Back to Journals » Therapeutics and Clinical Risk Management » Volume 15
Acute Intermittent Porphyria: Current Perspectives And Case Presentation
Authors Spiritos Z, Salvador S, Mosquera D, Wilder J ![]()
Received 4 April 2019
Accepted for publication 14 September 2019
Published 16 December 2019 Volume 2019:15 Pages 1443—1451
DOI https://doi.org/10.2147/TCRM.S180161
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Zachary Spiritos,1 Shakirat Salvador,2 Diana Mosquera,3 Julius Wilder1,4
1Department of Medicine, Division of Gastroenterology, Duke University School of Medicine, Durham, NC, USA; 2Department of Medicine, Division of Gastroenterology, Vanderbilt University Medical Center, Nashville, TN, USA; 3Department of Medicine, Duke University School of Medicine, Durham, NC, USA; 4Department of Medicine, Duke Clinical Research Institute, Durham, NC, USA
Correspondence: Julius Wilder
Department of Medicine, Division of Gastroenterology, Duke University School of Medicine, Durham, NC, USA
Tel +1 919 668 3063
Fax +1 919 668 7164
Email [email protected]
Abstract: Acute intermittent porphyria (AIP) is an autosomal dominant metabolic disorder characterized by a deficiency in heme biosynthesis. Heme biosynthesis occurs throughout the body, but it is most prominent in the erythroblastic system and liver. AIP is a hepatic porphyria whereby the liver is the source of toxic heme metabolites. Clinical manifestations of AIP result from a genetic mutation that leads to partial function of porphobiliogen deaminase (PBGD). This causes an accumulation of upstream, neurotoxic metabolites. Symptoms include but are not limited to peripheral neuropathies, autonomic neuropathies and psychiatric manifestations. AIP can be life threatening and clinical signs and symptoms are often heterogeneous and non-specific. Therefore, it is important to be able to recognize these patients to make a prudent diagnosis and offer appropriate therapy. Here, we review the epidemiology, pathophysiology, clinical presentation, diagnosis, and management of AIP including the role of liver transplantation.
Keywords: acute porphyria, acute intermittent porphyria, hepatic porphyria
Introduction
Acute intermittent porphyria (AIP) is a member of a rare family of diseases characterized by deficiencies in heme biosynthesis. It is caused by a mutation which leads to partial function of porphobiliogen deaminase (PBGD) and is inherited in autosomal dominant pattern with variable penetrance. Inadequate PBGD function leads to accumulation of upstream metabolites δ-aminolevulinic acid (ALA) and porphobilinogen (PBG) which cause toxicity to the neurologic system. Neurologic dysfunction is variable in anatomic location and severity. The central, peripheral, autonomic, sensory and motor systems can be involved. Patients have very heterogeneous presentations with non-specific physical exam findings, laboratory values, and imaging. This often leads to multiple re-admissions without a diagnosis which can be frustrating for both patients and medical providers. AIP can be life-threatening and treatment is very effective. Therefore, it is crucial to make an early diagnosis and offer therapy when there is sufficient clinical suspicion. Here, we review the epidemiology, pathophysiology, clinical presentation, diagnosis, and management of AIP.
Epidemiology
The prevalence of AIP is difficult to estimate due to its rarity as a disease, variability in phenotypic presentations, and penetrance. The three most common porphyrias are Porphyria cutanea tarda (PCT), Acute intermittent porphyria (AIP), and Erythropoeitic porphyria (EPP). AIP is the most common acute porphyria and is estimated to occur in 1 in 20,000.1 There are estimated to be less than 200,000 patients with AIP living in the United States. AIP is more frequent in Sweden where a founder effect has been described. Haplotype studies have identified a common origin for the AIP mutation in families located in the northern area of that country.2
Pathophysiology
AIP is an autosomal dominant metabolic disorder arising from a deficiency in the enzyme porphobilinogen deaminase (PBGD), one of the enzymes involved in the heme biosynthesis pathway (Figure 1). It occurs due to mutations in the hydroxymethylbilane synthesis (HMBS) gene and usually causes a partial deficiency of PBGD. Mutations of the PBGD gene result in an approximate 50% deficiency of enzymatic function, particularly within hepatocytes. To date there are over 200 recognized genetic mutations associated with the disease. Despite the pattern of inheritance, 90% of heterozygotic individuals do not exhibit manifestations of the disease.1,3 There is no described relationship between mutation and severity of AIP. This is presumably due to the relationship between the primary genetic defect as well as secondary acquired mutations and environmental factors.4
 |
Figure 1 Haem synthesis pathway.Note: Adapted from Stein PE, Badminton MN, Rees DC. Update review of the acute porphyrias. Br J Haematol. 2017;176(4):527–538. © 2016 John Wiley & Sons Ltd.5 |
Heme is essential for the synthesis of hemoglobin as well as the synthesis of the hepatic cytochrome P450 enzymes. Heme biosynthesis occurs throughout the body, but it is most prominent in the erythroblastic system (80%) and liver (15%).6 Most erythropoeitic porphyrias have a major cutaneous component often leading to photosensitivity like EPP.1 On the other hand, AIP is one of the hepatic porphyrias whereby the liver is the source of toxic heme biosynthesis metabolites. The heme pathway is comprised of 7 steps that occur in the mitochondria and the cytoplasm.7 In the first step, the substrates succinyl CoA and glycine are used by δ-aminolevulinic acid synthase (ALAS) to synthesize δ-aminolevulinic acid (ALA). ALA dehydratase (ALAD) then uses ALA to create porphobilinogen (PBG), which is then used as a substrate by PBGD to yield hydroxymethylbilane. Uroporphyrinogen III cosynthesase then uses Hydroxymethylbilane to synthesize uroporphyrinogen III. Decarboxylation of uroporphyrinogen III then yields Coprophyrinogen III, which undergoes 3 further steps in the mitochondria to finally synthesize heme.7
In AIP, the partial deficiency in PBGD leads to the accumulation of metabolites PBG and ALA due to lack of inhibition of ALAS.8 Acute attacks are precipitated by factors that induce ALAS transcription or function, including hormonal changes during the menstrual cycle, drugs, fasting, and infections. Accumulation of these metabolites lead to the neuropsychiatric and visceral symptoms seen in AIP.7
Clinical Manifestations
AIP leads to the toxic accumulation of the precursors PBG and ALA causing biochemical degenerative changes in the peripheral and central nervous systems. AIP is a neurovisceral disorder with multisystemic clinical manifestations and as such clinical presentation is non-specific and widely variable.9 Symptoms include but are not limited to peripheral neuropathies, autonomic neuropathies and psychiatric manifestations.10 Therefore clinicians must maintain a high index of suspicion for the disease, as patients can present with symptoms that can be attributed to an alternative disease process.
The majority of patients develop symptoms following puberty, specifically during the third to fourth decade of life. Women are more often affected than men. Alternatively, patients with homozygous genotype – which is rarer – can experience neurologic symptoms during childhood. Symptomatic patients experience independent attacks that can often be attributed to a specific trigger.10 Duration of symptoms are variable and can last between days to months especially if the patient is inappropriately or undertreated.9 Disease can be classified by four distinct sub-groups which are dependent on the number of annual exacerbations. These are listed in Table 1.
 |
Table 1 Disease States Of AIP |
Common Precipitants
There are several known precipitants of porphyria. The commonality between these triggers is excitation of the hepatic heme synthesis pathway which causes an accumulation of upstream neurotoxic precursors, specifically ALA and PBG.11 This can occur by increased transcription of ALAS which is the rate limiting step in hepatic heme synthesis or decreasing heme stores which stimulates the physiologic feedback loop.
Physiologic conditions that contribute to neurohormonal influx such as puberty have been implicated in precipitating porphyria flares.12 This is due to increased progesterone which induces ALAS production.3 The effect is also seen with administration of exogenous synthetic estrogens.11 Porphyria may worsen during pregnancy as female sex hormones are increased and can induce heme synthesis. However other studies have suggested that pregnancy seldom induces acute attacks. The association is controversial.13
Poor nutritional states can induce an acute episode as well.14 The mechanism of action occurs via the enzyme heme oxygenase-1 which both decreases hepatic heme production and induces ALAS synthesis leading to increased PBG and ALA production. Low carbohydrate states can stimulate co-activators that increase production of ALAS as well. Clinically this can be seen in patients who are starved, undergo lifestyle modifications, surgeries and in those with a prolonged illness.3 Conversely, high caloric diets result in decreased PBGD activity.11
Furthermore, certain pharmacotherapies and chemicals such as alcohol, drugs and tobacco can decrease hepatic heme stores and stimulate production of ALA and PBG (Table 2).11
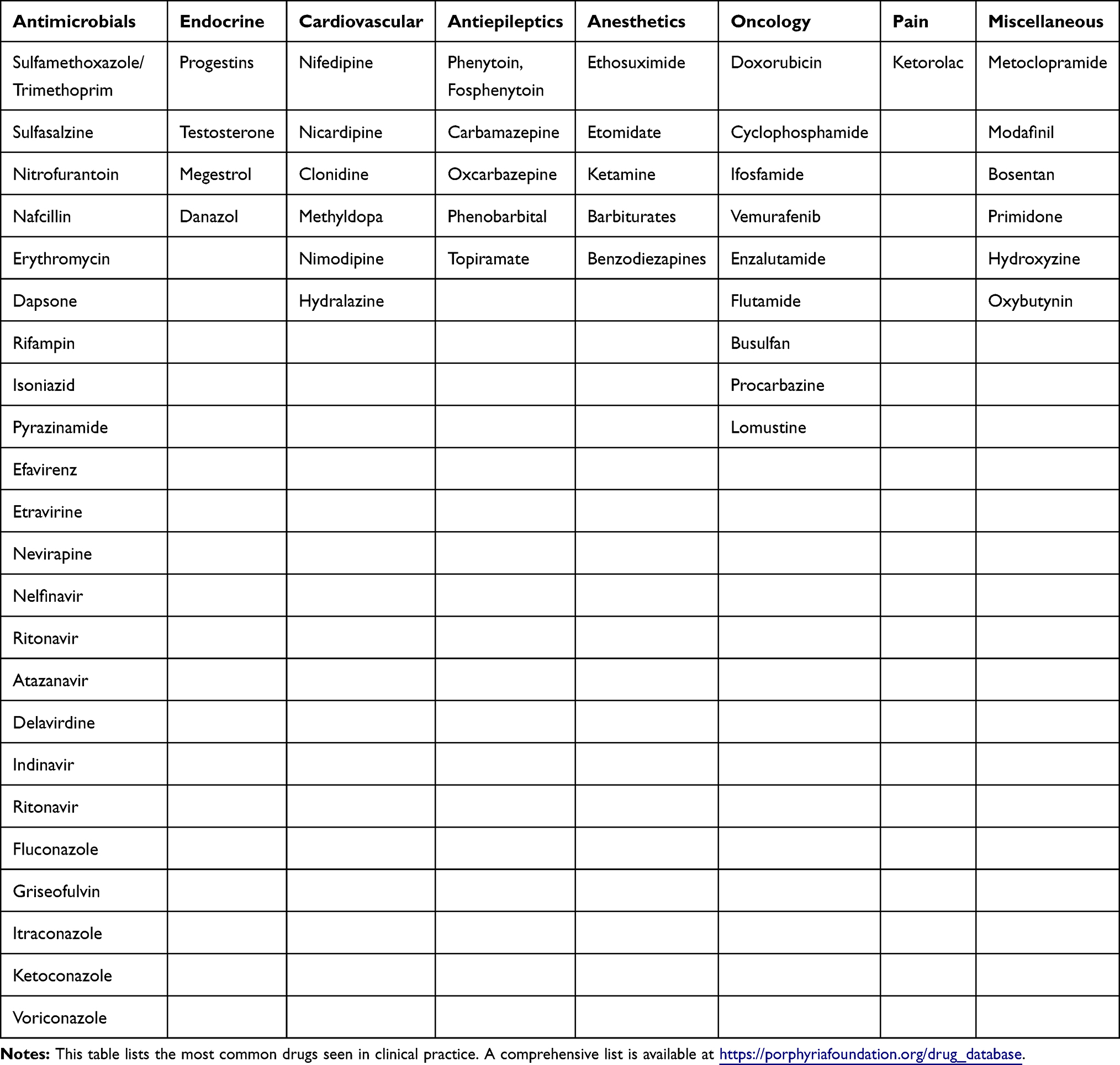 |
Table 2 Pharmacologic Therapies Implicated In Precipitating Episodes |
Gastrointestinal Manifestations
Autonomic nerve dysfunction contributes to the most common manifestation of the disease which is poorly localized abdominal pain. Abdominal pain is present in over 80% of patients.12 On physical examination, abdominal pain is typically out of proportion to the exam findings. Nonspecific symptoms such as nausea, vomiting, and poor appetite are also associated with acute episodes. Bowel habit dysfunction including constipation, abdominal distention, and ileus can also be seen.12 Without treatment patients have an increased risk of developing progressive liver disease, cirrhosis, and hepatocellular carcinoma.10
Cardiovascular Manifestations
Excitation of the sympathetic nervous system results in increased catecholamine production leading to cardiovascular manifestations such as arrhythmias, hypertension, and diaphoresis.9 Tachycardia occurs in over 80% of patients. If left untreated, tachycardia can progress to hemodynamic instability due to circulatory failure.3 Hypertension is a typical manifestation of acute episodes but may also persist following resolution of the acute episode in more than 40% of patients.11
Neuropsychiatric Manifestations
Neuropsychiatric manifestations are thought to be due to the structural similarities between ALA and gamma-aminobyturic acid (GABA). The excess of ALA may impair GABA function leading to central nervous system impairment. Other theories postulate that heme metabolites are directly neurotoxic and inadequate heme production has adverse effects on the brain.15
Both the peripheral and central nervous system can be affected. Peripheral involvement leads to pain in the extremities, back, and chest. Hyperesthesia is a common sensory manifestation. Motor neuropathies occur during the late stages of an acute attack. Bulbar paralysis with cranial nerve involvement is often severe. Symptoms include dysphagia, dysarthria and dysphonia. Diaphragmatic involvement may lead to respiratory compromise requiring intubation and mechanical ventilation. Seizures can occur in the setting of neurotoxicity or secondary to hyponatremia.11 Some theorize that excess PBG and ALA causes the syndrome of inappropriate antidiuretic hormone (SIADH) via neurotoxigenic mechanisms. Psychiatric symptoms may occur both during acute episodes or become chronic diagnoses in the form of depression, anxiety, hallucinations, psychosis, or delirium.14
Endocrine Manifestations
Hyponatremia is a common manifestation of AIP. This occurs due to hypothalamic involvement as well as SIADH, the latter of which occurs in 25–60% of patients. Hyponatremia may be exacerbated by poor oral intake and nausea with severe vomiting.3 Additionally, acute attacks increase thyroxin-binding globulin and patients may become hyperthyroid.3
Urologic Manifestations
ALAS and PBG are both colorless precursors. PBG degradation with subsequent oxidation leads to the formation of porphobilinogen which is a dark brown pigment. The combination of the red pigmented porphyrin leads to the characteristic deep red port wine colored urine which is a prominent finding in the disease process. Bladder dysfunction can present with urinary retention, urinary incontinence, or dysuria.9
Renal Manifestations
Porphyria related nephropathy is due to genetic variations in renal transporters. ALA is particularly nephrotoxic leading to chronic tubulostitial nephropathy or focal cortical atrophy. This can progress to chronic renal dysfunction without treatment. In addition, hypertension refractory to traditional standard therapy contributes to chronic kidney injury. In severe cases, individuals can progress to end stage kidney disease.9
Differential Diagnosis
As the manifestations of the disease can mimic other disease states, such emphasis must be made on distinguishing AIP from other diagnoses. The presentation of AIP is heterogeneous and the differential must be tailored to the patient’s symptoms. Alternate causes of acute abdominal pain, neuropathy, and neuropsychiatric symptoms must be considered. This includes but is not limited diverticulitis, appendicitis, pyelonephritis, inflammatory bowel disease, nephrolithiasis, Guillan-Barré, neurodegenerative diseases, or encephalitis.
Evaluation And Diagnosis
History And Physical Examination
All patients should have a complete history and physical examination. Emphasis should be taken on a thorough past medication history. A detailed social history including prior and on-going drug and alcohol use as well as a family history of health issues should be obtained. Physical examination is oftentimes unrevealing.10 Patients may complain of red port wine urine. A key distinguishing feature of AIP from other porphyrias is the lack of cutaneous involvement. Careful consideration should be given to the neurologic exam. Evaluation of strength testing, cranial nerve function, speech assessment, and detection of fasciculations can reveal early signs of neurologic and respiratory compromise.
Imaging
Cross sectional imaging is recommended to evaluate for causes of the abdominal symptoms which frequently occur during acute episodes of AIP. Magnetic resonance imaging of the brain during acute attacks may reveal cortical densities which can mimic posterior reversible encephalopathy syndrome (PRES).10
Laboratory Evaluation
Baseline laboratory evaluation should include complete blood count, metabolic panel, hepatic function evaluation and iron studies including ferritin. Electrolyte abnormalities such as hyponatremia are common which occurs in 25–60% of patients with acute episodes. Elevation aminotransferases are elevated in approximately 13% of cases. Hypomagnesemia and hypocalcemia can also be seen.9
Porphyrins are excreted in the urine and stool. However porphyrin fecal studies in AIP are routinely normal while they are elevated in hereditary coproporphyria and variegate porphyria.14 As such, urine and stool specimens are integral to establishing the diagnosis (Table 3). If the index of suspicion is high, first line testing includes spot urinary porphobilinogen (PBG). The Watson-Schwartz test is a qualitative test that can be used to detect elevated PBG levels. If positive, follow-up confirmatory quantitative testing should be performed which has a diagnostic sensitivity and specificity of 95% and 100%, respectively. Metabolite levels can be utilized to determine the response to therapy as well.3
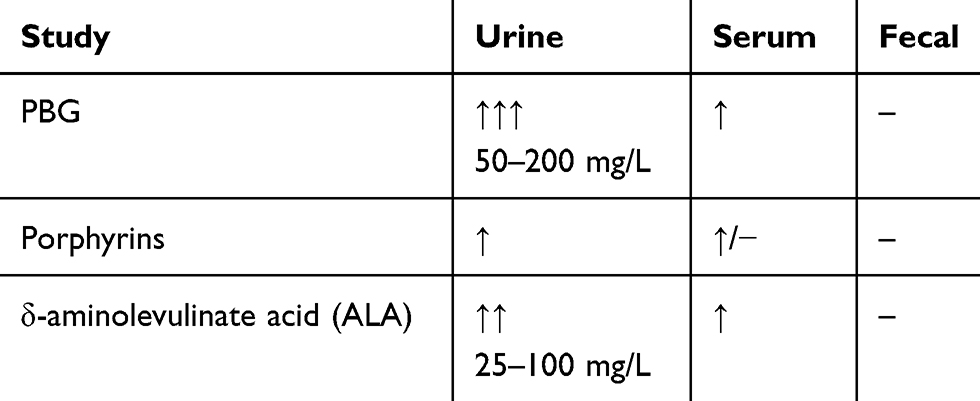 |
Table 3 Diagnostic Testing During Acute Episode Of AIP |
Evaluating PBG levels should be performed in all subtypes of porphyria.12 Normal PBG levels are 0–2 mg/L. During an acute attack, PBG levels in the range of 20–200 mg/L are diagnostic. Following remission, PBG levels can remain elevated between 10–20 mg/L.14 ALA levels are not necessary to establish the diagnosis. However, levels ranging 25−100 mg/L also support the diagnosis of AIP.3
Erythrocyte PBGD activity is not usually helpful to determine the diagnosis alone. The disease process is a partial deficiency therefore measurements are variable. Supplementary biochemical testing includes genetic analysis of HMBS with mutation analysis which has a detection rate of 98%.12 Molecular genetic testing is not necessary to confirm the disease.9 Subsequently, targeted mutation analysis should be offered for relatives of the index case.12
Treatment
The basis for treatment is the reduction of upstream heme intermediates. The rate limiting step in hepatic heme synthesis is the production of ALA which is catalyzed by ALAS. This process is tightly regulated by intracellular heme.1 Therefore treatment aim is to suppress the physiologic feedback mechanism driven by inadequate heme stores which in turn reduces levels of ALA and PBG by suppressing ALAS.
This underscores the utility of intravenous hemin as a treatment for AIP. Additionally, there are several medications that decrease hepatic heme stores which can lead to stimulate production of ALA and PBG. It is important to recognize these offending medications and discontinue them if thought to be contributing to an acute flare.18 If patients have uncontrolled disease despite therapy or experience significant side effects from the aforementioned interventions, then liver transplantation has been shown as definitive treatment.14
Management Of Acute Attacks
After diagnosis of a porphyria exacerbation, the most non-invasive intervention is to identify any offending medications or behaviors. In susceptible patients fasting, alcohol ingestion, smoking and stress from injury or illness may precipitate an event by activating the heme synthesis pathway.14 Several medications have been identified that increase production of porphyrins through ALAS induction (Table 2). The porphyrogenecity varies between agents and is dependent upon its tendency to decrease hepatic heme stores. The ability to do so is based upon the capacity for induction or destruction of cytochrome P450 (CYP) enzymes via two mechanisms.16
The first is based on the production and activity of ALAS. ALAS is required for the transcription of CYP enzymes. Medications that induce or destroy CYP enzymes increase production ALAS. Secondly heme is a cofactor for CYP P450. Increasing the demand for CYP P450 enzymes for drug metabolism in turn will initiate transcription of ALAS and stimulate the feedback loop through depletion of the intracellular hepatic heme reservoir.16
There are many medications that utilize the CYP P450 system and it is difficult to predict which will cause clinically relevant elevations in toxic metabolites. There are several online databases that classify drug safety based on clinical evidence and pathophysiologic mechanisms. Many of these medications are listed in Table 2. It is recommended that porphyronogenic medications be withheld if thought to exacerbate AIP. In cases where the role of a drug is unknown or is required for other reasons, then patients may be simply observed clinically or monitored with daily spot urine PBG levels after medication initiation.16
Symptoms may subside after identification and removal of any potential offending medication. If there are no culprit medications and the patient remains symptomatic then intravenous hemin is the cornerstone of therapy. This is the only medication that is approved for the treatment of porphyria exacerbations. As previously mentioned, the theory is that increased levels of heme will decrease circulating levels of ALA and PBG through suppression of ALAS feedback control. There is no standard dosing, however it is generally administered at 1 to 4 mg/kg with a maximum dose of 6 mg/kg. On average it takes 2 to 5 days for symptom resolution. However in situations where symptoms are progressing it can be administered up to 14 days. Resolution of symptoms typically coincides with reduction in systemic levels of porphyrin intermediates.17
The most common side effect of intravenous hemin is phlebitis as a result from the anticoagulant effects of heme degradation products.18 The risk of phlebitis can be minimized by obtaining central access and administering via slow infusion. Reconstituting hemin with albumin instead of sterile water further diminishes the incidence and severity of phlebitis by increasing the stability of lipophilic hemin.18 The vein should be washed with saline after infusion.19 Hematin is generally well tolerated however at supratherapeutic doses has been shown to cause nephrotoxicity due to dilation and congestion of small vessels.20
If hematin is not available, carbohydrate loading in the form of intravenous glucose has been shown to control symptoms by downregulating ALAS synthesis. Loading should occur with 10% glucose with a total of 300 to 500 gm daily. In fact, milder attacks may be controlled by glucose infusion alone. Milder attacks are characterized by mild pain with no paresis, seizures, or hyponatremia.19
After initiating therapy, the mainstay of patient care is recognizing sequelae of an acute attack and providing appropriate support. Patients often experience nausea and non-focal gastrointestinal discomfort which should be managed with appropriate anti-emetics and pain medications respectively. Correction of hemodynamic, fluid and electrolyte abnormalities is imperative to ensure patient safety. As a result of neurovisceral toxicity patients may experience significant sympathetic drive resulting in hypertension, tachycardia and fatal cardiac arrhythmias.9 Axonal degeneration of peripheral nerves can occur and lead to respiratory failure and bulbar paralysis.11 Additionally, patients may develop hyponatremia due to poor oral intake, SIADH, carbohydrate loading or IV hemin administration which is typically administered in a hypotonic solution. Clinically apparent hyponatremia can manifest as altered mental status or seizures. Seizures can be difficult to control as most antiepileptic medications are porphyrogenetic. Other common manifestations are urinary retention, encephalopathy, insomnia and anxiety.11
Prevention And Management Of Recurrent Attacks
After diagnosis and resolution of an acute attack, the focus should be on avoiding the aforementioned precipitating factors. This includes avoiding unsafe medications, fasting, alcohol use, smoking and encouraging adequate calorie and carbohydrate intake. Most patients will in fact never experience an attack and the minority of those that do can suffer from relapsing events that portend poor quality of life with associated high morbidity and mortality.18 Unfortunately, there are no predictive models to help delineate which patients are at higher risk for recurrence.19
Patients who suffer frequent recurrent attacks can benefit from routine administration of prophylactic hemin infusion given every 1–2 weeks.17 Routine monitoring of urine PBG can detect a flare prior to onset of clinical symptoms which can prompt pharmacologic intervention.18 Chronic administration of hemin has significant potential side effects. Each 200 mg dose of hemin contains 17 mg of iron which puts patients at risk for iron overload and subsequent hemosiderosis. This can lead to hepatopathy, cardiomyopathy and endocrinopathies. Patients also must have routine intravenous access in the form of a chronic indwelling central line or port which increases the risk of bloodstream infection.20
Progesterone in women has been implicated as a cause for porphyria flares due to its interaction with the CYP P450 system. Therefore women are five times more common to experience attacks then men. Oral contraceptive preparations are controversial in the prevention of attacks. For pre-menopausal women, ovarian suppression through GnRH analogs have been shown to decrease the severity and frequency of attacks. This can be co-administered with estrogen to avoid bone loss.19
Patients with uncontrolled AIP characterized by frequent and severe flares are at risk for chronic hypertension and subsequent nephropathy. This should be monitored closely and treated appropriately as an outpatient.
Role Of Liver Transplantation
The alternative to suppressing ALAS activation is correcting the genetic defect of PBG deaminase through liver transplantation. This method has been shown to be an effective treatment option with almost universal resolution of symptoms and normalization of biomarkers within 3 days of transplantation. Chronic neurologic manifestations such as psychiatric disease and axonal degeneration have not been shown to improve with transplantation.14
Patients who benefit from liver transplant include those who do not respond to pharmacologic therapy, have incomplete response to therapy defined by refractory episodes with poor quality of life, and those who cannot tolerate the effects of long-term hemin therapy. The latter subset of patients typically suffer from iron overload, end-organ dysfunction due to hypertension, and hospitalizations related to vascular events from chronic central venous access.18 Ideal timing for transplant involves assessing risk for future attacks that may result in neurologic relapse precluding transplant, current health status as a result of relapsing porphyria, and long term hemin therapy.18
Specific pre-operative considerations include chronic neurologic deficits, difficulty with vascular access, and hemosiderosis. Patients with poorly controlled porphyria can develop severe neuromuscular dysfunction such as respiratory distress and failure requiring ventilator support. Patients who cannot ventilate independently have been shown to have poor post-transplant outcomes.14 The recommendation is to delay transplant until patients are extubated and recover ability to ventilate independently. Alternatively, patients who have developed chronic psychiatric symptoms such as depression and anxiety must have these diagnoses addressed prior to consideration for transplant.16 Chronic intravenous hemin administration can lead to poor venous access due to venous thrombi which can preclude transplant. Lastly patients who have hepatic iron overload pre-transplant have been shown to have worse long-term outcomes following transplantation.18
Monitoring PBG levels after transplantation is unnecessary as urinary PBG levels should normalize after surgery. It is unclear whether porphyronogenic medications are safe in the post-transplant setting. The allograft has normal heme metabolism and therefore patients should not be at risk. However there are reports of peripheral neuropathy in patients with acute porphyria in the absence of attacks which may suggest that there still may be dysfunctional heme synthesis in the peripheral nervous system. Alternatively, this may be due residual complications from prior attacks.19 As already mentioned, pre-existing neurologic deficits have not been shown to improve after transplant.
There are reports of increased risk of hepatic artery thrombosis (HAT) in patients with AIP who undergo OLT. The incidence of HAT in the adult liver transplant population is 3% to 5%. The incidence is considerably higher in patients with AIP with rates as high as 40%. The pathophysiology is unclear. For this reason, routine anticoagulation is administered post-operatively.17
Emerging Therapies
A developing therapeutic approach for AIP is RNA interference therapy (Givosiran) which works to decrease ALAS messenger RNA (mRNA) levels. Givosiran is administered via monthly injection. Investigational studies in rodent models have successfully demonstrated its ability to lower ALAS mRNA, ALA and PBG levels in a dose dependent manner. Phase 1 trials in patients with severe disease have shown similar biochemical outcomes with concomitant reductions in clinical relapses and IV hemin use compared to placebo. The most common side effect was injection-site reactions which were mild-to-moderate in severity. Studies with more patients and longer follow-up intervals are required prior to widespread use.21
Another promising intervention is gene therapy that targets hepatocytes harboring dysfunctional PBGD genes. Recombinant adeno-associated vectors expressing PBGD with a liver-specific promotor helps repair the defective gene. Studies in murine models have demonstrated the therapy’s safety and ability to restore PBGD activity. A phase I trial in patients suffering recurrent attacks showed variable efficacy. There were no changes in urinary ALA and PBG levels after therapy with only a minority of patients experiencing clinical improvement.22
Screening
There is an association between AIP and hepatocellular carcinoma (HCC) that also exists between other porphyria disorders. The etiology is unclear however there is a 60- to 70-fold increase in the incidence of HCC in patients with AIP compared to the general population. This can occur in the absence of cirrhosis. It is recommended that patients have 6-month screening with ultrasound and alpha-fetoprotein (AFP) to screen for HCC.19
It is important that family members should undergo mutation screening as this is an autosomal dominant disease.
Case Presentation
A 16-year-old female patient was diagnosed with AIP after several re-admissions for generalized abdominal pain. She had also been suffering from chronic anxiety and depression. Diagnosis was suspected after a urinary PBG excretion was elevated to above 260 mg/L and supported by genetic testing which showed a c.517 C>T (exon 10) mutation in the PBGD-gene, which has been reported as a mutation associated with AIP.23 Family screening was not performed. She was treated with weekly intravenous hemin infusions given the frequency and severity of her attacks. Constant hemin infusions lead to hyperferritinemia which was co-managed with bimonthly phlebotomies. She endorsed such poor quality of life due to time spent in the hospital, chronic fatigue and debilitating abdominal discomfort that she was evaluated for orthotropic liver transplant as a potential curative option.
She was approved for transplantation and underwent surgery with split right lobe with middle hepatic vein and standard arterial anatomy. She received prophylactic antibiotics however Fluconazole was avoided due to its porphyrogenicity. Her immunosuppression regimen included tacrolimus, mycophenolate mofetil and prednisone. Nebulized pentamidine rather than sulfamethoxazole-trimethoprim was used for pneumocystis pneumonia prophylaxis as the latter medication is porphyrogenic as well. Additional post-operative care included enoxaparin for 9 months to reduce the risk of hepatic artery thrombosis.
One year after transplant she had no clinical or biochemical sign of porphyria, normal graft function and improved quality of life.24
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ramanujam VM, Anderson KE. Porphyria diagnostics-part 1: a brief overview of the porphyrias. Curr Protoc Hum Genet. 2015;86:
2. Lee J, Anvret M. Identification of the most common mutation within the porphobilinogen deaminase gene in swedish patients with acute intermittent porphyria. Proc Natl Acad Sci USA. 1991;88(23):10912–10915. doi:10.1073/pnas.88.23.10912
3. Sassa S. Modern diagnosis and management of the porphyrias. Br J Haematol. 2006;135:281–292. doi:10.1111/j.1365-2141.2006.06289.x
4. Besur S, Hou W, Schmeltzer P, Bonkovsky LH. Clinically important features of porphyrin and heme metabolism and the porphyrias. Metabolites. 2014;4(4):977–1006. doi:10.3390/metabo4040977
5. Stein PE, Badminton MNRees DC. Update review of the acute porphyrias. Br J Haematol. 2017;176(4):527–538. doi: 10.1111/bjh
6. Szlendak U, Bykowska K, Lipniacka A. Clinical, biochemical and molecular characteristics of the main types of porphyria. Adv Clin Exp Med. 2016;25(2):361–368. doi:10.17219/acem/58955
7. Puy H, Gouya L, Deybach J. Porphyrias. The Lancet. 2010;375(9718):924–937. doi:10.1016/S0140-6736(09)61925-5
8. Floderus Y, Sardh E, Möller C, et al. Variations in porphobilinogen and 5-aminolevulinic acid concentrations in plasma and urine from asymptomatic carriers of the acute intermittent porphyria gene with increased porphyrin precursor excretion. Clin Chem. 2006;52(4):701. doi:10.1373/clinchem.2006.073882
9. Balwani M, Wang B, Anderson KE, et al. Acute hepatic porphyrias: recommendations for evaluation and long-term management. Hepatology (Baltimore, Md). 2017;66:1314–1322. doi:10.1002/hep.29313
10. Whatley SD, Badminton MN. Acute intermittent porphyria. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews((R)). Seattle: University of Washington, Seattle University of Washington. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.; 1993.
11. Sassa S. Diagnosis and therapy of acute intermittent porphyria. Blood Rev. 1996;10:53–58.
12. Balwani M, Desnick RJ. The porphyrias: advances in diagnosis and treatment. ASH Educ Prog Book. 2012;120:4496–5504.
13. Farfaras A, Zaqouri F, Zografos G, Kostopoulou A, Sergentanis TN, Antoniou S. Acute intermittent porphyria in pregnancy: a common misdiagnosis. Clin Exp Obstetr Gynecol. 2010;37(4):256–260.
14. Ajayi T, Ward R, Summers B, et al. Pathophysiology, pharmacology and treatment of acute intermittent porphyria: a patient case description and recommendations from the current literature. J Explor Res Pharmacol. 2017;2:49–53. doi:10.14218/JERP.2016.00022
15. Tracy JA, Dyck PJ. Porphyria and its neurologic manifestations. Handb Clin Neurol. 2014;120:839–849. doi:10.1016/B978-0-7020-4087-0.00056-5
16. Thunell S, Pomp E, Brun A. Guide to drug porphyrogenicity prediction and drug prescription in the acute porphyrias. Br J Clin Pharmacol. 2007;64(5):668–679. doi:10.1111/j.0306-5251.2007.02955.x
17. Singal A, Parker C, Bowden C, Thapar M, Lawrence L, McGuire BM. Liver transplantation in the management of porphyria. Hepatology. 2014;60(3):1082–1089. doi:10.1002/hep.27086
18. Seth AK, Badminton MN, Mirza D, Russell S, Elias E. Liver transplantation for porphyria: who, when, and how? Liver Transpl. 2007;13(9):1219–1227. doi:10.1002/lt.21261
19. Pischik E, Kauppinen R. An update of clinical management of acute intermittent porphyria. Appl Clin Genet. 2015;1(8):201–214. doi:10.2147/TACG.S48605
20. Soonawakka ZF, Orug T, Badminton MN, et al. Liver transplantation as a cure for acute intermittent porphyria. The Lancet. 2004;363(9410):705–706. doi:10.1016/S0140-6736(04)15646-8
21. Sardh E, Harper P, Balwani M, et al. Phase 1 trial of an rna interference therapy for acute intermittent porphyria. N Engl J Med. 2019;380(6):549–558. doi:10.1056/NEJMoa1807838
22. D’Avola D, López-Franco E, Sangro B, et al. Phase I open label liver-directed gene therapy clinical trial for acute intermittent porphyria. J Hepatol. 2016;65:776–783. doi:10.1016/j.jhep.2016.05.012
23. Puy H, Deybach JC, Lamoril J, et al. Molecular epidemiology and diagnosis of pbg deaminase gene defects in acute intermittent porphyria. Am J Hum Genet. 1997;60(6):1373–1383. doi:10.1086/515455
24. Kappus MR, Summers BB, Byrns JS, Berg CL, Wilder JM. Liver transplantation for the management of acute intermittent porphyria: a case report. J Hepatol Gastroint Dis. 2017;3:141. doi:10.4172/2475-3181
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.