppressed by a knockdown of ADAMDEC1 in the surrounding normal epithelial cells.14 Interestingly, the catalytically inactive mutant ADAMDEC1E353A maintained its ability to promote the apical extrusion of the oncogene-expressing epithelial cells, which indicates a distinct role of the disintegrin-like domain that is independent of the metalloprotease activity. The exact mechanistic function of ADAMDEC1 and its disintegrin-like domain in the apical extrusion process, which has been termed epithelial defense against cancer (EDAC), is still unclear.
Physiological Expression of ADAMDEC1 in Healthy State
Highly Restricted Expression of ADAMDEC1 in the Gastrointestinal Tract
ADAMDEC1 is almost exclusively found in the gastrointestinal tract proximally from the duodenum to the rectum distally. The highest expression is seen in the small intestine, followed by the rectum and the colon.15 Outside of the gastrointestinal tract, ADAMDEC1 expression is also seen in the lymph nodes, tonsils, spleen, urinary bladder and placenta to a much lesser extent.1,15 This localised pattern of expression seems to be conserved and has been reported in human, pig and mouse.16 Within the gastrointestinal tract, at the cellular level, ADAMDEC1 is expressed predominantly in the macrophages in the lamina propria.17,18 Within spleen and tonsils, ADAMDEC1 is believed to be expressed by the CD40-activated dendritic cells.1,2
Induction of ADAMDEC1 During Monocyte to Macrophage Differentiation
It is widely accepted that the majority of the macrophages within the gastrointestinal tract are continuously replenished by migration and differentiation of circulating monocytes into the lamina propria.19 The circulating monocytes are believed not to express ADAMDEC1.17 On the other hand, under normal physiological condition, the resident macrophages of the organs, except for the gastrointestinal tract, are believed to be replenished predominantly by self-renewing yolk-sac and/or embryonic foetal liver precursors. In these organs, where ADAMDEC1 is not normally expressed under healthy status, the recruitment of circulating monocytes and monocyte-derived macrophages (MDMs) are thought to play a role in pathological situations such as inflammation and infection during which an upregulation of ADAMDEC1 has been reported (discussed in more detail below). Thus, based on these findings and the strikingly high abundance of ADAMDEC1 in the healthy gastrointestinal tract, it has been speculated that ADAMDEC1 is a marker of the circulating monocytes which enter the organs and differentiate into macrophages.
This hypothesis is supported by the microarray analysis of the tissue resident macrophages in heathy intestine and spleen which shows a markedly significant expression of ADAMDEC1 in the intestinal macrophages (Gene Expression Omnibus (GEO) data repository GEO ID GSE8868) (Figure 3).20 Early in vitro studies are also consistent with this hypothesis. Differentiation of peripheral blood-derived monocytes into macrophages in vitro by various stimuli, such as human serum, 1α-25-dihydroxyvitamin D3 or macrophage colony-stimulating factor (MCSF) led to induced expression of ADAMDEC1.17,21 An additional increase in the level of ADAMDEC1 was seen when the MDMs were stimulated with lipopolysaccharides (LPS), which is present in the lamina propria of the healthy intestine.17,22 Sirtuins 1 and 2 (SIRT1/2), two NAD-dependent deacetylases, were purported to be responsible for de-methylation and elevation in ADAMDEC1 expression in the LPS-stimulated macrophages.21
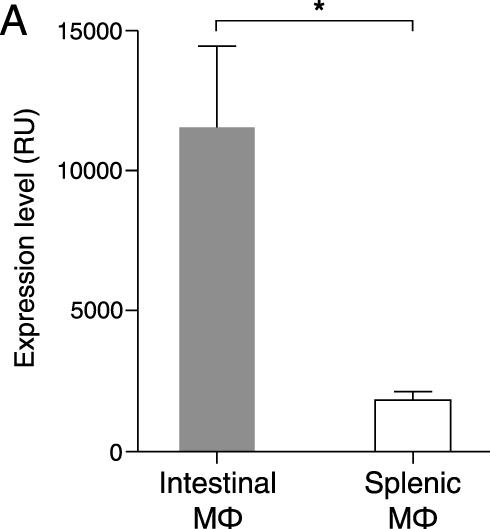 |
Figure 3 ADAMDEC1 gene expression in intestinal and splenic resident macrophages. Mouse intestinal macrophages express very high levels of ADAMDEC1 compared to splenic macrophages in a healthy state. *p<0.05. Data extracted from NCBI Gene Expression Omnibus (GEO) data repository GEO ID GSE 8868.20 |
More recently, a number of transcriptomic studies have also identified ADAMDEC1 expression in a range of MDM phenotypes in vitro.23–29 The consensus from these in vitro studies is that the ADAMDEC1 expression and secretion seem to be induced upon monocyte to macrophage differentiation irrespective of the final macrophage phenotype. However, not all extra-intestinal inflammatory conditions are associated with an induction of ADAMDEC1 in vivo (discussed in more detail below). The tissue macrophages are composed of highly heterogenous cells with a high plasticity, whose range of phenotypes depend on the surrounding microenvironment.30 Current models of in vitro monocyte to macrophage differentiation are unlikely to yield an exact replication of the intestinal macrophages or tissue-specific inflammatory macrophages. Further studies are needed to evaluate the in-depth mechanism of the regulation of ADAMDEC1 induction.
Expression of ADAMDEC1 in Self-Renewing Macrophages and Mesenchymal Cells
The recent development of single cell sequencing has provided an opportunity to further identify specific cellular populations within the gastrointestinal tract that express ADAMDEC1. These studies have demonstrated that ADAMDEC1 is expressed in a wider range of cells than originally thought within the intestine including evidence to suggest ADAMDEC1 is expressed in non-monocyte derived macrophages in healthy status.31,32 This is partially built on the findings from recent studies that challenged the long-believed concept of the resident intestinal macrophages being fully dependent on replenishment by recruitment and differentiation of circulating monocytes.31,33
De Schepper et al identified a population of self-renewing resident intestinal macrophages and additionally demonstrated that they were predominantly located in the submucosa and muscularis externa regions.31 Four subpopulations of the self-renewing macrophages with distinct anatomical localisation within the bowel were identified. Interestingly, ADAMDEC1 was identified as one of the marker genes for the second most abundant self-renewing macrophage subpopulation of the lamina propria and submucosa. These findings provide evidence that the expression of ADAMDEC1 is not limited to the macrophages of monocyte origin. Gene expression profiling of the ADAMDEC1+ self-renewing macrophage subpopulation revealed that they were enriched for vascular and immune response associated genes. In situ hybridisation further demonstrated a close proximity of these cells to the vasculature. In contrast, the ADAMDEC1− self-renewing macrophage subpopulation was enriched for the gene signature of neuron development and they were localised in close proximity with myenteric neuronal ganglia within the muscularis externa. Moreover, an ablation of the ADAMDEC1+ self-renewing macrophage subpopulation resulted in disruption of the vasculature with an increase in vascular permeability suggesting a role in support and maintenance of the local blood vessels. A precise role for ADAMDEC1 in vascular regulation has not been described, but Baran et al identified ADAMDEC1 expression within the placenta at mid-pregnancy, specifically at the foeto-maternal junction and around maternal vessels, with an increase in expression that mirrored angiogenesis.34 The expression of ADAMDEC1 is, however, unlikely to be critical for foetus development, as the Adamdec1−/- mice are fertile, born in expected Mendelian ratios and displayed no abnormal developmental phenotype or growth impairment.18 Finally, a recent study by Kinchen et al characterised a subpopulation of human colonic mesenchymal cells that expressed ADAMDEC1.32 These cells were the most abundant mesenchymal population in the colon and located throughout the lamina propria.
In summary, we now have evidence to show that the expression of ADAMDEC1 in the gastrointestinal tract is distributed across a number of cell types (Figure 4). The function of this unique metalloprotease is still proving elusive; however, its association with a number of diseases may provide some valuable information.
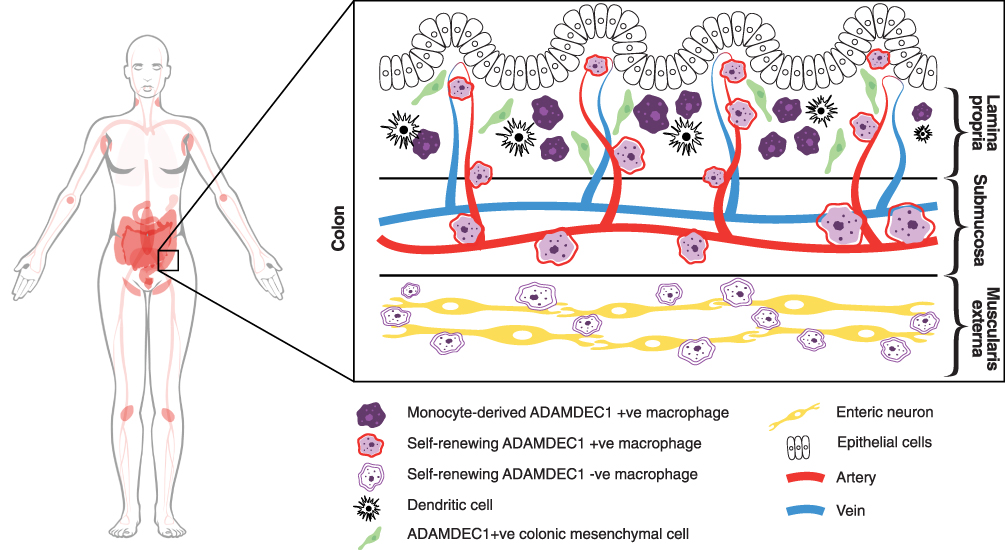 |
Figure 4 ADAMDEC1 gene expression in human tissue and organs is restricted to the lower gastrointestinal tract, lymph nodes, tonsils, spleen, urinary bladder and placenta (coloured in pink). (Image adapted from Human Protein Atlas available from http://www.proteinatlas.org/ENSG00000134028-ADAMDEC1/tissue).15 At the cellular level within the colon ADAMDEC1 expression has been identified in a number of macrophage populations and a subpopulation of colonic mesenchymal cells. |
Induction of ADAMDEC1 in CD40-Activated Dendritic Cells
Within the spleen and tonsils, the expression of ADAMDEC1 is believed to be in the CD40-activated dendritic cells. This finding is consistent with in vitro studies demonstrating the induction of ADAMDEC1 expression in the granulocyte-macrophage colony-stimulating factor (GMCSF) and IL-4 driven-monocyte-derived dendritic cells only upon further stimulation with LPS or CD40, not by simply differentiating them into dendritic cells.1 Studies focusing on the mechanism of its induction as well as the functional role of ADAMDEC1 in the dendritic cells are limited.
ADAMDEC1 and Inflammatory Diseases
Recent extensive usage of microarray technology and next-generation sequencing has allowed hypothesis-free, genome-wide transcriptomic analysis of many human diseases. This approach led to the identification of ADAMDEC1 as a differentially expressed transcript in various inflammatory diseases.
Gastrointestinal Tract
Crohn’s Disease
The association between ADAMDEC1 and Crohn’s disease was first reported in 2014 by de Bruyn et al.35 The study involved whole genome transcriptomic analysis of intestinal tissue and identified ADAMDEC1 to be a significantly downregulated gene in the inflamed ileal biopsy samples taken from patients with Crohn’s disease compared to healthy controls. Importantly, the low expression of ADAMDEC1 in the inflamed ileal tissue from the Crohn’s disease cohort was unaffected after the patients received Infliximab, anti-TNF-α antibody therapy. The lack of responsiveness of ADAMDEC1 expression was in contrast to other MMPs, TIMPs and ADAMs as the dysregulated expression levels of these proteins pre-Infliximab therapy were restored in the patients who responded to the therapy mirroring the resolution of inflammation. This suggests that the lower expression of ADAMDEC1 in the ileum of Crohn’s disease is independent of the degree of mucosal inflammation and thus might have an underlying role in the pathogenesis of the disease.
A further link between ADAMDEC1 and Crohn’s disease was reported in 2015 by Smith et al who demonstrated that MDMs generated from patients with Crohn’s disease displayed lower ADAMDEC1 expression compared to healthy controls.36 Furthermore, when these MDMs were stimulated with heat-killed Escherichia coli, the Crohn’s disease MDMs secreted reduced level of TNF-α and IFN-γ compared to the healthy control MDMs. Although the pathophysiology of Crohn’s disease is yet to be fully elucidated, one of the hypotheses is an inability of the innate immune system to elicit an effective inflammatory response at a time of mucosal barrier breakdown.37 This results in a defective initial clearance of invading bacteria and subsequent inappropriate activation of the adaptive immune response leading to the development chronic inflammation.37,38 The reduced level of ADAMDEC1 in the Crohn’s disease MDMs might have a role in the attenuated levels of TNF-α and IFN-γ secretion by these MDMs and contribute to the disease pathogenesis (Figure 5).
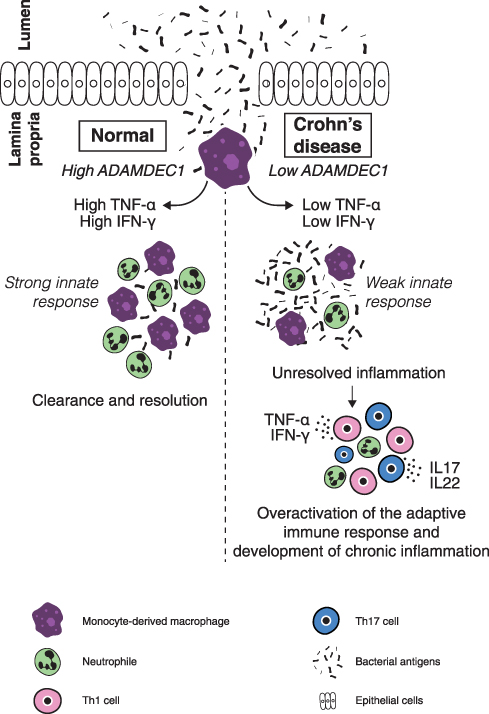 |
Figure 5 A possible mechanism through which a reduced expression of ADAMDEC1 in the monocyte-derived macrophages in the lamina propria could lead to chronic mucosal inflammation of Crohn’s disease.Notes: Data from Segal.37 |
The generation of a transgenic ADAMDEC1 knockout mouse (Adamdec1−/-) finally provided evidence to support the role of ADAMDEC1 in gastrointestinal immune regulation and how the loss in the gene expression resulted in the development of intestinal mucosal inflammation.18 It is worth noting that despite the high expression of ADAMDEC1 in the gastrointestinal tract, Adamdec1−/- mice did not exhibit any abnormality with regard to the development, growth, reproductivity and intestinal mucosal permeability and did not exhibit any signs of gastrointestinal pathologies under specific pathogen-free conditions. This suggests that the function of ADAMDEC1 is dispensable or substituted by an alternative mechanism under normal healthy status. However, when colonic inflammation was induced by oral administration of dextran sodium sulphate (DSS) over 7 days, Adamdec1−/- mice demonstrated more severe systemic sequala of inflammation with a greater loss of weight and a higher mortality rate compared to the wild type (WT) mice. The histological examination of the colonic tissue after the DSS administration also confirmed a greater degree of local mucosal inflammation in the Adamdec1−/- colonic tissue compared to the WT with increased cellular infiltration, crypt distortion and ulceration. Furthermore, Adamdec1−/- mice showed even greater degree of weight loss and mortality compared to the WT when challenged with live Citrobacter rodentium, which usually causes a self-limiting bacterial colitis predominantly affecting the caecum in WT animals. A similar exaggerated inflammatory response was also seen when Adamdec1−/- mice were challenged with Salmonella typhimurium, a pathogen that affects the distal portion of the ileum. These findings suggest that the exaggerated mucosal inflammation in the absence of ADAMDEC1 occurs irrespective of the mode of inflammation or the location along the small and large intestine. In the case of Citrobacter rodentium infection, it was also noted that a greater number of the bacteria were found in the spleen of the Adamdec1−/- mice compared to the WT indicating more severe breakdown of the mucosal barrier and increased bacteraemia in the absence of ADAMDEC1. This study also showed a significant upregulation of ADAMDEC1 in the colonic tissue of the WT mice during the DSS challenge and a return to the baseline as the mucosal inflammation resolved. The change in expression may result from the increased recruitment of the circulating monocytes into the inflamed tissue or an up-regulation in the expression in the tissue resident cells. These findings clearly demonstrated that the loss in ADAMDEC1 has a direct effect on the degree of mucosal inflammation in vivo and how individuals who express reduced levels may be more susceptible to developing Crohn’s disease.
Extra-Intestinal System
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a systemic chronic progressive immune-mediated condition of unknown cause that primarily affects joints leading to synovial hypertrophy and joint destruction.39 Osteoarthritis (OA), on the other hand, is a chronic degenerative inflammatory arthritis caused by abnormal tissue remodelling triggered by repetitive stress or joint injury.40 Although both of these diseases share a common eventual clinical manifestation with joint inflammation and destruction, the pathogenesis of RA and OA are distinctively different.
An association between ADAMDEC1 and RA was first reported in 2007 by Galligan et al who performed whole genome expression profiling of fibroblast-like synovial cells isolated from joint tissue taken from patients with an erosive end-stage RA undergoing joint replacement.41 ADAMDEC1 was the most upregulated gene, by 23.8 folds, in the samples taken from the RA patients compared to the non-RA patients undergoing joint replacement due to trauma. This upregulation was independent of disease activity, suggesting ADAMDEC1 might potentially have a mechanistic role in the disease pathogenesis rather than simply being a marker of circulating monocyte recruitment to the joint, or degree of inflammation. The expression of ADAMDEC1 was also upregulated in the samples taken from patients with end-stage OA compared to those from non-RA, non-OA trauma patients; however, this was 19-fold lower than that observed in the RA cohort. In this study by Galligan et al, the fibroblast-like synovial cells were cultured and expanded in vitro prior to RNA extraction. This step was shown to significantly alter gene expression, and it is not clear if they are a true representation of the tissue resident cells or to what extent the culturing influenced the results. Nevertheless, this study adds further proof that ADAMDEC1 can be expressed in cells other than macrophages. Evidence for the association between ADAMDEC1 and RA was also provided by Li et al who analysed three independent transcriptional datasets generated from the synovial tissue, rather than isolated cells, collected from patients with RA and OA.42 ADAMDEC1 was one of the 13 genes identified to demonstrate a significant change in the expression across all three datasets. In all cases the ADAMDEC1 expression was upregulated in RA compared to OA.
With regards to OA, two genome-wide expression studies which profiled biopsies from the subchondral bone and articular cartilage did not identify ADAMDEC1 as a differentially expressed gene compared with healthy controls.43,44 A comprehensive proteomic analysis of synovial fluid collected from patients with OA identified 677 proteins of which 545 proteins had not been reported previously, including ADAMDEC1.45 This study, however, did not use any comparison cohorts, such as synovial fluid collected from healthy controls or patients with RA; thus, it is impossible to conclude that the presence of ADAMDEC1 was unique to the synovial fluid of OA.
Current available evidence suggests that ADAMDEC1 is upregulated in the affected joints of RA and potentially has a mechanistic role, which does not seem to be the case in OA. This discrepancy of ADAMDEC1 expression in these two conditions, that both cause inflammation of joints, suggests that the induction of ADAMDEC1 in inflammatory diseases is not simply secondary to the recruitment and differentiation of circulating monocytes to macrophages. To date, there have not been any studies attempting to determine the potential functional role of ADAMDEC1 in RA.
Carotid Artery Atherosclerosis
Atherosclerosis begins with an accumulation of lipids in the intimal layer of the artery followed by an upregulation of adhesion molecules by the endothelial and smooth muscle cells, leading to recruitment of monocytes and T lymphocytes.46 MDMs upregulate scavenger receptors in response to the local MCSF and develop into foam cells, releasing inflammatory cytokines that stimulate proliferation and migration of the smooth muscle cells from the arterial media to the intima, driving formation of atherosclerotic plaques.47–49 These plaques can become unstable and rupture due to the proteolytic action of MMPs produced by the foam cells. This rupturing of the plaque causes platelet aggregation and stenosis of the artery which leads to ischaemia of tissues that manifests as clinical morbidity and mortality.
Papaspyridonos et al conducted whole transcriptome analysis of the unstable parts of atherosclerotic plaque taken from 27 patients who underwent carotid endarterectomy.50 In comparison with the stable regions within the same specimens, ADAMDEC1 was significantly upregulated, by 37 folds, along with 36 other upregulated genes. The analysis also showed an increase in CD68, a gene expression marker for macrophages; however, no statistical correlation between the increased expression of ADAMDEC1 was seen. Verdugo et al performed a large transcriptome analysis of circulating monocytes taken from 936 individuals who underwent common carotid artery assessment for the presence of atherosclerotic plaques.51 The expression of ADAMDEC1 in the circulating monocytes was found to have the third strongest positive association with the presence of atherosclerotic plaques in the common carotid artery.
These findings suggest a potential mechanistic role for ADAMDEC1 in the development of atherosclerosis and plaque instability. ADAMDEC1 may also have the potential to be a diagnostic marker for atherosclerosis.
Rosacea
Rosacea is a chronic inflammatory skin disease in which an upregulation of toll-like receptor-2 (TLR2) and an increased secretion of antimicrobial peptides such as cathelicidin lead to the subsequent secretion of proinflammatory cytokines and activation of the T helper 1 and T helper 17 adaptive immune response.52
A recent study has reported a potential mechanistic role of ADAMDEC1 in rosacea.28 ADAMDEC1 expression at both gene and protein levels in skin samples taken from 35 patients with rosacea was increased compared to 14 healthy controls. By immunohistochemistry, ADAMDEC1 was detected in the epidermis and was co-localised with macrophages. The authors went to performed rosacea-like mouse models and conducted a series of in vitro studies after which they concluded that ADAMDEC1 plays a pro-inflammatory role in rosacea via modulating the pro-inflammatory “M1” polarization of macrophages. The molecular mechanism for the modulation of M1 polarization by ADAMDEC1 in the skin remains to be determined and whether this applies to other tissue is still unclear.
ADAMDEC1 and Cancer
Gastrointestinal Tract
Colorectal Adenocarcinoma
Adenoma is considered to be a precursor lesion for the vast majority (approximately 95%) of colorectal adenocarcinoma (CRC), although less than 10% of adenoma are believed to undergo malignant transition.53,54 Metastasis, most commonly to the liver, accounts for the main mode of death in CRC.55,56 Thus, understanding the factors driving the development of adenoma, its malignant transition, tumourigenesis and metastatic progression is crucial in our search for treatments. The normally high expression of ADAMDEC1 along the healthy lower gastrointestinal tract was found to be drastically reduced in the colorectal adenoma compared to the normal colorectal tissue (GEO data repository GEO ID GSE100179) (Figure 6).57 The loss in ADAMDEC1 expression in the colorectal adenoma did not correlate with the loss in CD68 expression, which suggests that the tissue macrophages are still present although ADAMDEC1 is no longer expressed (Figure 6). Galamb et al also reported a reduction in the expression of ADAMDEC1 in the adenoma mucosa, as well as in the mucosal tissue biopsies taken from CRC compared to the non-matching, normal colorectal tissue biopsies taken from healthy controls.58 These findings suggest the involvement of ADAMDEC1 in the development of colorectal adenoma.
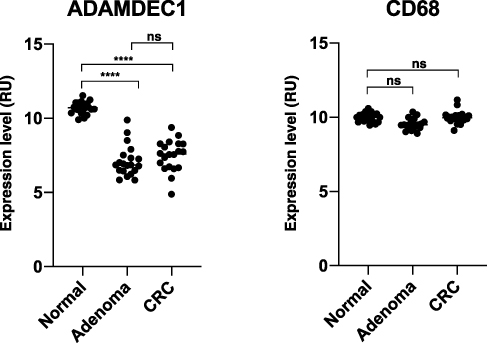 |
Figure 6 ADAMDEC1 and CD68 gene expression in normal colorectal tissue, colorectal adenoma and CRC. A highly significant reduction in the level of ADAMDEC1 expression is seen in the colorectal adenoma and CRC compared to the normal colorectal tissue. The CD68 expressions are comparable between the three different colorectal tissues suggesting that the number of intestinal macrophages remains unchanged. ****p<0.0001. Data extracted from Gene Expression Omnibus (GEO) data repository: GEO ID GSE 100179.57 |
Loss of heterozygosity (LOH) on chromosome 8p, where the locus of ADAMDEC1 is positioned, is well established in CRC. A study by Oh et al showed that the deletion was observed specifically in the region of 8p21-23 in both primary and metastatic adenocarcinoma tissue and demonstrated that the gene expression of ADAMDEC1 as well as all 11 other genes at this locus were reduced in both primary and metastatic tissues.59 Another study by Macartney-Coxson et al, using a larger sample population, reported a high frequency of LOH occurring at 8p21-22 in the metastatic liver tissue, which was not present in the primary tumour.60 The expression of ADAMDEC1 at the gene level was shown to be reduced in both the primary and matched secondary tumour sites, however, with a greater reduction seen in the metastatic tissue. In both studies, the gene expression of ADAMDEC1 was normalised to the normal colon, rather than the primary tumour tissues without metastasis. Thus, it is not certain whether the reduction of ADAMDEC1 is specific to the primary tumours with metastatic capability or to the primary tumour in general. Nevertheless, these findings suggest a role for ADAMDEC1 in the tumourigenesis and metastatic progression of CRC.
One possible mechanism responsible for the link between the adenocarcinoma formation and the loss in ADAMDEC1 expression could be its reported role in the EDAC response (Figure 7).14 The non-catalytic activity of ADAMDEC1 that facilitates the extrusion of transformed epithelial cells from a monolayer may be a crucial anti-cancer defence mechanism within the lower gastrointestinal tract. A loss in ADAMDEC1 expression as seen in colorectal adenoma could facilitate the survival of the transformed cells within the epithelium, allowing these cells to proliferate and acquire further oncogenic mutations. The role of ADAMDEC1 in the EDAC process is still not fully understood; however, in a study by Yako et al using MDCK cells, a knockdown of ADAMDEC1 in the normal epithelial cells resulted in the failure to accumulate filamin at the cell boundary between the transformed and surrounding normal epithelial cells, which suppressed apical extrusion.14 To date, however, ADAMDEC1 has not been reported to be expressed in normal human colonic epithelial cells.18,61 It might be possible that a loss of ADAMDEC1 expression in the macrophages within the lamina propria could lead to the same result of impaired EDAC (Figure 7). The relevance of EDAC within the gastrointestinal tract and a precise role for ADAMDEC1 in CRC development, nevertheless, are still unclear and further studies are warranted.
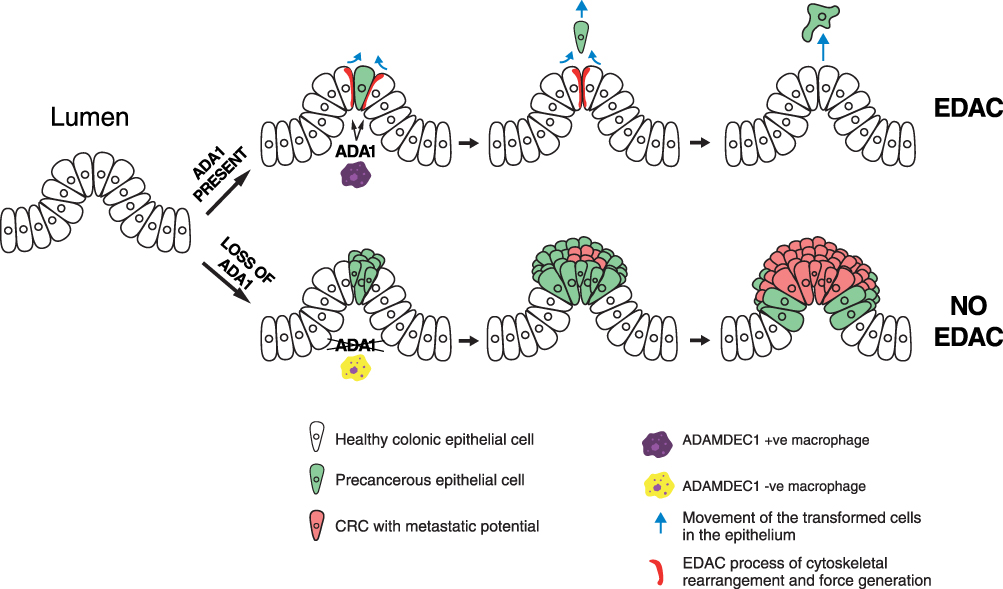 |
Figure 7 A possible mechanism in which a loss of ADAMDEC1 facilitates the development of colorectal adenoma and CRC by hindering EDAC. |
Gastric Adenocarcinoma
The expression of ADAMDEC1 in the stomach is much lower than in the lower gastrointestinal tract under healthy status; however, a reduction in its expression in gastric adenocarcinomas was shown to be significantly associated with poor prognosis.62 Poor prognosis was associated with a dampening of the inflammatory response in the tumour microenvironment.
Extra-Intestinal System
Glioblastoma
Glioblastoma (GBM) is the most common and also aggressive malignant lesion of the brain. The cellular origin of GBM is in fact unknown; however, neural stem cells, neural stem cell-derived astrocytes and glial progenitors are all believed to be the potential precursor cell.63 GBM’s aggressive nature, high recurrence rate and poor response to treatment are thought to be due to the resistant GBM cancer stem cells.
The association between ADAMDEC1 and GBM was first reported in a transcriptomic profiling study conducted by Vauléon et al focusing on the expression of immune-associated genes in the combined dataset of 101 specimens of GBM from published data.64 ADAMDEC1 was identified as one of the 108 immune-associated genes which were directly associated with overall survival in the patients with GBM.64 The involvement of FGF2, a reported substrate of ADAMDEC1, in promoting the proliferation of GBM cancer stem cells, tumour growth and vascularisation is well established although the mechanism underlying these effects remained uncertain.65,66 An intriguing study conducted by Jimenez-Pascual et al recently identified ADAMDEC1 as one of the top upregulated ADAM genes in GBM and showed that ADAMDEC1 was the only ADAM where its upregulation correlated with poor outcome.12 The study went on to show that ADAMDEC1 rapidly solubilized the membrane-bound FGF2 to stimulate FGF receptor-1 (FGFR1) expressed on the GBM stem cells. The FGFR1 signalling upregulated the transcription factor Zinc finger E-box-binding homeobox 1 (ZEB1), which induced ADAMDEC1 expression through the inhibition of the tumour-suppressor microRNA, miR-203, creating a positive feedback loop. The genetic or pharmacologic targeting of components of this pathway attenuated self-renewal and tumour growth. This study has unveiled a new pathological role for ADAMDEC1 and highlighted the potential of ADAMDEC1 as a therapeutic target.
Craniopharyngioma
Craniopharyngioma is a benign tumour which is believed to arise from squamous epithelial remnants of Rathke’s pouch.67 Although benign, it is a locally invasive tumour with a high recurrence rate and causes significant morbidity due to its space-occupying effect on the adjacent structures such as hypothalamus, pituitary gland and optic nerve, chiasm or tract. A study by Xu et al reported that ADAMDEC1 was present in the craniopharyngioma tissues resected from 4 patients; however, its expression was not detected in the normal brain tissues at either the gene or protein levels.68 By using the primary cell cultures prepared from the resected craniopharyngioma tissues, the authors went on to show that co-culturing the cells with tamoxifen reduced the tumour cell proliferation and also the expression of ADAMDEC1 at gene and protein level, thus suggesting that ADAMDEC1 might play a role in the tumour cell proliferation. This study adds further evidence to the potential tumour-promoting role of ADAMDEC1 in the development of non-gastrointestinal tumour.
Oral Squamous Cell Carcinoma
A potential mechanistic role for ADAMDEC1 in the development of oral squamous cell carcinoma (OSCC) has been described in two studies by Chen at al11,69 An overexpression of EGF-receptor (EGFR) is seen in approximately 90% of OSCC, which is believed to drive an aggressive phenotype as well as contributing to a poor response to radiotherapy.70–72 The thrombin-stimulated platelets were shown to secrete ADAMDEC1, resulting in the cleavage of the platelet membrane-bound pro-EGF. The soluble EGF then resulted in a migratory and invasive phenotypic shift in the ORCC via EGFR signalling.11,69 Although the authors focused on the thrombin-stimulated platelets within ORCC as the source of ADAMDEC1, various cell types present within the tumour could also be responsible, including macrophages.
Conclusion
ADAMDEC1 is an intriguing and idiosyncratic ADAM protein with many unique characteristics. Although its functional role is still far from fully resolved, evidence is emerging to support a potential involvement in regulation of the inflammatory process within the intestine and in tumourigenesis.
The initial studies showed that ADAMDEC1 was expressed in the macrophages and dendritic cells and suggested that the constitutively expressed high levels of ADAMDEC1 in the lower intestinal tract was due to the monocytic migration and differentiation into macrophages in the lamina propria. More recent evidence, however, identifies ADAMDEC1 in the self-renewing embryonic-derived macrophages, as well as the non-macrophage populations such as mesenchymal cells in the healthy gastrointestinal tract.
Within the intestinal tissue of Crohn’s disease, a chronic inflammatory bowel disease, the normally high expression of ADAMDEC1 is reduced which seems to have a mechanistic role in the disease pathogenesis. This is supported by the in vivo study demonstrating the loss of ADAMDEC1 leads to an exaggerated mucosal inflammation. The association between ADAMDEC1 and the vascular development and maintenance has also started to become apparent. On the other hand, outside of the gastrointestinal tract, where ADAMDEC1 is not normally expressed, an upregulation of ADAMDEC1 is observed in a selection of inflammatory conditions. The studies exploring the mechanistic role of ADAMDEC1 in these extra-intestinal inflammatory diseases are, however, still limited.
Finally, the potential role of ADAMDEC1 in the development and regulation of cancer is beginning to emerge, which once again seems to differ depending on the tissue affected. An anti-cancer role appears to predominate within the gastrointestinal tract, with a reduction in the gene expression, whereas a pro-oncogenic function occurs in the extra-intestinal tumours, with an increase in the gene expression.
In summary, these findings suggest the possibility of ADAMDEC1 to possess numerous functions within a wide range of physiological processes and disease states. In the majority of cases, these links have been forged through the use of hypothesis-free genome-wide expression profiling and very little is known about the relevance of the association or the mechanisms involved. The exceptions to this are the studies in Crohn’s disease and GBM, using an animal model and patient-derived cancer cells, respectively. It is clear that more studies focusing on the mechanistic involvement of ADAMDEC1 in disease pathogenesis are needed in order to take steps towards unravelling the function of this fascinating member of the ADAMs family.
Abbreviations
ADAMs, A disintegrin and metalloproteases; ADAMDEC1, A disintegrin and metalloprotease like decysin 1; CRC, colorectal cancer; DSS, dextran sodium sulphate; EDAC, epithelial defense against cancer; EGF, epidermal growth factor; RGFR, epidermal growth factor receptor; FGF2, fibroblast growth factor 2; GBM, glioblastoma; GMCSF, granulocyte-macrophage colony-stimulating factor; LOH, loss of heterozygosity; LPS, lipopolysaccharide; MCSF, macrophage colony-stimulating factor; MDCK, Madin-Darby Canine Kidney; MDMs, monocyte-derived macrophages; OA, osteoarthritis; OSCC, oral squamous cell carcinoma; RA, rheumatoid arthritis; SIRT1/2, sirtuins 1 and 2; TIMPs, tissue inhibitors of metalloproteases; TLR2, toll-like receptor 2; WT, wild type; ZEB1, zinc finger E-box-binding homeobox 1.
Acknowledgments
Simone Nunziato kindly created the graphical illustrations and Dr Claire Mulvey (Cancer Research UK Cambridge Institute, University of Cambridge, UK) for critical evaluation of the manuscript.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Mueller CG, Rissoan MC, Salinas B, et al. Polymerase chain reaction selects a novel disintegrin proteinase from CD40-activated germinal center dendritic cells. J Exp Med. 1997;186(5):655–663. doi:10.1084/jem.186.5.655
2. Mueller CGF, Cremer I, Paulet PE, et al. Mannose receptor ligand-positive cells express the metalloprotease decysin in the B cell follicle. J Immunol. 2001;167(9):5052–5060. doi:10.4049/jimmunol.167.9.5052
3. Edwards D, Handsley M, Penington C. The ADAM metalloproteinases. Mol Aspects Med. 2008;29(5):258–289. doi:10.1016/j.mam.2008.08.001
4. Häse CC, Finkelstein RA. Bacterial extracellular zinc-containing metalloproteases. Microbiol Rev. 1993;57(4):823–837. doi:10.1128/MMBR.57.4.823-837.1993
5. Pflughoeft KJ, Swick MC, Engler DA, Yeo H-J, Koehler TM. Modulation of the Bacillus anthracis secretome by the immune inhibitor A1 protease. J Bacteriol. 2014;196(2):424–435. doi:10.1128/JB.00690-13
6. Lund J, Olsen OH, Sørensen ES, Stennicke HR, Petersen HH, Overgaard MT. ADAMDEC1 is a metzincin metalloprotease with dampened proteolytic activity. J Biol Chem. 2013;288(29):21367–21375. doi:10.1074/jbc.M113.474536
7. Lund J, Troeberg L, Kjeldal H, et al. Evidence for restricted reactivity of ADAMDEC1 with protein substrates and endogenous inhibitors. J Biol Chem. 2015;290(10):6620–6629. doi:10.1074/jbc.M114.601724
8. Bates E, Fridman W, Mueller C. The ADAMDEC1 (decysin) gene structure: evolution by duplication in a metalloprotease gene cluster on Chromosome 8p12. Immunogenetics. 2002;54(2):96–105. doi:10.1007/s00251-002-0430-3
9. Bahudhanapati H, Bhattacharya S, Wei S. Evolution of vertebrate adam genes; Duplication of testicular adams from ancient Adam9/9-like Loci. PLoS One. 2015;10(8):e0136281. doi:10.1371/journal.pone.0136281
10. Wei S, Whittaker CA, Xu G, et al. Conservation and divergence of ADAM family proteins in the Xenopus genome. BMC Evol Biol. 2010;10(1):211. doi:10.1186/1471-2148-10-211
11. Chen R, Jin G, McIntyre TM. The soluble protease ADAMDEC1 released from activated platelets hydrolyzes platelet membrane pro-epidermal growth factor (EGF) to active high-molecular-weight EGF. J Biol Chem. 2017;292(24):10112–10122. doi:10.1074/jbc.M116.771642
12. Jimenez-Pascual A, Hale JS, Kordowski A, et al. ADAMDEC1 maintains a growth factor signaling loop in cancer stem cells. Cancer Discov. 2019;9(11):1574–1589. doi:10.1158/2159-8290.CD-18-1308
13. Houchen CW, George RJ, Sturmoski MA, Cohn SM. FGF-2 enhances intestinal stem cell survival and its expression is induced after radiation injury. Am J Physiol - Gastrointest Liver Physiol. 1999;276(1):
14. Yako Y, Hayashi T, Takeuchi Y, et al. ADAM-like Decysin-1 (ADAMDEC1) is a positive regulator of Epithelial Defense Against Cancer (EDAC) that promotes apical extrusion of RasV12-transformed cells. Sci Rep. 2018;8(1):9639. doi:10.1038/s41598-018-27469-z
15. Human Protein Atlas. Man. Available from: https://www.proteinatlas.org/ENSG00000134028-ADAMDEC1/tissue.
16. Wu C, Orozco C, Boyer J, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10(11):1–8. doi:10.1186/gb-2009-10-11-r130
17. Fritsche J, Müller A, Hausmann M, Rogler G, Andreesen R, Kreutz M. Inverse regulation of the ADAM-family members, decysin and MADDAM/ADAM19 during monocyte differentiation. Immunology. 2003;110(4):450–457. doi:10.1111/j.1365-2567.2003.01754.x
18. O’Shea NR, Chew TS, Dunne J, et al. Critical role of the disintegrin metalloprotease ADAM-like decysin-1 [ADAMDEC1] for intestinal immunity and inflammation. J Crohn’s Colitis. 2016;10(12):1417–1427. doi:10.1093/ecco-jcc/jjw111
19. Bain CC, Bravo-Blas A, Scott CL, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. 2014;15(10):929–937. doi:10.1038/ni.2967
20. Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8(10):1086–1094. doi:10.1038/ni1511
21. Li T, Garcia-Gomez A, Morante-Palacios O, et al. SIRT1/2 orchestrate acquisition of DNA methylation and loss of histone H3 activating marks to prevent premature activation of inflammatory genes in macrophages. Nucleic Acids Res. 2020;48(2):665–681. doi:10.1093/nar/gkz1127
22. Ravin HA, Rowley D, Jenkins C, Fine J. On the absorption of bacterial endotoxin from the gastro-intestinal tract of the normal and shocked animal. J Exp Med. 1960;112:783–792. doi:10.1084/jem.112.5.783
23. Brochériou I, Maouche S, Durand H, et al. Antagonistic regulation of macrophage phenotype by M-CSF and GM-CSF: implication in atherosclerosis. Atherosclerosis. 2011;214(2):316–324. doi:10.1016/j.atherosclerosis.2010.11.023
24. Bazzi S, El-Darzi E, McDowell T, et al. Defining genome-wide expression and phenotypic contextual cues in macrophages generated by granulocyte/macrophage colony-stimulating factor, macrophage colony-stimulating factor, and heat-killed mycobacteria. Front Immunol. 2017;8(OCT). doi:10.3389/fimmu.2017.01253
25. Becker M, De Bastiani MA, Parisi MM, et al. Integrated transcriptomics establish macrophage polarization signatures and have potential applications for clinical health and disease. Sci Rep. 2015;5. doi:10.1038/srep13351
26. Derlindati E, Cas AD, Montanini B, et al. Transcriptomic analysis of human polarized macrophages: more than one role of alternative activation? PLoS One. 2015;10(3):e0119751. doi:10.1371/journal.pone.0119751
27. Hans CP, Sharma N, Sen S, et al. Transcriptomics analysis reveals new insights into the roles of notch1 signaling on macrophage polarization. Sci Rep. 2019;9(1):1–21. doi:10.1038/s41598-019-44266-4
28. Liu T, Deng Z, Xie H, et al. ADAMDEC1 promotes skin inflammation in rosacea via modulating the polarization of M1 macrophages. Biochem Biophys Res Commun. 2020;521(1):64–71. doi:10.1016/j.bbrc.2019.10.073
29. Solinas G, Schiarea S, Liguori M, et al. Tumor-conditioned macrophages secrete migration-stimulating factor: a new marker for M2-polarization, influencing tumor cell motility. J Immunol. 2010;185(1):642–652. doi:10.4049/jimmunol.1000413
30. Xue J, Schmidt SV, Sander J, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40(2):274–288. doi:10.1016/j.immuni.2014.01.006
31. De Schepper S, Verheijden S, Aguilera-Lizarraga J, et al. Self-maintaining gut macrophages are essential for intestinal homeostasis. Cell. 2018;175(2):400–415.e13. doi:10.1016/j.cell.2018.07.048
32. Kinchen J, Chen HH, Parikh K, et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell. 2018;175(2):372–386. doi:10.1016/j.cell.2018.08.067
33. Shaw TN, Houston SA, Wemyss K, et al. Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J Exp Med. 2018;215(6):1507–1518. doi:10.1084/jem.20180019
34. Baran N, Kelly PA, Binart N. Decysin, a new member of the metalloproteinase family, is regulated by prolactin and steroids during mouse pregnancy. Biol Reprod. 2003;68(5):1787–1792. doi:10.1095/biolreprod.102.009761
35. de Bruyn M, Machiels K, Vandooren J, et al. Infliximab restores the dysfunctional matrix remodeling protein and growth factor gene expression in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2014;20(2):339–352. doi:10.1097/01.MIB.0000438430.15553.90
36. Smith AM, Sewell GW, Levine AP, et al. Disruption of macrophage pro-inflammatory cytokine release in Crohn’s disease is associated with reduced optineurin expression in a subset of patients. Immunology. 2015;144(1):45–55. doi:10.1111/imm.12338
37. Segal AW. Studies on patients establish Crohn’s disease as a manifestation of impaired innate immunity. J Intern Med. 2019;286(4):373–388. doi:10.1111/joim.12945
38. Marks DJB, Harbord MWN, MacAllister R, et al. Defective acute inflammation in Crohn’s disease: a clinical investigation. Lancet. 2006;367(9511):668–678. doi:10.1016/S0140-6736(06)68265-2
39. Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018;6(1). doi:10.1038/s41413-018-0016-9
40. Man GS, Mologhianu G. Osteoarthritis pathogenesis – a complex process that involves the entire joint. J Med Life. 2014;7(1):37–41.
41. Galligan CL, Baig E, Bykerk V, Keystone EC, Fish EN. Distinctive gene expression signatures in rheumatoid arthritis synovial tissue fibroblast cells: correlates with disease activity. Genes Immun. 2007;8(6):480–491. doi:10.1038/sj.gene.6364400
42. Li WC, Bai DL, Xu Y, et al. Identification of differentially expressed genes in synovial tissue of rheumatoid arthritis and osteoarthritis in patients. J Cell Biochem. 2019;120(3):4533–4544. doi:10.1002/jcb.27741
43. Karlsson C, Dehne T, Lindahl A, et al. Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthr Cartil. 2010;18(4):581–592. doi:10.1016/j.joca.2009.12.002
44. Sun J, Yan B, Yin W, Zhang X. Identification of genes associated with osteoarthritis by microarray analysis. Mol Med Rep. 2015;12(4):5211–5216. doi:10.3892/mmr.2015.4048
45. Balakrishnan L, Nirujogi RS, Ahmad S, et al. Proteomic analysis of human osteoarthritis synovial fluid. Clin Proteomics. 2014;11(1):6. doi:10.1186/1559-0275-11-6
46. Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012;111(2):245–259. doi:10.1161/CIRCRESAHA.111.261388
47. Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med. 2014;46(6). doi:10.1038/emm.2014.38
48. Clinton SK, Underwood R, Hayes L, Sherman ML, Kufe DW, Libby P. Macrophage colony-stimulating factor gene expression in vascular cells and in experimental and human atherosclerosis. Am J Pathol. 1992;140(2):301–316.
49. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118(4):692–702. doi:10.1161/CIRCRESAHA.115.306361
50. Papaspyridonos M, Smith A, Burnand KG, et al. Novel candidate genes in unstable areas of human atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2006;26(8):1837–1844. doi:10.1161/01.ATV.0000229695.68416.76
51. Verdugo RA, Zeller T, Rotival M, et al. Graphical modeling of gene expression in monocytes suggests molecular mechanisms explaining increased atherosclerosis in smokers. PLoS One. 2013;8(1):e50888. doi:10.1371/journal.pone.0050888
52. Yamasaki K, Gallo RL. Rosacea as a disease of cathelicidins and skin innate immunity. J Investig Dermatol Symp Proc. 2011;15:12–15. doi:10.1038/jidsymp.2011.4
53. Bond JH. Polyp guideline: diagnosis, treatment, and surveillance for patients with colorectal polyps. Am J Gastroenterol. 2000;95(11):3053–3063. doi:10.1111/j.1572-0241.2000.03434.x
54. Shinya H, Wolff WI. Morphology, anatomic distribution and cancer potential of colonic polyps. An analysis of 7,000 polyps endoscopically removed. Ann Surg. 1979;190(6):679–683. doi:10.1097/00000658-197912000-00001
55. Vatandoust S, Price TJ, Karapetis CS. Colorectal cancer: metastases to a single organ. World J Gastroenterol. 2015;21(41):11767–11776. doi:10.3748/wjg.v21.i41.11767
56. Riihimaki M, Hemminki A, Sundquist J, Hemminki K. Patterns of metastasis in colon and rectal cancer. Sci Rep. 2016;6:1–9. doi:10.1038/srep29765
57. Kalmár A, Nagy ZB, Galamb O, et al. Genome-wide expression profiling in colorectal cancer focusing on lncRNAs in the adenoma-carcinoma transition. BMC Cancer. 2019;19(1):1059. doi:10.1186/s12885-019-6180-5
58. Galamb O, Györffy B, Györffy G, et al. Inflammation, adenoma and cancer: objective classification of colon biopsy specimens with gene expression signature. Dis Markers. 2008;25:1–6. doi:10.1155/2008/586721
59. Oh BY, Cho J, Hong HK, et al. Exome and transcriptome sequencing identifies loss of PDLIM2 in metastatic colorectal cancers. Cancer Manag Res. 2017;9:581–589. doi:10.2147/CMAR.S149002
60. Macartney-Coxson DP, Hood KA, Shi H, et al. Metastatic susceptibility locus, an 8p hot-spot for tumour progression disrupted in colorectal liver metastases: 13 candidate genes examined at the DNA, mRNA and protein level. BMC Cancer. 2008;8(1):187. doi:10.1186/1471-2407-8-187
61. Sugimoto N, Nakayama T, Kasai Y, et al. The role of ADAM-like decysin 1 in non-eosinophilic chronic rhinosinusitis with nasal polyps. Acta Otolaryngol. 2018;138(9):830–836. doi:10.1080/00016489.2018.1481296
62. Pasini FS, Zilberstein B, Snitcovsky I, et al. A gene expression profile related to immune dampening in the tumor microenvironment is associated with poor prognosis in gastric adenocarcinoma. J Gastroenterol. 2014;49(11):1453–1466. doi:10.1007/s00535-013-0904-0
63. Yao M, Li S, Wu X, et al. Cellular origin of glioblastoma and its implication in precision therapy. Cell Mol Immunol. 2018;15(8):737–739. doi:10.1038/cmi.2017.159
64. Vauléon E, Tony A, Hamlat A, et al. Immune genes are associated with human glioblastoma pathology and patient survival. BMC Med Genomics. 2012;5:1–3. doi:10.1186/1755-8794-5-41
65. Kelly JJP, Stechishin O, Chojnacki A, et al. Proliferation of human glioblastoma stem cells occurs independently of exogenous mitogens. Stem Cells. 2009;27(8):1722–1733. doi:10.1002/stem.98
66. Haley EM, Kim Y. The role of basic fibroblast growth factor in glioblastoma multiforme and glioblastoma stem cells and in their in vitro culture. Cancer Lett. 2014;346(1):1–5. doi:10.1016/j.canlet.2013.12.003
67. Larkin SJ, Ansorge O. Pathology and pathogenesis of craniopharyngiomas. Pituitary. 2013;16(1):9–17. doi:10.1007/s11102-012-0418-4
68. Xu J, Liu L, Zheng X, You C, Li Q. Expression and inhibition of ADAMDEC1 in craniopharyngioma cells. Neurol Res. 2012;34(7):701–706. doi:10.1179/1743132812Y.0000000067
69. Chen R, Jin G, Li W, McIntyre TM. Epidermal growth factor (EGF) autocrine activation of human platelets promotes EGF receptor–dependent oral squamous cell carcinoma invasion, migration, and epithelial mesenchymal transition. J Immunol. 2018;201(7):2154–2164. doi:10.4049/jimmunol.1800124
70. Grandis JR, Tweardy,DJ. Elevated levels of transforming growth factor α and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53(15):3579–3584.
71. Grandis JR, Melhem MF, Gooding WE, et al. Levels of TGF-α and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998;90(11):824–832. doi:10.1093/jnci/90.11.824
72. Sheridan MT, O’Dwyer T, Seymour CB, Mothersill CE. Potential indicators of radiosensitivity in squamous cell carcinoma of the head and neck. Radiat Oncol Investig. 1997;5(4):180–186. doi:10.1002/(SICI)1520-6823(1997)5:4<180::AID-ROI3>3.0.CO;2-Up
 © 2020 The Author(s). This work is published by Dove Medical Press Limited, and licensed under a
Creative Commons Attribution License.
The full terms of the License are available at http://creativecommons.org/licenses/by/4.0/.
The license permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.
© 2020 The Author(s). This work is published by Dove Medical Press Limited, and licensed under a
Creative Commons Attribution License.
The full terms of the License are available at http://creativecommons.org/licenses/by/4.0/.
The license permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.