")
Back to Journals » International Medical Case Reports Journal » Volume 13
Adamantinomatous Craniopharyngioma in an Adult: A Case Report with NGS Analysis
Authors Jastania RA, Saeed M, Al-Khalidi H , AlQuthami K , Nageeti TH, Al-Allaf FA, Valerie K , Taher MM
Received 23 December 2019
Accepted for publication 25 March 2020
Published 21 April 2020 Volume 2020:13 Pages 123—137
DOI https://doi.org/10.2147/IMCRJ.S243405
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Raid A Jastania,1 Muhammad Saeed,2,3 Hisham Al-Khalidi,4 Khalid AlQuthami,5 Tahani H Nageeti,6 Faisal A Al-Allaf,7,8 Kristoffer Valerie,9 Mohiuddin M Taher7,8
1Department of Pathology, Faculty of Medicine, Umm-Al-Qura University, Makkah, Saudi Arabia; 2Department of Radiology, Faculty of Medicine, Umm-Al-Qura University, Makkah, Saudi Arabia; 3Department of Radiology, Al-Noor Specialty Hospital, Makkah, Saudi Arabia; 4Department of Pathology, King Saud University, Riyadh, Saudi Arabia; 5Division of Histopathology, Department of Laboratory Medicine and Blood Bank, Al-Noor Specialty Hospital, Makkah, Saudi Arabia; 6Department of Radiation Oncology, King Abdullah Medical City, Makkah, Saudi Arabia; 7Science and Technology Unit, Umm-Al-Qura University, Makkah, Saudi Arabia; 8Department of Medical Genetics, Faculty of Medicine, Umm-Al-Qura University, Makkah, Saudi Arabia; 9Department of Radiation Oncology and Massey Cancer Center, Virginia Commonwealth University, Richmond, VA, USA
Correspondence: Mohiuddin M Taher Email [email protected]
Purpose: Several recent studies have documented CTNNB1 and BRAF mutations which are mutually exclusive for adamantinomatous craniopharyngioma (ACP) and papillary craniopharyngioma (PCP) tumors. This discovery is helpful in the development of novel targeted therapies in successful clinical trials with BRAF mutations in PCP cases. However, no such targeted therapy is available yet for ACP. Here, we report novel mutations, which are not previously reported, in a case of an adult ACP using NGS analysis.
Results: Patient DNA was sequenced using Ion PI v3 chip on Ion Proton. A total of 16 variants were identified in this tumor by NGS analysis, out of which four were missense mutations, seven were synonymous mutations, and five were intronic variants. In CTNNB1 gene a known missense mutation in c.101G>T; in TP53 a known missense mutation in c.215C>G; and two known missense variants in PIK3CA, viz., in c.1173A>G; in exon 7, and in c.3128T>C; in exon 21, were found, respectively. Seven synonymous mutations were detected in this tumor, viz., in IDH1 (rs11554137), in FGFR3 (rs7688609), in PDGFRA (rs1873778), in APC (COSM3760869), in EGFR (rs1050171), in MET (rs35775721), and in RET (rs1800861), respectively. Three known, intronic variants were found in genes, such as PIK3CA, KDR, and JAK3, respectively. Also, a 3’-UTR and a splice site acceptor site variant in CSF1R and FLT3 genes were found in this tumor. We have shown allele coverage, allele ratio, and p-value, for all these mutations. The p-values and Phred quality score were significantly high for these variants.
Conclusion: As reported in previous studies, in ACP tumors we found a CTNNB1 mutation by NGS analysis. The PIK3CA variants we detected were not known previously in ACP tumors. Finding the PIK3CA mutations in the ACP tumors may help develop targeted therapy for a subset of craniopharyngiomas with PIK3CA activating mutations. Clinical trials are in progress with specific PIK3CA inhibitors in advanced stages of many cancers.
Keywords: craniopharyngioma, adamantinomatous, brain tumor, NGS, next generation DNA sequencing, CTNNB1
Introduction
Craniopharyngiomas (CP) account for 1–5% of all primary intracranial tumors.1,2 They are slow-growing non-glial tumors, most of the times they occur in children and adolescents, or adults over age 50.2,3 Craniopharyngioma tumors develop near the hypothalamus in the area of the brain near the pituitary gland that controls growth and many body functions. The etiology of these lesions is unknown, according to WHO classification, they are benign Grade-I tumors.4,5 However, they may sometimes be considered malignant because they can cause serious problems by interfering with neuroendocrine structures or can cause neuropsychological complications thus, posing serious challenges to an oncologist and the patients.6,7 Craniopharyngioma lesions are supposed to be originated in the suprasellar regions from Rathke pouch epithelium, above the sella turcica of the skull base of the brain.8,9 These tumors typically comprise a mixture of calcium deposits, and cysts containing protein, lipids, and cholesterol pockets. There are two main subtypes of CPs, adamantinomatous CP (ACP) which is more common in children, and papillary CP (PCP) which occur in adults.2–4,9 Specifically, PCPs consist of monomorphic squamous epithelium, and fibrovascular cores, with scattered immune cells, whereas ACPs consists of whorls, with stellate reticulum, palisading peripheral columnar epithelium, and “wet” keratin.10 Additionally, ACPs are known to have β-catenin gene (CTNNB1) mutations which are absent in PCP.11,12
Several recent studies have shown that CTNNB1 and BRAF (V600E) mutations were mutually exclusive for ACP and PCP tumors.13,14 In the majority of ACP tumors, somatic mutations in exon 3 of CTNNB1 prevents degradation of β-catenin protein, thus activating the Wnt signaling,15,16 whereas mutations in BRAF exon 15 in PCP tumors lead to MAP kinase pathway activation.17 This discovery of specific mutations became useful to distinguish between ACP and PCP, and leading to the development of novel targeted therapies for patients with BRAF mutations in PCP cases.18–20 In contrast, for ACP tumors, several studies documented mutations in the CTNNB1 gene, but no such targeted therapy is available yet. Also, now it is well documented that this kind of targeted BRAF and CTNNB1 sequencing by NGS or capillary method allows proper identification of ACPs from PCPs in tumor samples with limiting amounts of lesional cells when histopathology is not conclusive.21,22
In this case report, we present NGS data of the ACP tumor DNA sequenced on Ion Proton instrument, and finding of new variants that were not previously reported in this tumor. Clinical trials are being in progress with specific PIK3CA inhibitors in advanced stages of many cancers including gliomas.23–25 Thus, this kind of finding new activating mutations in the ACP tumors may help to develop targeted therapy with kinase inhibitors for a subset of craniopharyngiomas.
Case Report
Case History
This study was approved by the Institutional Review Board (IRB) for the bioethics committee of King Abdullah Medical City (KAMC), Makkah, Kingdom of Saudi Arabia (IRB number 14–140) and performed in accordance with the principles of the Declaration of Helsinki. Before starting the study, formal written informed consent was obtained from the patient for publishing the images and the case details. The male patient aged around 35 years of Yamani origin was referred to the Neurosurgery unit, with a history of RTA (Renal tubular acidosis), fits, headaches, vomiting and loss of consciousness. Generally, the patient was conscious and oriented, he was vitally stable. The coagulation profile, blood chemistry, CBC, and the pituitary function were performed. The patient’s pituitary hormones data are shown in Table 1. Pituitary hormones levels such as ACTH, TSH, FSH, LH, and Prolactin were low. Brain CT revealed the presence of a space-occupying lesion (SOL). Craniotomy was done by lateral supraorbital approach (LSO) for complete tumor excision.26 After surgery, and before discharging from hospital, the patient was re-examined with MRI and CT scan. Results of re-examination revealed that the tumor was totally removed. Tumour was classified histologically according to the WHO grading system.5 Final diagnosis was made following the radiological and histopathological reports.
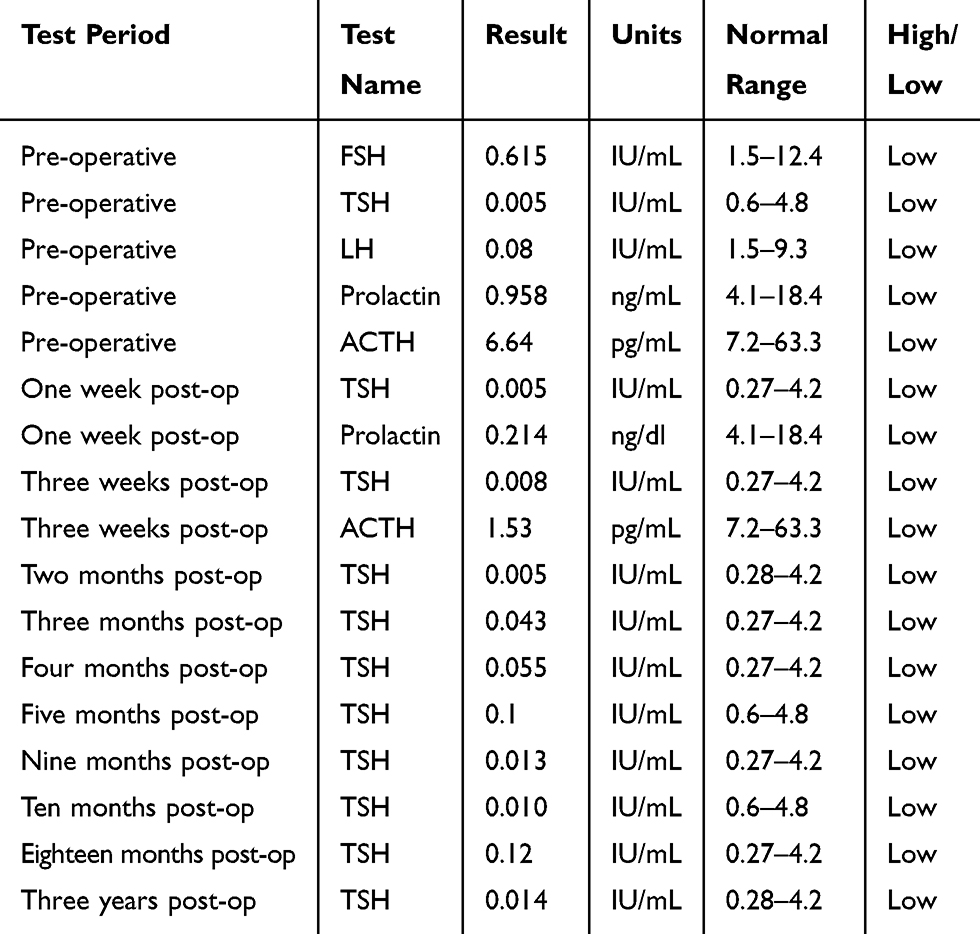 |
Table 1 Pituitary Hormones in the Craniopharyngioma Patient Before and After Surgery |
Histology and Radiology Examination
The MRI of the brain was performed by a 3 tesla Syngo MR D13, Siemens MRI scanner. Preoperative Magnetic Resonance Imaging (MRI) of the brain (Figure 1 A–C), and post-operative follow-ups MRI of the brain of this ACP patient is shown in Figure 1D–F.
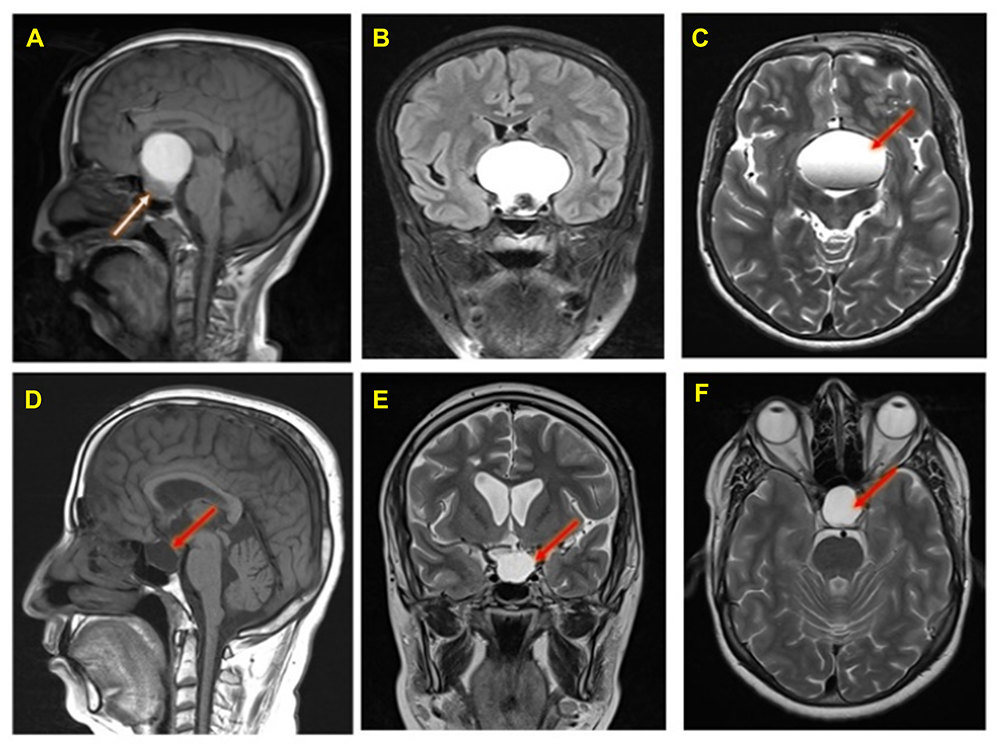 |
Figure 1 Preoperative MRI Brain of an adamantinomatous craniopharyngioma patient (A, B, and C): A large sellar and supra-sellar space-occupying lesion showing high Signal intensity in T1 (panel A), FLAIR (panel B) and T2 (panel C) images with fluid level (red arrow). The lesion compressing the pituitary gland downward (white arrow, panel A) and extending upward displacing the optic chiasma and no invasion of the sphenoid sinus or cavernous sinus. Post-operative follow-up MRI brain done after 4 months (panels D, E, and F): T1 sagittal (panel D) and T2 coronal and axial (panels E and F) show residual fluid collection (red arrows) at the operative bed with complete resolution of the previously reported seller and supra-sellar lesion. No evidence of residual or recurrent disease. |
Brain MRI with contrast showed a large sellar and suprasellar space-occupying lesion measuring 48 x 36 x 42 mm, this lesion showed high signal intensity in T1 (Figure 1A), FLAIR (Figure 1B) and T2 (Figure 1C) with fluid levels. The lesion compressing the pituitary gland downward and extending upward displacing the optic chiasma and no invasion of the sphenoid sinus or cavernous sinus is visible in Figure 1A. Normal MRI appearance of the basal ganglia, thalami, internal and external capsules on both sides were noted. Normal homogenous intensity of the periventricular white matter and centrum semi-ovale, and no midline shifts were also observed. Subarachnoid spaces were normal, including cisterns, fissures and sulci. Also, there were normal features of the cerebellar hemispheres, vermis, peduncles and brain stem with no abnormal intensities. Normal MRI features of the cerebellopontine angles and internal auditory canal were also found.
Post-operative MRI with contrast T1 sagittal (Figure 1D) and T2 coronal and axial (Figure 1E and F) show residual fluid collection at the operative bed with complete resolution of the previously reported seller and supra-sellar lesion, and stable appearance of the sellar and suprasellar region with widened sella turcica with no evidence of enhanced recurrent tumor was found. No interval changes were seen in the right frontal deep and periventricular white matter T2 hyperintensities with cystic areas denoting old insult. Unchanged scattered juxta cortical and deep white matter rounded T2 hyperintensities without restricted diffusion were noticed. The ventricular system remains prominent. The remainder of the brain parenchyma shows no abnormal signal intensity or post-contrast enhancement. The CT scan of the brain was performed by a multi-slice CT (MSCT), using a 64-detector-row scanner. Images were acquired with 5 mm slice thickness throughout on a GE Medical Systems, light speed VCT, 64-slice multidetector CT (MDCT), and processed at low dose performance on Volara™ digital DAS (Data Acquisition System). Pre- and post-operative CT images are shown in Figure 2. Pre-operative-CT-showed a large sellar and suprasellar space-occupying cystic lesion with marginal curvilinear calcification of the wall (Figure 2A and B), and post-operative-CT-showed no residual or recurrent lesion at the operative bed (Figure 2C and D).
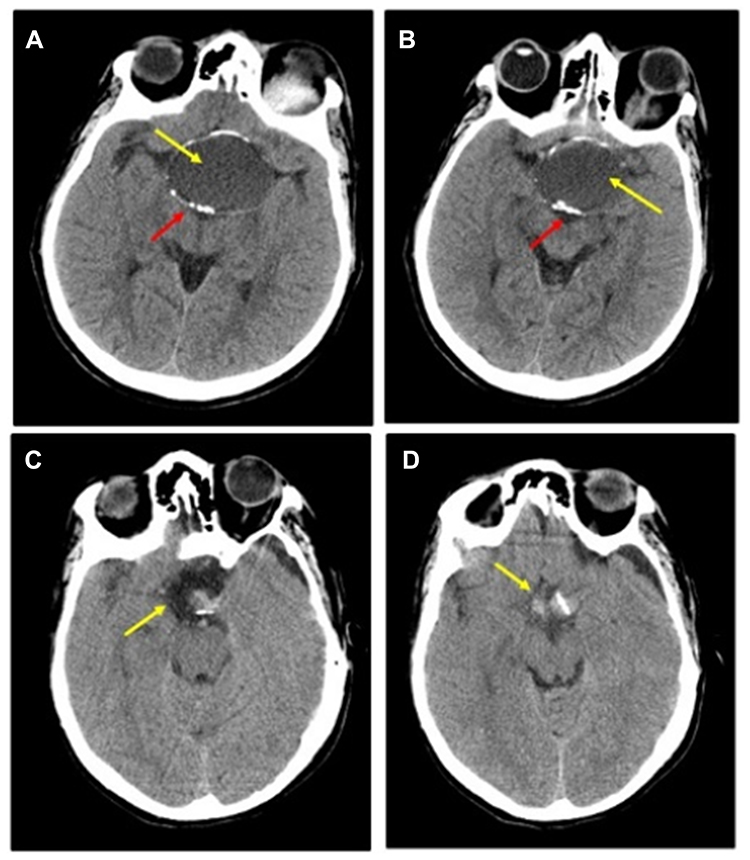 |
Figure 2 Preoperative and Post-operative CT: (A and B) Pre-operative-CT-showing a large sellar and supra-sellar space-occupying cystic lesion (yellow arrow) with marginal curvilinear calcification (red arrow) of the wall; (C and D) Post-operative-CT done after 2 months showing no residual or recurrent lesion at the operative bed (yellow arrow). |
A grayish white cystic lesion mass from the supra-sellar brain region was obtained after excision biopsy. The excised tumor was fixed in 4% buffered formaldehyde, processed and paraffin embedded. Four-micrometer-thick sections were prepared on clear ground glass microscope slides with ground edges and these sections were deparaffinized with EZ Prep, and routinely stained using Dako Reagent Management System (DakoRMS) with hematoxylin and eosin (H & E) on a Dako Coverstainer (Agilent). For immunohistochemistry, sections were collected on Citoglas adhesion microscope slides (Citotest) and then stained with antibodies according to standard protocols of Ventana BenchMark XT automated Stainer (Ventana, Tucson, AZ) as described previously.27,28 Images were acquired using NIKON Digital Microscope Camera - DS-Ri1, with image software NIS Elements v.4.0.
Histology data of the ACP tumor are shown in Figures 3 and 4. Hematoxylin and eosin (H&E) stained pictures reveal nests and cords of stratified squamous epithelium embedded within stroma. Squamous cells show peripheral palisading, with dystrophic calcification. Cholesterol clefts are also noted at different foci, and distinct and compact, lamellar keratin formations (wet keratin) are prominent in Figure 3A and B. In Figure 3C and D picket-fence like the pattern of palisading cells, and gliotic brain tissue, a large number of white spindle-shaped clefts representing cholesterol clefts are present. Hematoxylin and eosin staining also revealed basaloid cells in Figure 4A, and the tumor containing protrusion-forming palisading epithelium and a whorl like nodules with the presence of ghost cells, cystic changes and calcification (Figure 4B). Whorling and streaming spindle-cell fascicles are observed around the epithelial cell nests (Figure 4C), with stellate reticulum extending into the adjacent gliotic brain (Figure 4D). Immunohistochemistry of the ACP tumor is shown in Figure 5. This tumor is positive for Bcl2 in cytoplasm, and E-cadherin staining showed positive in membranes (Figure 5A and B); tumor showed membranous and cytoplasmic staining for β-catenin, peripherally palisading cells show stronger expression, whereas clusters of cells along the epithelial cell whorls show strong nuclear accumulation in Figure 5C. Also, spindle cells show moderate to strong cytoplasmic and nuclear expression and lack of membranous staining observed in squamous whorls. In Figure 5D, CK-8-18 staining shows membranal positive in all three layers squamous layer, stellate reticulum, and palisaded basal columnar cells, but negative in wet keratin. The final diagnosis was determined as ACP based upon histological, immunochemical, and radiological results.
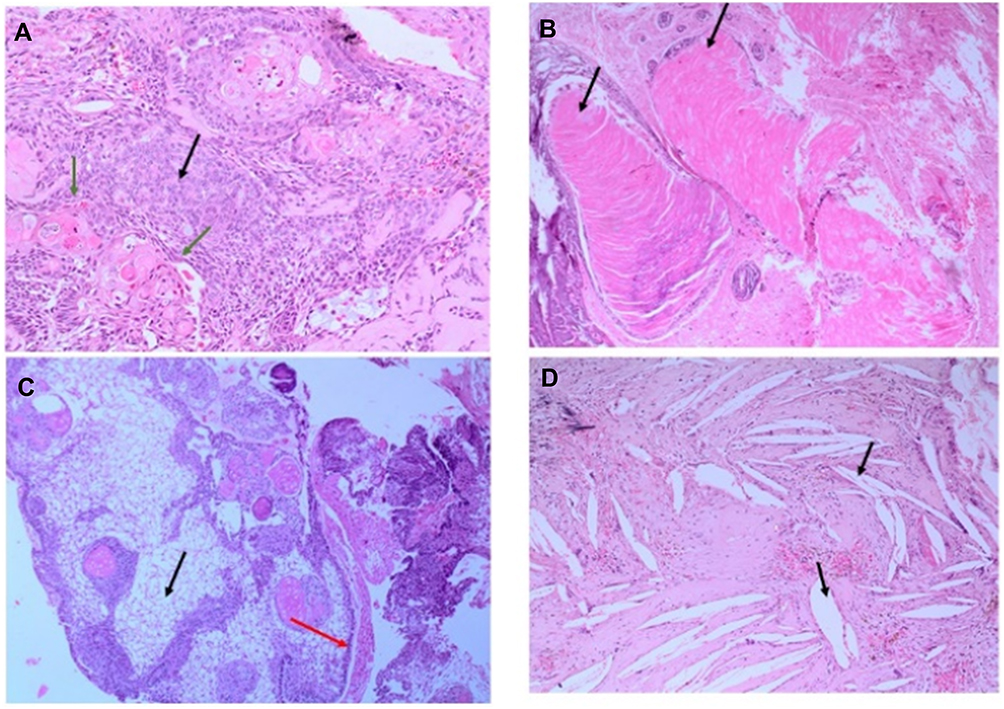 |
Figure 3 Hematoxylin and Eosin (H&E) stained pictures of ACP tumor. (A) Histology of ACP, showing loosely cohesive squamous cells surrounded by peripherally palisading cells with distinct wet keratin (green arrows), stellate reticulum (black arrow); (B) an aggregate of epithelium with squamous nodule multiple wet keratin (black arrows, 20x); (C) basal epithelial layer showing a pattern of picket-fence (red arrow), and gliotic brain tissue (black arrow); (D) large number of white clefts representing cholesterol deposits (black, arrow). |
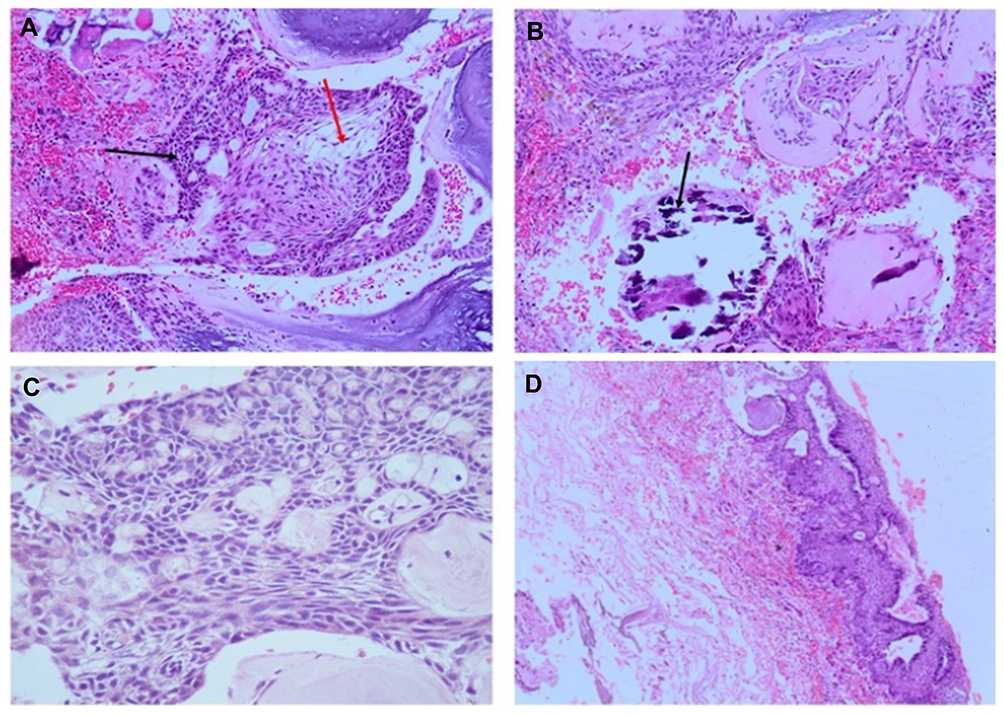 |
Figure 4 Hematoxylin and Eosin (H&E) stained pictures of ACP tumor. (A) Nests of epithelial cells with a basaloid appearance (blue arrow), and stellate reticulum (red arrow); (B) the ACP with palisading epithelium and a whorl like nodules, cystic changes and calcification (black arrow); (C) Whorl like and streaming fascicles are observed around the epithelial cell nests (10x); (D) stellate Epithelium extending into adjacent stroma. |
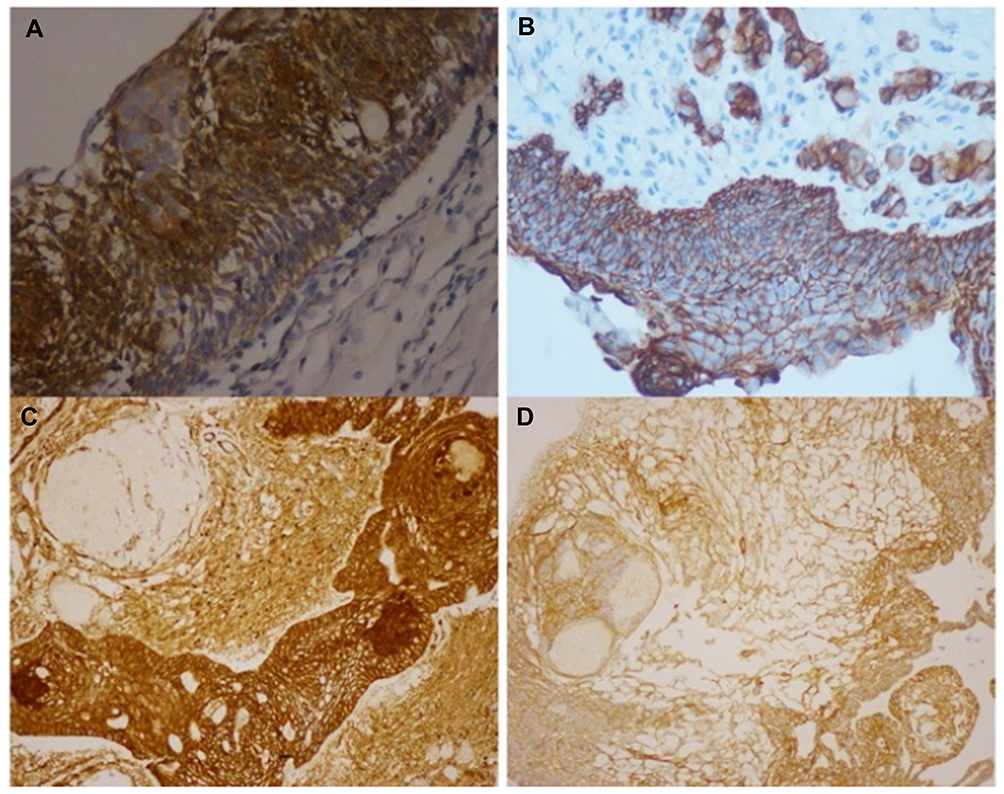 |
Figure 5 Immunohistochemistry of craniopharyngioma tumor. (A) Bcl2 staining shows positive in cytoplasm; (B) E-cadherin staining shows positive membranous stain; (C) β-catenin staining in squamous whorls, both intranuclear (translocated) and predominantly in cytoplasmic; (D) CK-8-18 staining shows positive in membranes. |
DNA Isolation and NGS Analysis on Ion Proton
Isolation of DNA was carried out by Qiagen FFPE Kits. FFPE sections (5–10; 5 microns) were deparaffinised using xylene, and then treated with ethanol and dried at 65°C for 5 mins. The pellets were digested with proteinase K in buffer ATL. Later, steps were carried out according to the user manuals. Ten ng of DNA was used for NGS analysis. Only tumor DNA was sequenced, a germline DNA was not sequenced.13,21 However, to minimize turnaround time and the cost of testing many times, tumor DNA sequencing by NGS does not currently involve sequencing of a matched germline sample. Sequencing was performed using Ion PI v3 chip on Ion Proton instrument.27,28 Libraries were prepared using Ion AmpliSeq Cancer HotSpot panel v.2 (P.No. 4471262) primer pools. The complete list of 50 genes is published previously.29 Ion AmpliSeq 2.0 library kit, and Ion PI Hi-Q OT2 200 kit was used for libraries and templates preparation, respectively. Sequencing was done using Ion PI Hi-Q Sequencing 200 kit, library was tagged with Ion Express barcode. After the sequencing, amplicon sequences were aligned to the human reference genome GRCh37 (hg19) in the target regions of the cancer panel using the Torrent Suite Software v.5.0.2 (Life Technologies). Variants call format (vcf) file was generated by running the Torrent Variant Caller Plugin v5.2. The vcf file data were analysed using Ion Reporter v5.6 (ThermoFisher Scientific, USA), which calculated allele coverage, allele frequency, allele ratio, variant impact, clinical significance, PolyPhen 2, Phred, SIFT and FATHMM scores.
Ion Torrent Suite software v 5.0.2 generated Ion PI chip run report metrics before and after alignment of sequences are shown in Figure 6. The Ion sphere particle (ISP) density image is showing the semi-conductor chip percent loading across the wells that contain a live ISP; the total number of bases reported in the output file is 10.2 G (Figure 6A); ISP summary showing loading, enrichment, clonality and final library quality is shown in Figure 6B; In the histogram, the first row shows 95% of loaded wells and empty wells are 5%, wells with the predicted number of live library ISPs (enrichment) are 100%, clonal ISPs are 74% and 26% ISPs are polyclonal (ISPs carrying clones from two or more templates). The mean read length of sequencing (trimmed lengths) of all reads is 110 bp (Figure 6C). In Figure 6D, after alignment analysis of the target regions of the cancer HotSpot panel used (CHP. 20131001.designed) to the reference genome GRCh37 hg19 (Human genome build 19, Accession no. GCA_000001405.1) and sample coverage overview is given. For this tumor NGS generated 6, 814, 682 mapped reads, with more than 96% reads were on target. Average base coverage depth, uniformity of base coverage, amplicon and target base read coverages of sequencing are shown in Table 2. Initial analysis by Ion Reporter 5.6 program found a total of 1652 variants passed all filters. We have considered true mutations based upon Phred score above 40, and significant of mutation call by Ion Reporter software when p-value with below 0.05.
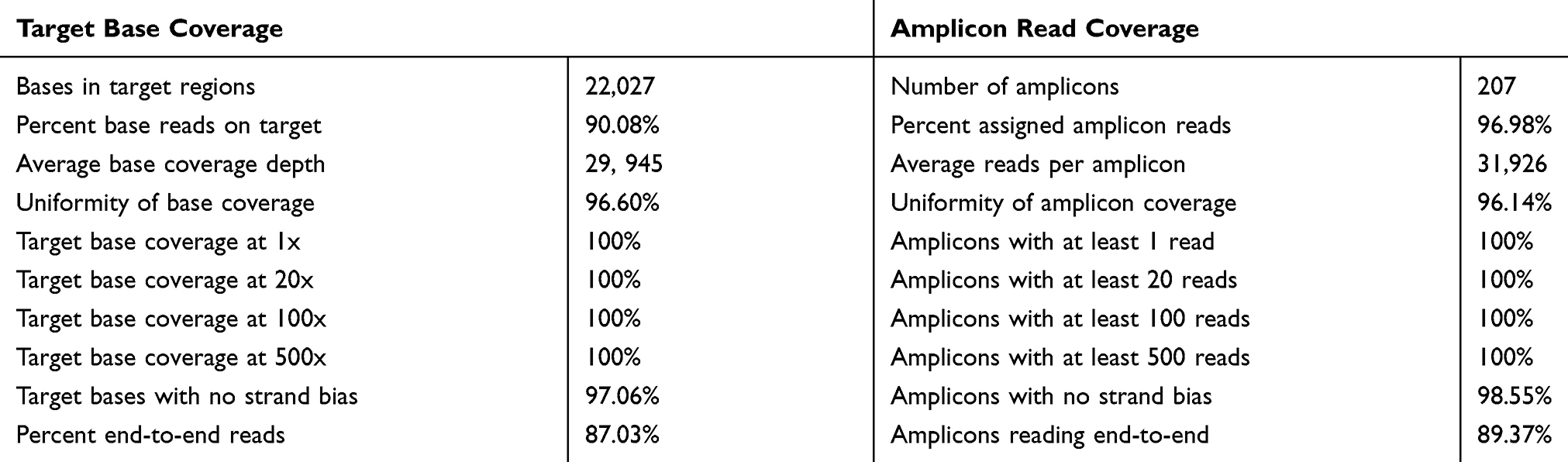 |
Table 2 Sequencing Coverage Analysis Report from Torrent Suite Software |
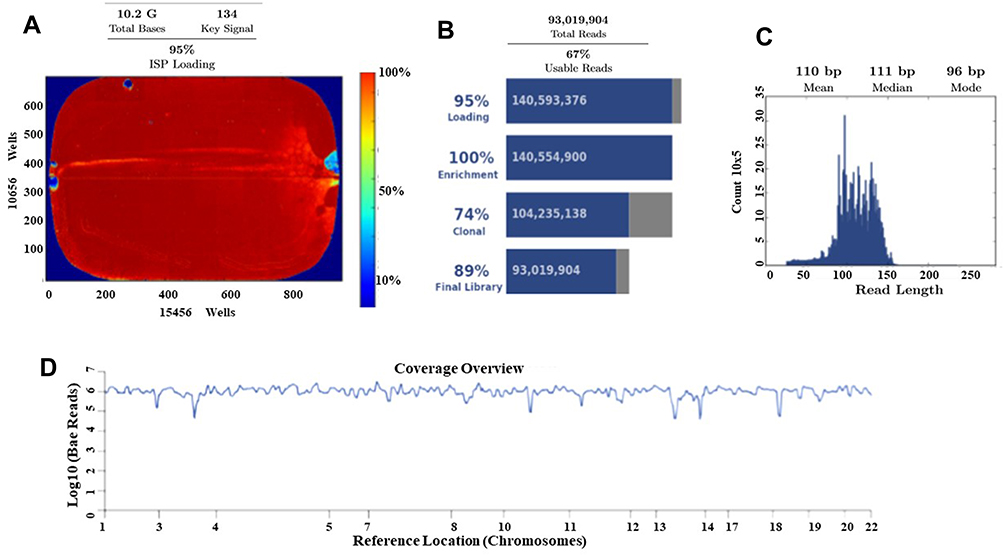 |
Figure 6 Torrent suite software generated Ion PI chip run report metrics before and after alignment of sequences. (A) The ISP density image is showing percent loading across the semi-conductor chip; (B) ISP Summary showing loading, enrichment, clonality and final library quality; (C) Read length (bases) histogram showing trimmed lengths of all reads. (D) After alignment analysis of the target regions (CHP. 20131001.designed) to the reference genome (Hg 19) and sample coverage overview as performed by the Ion Torrent Suite software v 5.0.2 for Ion Proton sequencing. |
Summary of all missense mutations found by NGS analysis is shown in Table 3. In this tumor NGS data analysis identified 16 variants out of which 4 are missense mutations, six were synonymous mutations, and five were intronic variants. None of these mutations were novel variants. In CTNNB1 gene a known missense mutation in exon 3, in TP53 in exon 4, and two known missense variants in PIK3CA, viz., in exon 7, and in exon 21 were found in this tumor, respectively. Seven synonymous mutations were found in this tumor viz., in IDH1, in FGFR, in PDGFRA, in APC, in EGFR, in MET, and in RET genes, respectively. Also, three known, intronic variants were found, one each in PIK3CA, in KDR, and in JAK3 genes, respectively, in this tumor (Table 3). A known splice site mutation at acceptor site in FLT3 c.1310-3T>C, and an SNV in 3’-UTR of CSF1R gene c.*1841TG>GA, were also detected. For each variant, the allele coverage, allele ratio and frequency, p-value, and Phred score are shown in Table 4. The p-values and Phred quality score were significantly high for these variants. Variants with Phred scores below 40 were discarded.
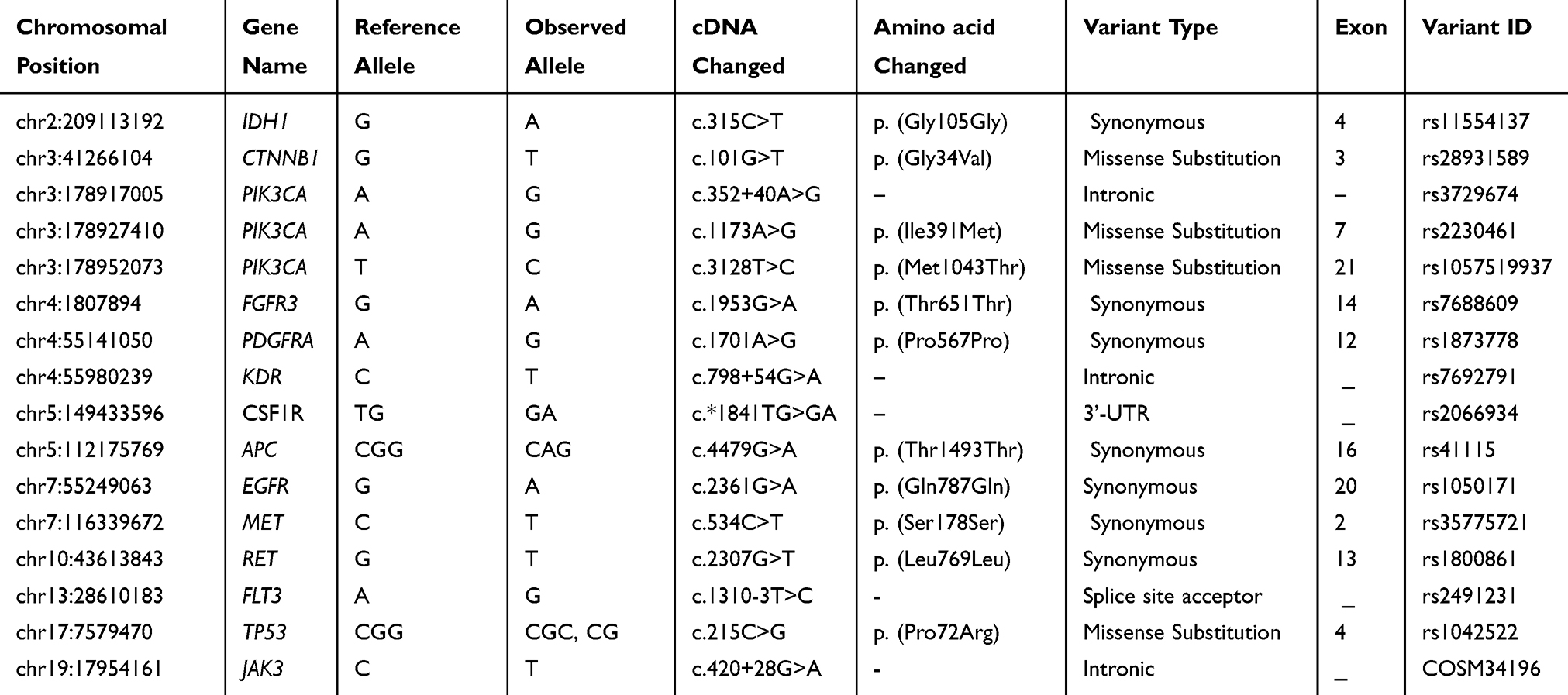 |
Table 3 Variants Found in the Craniopharyngioma Tumor by NGS Analysis |
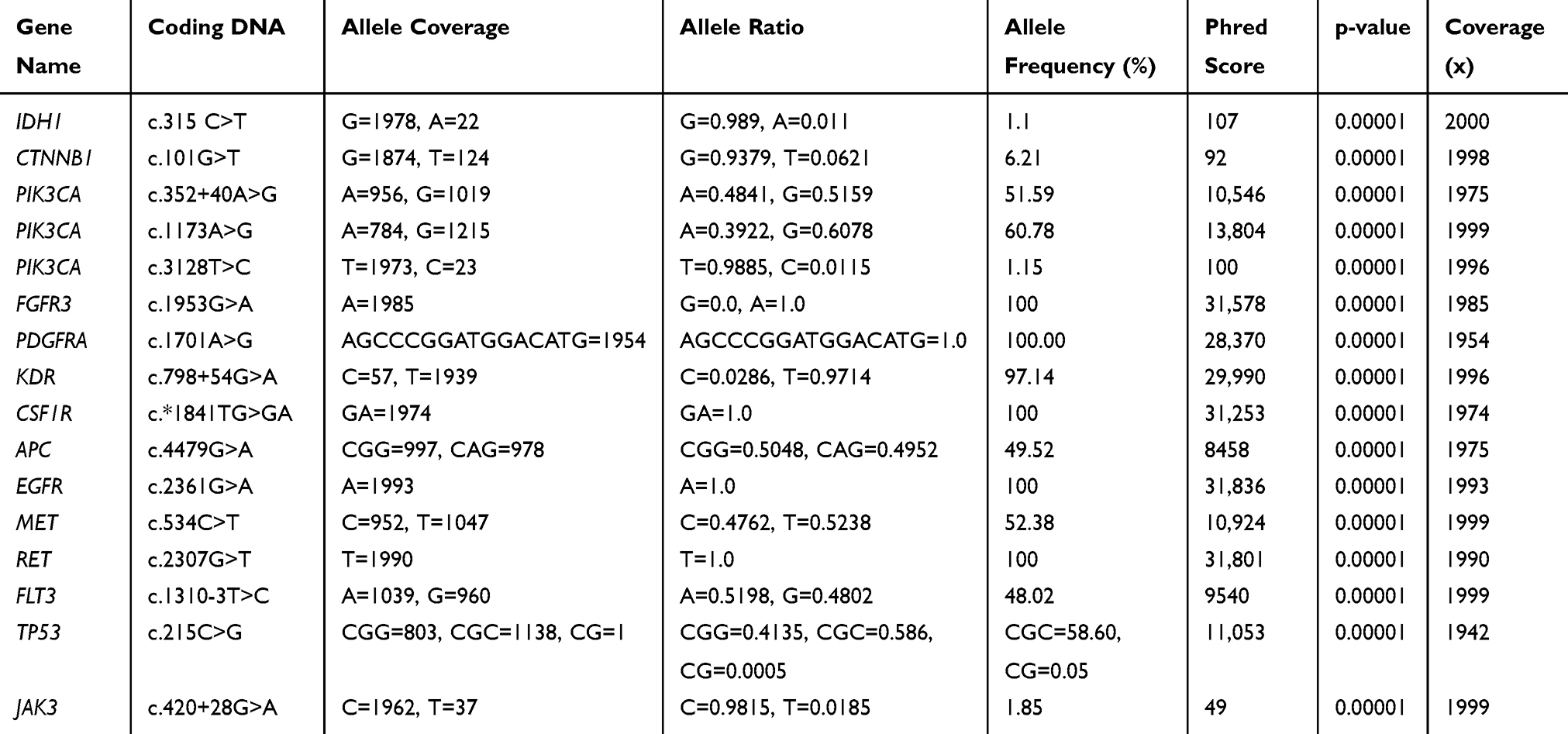 |
Table 4 Quality of DNA Sequencing on Ion Proton for the ACP Tumor |
As shown in Table 5, within the last 5 years, nine studies were published on the mutation profiling in ACP tumors using NGS methods.13,21,30-36 So far one hundred ACP tumor samples were sequenced by NGS, and 85 tumors (85%) had CTNNB1 mutations. In these studies, the majority of ACPs tumors have somatic activating mutations in the degradation-targeting box present between amino acids 32 to 45 in the exon 3 of CTNNB1 gene where casein kinase 1 (CK1 α) first phosphorylates on Ser-45.37 This phosphorylation event triggers subsequent phosphorylation by glycogen synthase kinase-3 β (GSK3 β) at Ser-33, Ser-37, and Thr-41. In Figure 7, a schematic of amino acid motifs in β-catenin phosphorylation domain, also ubiquitination, and various regulatory protein binding domains are shown. The phosphorylation of these four residues causes the binding of β-catenin to the destruction complex resulting in the degradation of β-catenin.38 Mutations adjacent to the CK1 α and GSK3 β phosphorylation sites prevent this destruction complex formation and β-catenin degradation, causing the translocation of β-catenin to the nucleus (Figure 7).38,39
 |
Table 5 Previously Published Reports of ACP Tumors Profiling by NGS Methods |
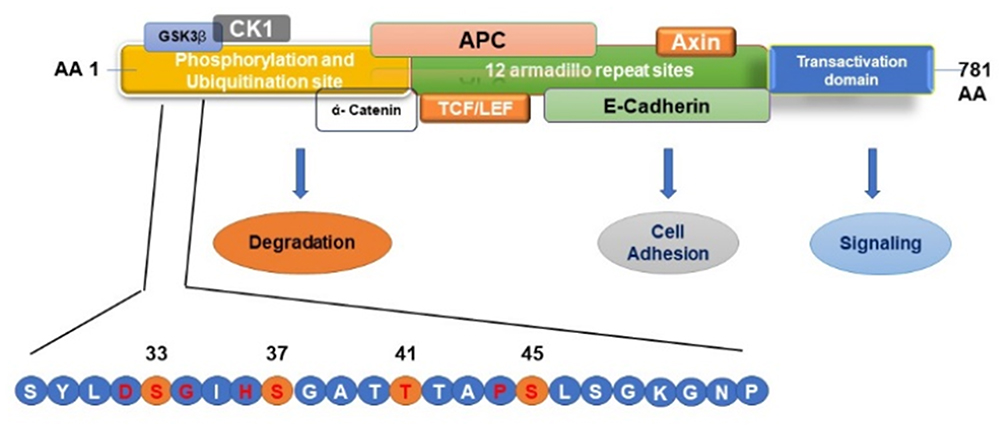 |
Figure 7 Schematic Representation of β–catenin phosphorylation, ubiquitination, and protein-binding domains. Mutations in the β–catenin are always found in the degradation-targeting box where CK1 α first phosphorylates Ser45 (shown in yellow circle). This phosphorylation event primes for subsequent phosphorylation by GSK3 β at Ser33, Ser37, and Thr41 (shown in yellow circles). The phosphorylation of these sites initiates the β–catenin degradation by the proteasome system by forming a destruction complex which is formed by axin, adenomatous polyposis coli (APC), and protein phosphatase 2A (PP2A) proteins resulting in the degradation of β-catenin by 26S proteasome system. Mutations in the codons adjacent to the CK1 α and GSK3 β phosphorylation sites (shown in red letter) prevent this destruction complex formation and β-catenin degradation, causing the translocation of β-catenin to the nucleus and activating the transcription of target genes. |
Discussion
While PCP tumors are predominantly found in adults, ACP tumors represent the vast majority of CP (9:1) and have a bimodal peak incidence (childhood and adults).40,41 The present ACP patient is 35 years old male. Hypopituitarism and hypothyroidism are common endocrine symptoms of craniopharyngioma.42 The leaking of fluid from the cystic component causes a significant problem in craniopharyngiomas, and it has been documented that cystic fluid is rich with necrotic and inflammatory components like cholesterol, cytokines and chemokines with particularly elevated concentrations of IL-6, IL-8, CXCL1, CXCL8 and the immunosuppressive cytokine IL-10.10,43 In our present case also, cholesterol crystals and calcification were present. The present case is positive for cytokeratin 8 and 18. ACP has distinctive patterns of immunostainings for different epithelial and squamous cells markers. Immunostaining for cytokeratin (CKs) is a valuable tool in distinguishing CP tumors. Cytokeratins are a complex family of many different polypeptides, are expressed in epithelial cells and their related neoplasms. Strong expression of cytokeratin 18, 19 and 20 are reported in ACP tumors.44
It is well established that in cells that lack mutations in CTNNB1 such as in PCP, the β-catenin is localized at the membranes, whereas mutations in CTNNB1 gene in ACP translocate β-catenin to both the cytoplasm and nucleus.45 In the present case, also β-catenin immunostaining has confirmed this observation. The E-cadherin- β-catenin complex plays an essential role in the cell-cell adhesion mechanism. Cadherins are transmembrane proteins whose extracellular domains promote calcium-dependent adhesion between neighboring cells, whereas their cytoplasmic domain is linked to the actin cytoskeleton by means of cytoplasmic catenin proteins.46 Its dysfunction is associated with a decrease in cell differentiation and with increased invasiveness and metastasis.47 β-Catenin expressing cells in ACP tumor in this study found to express Bcl2 protein. However, not many previous reports examined the immunostaining of Bcl2 in ACP tumors, one study described β-catenin expressing cells in ACP tumor were found to have high levels of anti-apoptotic proteins of the Bcl2/Bcl-xL family and were found to be actively dividing.48
The Ion AmpliSeq Cancer HotSpot panel we used in this study for the mutation analysis consists of 207 primers, targeting 50 oncogenes and tumor suppressor genes that are frequently mutated in several types of cancers.29 In the present tumor, not many actionable mutations were detected in cancer driver genes, this is in agreement with several studies previously published by NGS analysis of ACP tumors.21,30-34 Marucci et al (2015) have reported only CTNNB1 mutation using NGS method in 12 out of 15 cases (80.0%) of BRAF negative ACP tumors.21 Besides the CTNNB1 mutations in ACP and BRAF V600E mutations in PCP, no further mutations were detectable by NGS in both tumor subtypes in their studies. We have detected only synonymous and benign mutations in the cancer driver genes (APC, EGFR, MET, RET, FGFR3, IDH1 and PDGFRA) and the intronic variants in PIK3CA, in KDR, and in JAK3, a splice variant in FLT3, and a 3’-UTR of CSF1R gene. Although these variants are known in other tumors, for ACP tumors these variants reporting is new, we report this for the first time here. All these synonymous variants have high frequencies except IDH1 variant, suggesting these are germline variants. IDH1 mutations typically occur in low-grade gliomas; however, in the present case of ACP, we did not find IDH1 exon two and IDH2 exon four missense mutations.
The EGFR synonymous exon 20 mutation (rs1050171) we have reported in this ACP tumor also reported in a case of grade III anaplastic ependymoma.28 This synonymous EGFR SNP (p. Gln787Gln) was reported in 70% of lung adenocarcinoma tumors. This SNP was not a prognostic marker for anti-EGFR therapy; however, it is associated with improved overall survival (OS) in CRC patients.49,50 Also, this SNP showed a significant association with susceptibility to acute renal allograft rejection in a Korean population.51 In the present case, EGFRvIII variant was also not expressed as validated by Real-Time PCR assay which agrees with the previous report.52 The activated form of EGFR is reported in human ACP clusters cells and the activation of the EGFR pathway in β-catenin accumulating cells with activated Wnt signaling was demonstrated previously by others.52,53 Over-activating mutations in FGF receptors have been identified in several human cancers, including breast, bladder, prostate, endometrial and lung cancers as well as hematological malignancies.54 FGFR 1–3, PDGFRA, PIK3CA, and TP53 mutations are very rare in ACP tumors. In a previous study, two cases with CTNNB1 mutations tested positive by NGS analysis were reported to have FGFR3 mutations p. (Ile378Thr) and p. (Ala719Thr), respective, but not FGFR1 or FGFR2 mutations.32 In the present case only, a synonymous mutation was found in FGFR3. Very rarely, other mutations also reported in ACP tumors having CTNNB1 mutations, in one case with CTNNB1 mutation, a mutation in the NF2 gene, and a frameshift mutation in the SETD2 gene was shown previously.35 An intronic SNP (rs7692791) of Kinase insert domain receptor (KDR) was associated with poor overall survival (OS) among patients with gastric and biliary tract cancer who were treated with sunitinib, this SNP is also found in the present ACP tumor.55
Malignant transformation of ACP is extremely uncommon, increased expression of proliferation markers and TP53 have been shown in malignantly transformed CP tumors.56 We have found a missense mutation (rs1042522) in TP53 gene with a frequency of 47.94% which was not known previously in CP tumors. As reported by Zhang et al (2019), this SNP may be a potentially protective factor against neuroblastoma in Chinese children.57 However, this variant is reported to be associated with an increased risk of leiomyoma development.58 This mutation was detected in medulloblastoma tumor of a young patient and also in other cancers.59,60 This TP53 variant also frequently altered in meningioma and in astrocytoma tumors, and predicted to be neutral with FATHMM score of 0.36.61,62 One investigation showed that this TP53 polymorphism was effective in predicting the response to chemotherapy and correlated with PFS and OS in patients with advanced gastric cancer treated with paclitaxel and capecitabine chemotherapy.63 More studies related to this variant’s clinical significance were reviewed by “Expert Panel in ClinVar database”. However, the data were not statistically significant, and further studies are warranted.
Wnt/β-catenin and Sonic Hedgehog (SHH) pathways are defective in ACP tumors due to CTNNB1 mutations. In the present case, the nucleo-cytoplasmic β-catenin is only seen in a small proportion of cells, often correlating with epithelial whorls and referred to as “clusters” as reported in other studies.2,11 In ACP tumors, all exon 3 mutations of the β-catenin gene targeted only near the CK-1 and GSK-3 β phosphorylation site, therefore, these mutations are important in the etiology of an ACP.64 The CTNNB1 substitution – missense mutation (COSM5670) at codon Gly34Val we detected in this tumor is a pathogenic one (FATHMM score 0.98). This mutation prevents the phosphorylation of the serine residues by CK1 α and GSK3 β thus preventing the destruction of β-catenin degradation, causing the translocation of β-catenin to the nucleus for increased gene expression. The mutations in the APC gene also prevent the interaction of β-catenin with APC protein and prevent the formation of the β-catenin phosphorylation complex. This leads to speculations that somatic mutations of the APC gene also may have a role in the origin of ACP tumors. Surprisingly a WES study recently revealed a presence of CTNNB1 mutation and a germline APC frameshift mutation in a pediatric case of ACP in association with Familial adenomatous polyposis (FAP).36 However, it is not well established that patients with FAP are associated with ACP. Several investigations with NGS analysis of ACP tumors did not find any APC mutations so far. The synonymous APC mutation [c.4479G>A; p. (Thr1493Thr)] we found in the present case, was also reported in colorectal cancer (CRC), and breast cancer.65,66
Somatic mutations of PIK3CA are common in a variety of primary tumors such as those of the CRC, breast, brain, and stomach.25,66,67 Earlier investigation using NGS of 26 ACP tumors did not detect any recurrent mutations other than CTNNB1 mutations; however, in these customized panel of genes, PIK3CA was not present.33 In the WES panel, and the customized primer panels used in two other studies, PIK3CA primers were present, but no mutation was found in this gene in ACP tumors.13,30,32,34 Our NGS profiling showed that the PIK3CA gene, which is involved in cellular proliferation and inhibition of apoptosis, was mutated in exon 7 (C2 domain) and in exon 21 (kinase domain) and in an intronic site also. This exon 7 variant p. (Ile391Met) was reported in grade III anaplastic ependymoma.28 This variant is reported as a benign one in ClinVar and in SNP databases with reference numbers VCV000135038.1 and rs2230461, respectively. The intronic variant (rs3729674) also is a benign one, and it was reported in CRC and in endometrial cancer.65,68 The C2 domain in PIK3CA contains amino acid residues from 330 to 480, and kinase domain contains amino acids 696 to 1068.69 The p110α subunit C2 domain is adjacent to the Ras-binding domain (RBD), and mutations of this domain were reported to increase the positive surface charge of the protein, thus increases the binding of p110α subunit of PIK3CA to cellular membrane enhancing the kinase activity.70 The exon 21 mutation is present in kinase domain, and it is a pathogenic one, with FATHMM prediction score of 0.95. This exon 21, PIK3CA mutation p. (Met1043Thr) found in this present case also reported in astrocytoma.71 A different mutation p. (Met1043Val) also is known at this codon in endometrial cancer.72 Previous studies have not reported any PIK3CA mutations in ACP tumors; however, Kassab, et al, 2019, have reported in one PCP case a kinase domain mutation in exon 20 p. (His1047Arg) in this gene that is been reported to activate the PI3K/Akt/mTOR pathway.25,35,73
The NGS method detects mutations with high accuracy as evidenced by the p-values (0.00001) and the high Phred score for all the variants, indicating high confidence in the variants found in this tumor. The Phred quality score [Q] is a sequencing property which is logarithmically related to the base calling error probabilities [P]; ie, Q= −10log10P.74 That means if a score of 100 to a base, the chances that this base is called incorrectly are 1 in 10, 000 million. The frequencies of missense mutations found in the present study for CTNNB1, PIK3CA exon 7, and exon 21 missense variants, and PIK3CA intronic variant are 6.21%, 60.78%, 1.1.5%, and 51.59%, respectively, suggesting these are somatic variants. Although some variants have low frequencies, they all have Phred score of about 100 and high suggest they are true variants. Also, a high depth of coverage allowed us to reliably detect low-frequency mutations with high confidence. Allele coverage in most of the variants is around 2000x. We have verified all mutations in various databases (COSMIC, ExAc and dbSNP) to confirm whether variants are novel in this ACP tumor.
The patient in this study was regularly followed up by MRI examinations, and the pituitary functions were monitored and treated. His MRI of the brain was done, 4 years post-operatively revealed stable appearances of residual post-operative fluid collection on the pituitary bed, and no evidence of tumor recurrence. In general, this patient was doing well on follow up. However, the patient’s developed symptoms of hypothyroidism and osteoporosis 4 years post-operatively. His 25-hydroxy vitamin D, and testosterone levels were low. The patient started developing symptoms of diabetes, and cardiovascular issues, he has higher HBA1c levels (6.4), and blood glucose levels (168 mg/dl), and his cholesterol and LDL-cholesterol levels were also high. Overall 5-year and 10-year survival rates reported ranges are from 89% to 94% and from 85% to 90%, respectively, with follow-up.75 Survivors of craniopharyngioma known to exhibit several metabolic syndromes, endocrinopathy, pituitary insufficiency, hypothalamic dysfunction, cerebrovascular problems, and neurocognitive dysfunction.4,75-78
Conclusion
Further studies of the ACP tumors NGS profiling to confirm the PIK3CA mutations will help in developing targeted therapy, and for recruiting patients for clinical trials. Targeted therapy with BRAF inhibitors such as dabrafenib, trametinib and vemurafenib, or in combination BRAF and MEK inhibitors in PCP is giving promising responses in clinical trials.18–20 Clinical trials are being in progress with specific PIKCA3 inhibitors BKM120, in glioblastoma patients (Trail IDs: NCT01339052; NCT01349660; NCT01870726), and recently one clinical trial was completed with another inhibitor BYL719 (Trail ID: NCT01219699) with encouraging results in metastatic breast cancer.23,24 This kind of targeted therapy may be useful for a subset of craniopharyngiomas with PIK3CA activating mutations.
Data Sharing Statement
The Sequence file is deposited in SRA (Sequence Read Archive) database with access numbers SRA: SRP247830; BioProject: PRJNA605595.
Acknowledgment
The authors wish to thank Mrs. Hatoon Mohammed Badaawood of Histopathology Laboratory, at Al-Noor Specialist Hospital Makkah, for technical support.
Funding
This study was supported by the National plan for Science, Technology and Innovation (MAARIFAH) King Abdul Aziz City for Science and Technology that awarded to Dr MM. Taher (Grant Code. 12-MED 2961-10).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Nielsen EH, Feldt-Rasmussen U, Poulsgaard L, et al. Incidence of craniopharyngioma in Denmark (n = 189) and estimated world incidence of craniopharyngioma in children and adults. J Neurooncol. 2011;104(3):755–763. doi:10.1007/s11060-011-0540-6
2. Muller HL. Craniopharyngioma. Endocr Rev. 2014;35(3):513–543. doi:10.1210/er.2013-1115
3. Muller HL. Childhood craniopharyngioma–current concepts in diagnosis, therapy and follow-up. Nat Rev Endocrinol. 2010;6(11):609–618. doi:10.1038/nrendo.2010.168
4. Karavitaki N, Cudlip S, Adams CB, Wass JA. Craniopharyngiomas. Endocr Rev. 2006;27:371–397. doi:10.1210/er.2006-0002
5. Louis DN, Perry A, Reifenberger G, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. doi:10.1007/s00401-016-1545-1
6. Sterkenburg AS, Hoffmann A, Gebhardt U, Warmuth-Metz M, Daubenbuchel AM, Muller HL. Survival, hypothalamic obesity, and neuropsychological/psychosocial status after childhood-onset craniopharyngioma: newly reported long-term outcomes. Neuro Oncol. 2015;17(7):1029–1038. doi:10.1093/neuonc/nov044
7. Feng Y, Ni M, Wang YG, Zhong LY. Comparison of neuroendocrine dysfunction in patients with adamantinomatous and papillary craniopharyngiomas. Exp Ther Med. 2019;17(1):51–56. doi:10.3892/etm.2018.6953
8. Alomari AK, Kelley BJ, Damisah E, et al. Craniopharyngioma arising in a Rathke’s cleft cyst: case report. J Neurosurg Pediatr. 2015;15:250–254. doi:10.3171/2014.11.PEDS14370
9. Schlaffer SM, Buchfelder M, Stoehr R, Buslei R, Hölsken A. Rathke’s cleft cyst as origin of a pediatric papillary craniopharyngioma. Front Genet. 2018;22(9):49. doi:10.3389/fgene.2018.00049
10. Zada G, Lin N, Ojerholm E, Ramkissoon S, Laws ER. Laws ER: craniopharyngioma and other cystic epithelial lesions of the sellar region: a review of clinical, imaging, and histopathological relationships. Neurosurg Focus. 2010;28(4):E4. doi:10.3171/2010.2.FOCUS09318
11. Sekine S, Shibata T, Kokubu A, et al. Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am J Pathol. 2002;161:1997–2001. doi:10.1016/s0002-9440(10)64477-x
12. Martinez-Barbera JP. Invited review: molecular and cellular pathogenesis of adamantinomatous craniopharyngioma. Neuropathol Appl Neurobiol. 2015;41(6):721–732. doi:10.1111/nan.12226
13. Brastianos PK, Taylor-Weiner A, Manley PE, et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat Genet. 2014;46(2):161–165. doi:10.1038/ng.2868
14. Yoshimoto K, Hatae R, Suzuki SO, et al. High-resolution melting and immunohistochemical analysis efficiently detects mutually exclusive genetic alterations of adamantinomatous and papillary craniopharyngiomas. Neuropathology. 2018;38(1):3–10. doi:10.1111/neup.12408
15. Gao C, Wang Y, Broaddus R, Sun L, Xue F, Zhang W. Exon 3 mutations of CTNNB1 drive tumorigenesis: a review. Oncotarget. 2017;9(4):5492–5508. doi:10.18632/oncotarget.23695
16. Holsken A, Sill M, Merkle J, et al. Adamantinomatous and papillary craniopharyngiomas are characterized by distinct epigenomic as well as mutational and transcriptomic profiles. Acta Neuropathol Commun. 2016;4(1):20. doi:10.1186/s40478-016-0287-6
17. Brastianos PK, Dias-Santagata D. ENDOCRINE TUMORS: BRAF V600E mutations in papillary craniopharyngioma. Eur J Endocrinol. 2016;174(4):R139–R144. doi:10.1530/EJE-15-0957
18. Brastianos PK, Shankar GM, Gill CM, et al. Dramatic response of BRAF V600E mutant papillary craniopharyngioma to targeted therapy. J Natl Cancer Inst. 2016;108(2):djv310. doi:10.1093/jnci/djv310
19. Aylwin SJ, Bodi I, Beaney R. Pronounced response of papillary craniopharyngioma to treatment with vemurafenib, a BRAF inhibitor. Pituitary. 2016;19(5):544–546. doi:10.1007/s11102-015-0663-4
20. Roque A, Odia Y. BRAF-V600E mutant papillary craniopharyngioma dramatically responds to combination BRAF and MEK inhibitors. CNS Oncol. 2017;6(2):95–99. doi:10.2217/cns-2016-0034
21. Marucci G, de Biase D, Zoli M, et al. Targeted BRAF and CTNNB1 next-generation sequencing allows proper classification of nonadenomatous lesions of the sellar region in samples with limiting amounts of lesional cells. Pituitary. 2015;18(6):905–911. doi:10.1007/s11102-015-0669-y
22. Capper D, Preusser M, Habel A, et al. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathol. 2011;122(1):11–19. doi:10.1007/s00401-011-0841-z
23. Janku F, Tsimberidou AM, Garrido-Laguna I, et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther. 2011;10(3):558–565. doi:10.1158/1535-7163.MCT-10-0994
24. Juric D, Rodon J, Tabernero J, et al. Phosphatidylinositol 3-kinase alpha-selective inhibition with alpelisib (BYL719) in PIK3CA-altered solid tumors: results from the first-in-human study. J Clin Oncol. 2018;36(13):1291–1299. doi:10.1200/JCO.2017.72.7107
25. Li X, Wu C, Chen N, et al. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget. 2016;7(22):33440–33450. doi:10.18632/oncotarget.7961
26. Chen G, Wang Z, Zhou D. Lateral supraorbital approach applied to sellar tumors in 23 consecutive patients: the Suzhou experience from China. World J Surg Oncol. 2013;11:41. doi:110.1186/1477-7819-11-41
27. Taher MM, Hassan AA, Saeed M, et al. Next generation DNA sequencing of atypical choroid plexus papilloma of brain: identification of novel mutations in a female patient by Ion Proton. Oncol Lett. 2019. doi:10.3892/ol.2019.10882
28. Butt M, Alyami S, Nageeti TH, et al. Mutation profiling of anaplastic ependymoma grade III by Ion Proton by next generation DNA sequencing. [version 1; peer review: 1 approved, 1 approved with reservation]. F1000Research. 2019;8:613. doi:10.12688/f1000research.18721.1
29. Li SD, Ma M, Li H, et al. Cancer gene profiling in non-small cell lung cancers reveals activating mutations in JAK2 and JAK3 with therapeutic implications. Genome Med. 2017;9(1):89. doi:10.1186/s13073-017-0478-1
30. Ballester LY, Fuller GN, Powell SZ, et al. Retrospective analysis of molecular and immunohistochemical characterization of 381 primary brain tumors. J Neuropathol Exp Neurol. 2017;76(3):179–188. doi:10.1093/jnen/nlw119
31. Hara T, Akutsu H, Takano S, et al. Clinical and biological significance of adamantinomatous craniopharyngioma with CTNNB1 mutation. J Neurosurg. 2018;1–10. doi:10.3171/2018.3.JNS172528
32. Bartels S, Adisa A, Aladelusi T, et al. Molecular defects in BRAF wild-type ameloblastomas and craniopharyngiomas-differences in mutation profiles in epithelial-derived oropharyngeal neoplasms. Virchows Arch. 2018;472(6):1055–1059. doi:10.1007/s00428-018-2323-3
33. Goschzik T, Gessi M, Dreschmann V, et al. Genomic alterations of adamantinomatous and papillary craniopharyngioma. J Neuropathol Exp Neurol. 2017;76(2):126–134. doi:10.1093/jnen/nlw116
34. Bi WL, Greenwald NF, Ramkissoon SH, et al. Clinical identification of oncogenic drivers and copy-number alterations in pituitary tumors. Endocrinology. 2017;158(7):2284–2291. doi:10.1210/en.2016-1967
35. Kassab C, Zamler D, Kamiya-Matsuoka C, et al. Genetic and immune profiling for potential therapeutic targets in adult human craniopharyngioma. Clin Oncol Res. 2019;2(3):2–8. doi:10.31487/j.COR.2019.03.05
36. Dahl NA, Pratt D, Camelo-Piragua S, et al. Pediatric craniopharyngioma in association with familial adenomatous polyposis. Fam Cancer. 2019;18(3):327–330. doi:10.1007/s10689-019-00126-8
37. Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: wnt signaling in cancer. Biochim Biophys Acta. 2003;1653:1–24. doi:10.1016/s0304-419x(03)00005-2
38. Stamos JL, Weis WI. The β-catenin destruction complex. Cold Spring Harb Perspect Biol. 2013;5(1):a007898. doi:10.1101/cshperspect.a007898
39. Logan CY, Nusse R. The WNT signaling pathway in development and disease. Ann Rev Cell Dev Biol. 2004;20(1):781–810. doi:10.1146/annurev.cellbio.20.010403.113126
40. Pekmezci M, Louie J, Gupta N, Bloomer MM, Tihan T. Clinicopathological characteristics of adamantinomatous and papillary craniopharyngiomas: university of California, San Francisco experience 1985–2005. Neurosurgery. 2010;67(5):1341–1349. doi:10.1227/NEU.0b013e3181f2b583.
41. Capatina C, Vintila M, Gherlan I, et al. Craniopharyngioma - clinical and therapeutic outcome data in a mixed cohort of adult and paediatric cases. Acta Endocrinol. 2018;14(4):549–555. doi:10.4183/aeb.2018.549
42. Halac I, Zimmerman D. Endocrine manifestations of craniopharyngioma. Childs Nerv Syst. 2005;21(8–9):640–648. doi:10.1007/s00381-005-1246-x
43. Donson AM, Apps J, Griesinger AM, et al. Molecular analyses reveal inflammatory mediators in the solid component and cyst fluid of human adamantinomatous craniopharyngioma. J Neuropathol Exp Neurol. 2017;76(9):779–788. doi:10.1093/jnen/nlx061
44. Abu-Farsakh N, Sbeih I, Farsakh HA. Differential expression of immunohistochemistry markers for epithelial/squamous cells in adamantinomatous craniopharyngioma. Int Clin Pathol J. 2017;5(1):
45. Hofmann BM, Kreutzer J, Saeger W, et al. Nuclear beta-catenin accumulation as reliable marker for the differentiation between cystic craniopharyngiomas and Rathke cleft cysts: a clinico-pathologic approach. Am J Surg Pathol. 2006;30(12):1595–1603. doi:10.1097/01.pas.0000213328.64121.12
46. Aberle H, Schwartz H, Kemler R. Cadherin-catenin complex: protein interactions and their implications for cadherin function. J Cell Biochem. 1996;61(4):514–523. doi:10.1002/(SICI)1097-4644(19960616)61:4%3C514:AID-JCB4%3E3.0.CO;2-R
47. Behrens J, Vakaet L, Friis R, et al. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol. 1993;120:757–766. doi:10.1083/jcb.120.3.757
48. Andoniadou CL, Gaston-Massuet C, Reddy R, et al. Identification of novel pathways involved in the pathogenesis of human adamantinomatous craniopharyngioma. Acta Neuropathol. 2012;124(2):259–271. doi:10.1007/s00401-012-0957-9
49. Leichsenring J, Volckmar AL, Magios N, et al. Synonymous EGFR variant p.Q787Q is neither prognostic nor predictive in patients with lung adenocarcinoma. Genes Chromosomes Cancer. 2017;56(3):214–220. doi:10.1002/gcc.22427
50. Bonin S, Donada M, Bussolati G, et al. A synonymous EGFR polymorphism predicting responsiveness to anti-EGFR therapy in metastatic colorectal cancer patients. Tumour Biol. 2016;37(6):7295–7303. doi:10.1007/s13277-015-4543-3
51. Kim BW, Kim SK, Heo KW, et al. Association between epidermal growth factor (EGF) and EGF receptor gene polymorphisms and end-stage renal disease and acute renal allograft rejection in a Korean population. Ren Fail. 2020;42(1):98–106. doi:10.1080/0886022X.2019.1710535
52. Holsken A, Gebhardt M, Buchfelder M, Fahlbusch R, Blumcke I, Buslei R. EGFR signaling regulates tumor cell migration in craniopharyngiomas. Clin Cancer Res. 2011;17(13):4367–4377. doi:10.1158/1078-0432.CCR-10-2811
53. Holsken A, Stache C, Schlaffer SM, et al. Adamantinomatous craniopharyngiomas express tumor stem cell markers in cells with activated Wnt signaling: further evidence for the existence of a tumor stem cell niche? Pituitary. 2014;17(6):546–556. doi:10.1007/s11102-013-0543-8
54. Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J. 2011;437(2):199–213. doi:10.1042/BJ20101603
55. Maeng CH, Yi JH, Lee J, et al. Effects of single nucleotide polymorphisms on treatment outcomes and toxicity in patients treated with sunitinib. Anticancer Res. 2013;33(10):4619–4626.
56. Ishida M, Hotta M, Tsukamura A, et al. Malignant transformation in craniopharyngioma after radiation therapy: a case report and review of the literature. Clin Neuropathol. 2010;29(01):2–8. doi:10.5414/npp29002
57. Zhang J, Yang Y, Li W, et al. TP53 gene rs1042522 allele G decreases neuroblastoma risk: a two-centre case-control study. J Cancer. 2019;10(2):467–471. doi:10.7150/jca.27482
58. Altinkaya SO, Avcioglu SN, Sezer SD, Ceylaner S. Analysis of TP53 gene in uterine myomas: no mutations but P72R polymorphism is associated with myoma development. J Obstetrics Gynaecology Res. 2019;45(10):2088–2094. doi:10.1111/jog.14071
59. Sardi I, Giunti L, Donati P, et al. Loss of heterozygosity and p53 polymorphism Pro72Arg in a young patient with medulloblastoma. Oncol Rep. 2003;10(3):773–775.
60. Nigro JM, Baker SJ, Preisinger AC, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342(6250):705–708. doi:10.1038/342705a0
61. Bukovac A, Kafka A, Hrašćan R, Vladusic T, Pecina-Slaus N. Nucleotide variations of TP53 exon 4 found in intracranial meningioma and in silico prediction of their significance. Mol Clin Oncol. 2019;11(6):563–572. doi:10.3892/mco.2019.1936
62. Nickel GC, Barnholtz-Sloan J, Gould MP, et al. Characterizing mutational heterogeneity in a glioblastoma patient with double recurrence. PLoS One. 2012;7(4):e35262. doi:10.1371/journal.pone.0035262
63. Zha Y, Gan P, Liu Q, Yao Q. TP53 codon 72 polymorphism predicts efficacy of paclitaxel plus capecitabine chemotherapy in advanced gastric cancer patients. Arch Med Res. 2016;47(1):13–18. doi:10.1016/j.arcmed.2015.12.001
64. Kato K, Nakatani Y, Kanno H, et al. Possible linkage between specific histological structures and aberrant reactivation of the Wnt pathway in adamantinomatous craniopharyngioma. J Pathol. 2004;203(3):814–821. doi:10.1002/path.1562
65. Ashktorab H, Mokarram P, Azimi H, et al. Targeted exome sequencing reveals distinct pathogenic variants in Iranians with colorectal cancer. Oncotarget. 2017;8(5):7852–7866. doi:10.18632/oncotarget.13977
66. Chang YS, Lin CY, Yang SF, Ho CM, Chang JG. Analysing the mutational status of adenomatous polyposis coli (APC) gene in breast cancer. Cancer Cell Int. 2016;16(1):23. doi:10.1186/s12935-016-0297-2
67. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. doi:10.1126/science.1096502
68. Malentacchi F, Turrini I, Sorbi F, et al. Pilot investigation of the mutation profile of PIK3CA/PTEN genes (PI3K pathway) in grade 3 endometrial cancer. Oncol Rep. 2019;41(3):1560–1574. doi:10.3892/or.2018.6939
69. Gabelli SB, Huang CH, Mandelker D, Schmidt-Kittler O, Vogelstein B, Amzel LM. Structural effects of oncogenic PI3Kα mutations. Curr Top Microbiol Immunol. 2010;347:43–53. doi:10.1007/82_2010_53
70. Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A. 2007;104(13):5569–5574. doi:10.1073/pnas.0701005104
71. Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi:10.1126/science.1164382
72. Rudd ML, Price JC, Fogoros S, et al. A unique spectrum of somatic PIK3CA (p110α) mutations within primary endometrial carcinomas. Clin Cancer Res. 2011;17(6):1331–1340. doi:10.1158/1078-0432.CCR-10-0540
73. Samuels Y, Diaz LA
74. Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8(3):186–194. doi:10.1101/gr.8.3.186
75. Van Effenterre R, Boch AL. Craniopharyngioma in adults and children: a study of 122 surgical cases. J Neurosurg. 2002;97(1):3–11. doi:10.3171/jns.2002.97.1.0003
76. Karavitaki N, Brufani C, Warner JT, et al. Craniopharyngiomas in children and adults: systematic analysis of 121 cases with long-term follow-up. Clin Endocrinol (Oxf). 2005;62(4):397–409. doi:10.1111/j.1365-2265.2005.02231.x
77. Gleeson H, Amin R, Maghnie M. Do no harm’: management of craniopharyngioma. Eur J Endocrinol. 2008;159:S95–S99.
78. Srinivasan S, Ogle GD, Garnett SP, Briody JN, Lee JW, Cowell CT. Features of the metabolic syndrome after childhood craniopharyngioma. J Clin Endocrinol Metab. 2004;89:81–86.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.