Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Acute lipopolysaccharide exposure facilitates epileptiform activity via enhanced excitatory synaptic transmission and neuronal excitability in vitro
Authors Gao F, Liu Z, Ren W, Jiang W
Received 7 April 2014
Accepted for publication 5 June 2014
Published 16 August 2014 Volume 2014:10 Pages 1489—1495
DOI https://doi.org/10.2147/NDT.S65695
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Fei Gao,1,2 Zhiqiang Liu,3 Wei Ren,3 Wen Jiang1
1Department of Neurology, Xijing Hospital, Fourth Military Medical University, Xi’an 710032, People’s Republic of China; 2Department of Neurology, First Affiliated Hospital of Xi’an Medical University, Xi’an 710077, People’s Republic of China; 3College of Life Sciences, Shaanxi Normal University, Xi’an 710062, People’s Republic of China
Abstract: Growing evidence indicates brain inflammation has been involved in the genesis of seizures. However, the direct effect of acute inflammation on neuronal circuits is not well known. Lipopolysaccharide (LPS) has been used extensively to stimulate brain inflammatory responses both in vivo and in vitro. Here, we observed the contribution of inflammation induced by 10 μg/mL LPS to the excitability of neuronal circuits in acute hippocampal slices. When slices were incubated with LPS for 30 minutes, significant increased concentration of tumor necrosis factor α and interleukin 1β were detected by enzyme-linked immunosorbent assay. In electrophysiological recordings, we found that frequency of epileptiform discharges and spikes per burst increased 30 minutes after LPS application. LPS enhanced evoked excitatory postsynaptic currents but did not modify evoked inhibitory postsynaptic currents. In addition, exposure to LPS enhanced the excitability of CA1 pyramidal neurons, as demonstrated by a decrease in rheobase and an increase in action potential frequency elicited by depolarizing current injection. Our observations suggest that acute inflammation induced by LPS facilitates epileptiform activity in vitro and that enhancement of excitatory synaptic transmission and neuronal excitability may contribute to this facilitation. These results may provide new clues for treating seizures associated with brain inflammatory disease.
Keywords: lipopolysaccharide, hippocampus, inflammation, epileptiform activity, synaptic transmission, neuronal excitability
Introduction
Brain injuries such as trauma, stroke, and infection are often associated with acute occurrence of seizures.1,2 Although the underlying mechanism remains unclear, accumulating clinical and experimental evidence has suggested that inflammatory processes involved in these injuries may contribute to the genesis of seizures.3 Traditionally, the brain has been considered an immunoprivileged organ because of the presence of the blood–brain barrier and the lack of a conventional lymphatic system. Nevertheless, both the innate and adaptive immune responses are readily evoked within the brain after varied injuries. Resident cells in brain parenchyma such as microglia, astrocytes, and neurons can respond to these stimuli and create inflammatory molecules. In addition, peripheral immune cells extravasating from cerebral vascular can also produce inflammatory media and aggravate inflammatory surroundings.4 These inflammatory mediators, including tumor necrosis factor α (TNF-α), interleukin 1β (IL-1β), nitric oxide (NO), and reactive oxygen species, are reported to increase cellular excitability.5–7
In animal experiments, lipopolysaccharide (LPS), a major component of the outer membrane of gram-negative bacteria, has been used extensively in investigating mechanisms of brain inflammation both in vivo and in vitro. Mainly in microglia,8 by the stimulation of toll-like receptor 4 LPS results in the induction of transcriptional factors such as nuclear factor κB, which trigger various proinflammatory genes such as those encoding cytokines, chemokines, proteins of the complement system, and inducible nitric oxide.9,10 Furthermore, LPS induces a rapid glutamate (a major excitatory neurotransmitter in brain) release in rat cortex slices, which may lead to an imbalance between excitation and inhibition in the neuronal circuit.11 In vivo, previous studies showed that LPS intraperitoneal or intracerebroventricular injection enhanced seizure susceptibility through increasing IL-1β, cyclooxygenase 2, NO, or prostaglandins in different epilepsy models.12–15 However, little is known about the direct effects of the acute application of LPS on epileptiform activity, synaptic strength, and neuronal excitability in vitro and the possible underlying mechanisms.
In the electrophysiology of seizure, an imbalance between excitation and inhibition is thought to mediate seizure activity. Increased excitation and/or decreased inhibition can induce the initiation of a seizure. Neuronal excitability and synaptic transmission are both involved in maintaining this balance.16,17 Therefore, in the present study, we focused on the effects of acute inflammation induced by LPS on the epileptiform discharges, neuronal excitability, and synaptic transmission in hippocampal slices.
Materials and methods
Slices preparation
All procedures used were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Fourth Military Medical University Animal Care Committee. Sprague-Dawley rats (14–17 days old) were anesthetized with chloral hydrate (400 mg/kg intraperitoneally) and decapitated. Brain was removed rapidly, and transverse 400 μM hippocampal slices were obtained with a Vibratome 1,000 plus (Vibratome Company, St Louis, MO, USA) in ice-cold oxygenated (95% O2/5% CO2) artificial cerebrospinal fluid (ACSF) containing (in mM) 125 NaCl, 3 KCl, 2.4 CaCl2, 1.2 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, and 10 glucose at pH 7.4 when gassed with 95% O2 and 5% CO2. In Mg2+-free ACSF, MgCl2 was omitted, and the concentration of NaCl was adjusted to 126 mM for stable osmolarity. Slices were transferred to a holding chamber and incubated in oxygenated ACSF for at least 1 hour before electrophysiological recording.
All chemicals, unless otherwise stated, were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). LPS (Escherichia coli serotype O55:B5) was prepared in 18 MΩ water and added to ACSF as needed. The final concentration of LPS used was 10 μg/mL. This concentration was chosen because it was previously used in acute brain hippocampal slices in vitro.18
Enzyme-linked immunosorbent assay
After being incubated in oxygenated ACSF for 1 hour, slices were transferred to 12 multiwell plates and treated with 10 μg/mL LPS for 30 minutes. The treated slices and untreated control slices were homogenized and centrifuged at 8,000 rpm for 15 minutes at 4°C. The supernatant was collected for subsequent analysis of cytokine concentration. Commercially available enzyme-linked immunosorbent assay kits for IL-1β and TNF-α were used according to the manufacturer’s instructions (Westang Bio-tech Co, Ltd, Shanghai, People’s Republic of China).
Electrophysiological recordings
Slices were transferred to an immersion recording chamber and perfused with oxygenated ACSF continuously (flow rate, 1–2 mL/minute). Patch electrodes were pulled by a electrode puller (model P-97; Sutter Instrument Company, Novato, CA, USA) and had a resistance of 3–5 MΩ when filled with the internal solution that contained (in mM) 120 K-gluconate, 20 KCl, 0.2 ethylene glycol tetraacetic acid, 10 HEPES (N-2-hydroxyethylpiperazine-N-ethane-sulphonicacid), 2 MgCl2, 4 Na2-ATP (adenosine triphosphate), 0.3 Tris-GTP (guanosine triphosphte), and 7 phosphocreatine at pH adjusted to 7.2–7.3 with KOH. Whole-cell recordings of hippocampal CA1 pyramidal neurons in the current- or voltage-clamp modes were performed with a Multi-Clamp 700B amplifier (Axon Instruments, Union City, CA, USA) in all electrophysiological experiments. Epileptiform discharges were induced with 100 μM 4-aminopyridine (4-AP) added to Mg2+-free ACSF. Synaptic responses were evoked by Schaffer collaterals stimulation through a pair of Elgiloy electrodes (Elgin Specialty Metals, Elgin, ILL, USA) placed in the stratum radiatum near the border of the CA1 pyramidal layer. Stimuli were single delivered at 0.033 Hz via an isolated pulse stimulator (model 2100; AM System Inc, Billerica, MA, USA), and stimulation intensity (0.1 ms, 50–200 μA) was adjusted to produce evoked excitatory postsynaptic current (eEPSC) or evoked inhibitory postsynaptic current (eIPSC) amplitudes that were 60%–70% the maximal responses in voltage-clamp experiments. Membrane potential was held at -70 mV for recording eEPSCs and -40 mV for eIPSCs. For eIPSC recording, glutamatergic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid and N-methyl-D-aspartate receptor antagonists (6,7-dinitroquinoxaline-2,3-dione, 20 μM; aminophosphonovaleric acid, 50 μM) were added to ACSF; for eEPSCs recording, γ-aminobutyric acid type A receptor antagonist (Pierotoxin, 100 μM) was added. Action potential frequency was calculated in response to a series of current steps 1 second in duration (10 pA per step, 0–200 pA). Neuron was rejected if the resting membrane potential was more positive than -60 mV or if action potential amplitude was less than 60 mV. Data were filtered at 2 KHz and transferred to the hard disk of a Dell computer (Round Rock, TX, USA). The software pCLAMP 10.0 (Axon Instruments, Union City, CA, USA) was used for data analysis offline.
Statistics
SPSS 16.0 (IBM Inc, Chicago, IL, USA) for Windows was used for statistical analysis. Data were expressed as mean ± standard deviation. Statistical significance was assessed using paired-sample Student’s t-test or two-factor (current and treatment) analysis of variance for electrophysiological recordings and independent-samples Student’s t-test for cytokine concentration. P<0.05 was considered statistically significant.
Results
LPS induces significant increased concentration of TNF-α and IL-1β in brain slices
After treatment with LPS for 30 minutes, there was a significantly increased level of IL-1β in brain slices compared with in untreated slices (LPS, 67.2±10.43 pg/mg, versus control, 38.6±10.67 pg/mg; n=5; P<0.01; Figure 1A). Exposure to LPS for 30 minutes also elevated TNF-α concentration in brain slices (LPS, 30.4±7.96 pg/mg, versus control, 13.8±4.60 pg/mg; n=5; P<0.01; Figure 1B). These results suggest that LPS induced inflammatory response in brain slices.
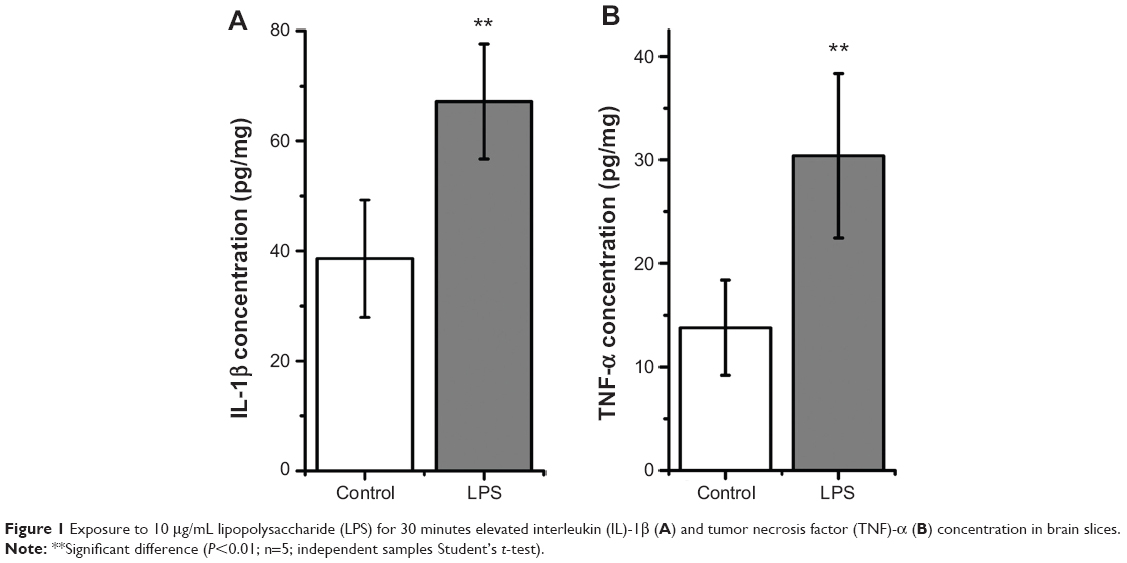 | Figure 1 Exposure to 10 μg/mL lipopolysaccharide (LPS) for 30 minutes elevated interleukin (IL)-1β (A) and tumor necrosis factor (TNF)-α (B) concentration in brain slices. |
LPS facilitates epileptiform discharge induced by Mg-free ACSF plus 4-AP
Under current-clamp conditions, slices were perfused with 100 μM 4-AP + Mg2+-free ACSF to induce epileptiform discharges. For hippocampal CA1 pyramidal neurons of rat in vitro, epileptiform discharges were characterized by spontaneous depolarization and bursts with multiple action potentials (Figure 2A), in agreement with a previous report.19 After about 10 minutes of stable epileptiform discharge recording, LPS was added. The counts of spontaneous bursts and spikes during bursts in 5 minutes were calculated before and 30 minutes after infusion of LPS. LPS statistically strengthened the frequency of epileptiform discharges (4-AP, 26.38±5.04, versus LPS, 45.38±6.74; n=8; P<0.001; Figure 2B and D) and also increased the number of spikes per burst (control, 5.01±0.95, versus LPS, 9.58±0.59; n=8; P<0.001; Figure 2E and F). After a 30 minute washout with 100 μM 4-AP + Mg2+-free ACSF, the augmentation of burst (LPS, 45.38±6.74, versus washout, 43.26±5.23; n=8; P>0.05) and spikes per burst (LPS, 9.58±0.59, versus washout, 10.16±0.51; n=8; P>0.05) remained. These data suggest LPS facilitates epileptiform activity in hippocampal CA1 pyramidal neurons in vitro. This effect has been maintained for at least 30 minutes after LPS washout.
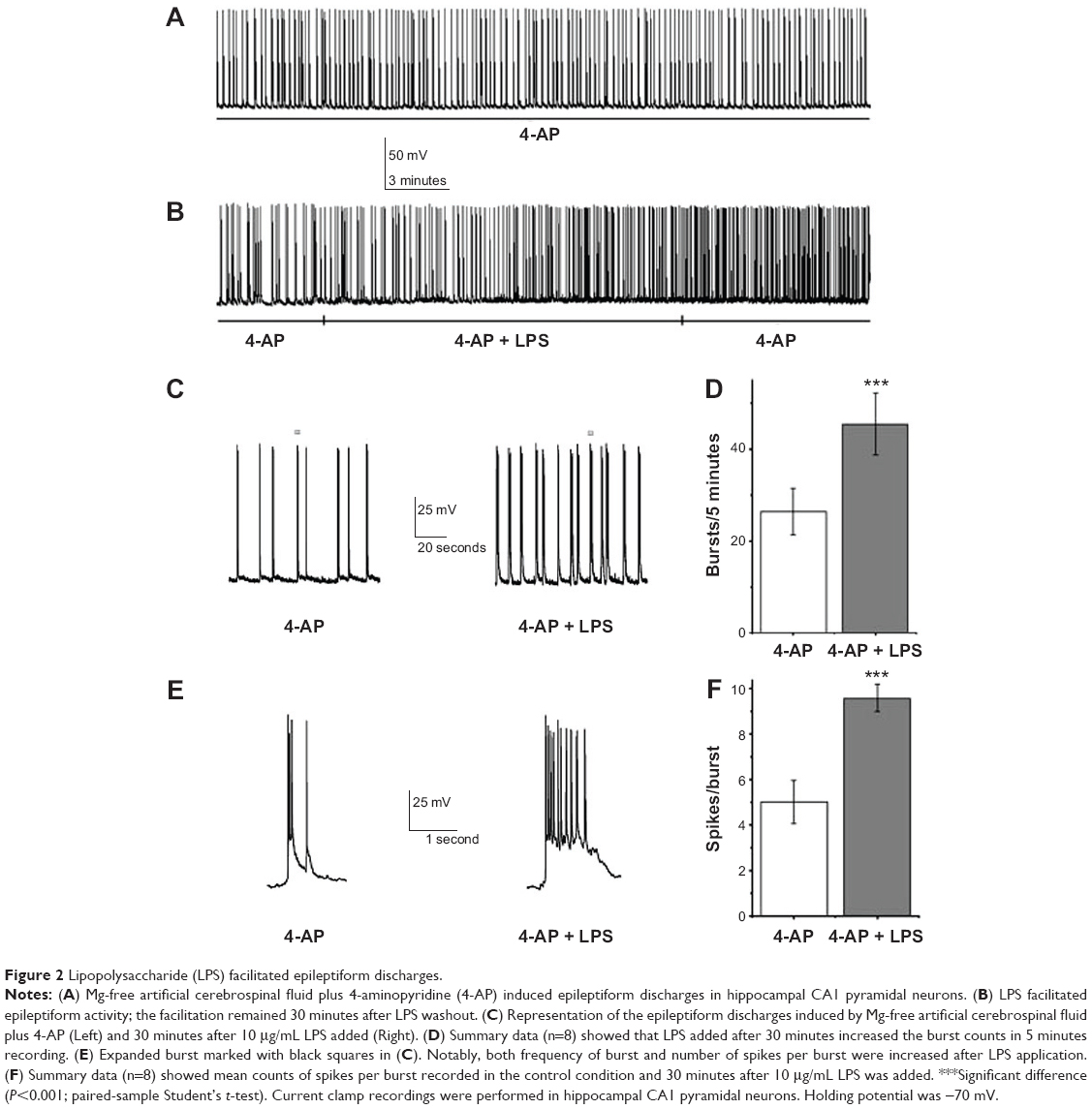 | Figure 2 Lipopolysaccharide (LPS) facilitated epileptiform discharges. |
LPS enhances eEPSCs and does not modify eIPSCs
To explore the underlying mechanisms mediating the facilitated epileptiform activity induced by LPS, we observed the effects of LPS on excitatory and inhibitory synaptic transmission. eEPSCs were recorded by stimuli of Schaffer collaterals in voltage-clamp mode. After 10 minutes, baseline was recorded and slices were perfused with LPS-containing ACSF. There was a significant increase in eEPSC amplitudes 30 minutes after LPS application (193.75%±31.98% control; n=5; P<0.01; Figure 3A and C). In addition to enhanced excitatory synaptic transmission, decreased inhibitory synaptic transmission may contribute to genesis of seizure. Therefore, we recorded eIPSCs to assess the change of inhibitory synaptic transmission. In this proceeding, holding potential was -40 mV to enlarge the amplitude of eIPSCs. Perfusion with ACSF containing LPS had no effects on inhibitory synaptic transmission. The difference between eIPSC amplitudes in control slices and slices bathed with ACSF containing LPS for 30 minutes was not significant (103.38%±10.87% control; n=5; P>0.05; Figure 3B and D).
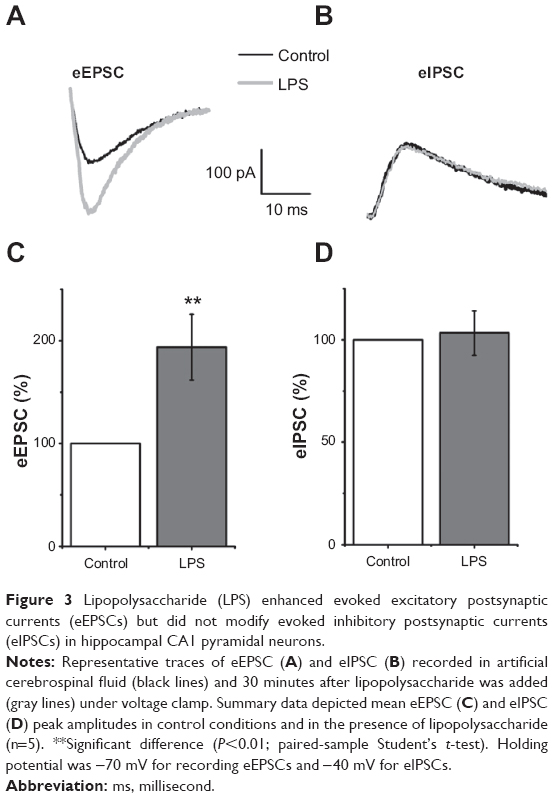 | Figure 3 Lipopolysaccharide (LPS) enhanced evoked excitatory postsynaptic currents (eEPSCs) but did not modify evoked inhibitory postsynaptic currents (eIPSCs) in hippocampal CA1 pyramidal neurons. |
LPS enhances the excitability of hippocampal CA1 pyramidal neurons
In addition to synaptic mechanisms, the facilitated epileptiform discharges by LPS may also be attributed to the increased excitability of hippocampal CA1 pyramidal neurons. To address this issue, we compared the responses evoked by incremental strength of injected depolarizing currents in ACSF with those elicited by identical currents in ACSF containing LPS. After a 30 minute application of LPS, the frequency of action potential was increased when injected currents were 90–200 pA (Figure 4A and B). LPS significantly increased action potential frequency compared with control (F[1, 14]=54.35; P<0.001; two-way analysis of variance; Figure 4B). There was a significantly decreased rheobase current amplitude (control, 100±17.73 pA, versus LPS, 78.75±25.88 pA; P<0.05; n=8; paired-sample Student’s t-test) after LPS was added (Figure 4C). LPS did not change the amplitude (control, 81.09±9.21 mV, versus LPS, 82.45±5.12 mV; P>0.05; n=8; paired-sample Student’s t-test) or half-width of the first action potential (control, 1.70±0.17 ms, versus LPS, 1.62±0.28 ms; P>0.05; n=8; paired-sample Student’s t-test) evoked by rheobase current (Figure 4D and E).
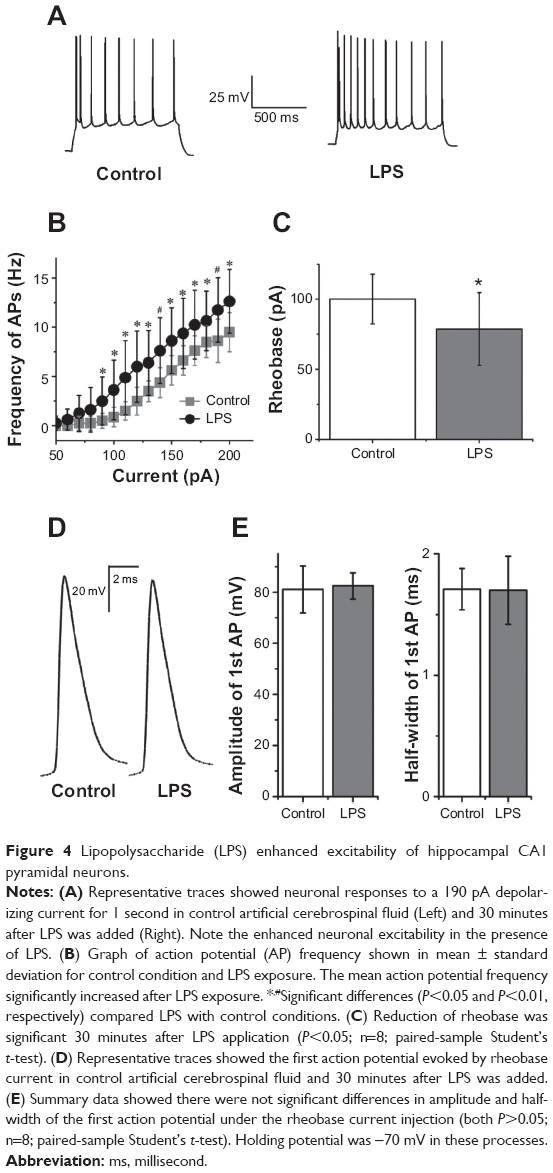 | Figure 4 Lipopolysaccharide (LPS) enhanced excitability of hippocampal CA1 pyramidal neurons. |
Discussion
Our data indicate that acute administration of LPS induces increased concentration of TNF-α and IL-1β and facilitates the epileptiform discharges in brain slices in vitro. Exposure to LPS enhanced eEPSC and neuronal excitability but did not modify eIPSC.
The important role of inflammatory processes in epilepsy is increasingly recognized. In the previous studies in vivo, it has been found that injection of LPS increases seizure susceptibility in different epileptic models.13–15 In addition, cortex neuronal excitability increases when LPS is directly applied to rat cortex.20 Here, our data suggest that acute administration of LPS induced brain slice inflammatory response, demonstrated by an elevated level of TNF-α and IL-1β after 30 minutes of incubation with LPS. Exposure to LPS exacerbated the epileptiform discharges of CA1 pyramidal neurons in hippocampal slices. These results are in accordance with those in vivo. In our experiments, the effects of LPS on epileptiform activity had remained at least 30 minutes after LPS withdrawal. Transient LPS application might alter epileptiform activity for a long time. It is in agreement with the results in vivo that a single LPS injection causes a long-lasting increase in seizure susceptibility.21 The exact mechanism is still unknown and should be clarified in the future.
A balance between excitatory and inhibitory synapse plays an important role in the control of neuronal excitability. Increase of excitatory synaptic strength and/or decrease of inhibitory synaptic strength may contribute to hyperexcitability of neuronal circuits, leading to onset of seizure. We observed that LPS enhanced eEPSCs and did not modify eIPSCs in hippocampal slices. This suggests that increasing excitatory synaptic strength, rather than decreasing inhibitory synaptic strength, is involved in facilitatory effects of LPS on epileptiform activity in this condition. Wang and White report that LPS induces glutamate release, and this excitatory transmitter can directly increase excitatory synaptic transmission.11 It is possible that an increase of excitatory synaptic strength is mediated by other factors associated with immune/inflammatory responses induced by LPS. IL-1β, an important cytokine involved in brain inflammation by stimulus of LPS, inhibits glutamate reuptake by astrocytes and enhances astrocytic glutamate release.22 In addition, IL-1β can directly activate N-methyl-D-aspartate receptor and strengthen excitatory synaptic transmission.23 TNF-α, another important factor released by LPS, is found to enhance the frequency of spontaneous EPSCs in hippocampal and spinal neurons.24–27 An increased level of IL-1β and TNF-α may partly lead to hyperexcitability of neuronal circuits after LPS application in our experiment. The effects of acute exposure of LPS on inhibitory synapse have not been reported previously. Acute application of LPS did not alter inhibitory transmission in our study, thereby excluding the participation of inhibitory synapse in the facilitated effects of LPS on epileptiform discharges. However, in organotypic slices, inhibitory synaptic strength is potentiated after 7 days of exposure to 100 ng/mL LPS.28 In addition to concentration, this different result may stem from the duration of time that brain slices are exposed to LPS.
Alterations of neuronal properties control cellular excitability and also play important roles in seizure occurrence. We found that acute LPS application enhanced neuronal excitability but did not alter the amplitude and half-width of action potential in hippocampal CA1 pyramidal neurons. Ion fluxes across the cellular membrane control neuronal excitability. Inflammatory mediators yielded after LPS application are various and have complicated effects on ion channels. For example, IL-1β can activate voltage-gated Ca2+ channels29 and inhibit voltage-gated Na+ channels.30 TNF-α can up-regulate voltage-gated sodium channel (Nav)1.3 and Nav1.8, which increase sodium ion influx.31 Therefore, our observations might reveal the collective effects of all these factors on neuronal excitability. Brain responses to LPS challenge induce the production of various inflammatory media such as IL-1β, NO, and prostaglandins. Our present study in vitro did not indicate which factor was involved in the change of synaptic transmission and neuronal excitability, or whether these changes were ascribed to LPS itself. Further study should clarify these problems.
Our findings demonstrate that acute LPS application induces inflammatory responses and facilitates the epileptiform activity of CA1 pyramidal neurons in hippocampal slices. Strengthened excitatory synaptic transmission and neuronal excitability are most likely attributable to this facilitation, whereas inhibitory synaptic transmission does not alter in this process. It may provide new clues for treating seizures associated with brain inflammatory disease.
Acknowledgment
This work was supported by a grant from the Natural Science Foundation of China (No. 30870840).
Disclosure
The authors report no conflicts of interest in this work.
References
Bartfai T, Sanchez-Alavez M, Andell-Jonsson S, et al. Interleukin-1 system in CNS stress: seizures, fever, and neurotrauma. Ann N Y Acad Sci. 2007;1113(1):173–177. | ||
Pitkänen A, Sutula TP. Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol. 2002;1(3):173–181. | ||
Wirrell E, Farrell K, Whiting S. The epileptic encephalopathies of infancy and childhood. Can J Neurol Sci. 2005;32(4):409–418. | ||
Banks WA, Erickson MA. The blood–brain barrier and immune function and dysfunction. Neurobiol Dis. 2010;37(1):26–32. | ||
Galic MA, Riazi K, Pittman QJ. Cytokines and brain excitability. Front Neuroendocrinol. 2012;33(1):116–125. | ||
Nasif FJ, Hu XT, Ramirez OA, Perez MF. Inhibition of neuronal nitric oxide synthase prevents alterations in medial prefrontal cortex excitability induced by repeated cocaine administration. Psychopharmacology (Berl). 2011;218(2):323–330. | ||
Li Z, Ji G, Neugebauer V. Mitochondrial reactive oxygen species are activated by mGluR5 through IP3 and activate ERK and PKA to increase excitability of amygdala neurons and pain behavior. J Neurosci. 2011;31(3):1114–1127. | ||
Lehnardt S, Massillon L, Follett P, et al. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100(14):8514–8519. | ||
Rivest S. Molecular insights on the cerebral innate immune system. Brain Behav Immun. 2003;17(1):13–19. | ||
Laflamme N, Echchannaoui H, Landmann R, Rivest S. Cooperation between toll-like receptor 2 and 4 in the brain of mice challenged with cell wall components derived from gram-negative and gram-positive bacteria. Eur J Immunol. 2003;33(4):1127–1138. | ||
Wang YS, White TD. The bacterial endotoxin lipopolysaccharide causes rapid inappropriate excitation in rat cortex. J Neurochem. 1999;72(2):652–660. | ||
Sayyah M, Najafabadi IT, Beheshti S, Majzoob S. Lipopolysaccharide retards development of amygdala kindling but does not affect fully-kindled seizures in rats. Epilepsy Res. 2003;57(2–3):175–180. | ||
Magni DV, Souza MA, Oliveira AP, et al. Lipopolysaccharide enhances glutaric acid-induced seizure susceptibility in rat pups: behavioral and electroencephalographic approach. Epilepsy Res. 2011;93(2–3):138–148. | ||
Sayyah M, Javad-Pour M, Ghazi-Khansari M. The bacterial endotoxin lipopolysaccharide enhances seizure susceptibility in mice: involvement of proinflammatory factors: nitric oxide and prostaglandins. Neuroscience. 2003;122(4):1073–1080. | ||
Auvin S, Shin D, Mazarati A, Sankar R. Inflammation induced by LPS enhances epileptogenesis in immature rat and may be partially reversed by IL1RA. Epilepsia. 2010;51(suppl 3):34–38. | ||
Scharfman HE. The neurobiology of epilepsy. Curr Neurol Neurosci Rep. 2007;7(4):348–354. | ||
Jefferys JG. Advances in understanding basic mechanisms of epilepsy and seizures. Seizure. 2010;19(10):638–646. | ||
Jo JH, Park EJ, Lee JK, Jung MW, Lee CJ. Lipopolysaccharide inhibits induction of long-term potentiation and depression in the rat hippocampal CA1 area. Eur J Pharmacol. 2001;422(1–3):69–76. | ||
Fernández M, Lao-Peregrín C, Martín ED. Flufenamic acid suppresses epileptiform activity in hippocampus by reducing excitatory synaptic transmission and neuronal excitability. Epilepsia. 2010;51(3):384–390. | ||
Rodgers KM, Hutchinson MR, Northcutt A, Maier SF, Watkins LR, Barth DS. The cortical innate immune response increases local neuronal excitability leading to seizures. Brain. 2009;132(Pt 9):2478–2486. | ||
Galic MA, Riazi K, Heida JG, et al. Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci. 2008;28(27):6904–6913. | ||
Ye ZC, Sontheimer H. Cytokine modulation of glial glutamate uptake: a possible involvement of nitric oxide. Neuroreport. 1996;7(13):2181–2185. | ||
Vezzani A, Conti M, De Luigi A, et al. Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J Neurosci. 1999;19(12):5054–5065. | ||
Beattie EC, Stellwagen D, Morishita W, et al. Control of synaptic strength by glial TNFalpha. Science. 2002;295(5563):2282–2285. | ||
Zhang H, Nei H, Dougherty PM. A p38 mitogen-activated protein kinase-dependent mechanism of disinhibition in spinal synaptic transmission induced by tumor necrosis factor-alpha. J Neurosci. 2010;30(38):12844–12855. | ||
Jakubs K, Bonde S, Iosif RE, et al. Inflammation regulates functional integration of neurons born in adult brain. J Neurosci. 2008;28(47):12477–12488. | ||
Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008;28(20):5189–5194. | ||
Hellstrom IC, Danik M, Luheshi GN, Williams S. Chronic LPS exposure produces changes in intrinsic membrane properties and a sustained IL-beta-dependent increase in GABAergic inhibition in hippocampal CA1 pyramidal neurons. Hippocampus. 2005;15(5):656–664. | ||
Zhang R, Yamada J, Hayashi Y, Wu Z, Koyama S, Nakanishi H. Inhibition of NMDA-induced outward currents by interleukin-1beta in hippocampal neurons. Biochem Biophys Res Commun. 2008;372(4):816–820. | ||
Zhou C, Qi C, Zhao J, et al. Interleukin-1β inhibits voltage-gated sodium currents in a time- and dose-dependent manner in cortical neurons. Neurochem Res. 2011;36(6):1116–1123. | ||
He XH, Zang Y, Chen X, et al. TNF-α contributes to up-regulation of Nav1.3 and Nav1.8 in DRG neurons following motor fiber injury. Pain. 2010;151(2):266–279. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.