Back to Journals » Journal of Hepatocellular Carcinoma » Volume 8
Acyl-CoA Thioesterase 7 is Transcriptionally Activated by Krüppel-Like Factor 13 and Promotes the Progression of Hepatocellular Carcinoma
Authors Xie X, Chen C, Feng S, Zuo S, Zhao X, Li H ![]()
Received 9 September 2021
Accepted for publication 1 December 2021
Published 23 December 2021 Volume 2021:8 Pages 1623—1641
DOI https://doi.org/10.2147/JHC.S338353
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Laura A. Dawson
Xingming Xie,1,2 Chaochun Chen,1,2 Shu Feng,1,3 Shi Zuo,2 Xueke Zhao,3 Haiyang Li1,2
1School of Clinical Medicine, Guizhou Medical University, Guiyang, Guizhou, People’s Republic of China; 2Department of Hepatobiliary Surgery, The Affiliated Hospital of Guizhou Medical University, Guiyang, Guizhou, People’s Republic of China; 3Department of Infectious Diseases, The Affiliated Hospital of Guizhou Medical University, Guiyang, Guizhou, People’s Republic of China
Correspondence: Haiyang Li
Department of Hepatobiliary Surgery, The Affiliated Hospital of Guizhou Medical University, 28 Guiyi Street, Guiyang, Guizhou, People’s Republic of China
Tel/Fax +86 851-6855119
Email [email protected]
Purpose: Acyl-CoA thioesterase 7(ACOT7) plays an important role in the metabolism of fatty acids. Hepatocellular carcinoma (HCC) has an abnormal lipid profile, and the role of ACOT7 in hepatocellular carcinoma has not been detailedly elucidated. Therefore, we conducted the study to explore the role of ACOT7 in HCC.
Materials and Methods: The ACOT7 and Krüppel-like factor 13 (KLF13) mRNA expression levels were obtained from The Cancer Genome Atlas (TCGA) database. Bioinformatics analysis identified the underlying upstream regulator of ACOT7. Quantitative real-time PCR was used to detect the expression of mRNA, and immunohistochemical staining and Western blotting were used to detect the expression of protein. Cell Counting Kit-8 and EdU assays were employed to assess the proliferation of HCC cells. Wound-healing and Transwell migration assays were utilized to test the migration ability of HCC cells. Dual-luciferase reporter assay and ChIP assay were used to explore the potential mechanism. Gas chromatography-mass spectrometer was used to analyze the content of free fatty acids. Xenograft tumour growth was used to evaluate the effect of ACOT7 in vivo.
Results: According to The Cancer Genome Atlas (TCGA) database, ACOT7 mRNA was found to be upregulated and predicted the poor prognosis. Overexpression of ACOT7 enhanced the proliferation, migration and invasion abilities of HCC cells in vitro, as well as the HCC cells proliferation in vivo. Moreover, ACOT7 overexpression increased the yield of the monounsaturated fatty acid Oleic acid (C18:1), which strengthened the proliferation and migration abilities of HCC cells. Mechanistically, KLF13 transcriptionally promoted ACOT7 expression. Further, KLF13 was also overexpressed in HCC tissues and facilitated HCC progression.
Conclusion: Acyl-CoA thioesterase 7 is transcriptionally activated by Krüppel-like factor 13 and promotes the progression of hepatocellular carcinoma.
Keywords: acyl-CoA thioesterase 7, Krüppel-like factor 13, hepatocellular carcinoma, unsaturated fatty acid, prognosis
Introduction
Liver cancer is one of the most common malignant tumors with the sixth highest morbidity and the third leading cause of cancer death worldwide in 2020, with approximately 906,000 new cases and 830,000 deaths, and the difficulty of early diagnosis, poor prognosis and limited treatment for liver cancer seriously threatens the physical and mental health of humans.1 Therefore, elucidation of the underlying molecular mechanism of HCC is urgently needed to discover novel biomarkers for diagnosis and treatment.
Acyl-CoA thioesterase 7 (ACOT7) (also known as BACH, CTE-II, ACT, ACH1, and BACHa), a major isoform of the ACOT family, is one of the most extensively studied ACOTs.2 ACOT7 is located in the cytosol and is abundant in brain tissue and testis3,4 and it has a vital role in neurotoxic prevention through regulation of neuronal fatty acid metabolism.5 Fatty acid alterations are involved in ovarian cancer,6 prostate cancer,7 colon cancer,8 and hepatocellular carcinoma.9,10 Acyl-CoA thioesterases hydrolyse fatty acyl-CoAs into free fatty acids and CoA-SH. ACOT7 is responsible for the hydrolysis of arachidonoyl-CoA (AA-CoA) to arachidonic acid (AA) and CoA. In addition, ACOT7 has been demonstrated to alter the abundance of prostaglandins D2 and E2 in a macrophage cell line.2 Jung et al11 reported that ACOT7 was highly expressed in lung cancer tissues and promoted cell cycle progression in breast and lung cancer cells. Cai et al12 reported that ACOT7 was upregulated and was correlated with a poor prognosis for HCC patients. However, the role of ACOT7 and its effect on fatty acid metabolism in hepatocellular carcinoma are still unclear.
Krüppel-like factors (KLFs), the transcription factors with a zinc-finger motif, regulate gene expression by binding to the promoter.13 KLFs are also involved in tumor development. KLF7 promoted ovarian cancer growth and dissemination.14 KLF8 bond to the promoter of GLUT4 to regulate glycolysis and was responsible for poor prognosis in gastric cancer.15 KLF16 activated MYC signaling and promoted tumor cell growth in prostate cancer.16 KLF2, a tumor suppressor, was inactivated by up-regulated long non-coding RNA DUXAP8 in hepatocellular carcinoma.17 KLF17, which inhibited EMT-related genes (ZO-1, Vimentin and Fibronectin), was inactivated by microRNA-9 in hepatocellular carcinoma.18 KLF13, a transcription factor, is abnormally expressed in a variety of cancers. KLF13 was downregulated in prostate carcinoma and inhibited prostate carcinoma cell proliferation by suppressing the AKT pathway.19 However, KLF13 was overexpressed in oral cancer cells, and knockdown of KLF13 decreased oral cancer cell proliferation and increased sensitivity to ionizing radiation.20 Recent studies have indicated that KLF13 plays a distinct role in cancer. However, the role of KLF13 in hepatocellular carcinoma remains unknown. In addition, KLF13 is involved in lipid metabolism, and it transcriptionally inhibited HMGCS1-mediated cholesterol biosynthesis in colorectal cancer.21 In the present study, we performed bioinformatics analysis and identified that ACOT7 was transcriptionally regulated by KLF13.
In the present study, in vitro and in vivo experiments demonstrated that ACOT7 acted as a proto-oncogene and promoted C18:1 free fatty acid production, which enhanced HCC cell proliferation and migration. Moreover, the expression of ACOT7 was transcriptionally activated by KLF13. Therefore, the KLF13/ACOT7/monounsaturated fatty acid axis may be a novel target for the diagnosis and treatment of hepatocellular carcinoma.
Materials and Methods
Patient Information
The ACOT7 or KLF13 mRNA differential expression was analyzed between normal and primary HCC in UALCAN website (http://ualcan.path.uab.edu/) based on The Cancer Genome Atlas (TCGA) database. And the survival analysis with ACOT7 was performed using the Kaplan–Meier method in UALCAN website. Twenty HCC tissues and corresponding adjacent noncancerous tissues were used for immunohistochemical staining, and 26 HCC tissues and corresponding adjacent noncancerous tissues were used for qRT-PCR. Patients’ tissues were collected from the Affiliated Hospital of Guizhou Medical University between January 2020 and August 2021. All patients were diagnosed with primary HCC. No treatment had been received for these patients before surgery. Patients had complete medical records. Patient consent was obtained for use of their tissues and medical records. Patients had no metastasis. The clinicopathologic characteristics of 26 HCC patients are summarized in Table 1. This study was approved by the Ethics Committee of the Affiliated Hospital of Guizhou Medical University (NO.2019-231). This research complied with the provisions of the Declaration of Helsinki.
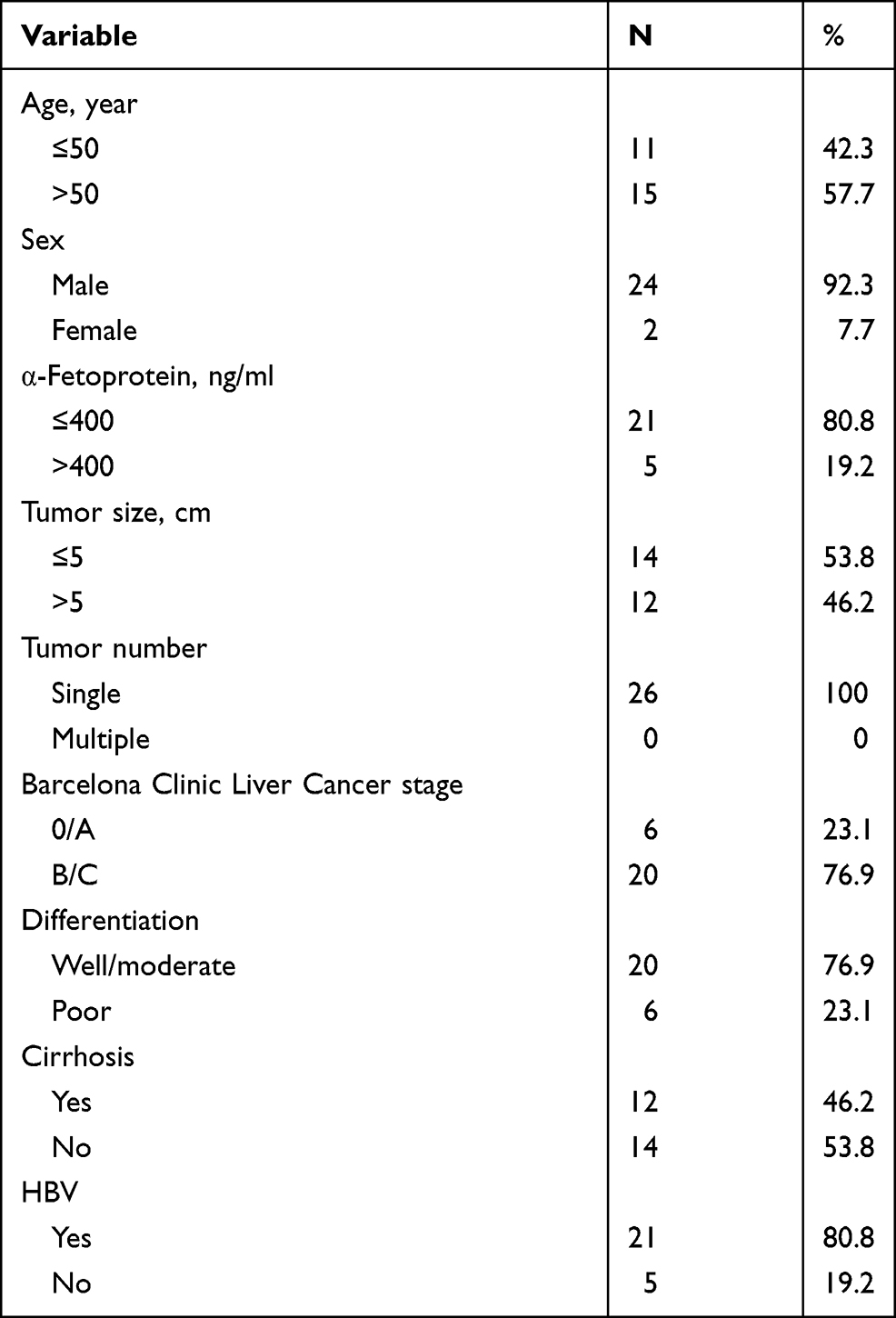 |
Table 1 Patient Information |
Hepatocellular Carcinoma Cell Lines
HCC cell lines (HepG2, SK-Hep1, HCCLM3, MHCC97H and Huh7) and 293T cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All cell lines were cultured in DMEM (GibcoBRL, Rockville, MD, USA) with 10% foetal bovine serum (04-001-1A, Biological Industries) and incubated at 37 °C with 5% CO2.
Plasmids, Transfection and Infection
ACOT7 and KLF13 were overexpressed in cells with lentiviruses constructed by GENECHEM (Shanghai, China). Knockdown of ACOT7 and KLF13 was conducted by shRNA in cells. The shRNA sequences were as follows: ACOT7 sh #1, GTCGCTGAAGAATGTGGACAA; ACOT7 sh #2, CCGGCATTGCAACAGCCAGAA; KLF13 sh #1, CGGGAAATCTTCGCACCTCAA; and KLF13 sh #2, CTAGCGGACCTCAACCAGCAA.
Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction
TRIzol reagent (Invitrogen) was used to extract mRNA. The cDNA was reverse transcribed using the PrimeScriptTM RT reagent Kit with gDNA Eraser (Perfect Real Time) (Cat# RR047A, TaKaRa), and TB Green Premix Ex TaqTM II (Tli RNaseH Plus) (Cat# RR820A TaKaRa) was used for qRT-PCR. The primer sequences were as follows: ACOT7 (forward primer, GCCTCCTGTTGTGTATTC; reverse primer, GTTCCTCCACTTGGTCTC); KLF13 (forward primer, CTCACACAGGTGAGAGGC; reverse primer, GTGCTTGGTCAGGTGGTC); and GAPDH (forward primer, GATCATCAGCAATGCCTC; reverse primer, GTCCTTCCACGATACCAA). All experiments were performed in triplicate, and the relative expression of ACOT7 and KLF13 was normalized to the expression of GAPDH using the 2‒ΔΔct method.
Cell Proliferation Assays
The Cell Counting Kit-8 (CCK-8, Dojindo) and EdU (BeyoClick™ EdU-594 Cell Proliferation Kit, Beyotime, China) assays were utilized for cell proliferation analysis according to the manufacturer’s instructions. EdU cells were imaged by a Zeiss laser scanning microscope.
Cell Migration and Invasion Assays
Wound-healing and transwell assays were used to evaluate migration. Cells were seeded into 6-well plates, and a 200 µL pipette tip was applied to wound the cell monolayer when grown to 100% confluency. The wound was photographed, which served as the 0 hour time point. Cells were incubated in medium without serum for 24 or 48 hours, and wound closure was photographed. For the transwell assay, cells (2×104 cells/200 µL) were seeded into the top chamber without serum in a 24-well polycarbonate transwell filter (8 µm pore size, Corning Incorporated, USA), which was precoated with (for invasion assay) or without (for migration assay) 30μL matrigel, and 20% foetal bovine serum (700 µL) was placed into the lower chamber. After incubation for 24 or 48 hours, cells grown in the polycarbonate transwell filter were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. A cotton swab was used to wipe cells from the top chamber, and migrated cells were photographed with an inverted microscope.
Apoptosis and Cell Cycle Analysis
An Annexin V-APC/7-AAD apoptosis kit (Multisciences Biotech Co., Ltd.) was used to detect cell apoptosis. The cell cycle was analysed by propidium iodide (PI) staining. A NAVIOS flow cytometer (Beckman Navios) was used to detect cell apoptosis and cycle distribution.
Lipid Extraction for GC-MS Analysis
For each sample, 1 mL of chloroform methanol (2:1) solution was added to a 2 mL centrifuge tube, and 100 mg glass beads were added to the sample followed by vortex oscillation for 30 seconds. The tube was then incubated in liquid nitrogen for 5 minutes, and the centrifuge tube was vibrated at 50 Hz for 1 minute. The procedure was repeated twice. Next, the samples were centrifuged at 12,000 rpm for 5 minutes, and the supernatants were extracted in a 15 mL centrifuge tube followed by the addition of 2 mL of 1% methanol sulfate solution and shaking for 1 minute. Subsequently, the extracted samples were esterified in 80 °C water for 30 minutes. After cooling, 1 mL of N-hexane was added to the extracted samples followed by vortexing for 30 seconds and left for 5 minutes. Then, 5 mL of H2O (4 °C) was added to the tube, and the extracted samples were washed followed by centrifugation at 3500 rpm for 10 min at 4 °C. All supernatants were removed and pipetted into a 2 mL centrifuge tube, and anhydrous sodium sulfate powder (100 mg) was added to remove excess water. The mixture was oscillated for 30 seconds followed by centrifugation at 12,000 rpm for 5 minutes. Finally, 300 µL of supernatants was pipetted into a 2 mL centrifuge tube with 15 µL of 500 ppm methyl salicylate as an internal standard for testing.
The following chromatographic conditions were used: the chromatographic column was a Thermo-TG-FAME capillary column (50 m × 0.25 mm ID × 0.20 µm) with a sample injection volume of 1 μL; the sample inlet temperature was 250 °C; the ion source temperature was 230 °C; the transmission line temperature was 250 °C; the quadrupole temperature was 150 °C; the temperature of the procedure was set to 80 °C for 1 minute and then increased to 160 °C at a rate of 20 °C/minute; the temperature of the procedure was increased to 196 °C at a rate of 3 °C/minute, kept at 196 °C for 8.5 minutes, increased to 250 °C (20 °C/minute) and held at 250 °C for 3 minutes; the carrier gas was helium; and the carrier gas flow rate was 0.63 mL/minute. MS spectra were acquired with an EI voltage of 70 eV and a m/z range of 45–450.
Cellular Immunofluorescence Assay
KLF13 localization was determined by a cellular immunofluorescence assay. Cells were seeded on coverslips in a 6-well plate. After 24 hours, coverslips were fixed with 4% paraformaldehyde for 30 minutes and permeabilized with 0.3% Triton X-100 for 30 minutes. Fixed cells were blocked with 3% goat serum albumin and incubated with rabbit polyclonal KLF13 antibody (1:100 dilution, 18352-1-AP, Proteintech) overnight at 4 °C. After washing three times with PBS, fixed cells were incubated with CoraLite594-conjugated goat anti-rabbit IgG (H+L) (1:100 dilution, SA00013-4, Proteintech) for 1 hour. Fixed cells were then washed three times with PBS and stained with DAPI. Images were subsequently captured by a Zeiss laser scanning microscope.
Luciferase Reporter Assay
293T cells were inoculated into a 96-well plate, and when the confluence reached 70%, the Renilla luciferase reporter plasmid, GV657–KLF13 or GV657–vector and GV238-ACOT7-WT or GV238-ACOT7-MUT promoter plasmid were contransfected with Lipofectamine™ 3000. After 36 hours, cells were lysed and measured by Dual–Glo luciferase Assay System(Promega) and the luciferase activity was obtained by normalizing with Renilla for each sample.
Chromatin Immunoprecipitation (ChIP) PCR
To investigate whether KLF13 binds to the promoter of the ACOT7 gene, a ChIP assay was conducted in Flag-KLF13-overexpressing HepG2 and Huh7 cells using a simple ChIP enzymatic chromatin IP kit (Cell Signaling) according to the manufacturer’s protocols. The qPCR was utilized to verify the presence of a KLF13-binding region in the ACOT7 promoter. The following qPCR primer sequences were used: forward, 5ʹ GAAGGCAGCTAAGGCCCTG −3ʹ and reverse, 5ʹ-GAGAGTCGTGGGCGGAAC −3ʹ. The antibodies included anti-normal rabbit IgG (Cell Signaling Technology, #2729) and anti-Flag (Cell Signaling Technology, #14793).
Western Blotting
Western blot protocol was performed as previously described.22 Primary antibodies included anti-ACOT7 (1:7000, 15972-1-AP, Proteintech, China), anti-KLF13 (1:1000, 18352-1-AP, Proteintech, China), anti-GAPDH (1:7000, 10494-1-AP, Proteintech, China), anti-β-tubulin (1:5000, #M20005, Abmart), anti-cyclin D1 (1:1000, 26939-1-AP, Proteintech, China), anti-CDK2 (1:1000, 10122-1-AP, Proteintech, China), anti-CDK4 (1:1000, 11026-1-AP, Proteintech, China), anti-E-cadherin (1:7000, 20874-1-AP, Proteintech, China), anti-N-cadherin (1:7000, 22018-1-AP, Proteintech, China) and anti-vimentin (1:5000, ab92547, Abcam). The secondary antibodies included HRP-conjugated Affinipure goat anti-rabbit IgG (H+L) (1:7000, SA00001-2, Proteintech, China) and HRP-conjugated Affinipure goat anti-mouse IgG (H+L) (1:7000, SA00001-1, Proteintech, China).
Immunohistochemical Staining
First, the tumour tissue sections were deparaffinized and rehydrated. Second, antigen retrieval was performed. Third, nonspecific binding sites were blocked with 5% BSA for 20 minutes. Fourth, the sections were incubated with primary antibodies against ACOT7 (1:400, 15972-1-AP, Proteintech, China), Ki-67 (1:10,000, 27309-1-AP, Proteintech, China) and PCNA (1:100, 60097-1-Ig, Proteintech, China) at 4 °C overnight. The sections were then incubated with the secondary antibody for 60 minutes at room temperature. Finally, the sections were photographed under a microscope after DAB staining and haematoxylin staining.
Animal Study
Animal study was approved by the Animal Ethics Committee of the Hospital Affiliated to Guizhou Medical University (NO.1901049). We complied with the guideline of Laboratory animal-General requirements (GB/T 35823-2018) issued by People’s Republic of China for the welfare and treatment of the laboratory animals. Four-week-old female BALB/c nude mice (each group n=5) obtained from Beijing Huafukang Co., Ltd. were subcutaneously injected with 1×107 ACOT7-SK-Hep1 cells or control SK-Hep1 cells in 150 µL of PBS. The tumour volume was recorded every 4 days. After 4 weeks, mice were sacrificed, and the tumours were harvested. Subsequently, HE and IHC were conducted.
Statistical Analysis
Student’s t-test was used for two groups, and one-way ANOVA was performed for more than two groups. GraphPad Prism 8 (GraphPad Software, USA) was utilized for statistical analysis and to generate graphs. All experiments were conducted three times. p < 0.05 was considered statistically significant.
Results
ACOT7 is Overexpressed in HCC Tissues
According to the analysis of the TCGA database, the ACOT7 transcript was upregulated and predicted poor prognosis (Figure 1A and B). To validate this result from TCGA database, we performed immunohistochemical staining, qRT-PCR and Western blotting using our samples. ACOT7 was highly expressed (65%, 13/20) in HCC tissues compared to adjacent noncancerous tissues (35%, 7/20) (Figure 1C). Western blotting and qRT-PCR showed that ACOT7 was overexpressed in HCC tissues compared to adjacent noncancerous tissues (Figure 1D). Taken together, the results suggested that HCC patients with high ACOT7 expression had a lower overall survival than those with relatively low expression.
 |
Figure 1 The ACOT7 expression in HCC samples. (A) The expression of ACOT7 in HCC samples in UALCAN website (http://ualcan.path.uab.edu/) based on The Cancer Genome Atlas (TCGA) database. (B) The Kaplan–Meier analysis of the overall survival rate of ACOT7 for HCC patients based on the TCGA database. (C) The protein expression of ACOT7 tested by immunohistochemistry in HCC and adjacent noncancerous tissues. (D) The ACOT7 expression by qRT-PCR and Western blotting in patients’ tissues. ***p<0.001. |
ACOT7 Enhances HCC Cell Proliferation
Stable models of ACOT7 overexpression in SK-Hep1 and MHCC97H cell lines and transient models of ACOT7 knockdown in HepG2 and Huh7 cell lines were established based on the expression levels of ACOT7 in HCC cells (Figure 2A and B). CCK-8 and EdU assays showed that ACOT7 overexpression significantly enhanced proliferation in SK-Hep1 and MHCC97H cell lines, while suppressing ACOT7 had the opposite effects (Figure 2C and D). Thus, these results indicated that ACOT7 promoted HCC cells proliferation in vitro.
 |
Figure 2 ACOT7 promoted HCC cells proliferation in vitro. (A) The expression levels of ACOT7 in HCC cell lines and overexpression of ACOT7 in SK-Hep1 and MHCC97H were verified by qRT-PCR and Western blotting. (B) The knockdown of ACOT7 was verified in HepG2 and Huh7 by Western blotting. (C) The CCK8 assay for proliferation in HCC cells with ACOT7 overexpression or knockdown. (D) EdU assay for proliferation. Unpaired two-tailed Student’s t-test was adopted. All data are shown as the mean±SD of 3 independent experiments. *p <0.05, **p<0.01, ***p<0.001. |
ACOT7 Promotes HCC Cell Migration and Invasion
To observe the effect of ACOT7 on the migration and invasion of HCC cells, wound-healing assay and transwell migration assays were performed. Compared to the control group, the wound-healing areas were greater in the ACOT7 overexpression groups but less in the ACOT7 shRNA groups (Figure 3A). Moreover, transwell migration assay and Matrigel invasion assay demonstrated that ACOT7 promoted SK-Hep1 and MHCC97H cells to pass through the translucent polycarbonate membrane, while ACOT7 shRNA decreased migratory cells (Figure 3B). In addition, Western blot analysis demonstrated decreased expression of E-cadherin but increased expression of N-cadherin and vimentin, epithelial–mesenchymal transition (EMT) markers, were observed in SK-Hep1 and MHCC97H cells with ACOT7 overexpression, and the opposite results were observed in HepG2 and Huh7 cells with ACOT7 knockdown (Figure 3C). These findings indicated that ACOT7 promoted the migration and invasion of HCC cells.
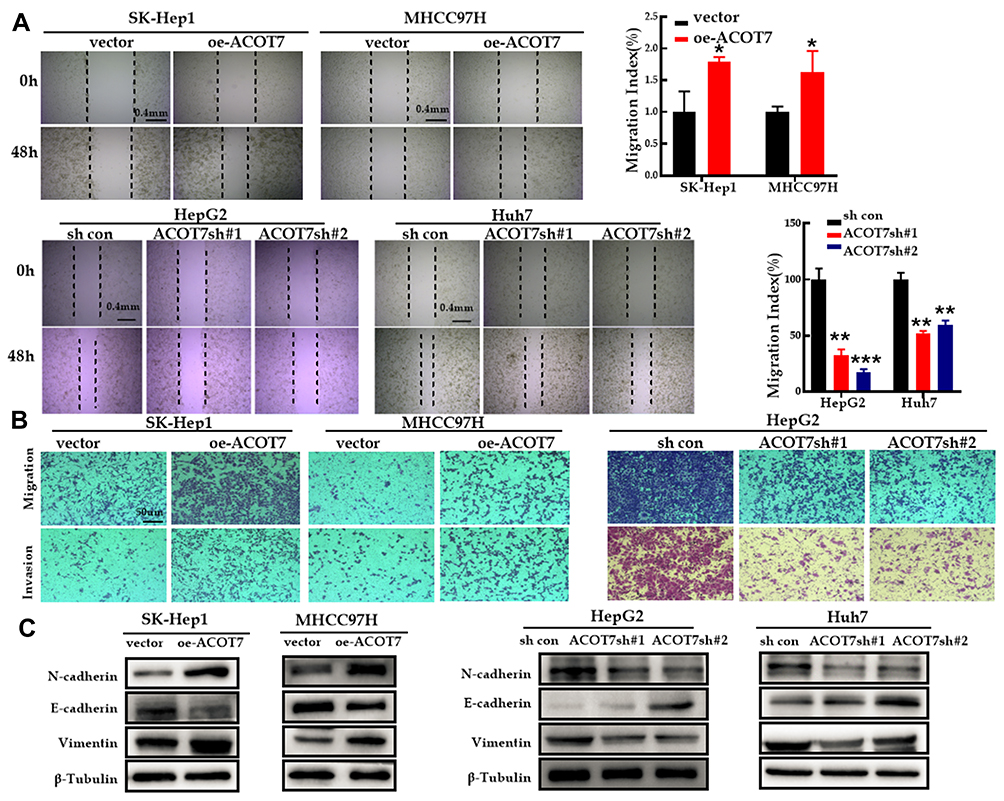 |
Figure 3 ACOT7 promoted migration and invasion of HCC cells. (A) Migratory properties of HCC cells with ACOT7 overexpression and knockdown were tested with wound healing assay. (B) Invasive properties of HCC cells with ACOT7 overexpression and knockdown were tested with transwell assay. (C) The expression of EMT-specific markers after ACOT7 overexpression and knockdown was verified by Western blotting. Unpaired two-tailed Student’s t-test was adopted. All data are shown as the mean±SD of 3 independent experiments. *p <0.05, **p<0.01, ***p<0.001. |
ACOT7 Promotes HCC Cell Cycle Progression and Reduces Apoptosis
Western blot analysis indicated that the expression of the G1 phase regulatory proteins, cyclin D1 and CDK2, but not CDK4, was significantly higher following ACOT7 overexpression, while the protein expression of cyclin D1 and CDK2 was downregulated with ACOT7 shRNA (Figure 4A). Furthermore, flow cell cycle analysis of SK-Hep1 and MHCC97H cells showed that ACOT7 overexpression promoted G1/S transition, decreased the number of cells in G1/G0 phase and increased the number of cells in S phase. In HepG2 and Huh7 cells, ACOT7shRNA inhibited G1/S transition (Figure 4B). For flow cytometry apoptosis analysis, ACOT7 reduced the apoptosis of HCC cells (Figure 4C). Thus, these results indicated that ACOT7 promoted G1/S transition and reduced apoptosis in HCC cells.
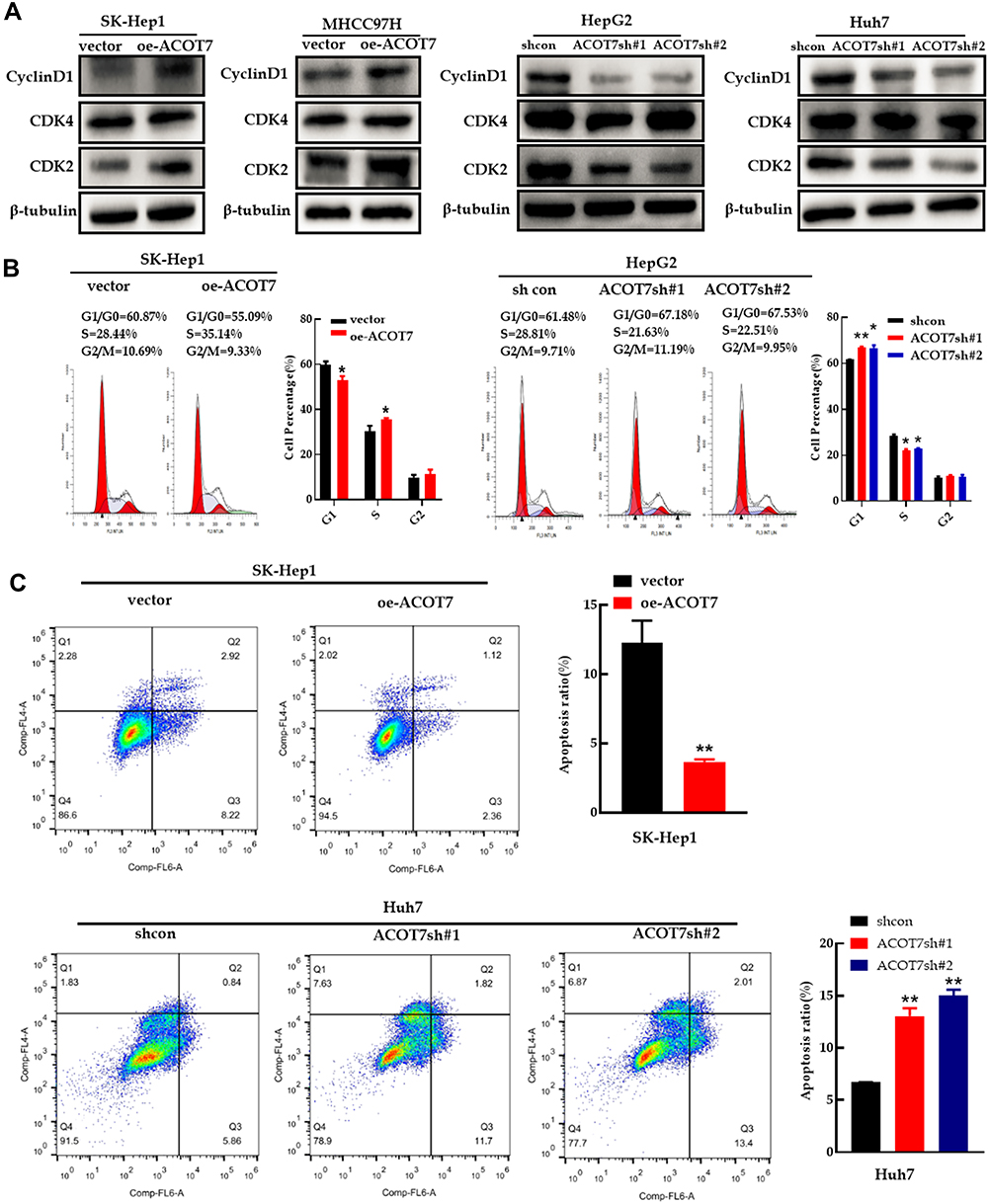 |
Figure 4 ACOT7 promoted cell cycle progression and inhibited apoptosis in HCC cells. (A) The expression of cell cycle-related proteins was verified by Western blotting. (B) Cell cycle analysis with flow cytometry in SK-Hep1 and HepG2 cells. (C) Apoptosis results of Annexin V-APC and 7-AAD staining by flow cytometry in SK-Hep1 and Huh7 cells. Unpaired two-tailed Student’s t-test was adopted. All data are shown as the mean±SD of 3 independent experiments. *p <0.05, **p<0.01. |
ACOT7 Upregulates the C18:1 Monounsaturated Fatty Acid Content and C18:1 Enhances the Proliferation and Migration of HCC Cells
ACOT7, one of the acyl-CoA thioesterases, mainly functions in the hydrolysis of long-chain acyl-CoAs. Budhu et al23 discovered that the content of monounsaturated fatty acids is increased in aggressive HCCs and promotes HCC cell proliferation, migration and invasion. To explore the role of ACOT7 in long-chain monounsaturated fatty acid metabolism in HCC cells, we determined the content of free fatty acids after overexpression of ACOT7 in SK-Hep1 cells and knockdown of ACOT7 in Huh7 cells via GC-MS analysis. We found that the content of oleic acid (C18:1) was largely regulated by overexpression or knockdown of ACOT7 (Figure 5A–C).
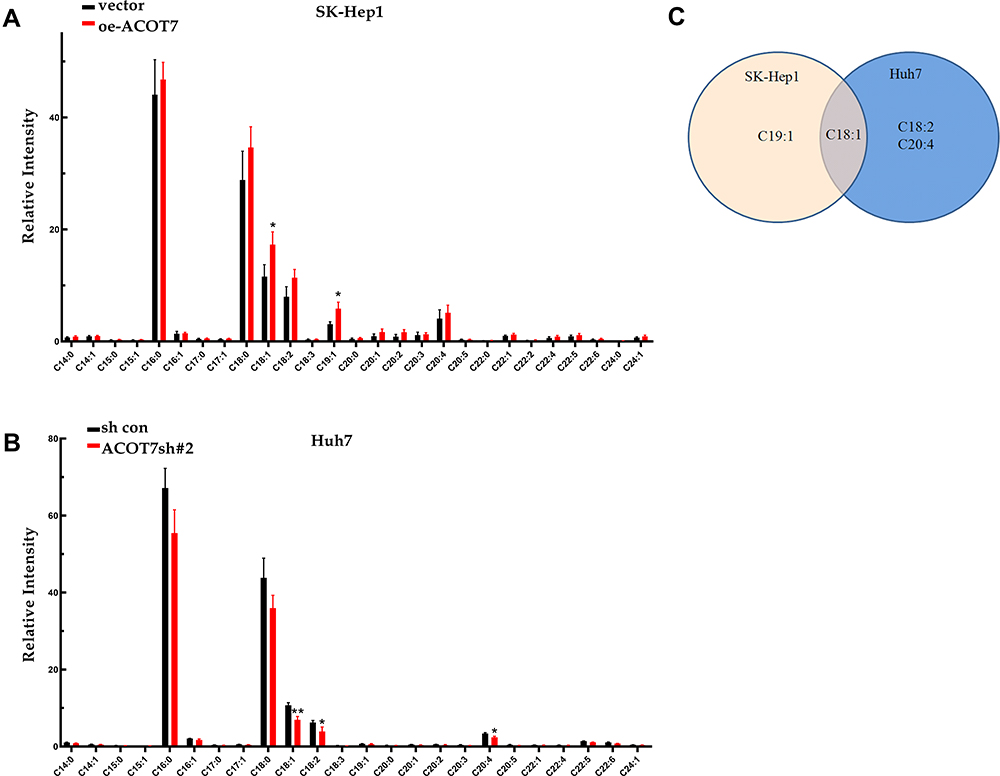 |
Figure 5 Changed Lipid profiles of HCC cell lines after ACOT7 overexpression and knockdown with GC-MS analysis. (A) Free fatty acid distributions in SK-Hep1 cells after ACOT7 overexpression. (B) Free fatty acid distributions in Huh7 cells after ACOT7 knockdown. (C) Overlapped changed free fatty acid after ACOT7 overexpression or knockdown. Unpaired two-tailed Student’s t-test was adopted. All data are shown as the mean±SD of 3 independent experiments. *p <0.05, **p<0.01. |
To elucidate the role of C18:1, CCK-8, EdU, wound-healing and transwell migration assays were employed. The CCK-8 assay indicated that increasing the C18:1 concentration (0, 50, 100, 150 and 200 µM) promoted SK-Hep1, HepG2, except for Huh7, cell proliferation in a dose-dependent manner (Figure 6A). EdU assay showed that C18:1(100 µM) significantly enhanced proliferation (Figure 6B). Wound-healing assay showed that C18:1 (100 µM) enhanced the healing ability of HCC cells (Figure 6C). In addition, the number of migrated cells after treatment with 100 µM C18:1 was increased compared to that in the control group (Figure 6D). Therefore, these results showed that ACOT7 upregulated the content of the C18:1 monounsaturated fatty acid, which enhanced the proliferation and migration of HCC cells.
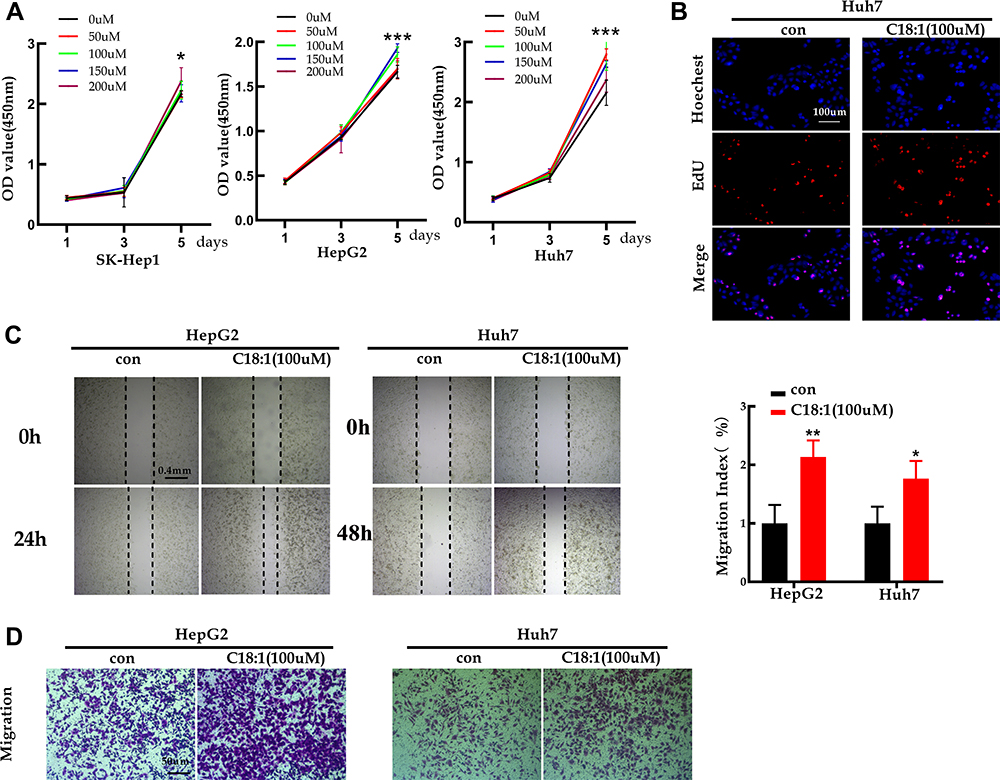 |
Figure 6 Monounsaturated fatty acid C18:1 enhanced the abilities of proliferation, migration for the HCC cells. (A) CCK8 for proliferation in C18:1-treated cells. (B) EdU assay for proliferation in C18:1-treated cells (100 µM). (C) Wound healing assay of C18:1-treated cells with a concentration 100 µM. (D) Migratory ability was detected for C18:1-treated cells in a concentration 100 µM by transwell. *p <0.05, **p<0.01, ***p<0.001. |
ACOT7 Facilitates Cell Proliferation in vivo
To further verify the effect of ACOT7 on cell proliferation in vivo, BALB/c nude mice were subcutaneously injected with 1×107 ACOT7-SK-Hep1 cells or control SK-Hep1 cells in 150 µL of PBS. The results indicated that ACOT7 overexpression significantly promoted cell proliferation (Figure 7A). Further analysis of xenografted tissues by immunohistochemical staining revealed that ACOT7 increased the protein levels of Ki-67 and PCNA (Figure 7B). Collectively, these results indicated that ACOT7 played a critical role in HCC cells proliferation in vivo.
 |
Figure 7 ACOT7 facilitated proliferation in vivo. (A) The image of tumors and tumor volume of the nude mice subcutaneously injected with 1×107 ACOT7-SK-Hep1 cells or control SK-Hep1 cells. (B) Representative HE staining and expression of ACOT7, Ki67 and PCNA determined by IHC. Unpaired two-tailed Student’s t-test was adopted. All data are shown as the mean±SD. ***p<0.001. |
The KLF13 Transcription Factor Induces ACOT7 Expression
ACOT7 enhances the content of C18:1 monounsaturated fatty acid, while KLF13 is involved in lipid metabolism, which transcriptionally inhibits HMGCS1-mediated cholesterol biosynthesis in colorectal cancer.21 Through JASPAR database analysis (http://jaspar.genereg.net/), we found that the transcription factor KLF13 binds to the promoter region of ACOT7 (Figure 8A). To validate the regulation of ACOT7 by KLF13 in HCC, we first investigated the expression profile of KLF13 in UALCAN website based on TCGA database. The level of KLF13 mRNA was upregulated in HCC tissues compared to normal tissues (Figure 8B). We further explored KLF13 expression in our patient’s tissues, and we observed that the protein level of KLF13 in HCC tissues was higher than that in adjacent nontumour tissues (Figure 8C). For the KLF13 transcription factor, immunofluorescence assay showed that KLF13 was mainly located in the nucleus (Figure 8D). Subsequently, we further investigated whether KLF13 affects the expression of ACOT7. First, we performed qRT-PCR for the mRNA expression of ACOT7 and KLF13 in the same HCC tissues. We observed that there was a positive correlation between ACOT7 mRNA and KLF13 mRNA level (Figure 8E). The upregulation of ACOT7 mRNA was observed in overexpressed KLF13-HCC cells (Figure 8F). Second, we generated HCC cells with stable and transient expression of KLF13 and found that the protein level of ACOT7 was upregulated or downregulated in HCC cells following KLF13 overexpression or knockdown (Figure 8G and H). Furthermore, luciferase reporter assay confirmed that KLF13 increased luciferase activity in ACOT7-WT group not in ACOT7-MUT group in 293T cell (Figure 8I), and the Chip-qPCR assay revealed that KLF13 bound to the promoter of ACOT7 (Figure 8J). Thus, these results indicated that KLF13 served as an oncogenic gene and KLF13 transcriptionally activated ACOT7 expression.
 |
Figure 8 The KLF13 transcription factor induced ACOT7 expression. (A) The binding motif of KLF13 in the promoter of ACOT7 from the JASPAR database analysis and the mutated binding sequence in the promoter of ACOT7. (B) The expression profile of KLF13 in UALCAN website (http://ualcan.path.uab.edu/) based on The Cancer Genome Atlas (TCGA) database. (C) The protein levels of KLF13 in HCC patients were verified by Western blotting. (D) KLF13 mainly was located in nuclear by immunofluorescence. (E) The correlation between KLF13 mRNA and ACOT7 mRNA. (F) The regulation of KLF13 overexpression for ACOT7 mRNA. (G) The expression levels of KLF13 in HCC cell lines. (H) ACOT7 was regulated by KLF13 in protein level. (I) The relative luciferase intensity in 293T cells cotransfected with the KLF13 and ACOT7-WT (containing a wild type (WT) KLF13-binding sequence) or ACOT7-MUT (containing a mutated (MUT) KLF13 binding sequence). Firefly luciferase activity was obtained by normalizing with Renilla for each sample. (J) ChIP-qPCR validation of Flag-KLF13 binding to the ACOT7 promoter. **p<0.01, ***p<0.001. |
KLF13 Enhances HCC Cell Proliferation
To understand the biological function of KLF13 in HCC cells, stable models of KLF13 overexpression in SK-Hep1 and MHCC97H cell lines and transient models of KLF13 knockdown in HepG2 and Huh7 cell lines were established based on the expression levels of KLF13 in HCC cells (Figure 8G and H). KLF13 overexpression significantly promoted the proliferation of SK-Hep1 and MHCC97H cells, while the opposite effects were observed in KLF13 knockdown HepG2 and Huh7 cells (Figure 9A). In addition, EdU assay also showed identical effects of KLF13 on HCC cell proliferation capacity (Figure 9B and C). Collectively, these results suggested that KLF13 promoted HCC cell proliferation in vitro.
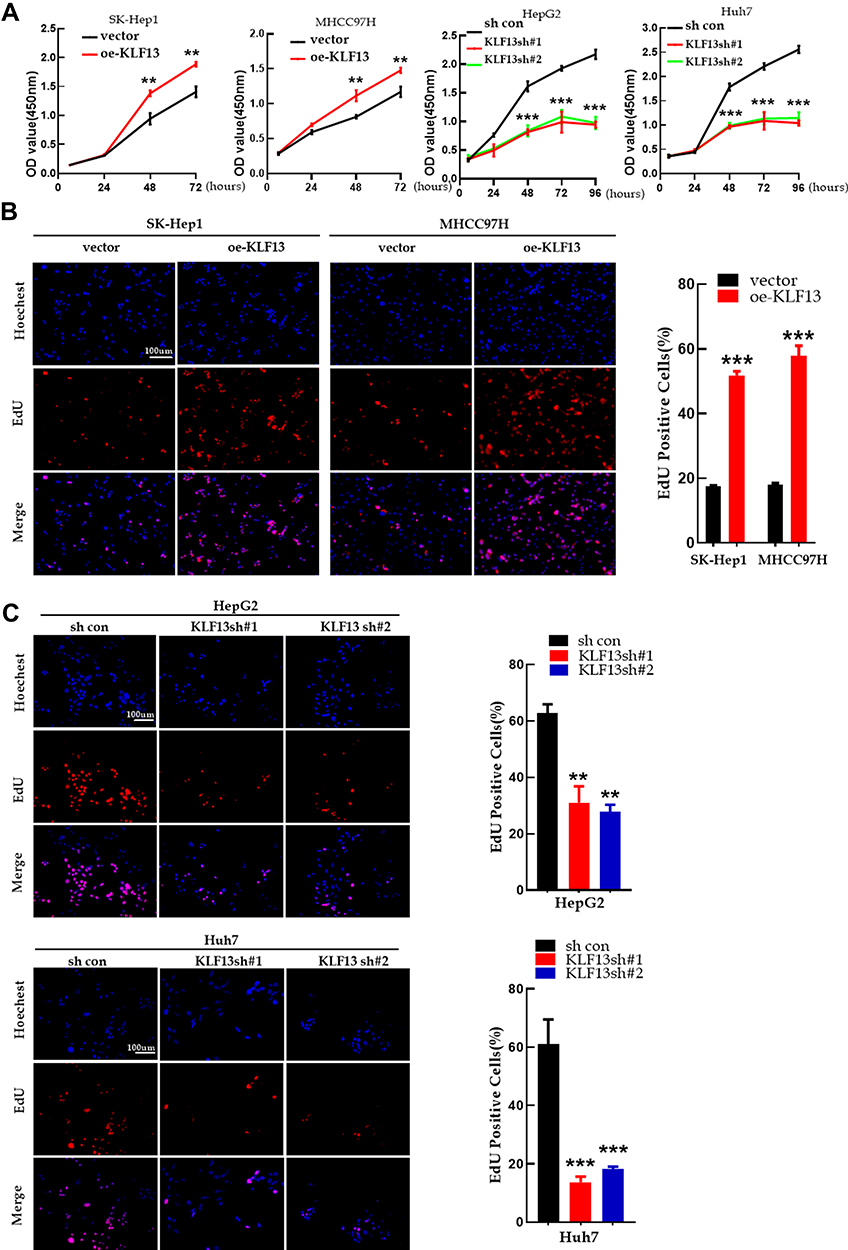 |
Figure 9 KLF13 promoted HCC cell proliferation in vitro. (A) The CCK8 assay for proliferation of KLF13 overexpression and knockdown. (B) EdU assay for proliferation of KLF13 overexpression and knockdown. (C) EdU assay for proliferation of KLF13 knockdown. Unpaired two-tailed Student’s t-test was adopted. All data are shown as the mean±SD of 3 independent experiments. **p<0.01, ***p<0.001. |
KLF13 Promotes HCC Cell Migration and Invasion
KLF13 overexpression remarkably increased the migration ability, but the migration ability was weakened in the KLF13 knockdown groups (Figure 10A). Transwell migration assay and matrigel invasion assay indicated that KLF13 promoted HCC cell migration and invasion (Figure 10B). In addition, Western blot analysis also demonstrated that decreased E-cadherin expression and increased vimentin and N-cadherin expression were observed in KLF13-overexpressing HCC cells, and the opposite results were observed in KLF13 knockdown cells (Figure 10C). These findings indicated that KLF13 promoted the migration and invasion of HCC cells.
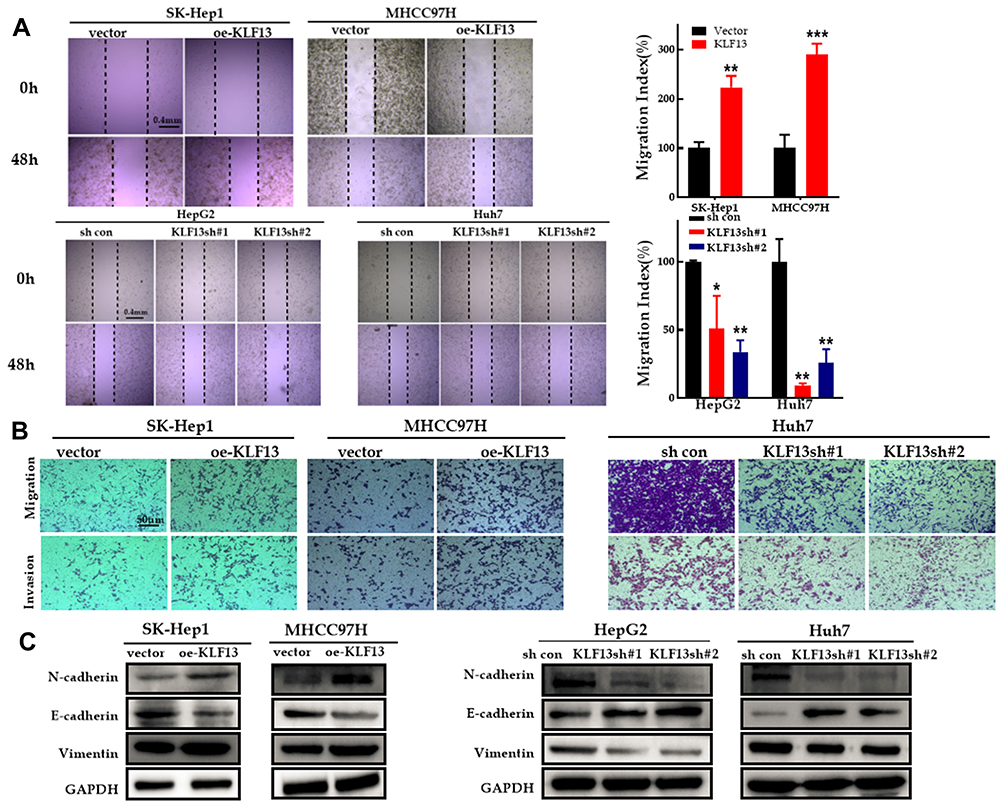 |
Figure 10 KLF13 promoted migration and invasion of HCC cells. (A) Migratory properties of HCC cells with KLF13 overexpression or knockdown were tested with wound healing assay. (B) Invasive properties of HCC cells with KLF13 overexpression or knockdown were tested with transwell assay. (C) The expression of EMT-specific markers after KLF13 overexpression or knockdown. Unpaired two-tailed Student’s t-test was adopted. All data are shown as the mean±SD of 3 independent experiments. *p <0.05, **p<0.01, ***p<0.001. |
KLF13 Promotes Cell Cycle Progression and Decreases Apoptosis in HCC Cells
For analysis of cell cycle progression, the G1 phase regulatory proteins, cyclin D1, CDK2 and CDK4, were increased in KLF13-overexpressing SK-Hep1 and MHCC97H cells, and the opposite results were observed in KLF13-deficient cells (Figure 11A). Similar results were concluded in the flow cell cycle analysis (Figure 11B). The flow cytometry apoptosis analysis indicated that KLF13 suppressed HCC cells apoptosis (Figure 11C). These results indicated that KLF13 decreased apoptosis and promoted cell cycle progression in HCC cells.
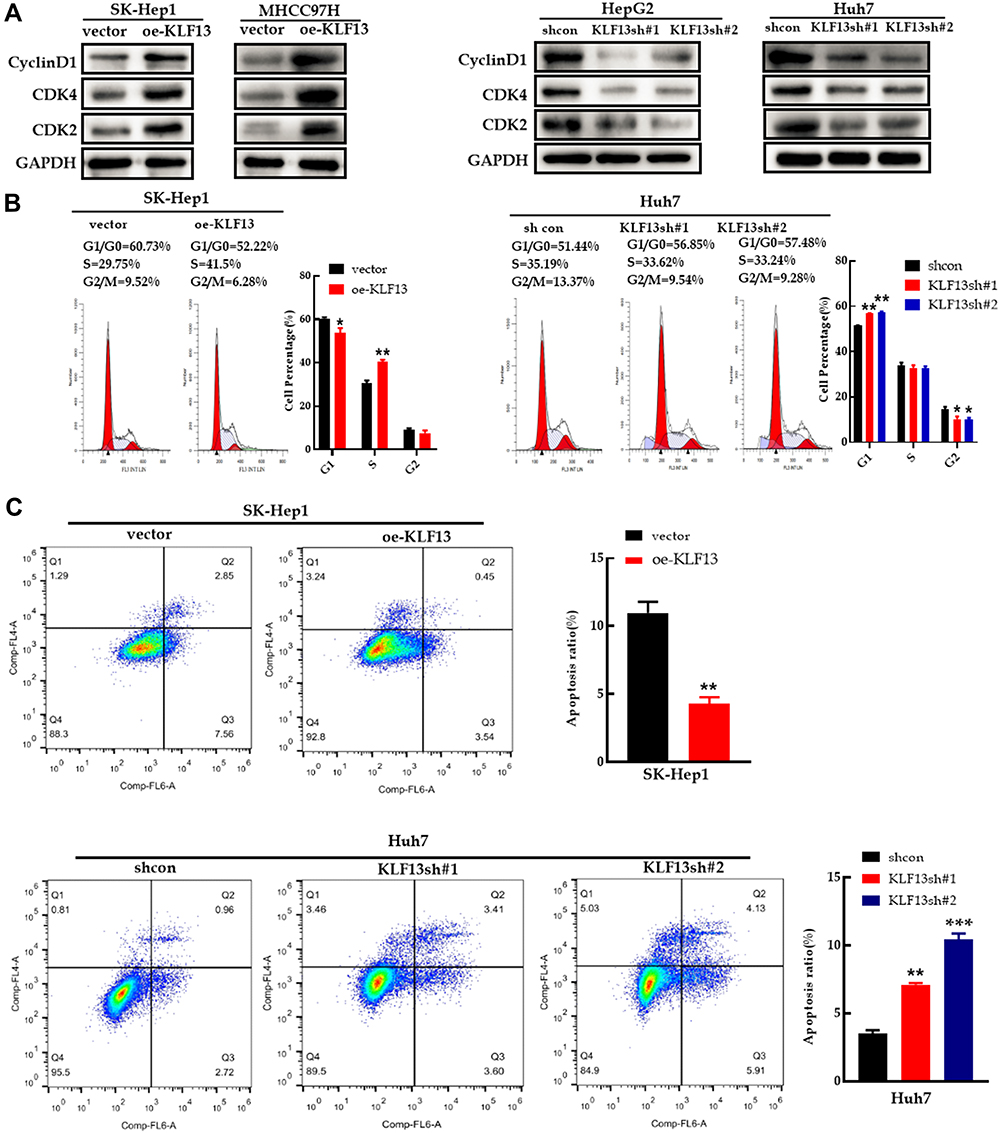 |
Figure 11 KLF13 promoted cell cycle progression and inhibited apoptosis in HCC cells. (A) The expression of cell cycle-related proteins in HCC cells with KLF13 overexpression or knockdown. (B) Cell cycle analysis with flow cytometry in HCC cells with KLF13 overexpression or knockdown. (C) Results of Annexin V-APC and 7-AAD staining by flow cytometry. Unpaired two-tailed Student’s t-test was adopted. All data are shown as the mean±SD of 3 independent experiments. *p <0.05, **p<0.01, ***p<0.001. |
ACOT7 Knockdown Inhibits KLF13-Mediated Proliferation and Migration
To further elucidate the regulation of ACOT7 by KLF13, functional inhibitory experiments were performed. We transfected ACOT7sh# 1 or ACOT7sh# 2 plasmid into lentivirus-KLF13 SK-Hep1 cells. First, KLF13 overexpression was verified in SK-Hep1 cells, and ACOT7 knockdown was confirmed in KLF13-overexpressing SK-Hep1 cells (Figure 12A). We then observed that ACOT7sh #1 or ACOT7sh #2 suppressed the proliferation of lentivirus-KLF13 SK-Hep1 cells (Figure 12B). In addition, ACOT7sh #1 or ACOT7sh #2 also suppressed the migration of lentivirus-KLF13 SK-Hep1 cells (Figure 12C and D). These data confirmed that KLF13 promoted HCC proliferation and migration via ACOT7.
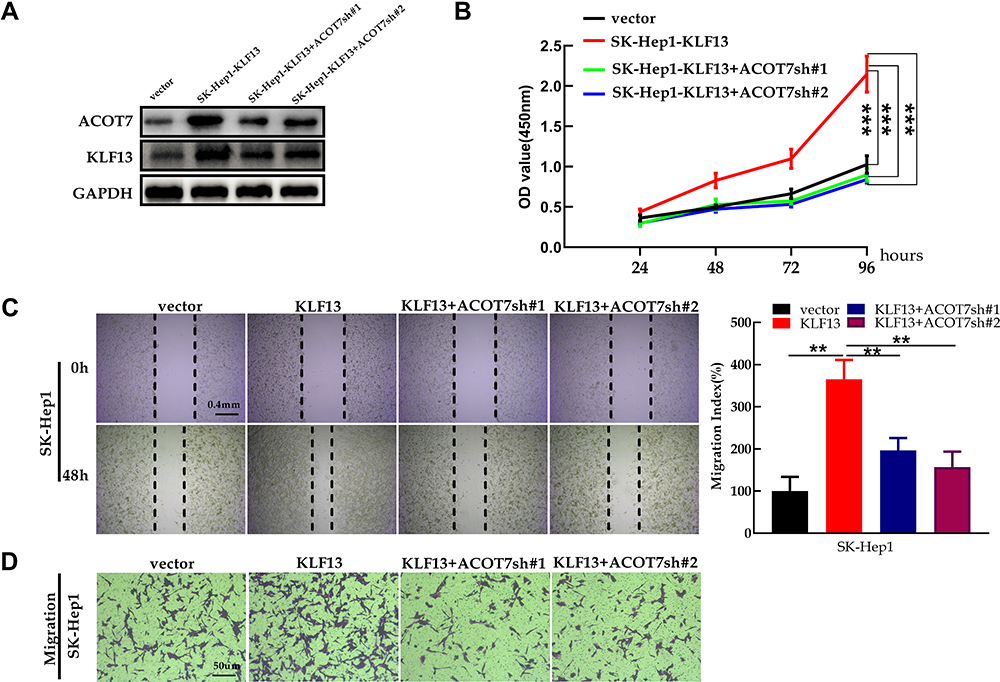 |
Figure 12 ACOT7 knockdown inhibited KLF13-mediated proliferation and migration. (A) The verification of KLF13 overexpression in SK-Hep1 and the confirmation of ACOT7 knockdown in KLF13- overexpressing SK-Hep1. (B) CCK8 assay for proliferation. (C) Cell migration was assessed by wound healing assay. (D) Cell migration was assessed by transwell assay. Unpaired two-tailed Student’s t-test was adopted. All data are shown as the mean±SD of 3 independent experiments. **p<0.01, ***p<0.001. |
Discussion
In the present study, we reported that acyl-CoA thioesterase 7 (ACOT7) promoted HCC cell progression via the elevated production of the C18:1 monounsaturated fatty acid. In addition, we discovered that the upstream gene, KLF13, a transcription factor, bound to the promoter of ACOT7 to promote the expression of ACOT7. Thus, these findings demonstrated that the KLF13/ACOT7/monounsaturated fatty acid axis plays an important role in the development of HCC and this axis maybe an underlying target for HCC treatment.
ACOT7 overexpression impairs glucose-stimulated insulin secretion in pancreatic β-cells, and ACOT7 mRNA is highly expressed in human type 2 diabetic islets.24 In acute myeloid leukaemia, ACOT7 is highly expressed, and ACOT7 overexpression predicts poor outcome for patients.25 In human lung carcinoma cells, ACOT7 promotes cell cycle progression.11 Through TCGA database analysis, we discovered that ACOT7 was upregulated in HCC tissues compared to normal tissues, and high ACOT7 expression was a risk factor for the survival time of HCC patients. In another study, it revealed that acyl-CoA thioesterase 7, a metabolic enzyme in the plasma, was upregulated and correlated with a poor prognosis for HCC patients.12 In the present study, we also revealed that ACOT7 was overexpressed in our HCC tissues. Next, we investigated the function of ACOT7 in HCC cells, which revealed that ACOT7 promoted HCC cell proliferation, migration and invasion as well as cell cycle progression. These results showed that ACOT7 serves as an oncogenic gene in HCC. Acyl-CoA thioesterases (ACOTs) possess the ability to hydrolyse acyl-CoAs to free fatty acids (FFAs) and free CoA.26–29 ACOT7, an acyl-CoA thioesterase, prevents neurotoxicity from fatty acid metabolism.5 In inflammatory disease, ACOT7 expression is induced by lipopolysaccharide, and upregulated ACOT7 elevates the abundance of unsaturated long (≥C20)-acyl chains in macrophages.2,30 The present study demonstrated that ACOT7 enhanced the content of the C18:1 monounsaturated fatty acid in HCC cells, and C18:1 strengthened the abilities of proliferation and migration for HCC cells. In non-small-cell lung cancer cells, the C18:1 monounsaturated fatty acid enhances the gefitinib-resistant and osimertinib-resistant ability.31 In colon cancer, C18:1 is critical for CSC stem cell maintenance.32 In clear cell renal cell carcinoma cells, C18:1 stimulates cell proliferation through stabilization of β-catenin.33 Clearly, the C18:1 monounsaturated fatty acid plays an important role in tumour progression. In addition, it has been reported that the content of C16:1, a monounsaturated fatty acid, is significantly decreased following overexpression of PTEN, a well-known tumour suppressor in goat mammary epithelial cells.34 Above all, monounsaturated fatty acids have a vital role in tumor progression. In HCC, the mechanism of C18:1 for the tumor progression needs to be further investigated. Collectively, ACOT7 overexpression was positively associated with the development of HCC and plays an important role in the unbalance of lipid metabolism.
KLF1–17 constitutes the Kruppel-like family (KLFs), which are zinc-finger transcription factors that bind to the gene promoter.35 For example, KLF7 promotes tumour growth and maintains the self-renewal of cancer stem cells in ovarian cancer.14 KLF8 enhances glycolysis by transcriptionally activating GLUT4 in gastric cancer.15 KLF13 suppresses proliferation and growth in colorectal cancer by transcriptionally inhibiting HMGCS1-mediated cholesterol biosynthesis.21 KLF13 is involved in lipid metabolism. In the present study, we found the KLF13-binding site in ACOT7 promoter through the JASPAR database, suggesting that it is theoretically feasible for KLF13 to modulate ACOT7 expression through binding to the ACOT7 promoter. First, we identified the effect of KLF13 on ACOT7 protein expression. Exogenous KLF13 overexpression increased ACOT7 expression, and KLF13 shRNA also reduced the expression of ACOT7. Through qRT-PCR, there was the evidence that KLF13 regulated the transcriptional expression of ACOT7. Further, we performed luciferase reporter assay and ChIP-qPCR and found that KLF13 bound to the promoter of ACOT7. Collectively, our study reveals the transcriptional regulation of ACOT7 by KLF13 in HCC.
Finally, we knocked down ACOT7 in KLF13-overexpressing SK-Hep1 cells and discovered that there was a remarkable weakness of KLF13 to promote HCC cell proliferation or migration. Henson et al20 discovered that KLF13 plays an oncogenic role in oral cancer cells. In this study, we also demonstrated that KLF13 is highly expressed in HCC tissues and presented an oncogenic role. These results indicated that KLF13 exerts its oncogenic function, at least in part, by modulating ACOT7 expression.
Conclusions
In summary, ACOT7 serves as an oncogene, and ACOT7-mediated monounsaturated fatty acids promote HCC development. Second, ACOT7 promotes the progression of HCC by KLF13 transcriptional activation. Third, KLF13 promotes HCC progression. In the future, KLF13/ACOT7/monounsaturated fatty acid axis may be a novel target for the diagnosis and treatment of hepatocellular carcinoma.
Abbreviations
ACOT7, Acyl-CoA thioesterase 7; TCGA, The Cancer Genome Atlas; HCC, Hepatocellular carcinoma; KLF13, Krüppel-like factor 13; qRT-PCR, quantitative Real-Time Reverse Transcription Polymerase Chain Reaction; CCK8, Cell Counting Kit-8; Chip PCR, Chromatin immunoprecipitation PCR; shRNA, short hairpin RNA.
Data Sharing Statement
The data described in this manuscript are contained in published articles or the experiment data are available upon reasonable request.
Ethics Statement
This study was approved by the Ethics Committee of the Affiliated Hospital of Guizhou Medical University (NO.2019-231) with written informed consent obtained from all patients. The animal study was approved by the Animal Ethics Committee of the Hospital Affiliated to Guizhou Medical University (NO.1901049).
Funding
This work is supported by the Science and Technology Program of Guizhou Province (No. [2020]4Y232) and the Cultivate project 2021 for National Natural Science Foundation of China, Affiliated Hospital of Guizhou Medical University.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hyuna S, Jacques F, Rebecca LS, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:3. doi:10.3322/caac.21660
2. Forwood JK, Thakur AS, Guncar G, et al. Structural basis for recruitment of tandem hotdog domains in acyl-CoA thioesterase 7 and its role in inflammation. Proc Natl Acad Sci USA. 2007;104(25):10382–10387. doi:10.1073/pnas.0700974104
3. Yamada J. Long-chain acyl-CoA hydrolase in the brain. Amino Acids. 2005;28:273–278. doi:10.1007/s00726-005-0181-1
4. Hunt MC, Greene S, Hultenby K, Svensson LT, Engberg S, Alexson SE. Alternative exon usage selectively determines both tissue distribution and subcellular localization of the acyl-CoA thioesterase 7 gene products. Cell Mol Life Sci. 2007;64(12):1558–1570. doi:10.1007/s00018-007-7062-6
5. Ellis JM, Wong GW, Wolfgang MJ. Acyl coenzyme A thioesterase 7 regulates neuronal fatty acid metabolism to prevent neurotoxicity. Mol Cell Biol. 2013;33:1869–1882. doi:10.1128/MCB.01548-12
6. West L, Yin Y, Pierce SR, et al. Docosahexaenoic acid (DHA), an omega-3 fatty acid, inhibits tumor growth and metastatic potential of ovarian cancer. Am J Cancer Res. 2020;10:4450–4463.
7. Ma Y, Zha J, Yang X, et al. Long-chain fatty acyl-CoA synthetase 1 promotes prostate cancer progression by elevation of lipogenesis and fatty acid beta-oxidation. Oncogene. 2021;40(10):1806–1820. doi:10.1038/s41388-021-01667-y
8. Peng S, Chen D, Cai J, et al. Enhancing cancer-associated fibroblast fatty acid catabolism within a metabolically challenging tumor microenvironment drives colon cancer peritoneal metastasis. Mol Oncol. 2021;15(5):1391–1411. doi:10.1002/1878-0261.12917
9. Yuan P, Mu J, Wang Z, et al. Down-regulation of SLC25A20 promotes hepatocellular carcinoma growth and metastasis through suppression of fatty-acid oxidation. Cell Death Dis. 2021;12:361. doi:10.1038/s41419-021-03648-1
10. Lin L, Ding Y, Wang Y, et al. Functional lipidomics: palmitic acid impairs hepatocellular carcinoma development by modulating membrane fluidity and glucose metabolism. Hepatology. 2017;66(2):432–448. doi:10.1002/hep.29033
11. Jung SH, Lee HC, Hwang HJ, et al. Acyl-CoA thioesterase 7 is involved in cell cycle progression via regulation of PKCζ-p53-p21 signaling pathway. Cell Death Dis. 2017;8:e2793. doi:10.1038/cddis.2017.202
12. Cai F, Song Y, Lu Y, Zhang Y, Hu Y, Su S. Analysis of plasma metabolic profile, characteristics and enzymes in the progression from chronic hepatitis B to hepatocellular carcinoma. Aging. 2020;12(14):14949–14965. doi:10.18632/aging.103554
13. Kaczynski J, Cook T, Urrutia R. Sp1- and krüppel-like transcription factors. Genome Biol. 2003;4(2):206. doi:10.1186/gb-2003-4-2-206
14. De Donato M, Babini G, Mozzetti S, et al. KLF7: a new candidate biomarker and therapeutic target for high-grade serous ovarian cancer. J Exp Clin Cancer Res. 2020;39(1):265. doi:10.1186/s13046-020-01775-9
15. Mao A, Zhou X, Liu Y, Ding J, Miao A, Pan G. KLF8 is associated with poor prognosis and regulates glycolysis by targeting GLUT4 in gastric cancer. J Cell Mol Med. 2019;23(8):5087–5097. doi:10.1111/jcmm.14378
16. Zhang J, Yu W, Wang X, Hu B, Wu D, Shi G. KLF16 affects the MYC signature and tumor growth in prostate cancer. Onco Targets Ther. 2020;13:1303–1310. doi:10.2147/OTT.S233495
17. Hao J, Xuefei S, Guochao Y, et al. Up-regulated long non-coding RNA DUXAP8 promotes cell growth through repressing krüppel-like factor 2 expression in human hepatocellular carcinoma. Onco Targets Ther. 2019;11:7429–7436. doi:10.2147/OTT.S214336
18. Zhao S, Qin H, Na Z, et al. MicroRNA-9 enhances migration and invasion through KLF17 in hepatocellular carcinoma. Mol Oncol. 2013;7:884–894. doi:10.1016/j.molonc.2013.04.007
19. Wang Q, Peng R, Wang B, et al. Transcription factor KLF13 inhibits AKT activation and suppresses the growth of prostate carcinoma cells. Cancer Biomark. 2018;22:533–541. doi:10.3233/CBM-181196
20. Henson BJ, Gollin SM. Overexpression of KLF13 and FGFR3 in oral cancer cells. Cytogenet Genome Res. 2010;128:192–198. doi:10.1159/000308303
21. Yao W, Jiao Y, Zhou Y, Luo X. KLF13 suppresses the proliferation and growth of colorectal cancer cells through transcriptionally inhibiting HMGCS1-mediated cholesterol biosynthesis. Cell Biosci. 2020;10:76. doi:10.1186/s13578-020-00440-0
22. Li Y, Liu J, Xiao Q, et al. EN2 as an oncogene promotes tumor progression via regulating CCL20 in colorectal cancer. Cell Death Dis. 2020;11:604. doi:10.1038/s41419-020-02804-3
23. Budhu A, Roessler S, Zhao X, et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology. 2013;144:1066–1075. doi:10.1053/j.gastro.2013.01.054
24. Martinez-Sanchez A, Pullen TJ, Chabosseau P, et al. Disallowance of Acot7 in β-cells is required for normal glucose tolerance and insulin secretion. Diabetes. 2016;65:1268–1282. doi:10.2337/db15-1240
25. Zhang X, Liu B, Zhang J, et al. Expression level of ACOT7 influences the prognosis in acute myeloid leukemia patients. Cancer Biomark. 2019;26:441–449. doi:10.3233/CBM-182287
26. Kirkby B, Roman N, Kobe B, Kellie S, Forwood JK. Functional and structural properties of mammalian acyl-coenzyme A thioesterases. Prog Lipid Res. 2010;49:366–377. doi:10.1016/j.plipres.2010.04.001
27. Kang HW, Niepel MW, Han S, Kawano Y, Cohen DE. Thioesterase superfamily member 2/acyl-CoA thioesterase 13 (Them2/Acot13) regulates hepatic lipid and glucose metabolism. FASEB J. 2012;26:2209–2221. doi:10.1096/fj.11-202853
28. Zhang Y, Li Y, Niepel MW, et al. Targeted deletion of thioesterase superfamily member 1 promotes energy expenditure and protects against obesity and insulin resistance. Proc Natl Acad Sci USA. 2012;109:5417–5422. doi:10.1073/pnas.1116011109
29. Hunt MC, Alexson SE. The role Acyl-CoA thioesterases play in mediating intracellular lipid metabolism. Prog Lipid Res. 2002;41:99–130. doi:10.1016/s0163-7827(01)00017-0
30. Wall VZ, Barnhart S, Kramer F, et al. Inflammatory stimuli induce acyl-CoA thioesterase 7 and remodeling of phospholipids containing unsaturated long (≥C20)-acyl chains in macrophages. J Lipid Res. 2017;58:1174–1185. doi:10.1194/jlr.M076489
31. Huang Q, Wang Q, Li D, et al. Co-administration of 20(S)-protopanaxatriol (g-PPT) and EGFR-TKI overcomes EGFR-TKI resistance by decreasing SCD1 induced lipid accumulation in non-small cell lung cancer. J Exp Clin Cancer Res. 2019;38:129. doi:10.1186/s13046-019-1120-4
32. Choi S, Yoo YJ, Kim H, et al. Clinical and biochemical relevance of monounsaturated fatty acid metabolism targeting strategy for cancer stem cell elimination in colon cancer. Biochem Biophys Res Commun. 2019;519:100–105. doi:10.1016/j.bbrc.2019.08.137
33. Kim H, Rodriguez-Navas C, Kollipara RK, et al. Unsaturated fatty acids stimulate tumor growth through stabilization of β-catenin. Cell Rep. 2015;13:495–503. doi:10.1016/j.celrep.2015.09.010
34. Yao DW, Ma J, Yang CL, et al. Phosphatase and tensin homolog (PTEN) suppresses triacylglycerol accumulation and monounsaturated fatty acid synthesis in goat mammary epithelial cells. J Dairy Sci. 2021;104:7283–7294. doi:10.3168/jds.2020-18784
35. McConnell BB, Yang VW. Mammalian krüppel-like factors in health and diseases. Physiol Rev. 2010;90:1337–1381. doi:10.1152/physrev.00058.2009
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.