Back to Journals » International Journal of Nanomedicine » Volume 13
Activation of polymeric nanoparticle intracellular targeting overcomes chemodrug resistance in human primary patient breast cancer cells
Authors Abou-El-Naga AM, Mutawa G ![]() , El-Sherbiny IM
, El-Sherbiny IM ![]() , Mousa SA
, Mousa SA ![]()
Received 1 August 2018
Accepted for publication 26 September 2018
Published 29 November 2018 Volume 2018:13 Pages 8153—8164
DOI https://doi.org/10.2147/IJN.S182184
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
This paper has been retracted.
Amoura M Abou-El-Naga,1 Ghada Mutawa,2 Ibrahim M El-Sherbiny,3 Shaker A Mousa4
1Zoology Department, Faculty of Sciences, Mansoura University, Mansoura 35516, Egypt; 2Department of Basic Science, Faculty of Dentistry, Horus University in Egypt (HUE), New Damietta 34517, Egypt; 3Center for Materials Science, Zewail City of Science and Technology, Cairo 12588, Egypt; 4The Pharmaceutical Research Institute, Albany College of Pharmacy and Health Sciences, Rensselaer, NY 12144, USA
Background: Successfully overcoming obstacles due to anticancer drugs’ toxicity and achieving effective treatment using unique nanotechnology is challenging. The complex nature of breast tumors is mainly due to chemoresistance. Successful docetaxel (DTX) delivery by nanoparticles (NPs) through inhibition of multidrug resistance (MDR) can be a bridge to enhance intracellular dose and achieve higher cytotoxicity for cancer cells.
Purpose: This study tested primary patient breast cancer cells in vitro with traditional free DTX in comparison with polymeric nanocarriers based on poly lactic co-glycolic acid (PLGA) NPs.
Materials and methods: Establishment of primary cell line from breast malignant tumor depends on enzymatic digestion. Designed DTX-loaded PLGA NPs were prepared with a solvent evaporation method; one design was supported by the use of folic acid (FA) conjugated to PLGA. The physical properties of NPs were characterized as size, charge potential, surface morphology, DTX loading, and encapsulation efficiency. In vitro cellular uptake of fluorescent NPs was examined visually with confocal fluorescence microscopy and quantitatively with flow cytometry. In vitro cytotoxicity of all DTX designed NPs against cancer cells was investigated with MTT assay. RT-PCR measurements were done to examine the expression of chemoresistant and apoptotic genes of the tested DTX NPs.
Results: Cellular uptake of DTX was time dependent and reached the maximum after loading on PLGA NPs and with FA incorporation, which activated the endocytosis mechanism. MTT assay revealed significant higher cytotoxicity of DTX-loaded FA/PLGA NPs with higher reduction of IC50 (8.29 nM). In addition, PLGA NPs, especially FA incorporated, limited DTX efflux by reducing expression of ABCG2 (3.2-fold) and MDR1 (2.86-fold), which were highly activated by free DTX. DTX-loaded FA/PLGA NPs showed the highest apoptotic effect through the activation of Caspase-9, Caspase-3, and TP53 genes by 2.8-, 1.6-, and 1.86-fold, respectively.
Conclusion: FA/PLGA NPs could be a hopeful drug delivery system for DTX in breast cancer treatment.
Keywords: PLGA NPs, chemoresistance, endocytosis, drug delivery system, active targeting, human breast cancer, DTX loaded PLGA NPs
Plain language summary
Worldwide, breast cancer can be a lethal disease, and patients can face a poor prognosis resulting from the resistance of cancer cells against chemodrugs. Loading the chemodrug docetaxel (DTX), a first-line treatment drug for breast cancer, into polymeric nanoscaled particles and adding a targeted breast cancer cell label “hides” the DTX from noncancerous cells. This reduces unwanted toxic side effects and so gives hope for a new and safe drug delivery system. Poly lactic co-glycolic acid nanoparticles (PLGA NPs) are one type of NPs that can be used. For breast cancer treatment, folic acid (FA) can be grafted as a label to the PLGA NPs because breast cancer cells express a receptor for FA. Therefore, FA/PLGA NPs can target breast cancer cells while largely sparing normal cells. Such FA/PLGA NPs can be loaded with DTX, and the breast cancer cellular uptake of the NPs is enhanced compared to free DTX as a treatment. Here, we used FA/PLGA NPS to deliver DTX into breast cancer cells from a primary breast cancer cell line derived from the breast cancer tumor of a patient. We observed significant breast cancer cellular toxicity. Loading PLGA NPs with DTX and using FA as a targeting label may give us a chance to treat breast cancer in future clinical trials.
Introduction
Breast cancer is a leading cause of cancer deaths affecting women in economically developed countries.1 Poor prognosis and the lack of targeted therapy are challenges in malignant breast cancer treatment. Making sure that the chemotreatment preserves the quality of life for a woman with breast cancer could be a major challenge.2
Recently, there has been great progress in application of nanotechnology in the treatment of malignant breast cancer.3 The field of nanomedicine uses nanoparticles (NPs) with dimension around 100 nm as a drug delivery system (DDS). Nanoscaled particles introduce optimal properties compared to bulk particles due to their higher solubility and absorption by neoplastic tissue. These unique properties are related to the large surface area-to-volume ratio that increases their reactivity to human cells.2
Various synthetic biopolymers have been applied in nanomedicine as DDS in cancer treatment.4 NPs synthesized from biodegradable polymers have unique properties such as sustained chemodrug release with minimized systemic side effects.5 Poly lactic co-glycolic acid (PLGA) is one of the most successful polymers due to its full biodegradability and biocompatibility and its ability to target the tumor-specific action of the chemodrug.6 PLGA NPs have been designed as a DDS for breast malignancy because they can passively accumulate in neoplastic tissues through a phenomenon known as the enhanced permeability and retention effect, which depends on the nanometer size of the particles and other architectural properties of the malignant tissues, mainly the permeable vasculature and insufficient lymphatic drainage.7
Nevertheless, targeting by passive mechanism only supports the sufficient accumulation of NPs in the tumor interstitial space. This mechanism cannot also enhance NPs’ cellular uptake. So, an active targeting mechanism is used for NPs and affords a greater selectivity and efficiency in achieving endocytosis through the specific interaction between the ligand loaded on the NPs and the receptor overexpressed on the cancer cells, which may promote endocytosis.8
Traditional chemotherapy drugs cannot specifically target breast tumors with sufficient dose-induced cancer cell killing. Docetaxel (DTX) is the most effective antineoplastic drug and is used in the therapy of many types of cancer including ovarian, prostate, and especially breast cancer. However, DTX is not considered ideal because it lacks targeting specificity. Synthesized polymeric NPs grafted with a specific ligand on their surface can be applied as DDS for breast cancer.9 Such a grafted selective ligand can achieve efficient intracellular delivery of DTX to the breast cancer cells. Because folic acid (FA) receptor is overexpressed on breast cancer tissue, it is considered the best choice as a ligand incorporated on the surface of PLGA NPs for breast cancer.10
Because the active mechanism of PLGA NPs enhances endocytosis of the DTX, it can resist cancer cellular drug effluxing. The powerful efflux of toxins is essentially controlled by the ATP-binding cassette superfamily (ABC). Most ABC carriers contribute to MDR genes that are expressed on the cancer cell membrane to eject chemodrugs and other toxins out of the cell. The expression of MDR after DTX treatment has been estimated as a main cause for chemoresistance in breast cancer,11 and the overexpression of MDR genes in breast cancer cells by treatment with chemodrugs has been detected in 52% of chemotreated patients.12 The first one identified was the MDR1 gene, which is implicated in cellular chemoresistance and drug endocytosis resistance. Also, breast cancer resistance gene BCRP/ABCG2 is activated in breast cancer tissue and effluxes toxic agents, which decreases their cellular uptake.13
The silencing of the MDR-1 and ABCG2 genes’ expression by siRNA is an effective method to overcome chemoresistance in breast cancer cells.14 But despite the advantages of the siRNA technique in minimizing toxicity toward healthy cells and its high selectivity, siRNA has poor characteristics such as fasting degradation and limited cellular uptake, which have minimized its use in clinical trials to date.15 A promising nanoapproach could be the solution by delivering the chemodrug without triggering chemoresistant genes.
Research applying nanotechnology as a DDS in breast cancer treatment faces the disappointing issue of results that differ between in vitro experimental trials and clinical studies.16 This may be explained due to using breast cancer cell lines that have different cellular properties as compared to the original tumor cells.17
Our focus here is to mimic original tumor cells by using a primary established breast cancer cell line and apply one of the most promising alternatives for the treatment of breast cancer, by nanoconjugating DTX in PLGA NPs and achieving active targeting by grafting FA as ligand. Using FA/PLGA NPs as a vehicle for DTX may not only achieve better selectivity but may also inhibit drug efflux by ABC pumps.
Materials and methods
Chemicals
PLGA (75% lactic, 25% glycolic), DTX, polyvinyl alcohol (PVA) and FA were purchased from Fermentas Thermo Fisher Scientific, Waltham, MA, USA. MTT was purchased from Miltenyi Biotec Inc., Auburn, CA, USA. Nile Red dye was purchased from Sigma-Aldrich Co., St Louis, MO, USA. Deionized water was used throughout the experiments. Culture media including DMEM, FBS, L-glutamine, penicillin–streptomycin, Geneticin 418, and 0.05% trypsin-EDTA were used in culturing the human breast cancer cells. Cell culture medium and collagenase-A were obtained from Miltenyi Biotec Inc.
Sample collection
A tissue sample isolated from a breast cancer tumor was acquired from the Department of Surgery, Mansoura University, Mansoura, Egypt, from one patient (female; 62 years old), who was diagnosed with malignant breast cancer. We worked under an IRB approved protocol (MU_SCI_16_8), which was in accordance with the Declaration of Helsinki, and informed written consent was obtained from the patient. Tumor tissue was biopsied from regions detected as invasive cancer. The tumor sample was placed into a sterile conical vial containing DMEM supplemented with 5% penicillin–streptomycin. The sample was delivered to the culture laboratory within a few minutes. A piece of the tumor sample was fixed by formalin and embedded with paraffin, then followed by histopathological analysis of section to determine the grade and stage of breast tumor (mucinous carcinoma; II) as examined by a pathologist at Mansoura University.
Establishment of primary human breast cancer cell line
Tumor sample (~8 g) was cut with a scalpel into smaller fragments in a Petri dish containing 7 mL of DMEM supplemented by 1% penicillin–streptomycin, then further cut into minute pieces with scissors. The sample slurry was placed into a Falcon tube and mechanically disrupted by a vortex for 15 minutes. This was followed by enzymatic digestion by mixing with collagenase-A diluted in PBS, stored at −20°C, and warmed to 37°C before using, and placed into a water bath for 1 hour (125 rpm), until the cells could be passed through 70 μL mesh. Finally, cells were suspended in DMEM and centrifuged at 1,500 × g for 10 minutes. DMEM was aspirated, and the cell pellet was collected.
Cell culture
The primary human breast cancer cell line was cultured by supplemented DMEM (supplemented with 10% FBS, 1% penicillin–streptomycin, and 1% L-glutamine; stored at 4°C and warmed to 37°C prior to use) and incubated in a 37°C, 5% CO2 incubator. The first culture was supported by Geneticin 418 (25 μg/mL) to kill the fibroblasts. Spent medium was aspirated and fresh medium was placed with every plating. As cells became confluent, they were detached with cold trypsin-EDTA to detach the cancer cells only without any remaining fibroblasts and subcultured into new flasks to allow more space for continuous proliferation. Cancer cells before each passage were counted and tested for viability with trypan blue dye exclusion assay.
Synthesis of different designs of DTX-loaded PLGA NPs
Different designs of DTX-loaded PLGA NPs were prepared by single emulsion solvent evaporation method (physical capsulation).18,19 These NP designs were DTX-loaded PLGA, DTX-loaded fluorescent (FL)/PLGA, DTX-loaded FA/PLGA, and DTX-loaded FL-FA/PLGA. Other NPs were prepared as control: PLGA and FL/PLGA, and all FL NPs were physically encapsulated with Nile Red as hydrophobic fluorescent dye.
For the oil phase, PLGA (1 g) was dissolved in 40 mL acetone, and DTX (20 mg) was added. The hydrophobic solution was homogenized for 15 minutes at room temperature (RT) (drug:polymer ratio =1:50). In the case of fluorescent NPs, Nile Red (10 mg) was dissolved in the oil solution (Nile Red:polymer ratio =1:100). For the water phase, FA (5 mg) was added into 80 mL of PVA (0.05%) solution and stirred for 20 minutes at 70°C. The oil phase was emulsified in the water phase (O/W single emulsion) using a probe sonicator (VCX 130, Sonic and Materials, Newtown, CT, USA), 60 W, cycles of 5 seconds sonication followed by 1 second of pause, total time 10 minutes (oil phase:water phase ratio =1:2). Solvent was evaporated by continuous stirring for 3 hours and then centrifuged for 15 minutes at 30,000 × g at 4°C. The supernatant was subsequently discarded and the pellet was freeze dried for 48 hours (Free Zone 2.5-L freeze-dry system; Labconco, Kansas City, MO, USA).
Characterization of PLGA NPs
Particle size and zeta potential measurements
Three milligrams of freeze-dried synthesized PLGA NPs were pipetted in 3 mL distilled H2O. The mean size of PLGA NPs was detected with photon correlation spectroscopy using a Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK). Zeta potential measurements were based on electrophoretic mobility of the PLGA NPs in double-distilled H2O.
Surface morphology
The surface morphology of PLGA NPs was detected with scanning electron microscopy (SEM, Philips XL 30S, Amsterdam, The Netherlands) and transmission electron microscopy (TEM) (CM-10; Philips). For SEM, a few freeze-dried PLGA NPs were collected onto metallic stud with conductive tapes. The NPs were coated with gold thin film (20 nm) using a sputter coater for SEM. In TEM, PLGA NPs were dispersed in distilled H2O and sonicated for 3 minutes. A drop of the sample suspension was placed on a grid and dried at RT.
Calculations of DTX loading and encapsulation efficiency
Twenty milligrams of synthesized DTX-loaded PLGA NPs were pipetted in 1 mL distilled H2O and placed on a shaker for 24 hours. The suspension was then centrifuged at 10,000 × g and the DTX content in the solution was measured with HPLC (Agilent 1200 Compact LC; Agilent Technologies, Santa Clara, CA, USA). DTX loading and encapsulation efficiency were calculated using equations (1) and (2):20
|
|
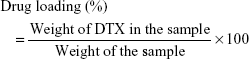
|
|

In vitro internalization
The in vitro cellular internalization of FL-PLGA NPs was detected with confocal laser scanning fluorescence microscopy. Breast cancer cells were seeded in 35 mm plates with density of 1×105 cells in DMEM medium (300 μL). After 24 hours incubation, the cells were treated with 200 μL of fresh medium containing FL-PLGA and FL-FA/PLGA NPs (500 nM Nile Red). After incubation time periods of 30, 60, and 180 minutes with PLGA NPs, the old medium was replaced with fresh DMEM medium and directly observed with confocal laser scanning fluorescence microscopy (DFC420C; Leica Microsystems GmbH, Wetzlar, Germany; with Leica application suite software). The signal of Nile Red fluorescence was detected using a long-pass filter (>590 nm) and appeared in red color.
In vitro quantitative cellular uptake
The breast cancer cellular uptake of FL-PLGA NPs was detected with flow cytometry. Breast cancer cells were seeded in six-well plates at a density of 2×105 cells/mL and incubated in DMEM medium for 48 hours. The cells were treated with FL-PLGA and FL-FA/PLGA NPs (200 mg/mL), triplicate for each design (DMEM as control), and incubated for 1, 2, and 3 hours. After incubation, the cells were washed twice with ice-cold PBS to eliminate the free PLGA NPs that did not bind with cells. Then each well was separately trypsinized and centrifuged for 2 minutes at 4°C (100 × g). Finally, cell pellets were suspended in PBS (0.5 mL) and florescence quantity (Nile Red) was measured with flow cytometer (FACS Aria Cell Sorter; BD Biosciences, Billerica, MA, USA). The quantity of Nile Red in the treated cells was compared to the control cells to obtain mean fluorescence intensity relative to control.
In vitro cytotoxicity
The cytotoxicities of DTX-loaded PLGA NPs and DTX-loaded FA/PLGA NPs were examined against human breast cancer cells with MTT assay. Breast cancer cells were plated in 96-well plates at a density of 1×104 cells per well and incubated overnight. The DMEM medium was changed with fresh media (100 μL) containing drug in various forms (free DTX, DTX-loaded PLGA, and DTX-loaded FA/PLGA NPs) with concentrations (1–25 nM) for 24 hours. For control, cells were treated with plain PLGA NPs by the same concentrations relative to the different designs of DTX. For free DTX, a stock concentration was prepared in DMSO (1 mg/mL). DMEM was used as diluent for preparing the specific concentrations of free DTX. After incubation, medium was removed and each well was washed with PBS (100 μL). MTT was prepared with stock (1 mg/mL PBS), and 75 μL of MTT solution was added to each well and incubated for 2–4 hours. Finally, DMSO was added (75 μL per well) to dissolve the formazan crystals and read at 570 nm with a microplate reader. In vitro cytotoxicity testing was done in triplicate.
Gene expression analysis
Real-time quantitative PCR (RT-qPCR) analysis was performed on 1 g of RNA that was extracted from each treated sample of human breast cancer cells using TRIzol Reagent and purified with GeneJET™ RNA Purification Kit (Fermentas Thermo Fisher Scientific). Then RNA was reverse transcripted into cDNA using the Maxima® First Strand cDNA Synthesis Kit (Fermentas Thermo Fisher Scientific). RT-qPCR was done with the Maxima® SYBR Green qPCR Master Mix (Fermentas Thermo Fisher Scientific). The test was done in a 20 μL volume with primer concentration 20 pmol. The primers for the chemoresistant, apoptotic, and tumor suppressor genes are listed in Table S1; GAPDH was used as a control. Quantitative analysis was applied with an RT-PCR detection system (Agilent Technologies) with an initial denaturation at 95°C (10 minutes), then 40 cycles at 95°C (15 seconds), annealing temperature 60°C–65°C (30 seconds), and extension at 72°C (30 seconds). Specificity was detected with melting curve analysis. RT-qPCR products were electrophoresed on 2% agarose gels. The Ct values of samples were used in the qPCR data analysis. Quantitative analysis was applied to evaluate the ABCG2, MDR1, apoptosis-related cysteine peptidase-9 (Caspase-9), apoptosis-related cysteine peptidase-3 (Caspase-3), tumor suppressor 53 (TP 53′) fold change for the different designs of DTX (free DTX, DTX-loaded PLGA, and DTX-loaded FA/PLGA NPs). For free DTX, cells treated with DMSO:PBS =1:1 were used as a control for free DTX, whereas plain PLGA NPs treated cells were used as a control for DTX-loaded PLGA and FA/PLGA NPs.
Statistical analysis
Half-maximal inhibitory concentration (IC50) value was calculated with GraphPad Prism 5 software (GraphPad, San Diego, CA, USA). The fold change of the tested genes was measured and analyzed with the 2−ΔΔCt method from RT-qPCR experiments. Final (IC50) values and fold change of tested genes were organized and statistically analyzed using one-way ANOVA, followed by the Tukey’s post hoc test with statistical significance defined as P<0.05.
Results
Characterization of PLGA NPs
In this work, the variables such as drug:polymer ratio, oil:water ratio, and fluorescent dye:polymer ratio were studied during the NPs’ preparation. Preliminary work was carried out to select the best ratios for the study, which affected size, polydispersity, and loading efficiency.
Different designs of DTX-loaded PLGA NPs were synthesized by emulsion solvent evaporation method and the optimum conditions were drug:polymer ratio =1:50, oil:water ratio =1:2, and Nile Red:polymer ratio =1:100.
Particle size and zeta potential measurements
Particle size and zeta potential of PLGA NPs are shown in Figure 1A and B, respectively. The average diameter of prepared PLGA NPs was 201.4 nm. The zeta potential of PLGA NPs was −8.63 mV.
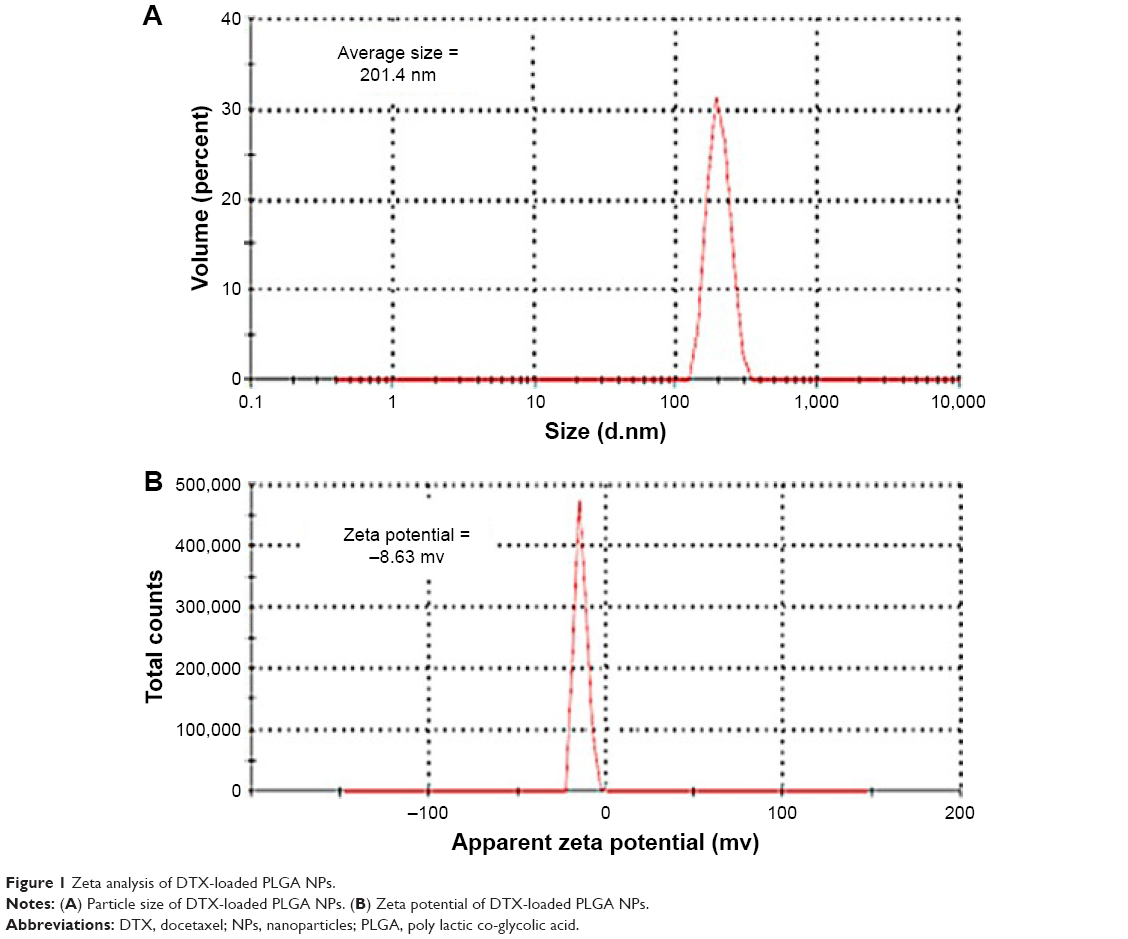 | Figure 1 Zeta analysis of DTX-loaded PLGA NPs. |
Surface morphology
The morphology and size of synthesized PLGA NPs were detected using SEM and TEM. SEM photographs of obtained NPs showed separated and homodispersed particles, with a downy surface appearance (Figure 2A). TEM photographs showed the NPs dispersed as singular particles with obvious globular shape and distributed without wide variations in the size and shape (Figure 2B and C).
 | Figure 2 Surface morphology of DTX-loaded PLGA NPs. |
Encapsulation efficiency and drug loading of DTX on PLGA NPs were measured to be 76% and 1.52%, respectively; that was the maximum encapsulation efficiency of the PLGA NPs depending on the saturation of the polymer.
In vitro internalization
To evaluate the in vitro internalization of the PLGA NPs in breast cancer cells, in vitro endocytosis test was applied using FL-PLGA and FL-FA/PLGA NPs while Nile Red was physically loaded as the fluorescent dye and detected with confocal laser scanning fluorescence microscopy. Results showed that the spots of red fluorescence started to appear outside of the cancer cells on the cell membrane (arrows on Figure 3A and B). After longer incubation, the internalization was visualized by the presence of small florescent red spots mostly colocalized in the cytoplasm. These results demonstrate time-dependent endocytosis of PLGA NPs in the breast cancer cells. Because variation between the two designs of PLGA NPs is visually observed, further confirmation of this result needs to be measured quantitatively.
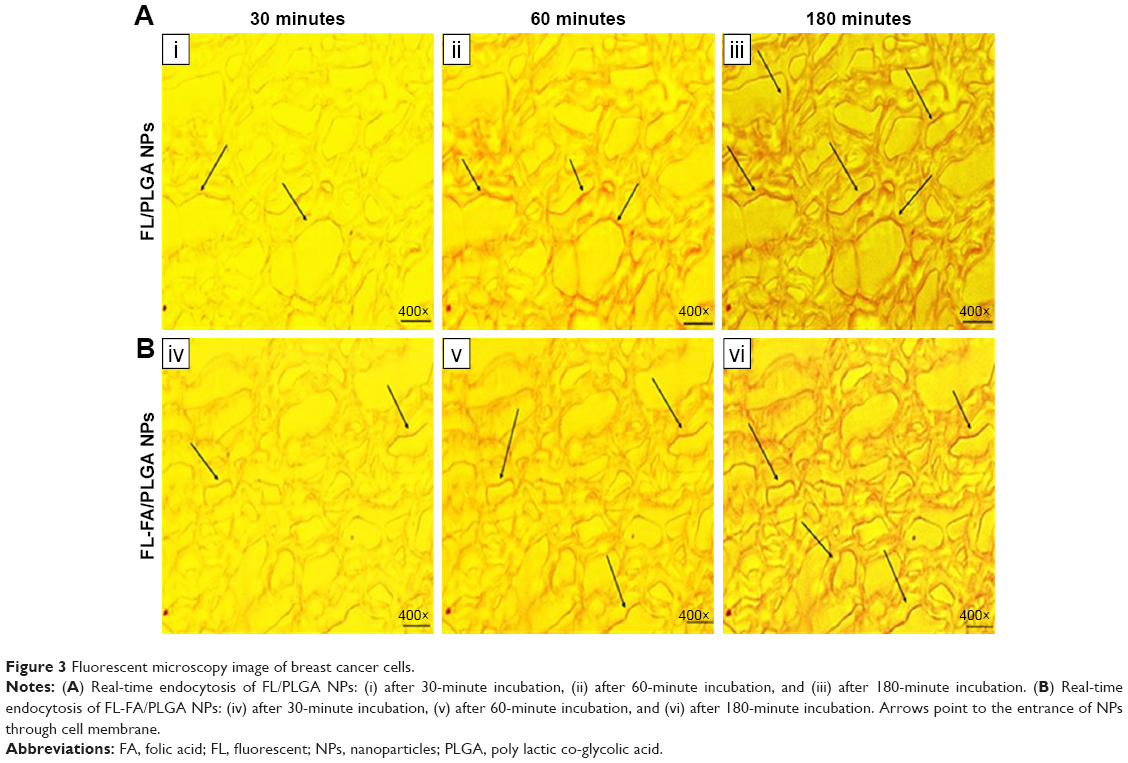 | Figure 3 Fluorescent microscopy image of breast cancer cells. |
In vitro cellular uptake
The breast cancer cell line was tested to confirm the quantitative intracellular uptake of FL-PLGA NPs and FL-FA/PLGA NPs using flow cytometry. The result showed a time-dependent increase in Nile Red intensity at ascending time intervals (1, 2, and 3 hours) for FL-PLGA and FL-FA/PLGA NPs treated cells (Figure 4). After an equal time interval, the Nile Red was taken up by breast cancer cells at higher levels in FL-FA/PLGA than in FL-PLGA NPs. Cellular uptake of PLGA NPs is a critical factor for more effective DDS. PLGA NPs’ uptake by breast cancer cells was improved through activation of endocytosis after loading FA as ligand. In these studies, maximum cellular uptake was detected by FL-FA/PLGA NPs at 3 hours.
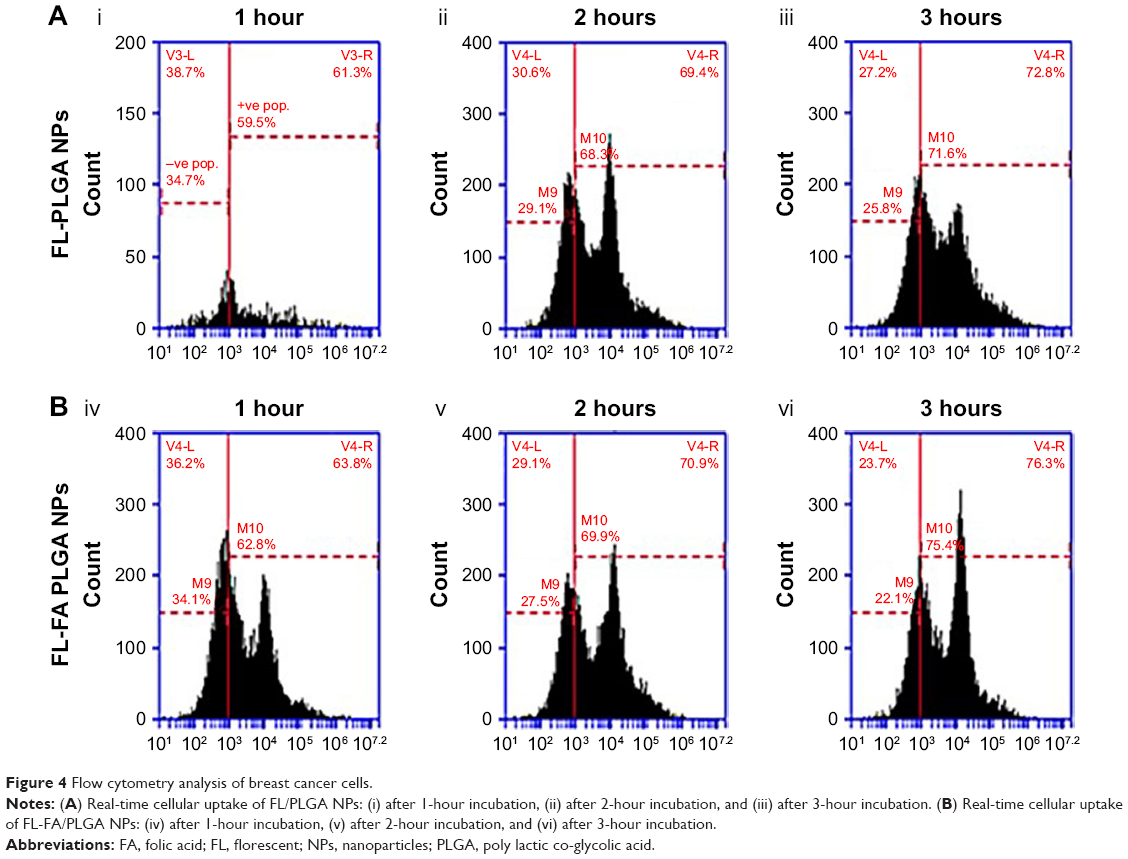 | Figure 4 Flow cytometry analysis of breast cancer cells. |
In vitro cytotoxicity
The viability of breast cancer cells was tested using MTT assay after 24 hours of incubation with free DTX, DTX-loaded PLGA, and DTX-loaded FA/PLGA NPs (Figure 5A–C). Plain PLGA NPs were also incubated with the tested cells as a control and used to normalize the results. The cytotoxic efficiency of all DTX forms was observed to be increased by gradually increasing the DTX concentration from 1 to 25 nM, so different forms of DTX were detected to have concentration-dependent cytotoxicity. For an equal dose of all forms of DTX, DTX-loaded FA/PLGA showed significantly the highest toxic effect against the breast cancer cells (P<0.05), while IC50 values of free DTX, DTX-loaded PLGA, and DTX-loaded FA/PLGA NPs were 12.09 nM, 9.39 nM, and 8.29 nM, respectively. DTX showed the maximum in vitro cytotoxicity after loading on FA/PLGA NPs.
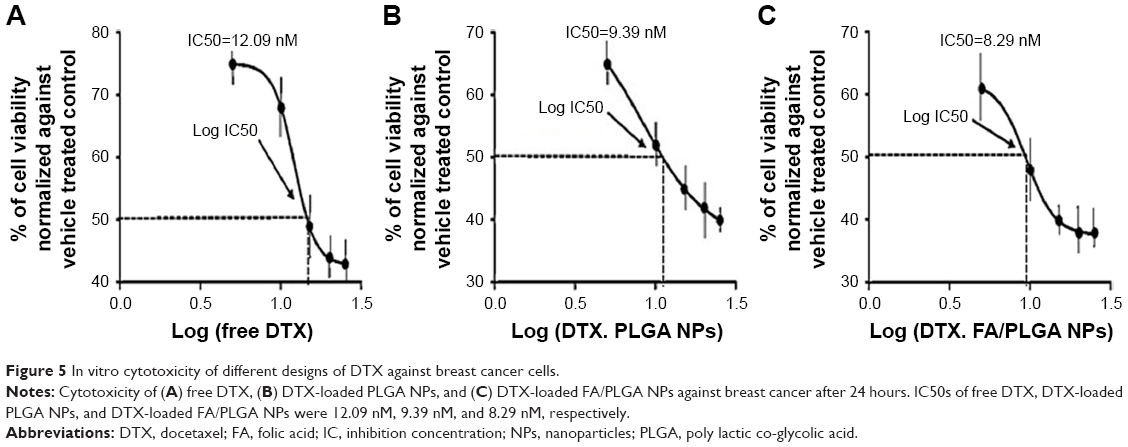 | Figure 5 In vitro cytotoxicity of different designs of DTX against breast cancer cells. |
Gene expression analysis
RT-qPCR analysis was applied to calculate the fold changes in expression of ABCG2, MDR1, Caspase-9, Caspase-3, and TP53 by breast cancer cells treated with free DTX, DTX-loaded PLGA NPs, and DTX-loaded FA/PLGA NPs (Figure 6). Free DTX highly activated the expression of chemoresistant genes in the breast cancer cells in vitro. This traditional design of the free DTX expressed chemoresistant genes ABCG2 and MDR1 3.2-fold and 2.86-fold, respectively, which are greater than DTX after loading on the PLGA NPs and FA/PLGA NPs, but not significant (P=0.18). There were no clear variations in the fold-change expression of apoptotic and tumor suppressor genes between the three different designs of DTX, as DTX-loaded FA/PLGA NPs expressed Caspase-9, Caspase-3, and TP53 2.8-fold, 1.6-fold, and 1.86-fold, respectively. DTX after loading on PLGA NPs and incorporation of FA as a ligand slightly activated the expressions of these genes more than the other two designs (P=0.9).
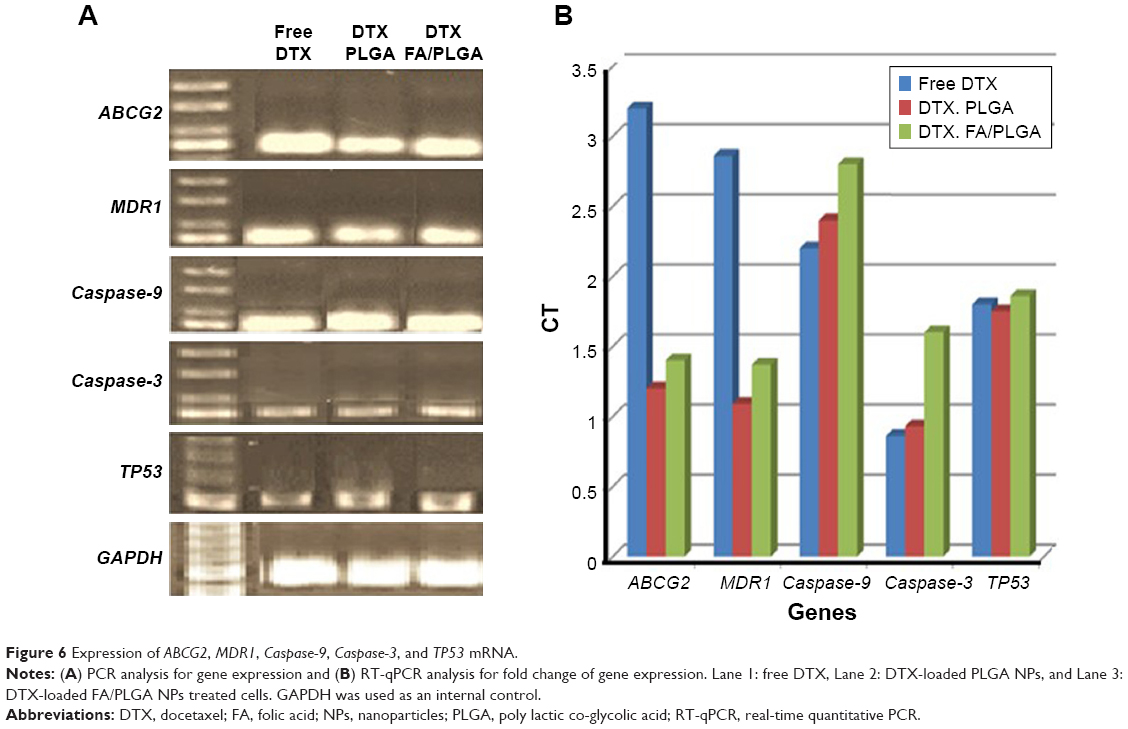 | Figure 6 Expression of ABCG2, MDR1, Caspase-9, Caspase-3, and TP53 mRNA. |
Discussion
Nanotechnology methodologies have emerged as a unique technique to treat breast cancer.21 In this study, we addressed the serious challenges in applying nanomedicine in breast cancer treatment such as endocytosis drug delivery, chemodrug escape from cellular gateways and enhancement of intracellular dose. In vitro studies using nanomedicine can successfully mimic in vivo methods while also minimizing animal model use,22 but many previous studies lacked exactness in using a well-established breast cancer cell line.
Using primary cells could solve many issues related to using cell lines in research because cell lines cannot exhibit and preserve functional characters of the original tumor. Previous genetic examination of these cell lines showed the alternation of their morphological characters, original functions, and even the response to the drugs. So breast cancer cell lines cannot sufficiently represent the original properties of breast tumor that has been biopsied and may lead to different results.23
The other main issue linked with these traditional cell lines is contamination with different cell lines and mycoplasma. Many cell lines that are produced by cell banks are contaminated with HeLa cells. Based on submissions to cell banks, 15%–35% of cell lines are estimated to be contaminated.24 So in our study, we used a well-established primary cancer cell line from a patient’s breast solid tumor.
PLGA is a unique polymer for chemo-DDS and is the foundation that we designed the NPs used here on. But PLGA NPs are considered as a double-edged weapon because of the biotoxicity of the chemicals that NPs are manufactured from. So, this study used the safest version of NPs’ synthesis, by applying physical incorporation of the DTX into the PLGA NPs using an emulsion solvent evaporation method that minimizes chemicals used.25
Enhancing cellular uptake and biodistribution of NPs can be effectively achieved through controlling their size, potentiality, and shape. So, in this study, these properties were evaluated and measured on synthesized DTX-loaded FA/PLGA NPs. Physiochemical characterization showed the negatively charged nanosized particles were in the optimum range in order to enhance cellular drug accumulation.26 The morphology of NPs also has a critical role on cellular fate; globular shaped particles as observed in SEM and TEM photographs improve their activity through greater surface area, and the soft surface appearance indicates the complete saturation of NPs with the drug and the surfactant.27
Enhancement of the endocytosis of a chemodrug is a main factor to consider when choosing the best design of PLGA NP-based DDS. Designs of DTX-loaded PLGA NPs showed time-dependent internalization in breast cancer cells. Physically loaded Nile Red dye is not an optimum marker for cellular internalization of PLGA NPs because the red fluorescent color on the cell membrane could be explained by PLGA NPs escaping from the cell membrane into the cytoplasm and effluxing the Nile Red outside the cells.28 So, for further investigation, our study was supported by in vitro cellular uptake testing with flow cytometry. Demonstration of the comparative cellular uptake of PLGA NPs with and without loading FA showed that the better result could be reached with time. Also, the active mechanism of PLGA NPs as DDS was achieved by loading FA as ligand, which enhances the quantitative cellular uptake. Time-dependent cellular uptake of FA/PLGA NPs will support the intracellular accumulation of DTX, which resulted in an effective chemotherapy.
Measurement of the IC50 of the studied designs of DTX against primary breast cancer cell line is an essential parameter in determining the best DDS of the chemodrug.29 Here, DTX showed higher in vitro anticancer efficiency with IC50=8.29 after loading on PLGA NPs as DDS and activating its mechanism of action by incorporation of FA as ligand. The higher cytotoxicity efficiency of this design against the breast cancer cells may be related to applying PLGA NPs as a carrier to DTX, which protects the drug from degradation until it reaches the targeted cells and hides the drug from being effluxed by cell membrane transporter.30 Also, this point is proved by the other two designs of DTX, free DTX, and DTX loaded on PLGA NPs, where the latter showed statistically significant greater in vitro cytotoxicity through minimized IC50. On the other hand, the active mechanism of the DDS triggered by FA could enhance the intracellular drug dose by improving the endocytosis through FA–FA receptor interaction.31
MDR in breast cancer is the most significant obstacle in chemotherapy. So avoiding MDR action has high potential in enhancing chemotherapeutic effect. Encapsulating the DTX by incorporating it into PLGA NPs may overcome cancer drug resistance through escaping from the efflux cell membrane transporters. In this study, the expression of chemoresistant genes reached maximum inhibition by hiding the DTX after loading on PLGA NPs that overcome the chemoefflux mechanism. Also FA receptor interaction supported this technique by stimulation of the active mechanism of DDS. Active mechanism enhanced more cellular uptake of NPs, which worked against the action of MDR effluxing. The molecular study showed that the higher expression of MDR genes was stimulated by free DTX, which lacked a supportive delivery system.
Collectively, the results of this study recommend that DTX-based treatment can be supported by applying nanoscale DDS as a unique technique to improve properties of the chemodrug, while polymeric NPs in the form of PLGA NPs after activation by loading FA as ligand is a competitive choice in delivering the DTX into breast cancer cells.
The brilliant combination of chemotherapy and nanotechnology promises a hopeful future in cancer treatment. Despite the challenges, in vitro studies should be supported by using the primary human cancer cells to mimic the reality of the cancer tumor. Overcoming the variation of results that are due to the difference between the original tumor cells and laboratory cancer cells could help shorten the long gap between experimental trials and pharmaceutical products.
Conclusion
This study concludes that applying nanoscaled particles with polymeric nature as a DDS can solve the obstacles that face chemodrugs in achieving better cancer treatment. Successful FA/PLGA NPs could overcome the DTX-resistant character of the primary breast malignant cells that actively enhanced cellular uptake of DTX and achieved higher cytotoxicity. Therefore, DTX-loaded FA/PLGA NPs could be recommended as a promising treatment line against human breast cancer, which deserves to be continuously studied in clinical trials.
Acknowledgments
This work was supported by Competitive Funding Projects Post graduate Research and Cultural Affairs Sector Mansoura University. We thank Zewail City of Science and Technology for their support. We appreciate the excellent editorial assistance of Kelly Keating (The Pharmaceutical Research Institute, Rensselaer, NY, USA).
Author contributions
AMA directed the research and the experiments from conception to production. GM conceived the main idea of the research, performed the experiments, and analyzed the data. IMES designed and managed the experiments of the synthesis and characterization of nanoparticles. SAM supported the research with new ideas and the design of the NPs. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Lee J, Chatterjee DK, Lee MH, Krishnan S. Gold nanoparticles in breast cancer treatment: promise and potential pitfalls. Cancer Lett. 2014;347(1):46–53. | ||
Yezhelyev MV, Gao X, Xing Y, Al-Hajj A, Nie S, O’Regan RM. Emerging use of nanoparticles in diagnosis and treatment of breast cancer. Lancet Oncol. 2006;7(8):657–667. | ||
Locatelli E, Comes Franchini M, Franchini MC. Biodegradable PLGA-b-PEG polymeric nanoparticles: synthesis, properties, and nanomedical applications as drug delivery system. J Nanoparticle Res. 2012;14(12):17. | ||
Feng S-S, Chien S. Chemotherapeutic engineering: application and further development of chemical engineering principles for chemotherapy of cancer and other diseases. Chem Eng Sci. 2003;58(18):4087–4114. | ||
Sadat Tabatabaei Mirakabad F, Nejati-Koshki K, Akbarzadeh A, et al. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac J Cancer Prev. 2014;15(2):517–535. | ||
Bazak R, Houri M, Achy SE, Hussein W, Refaat T. Passive targeting of nanoparticles to cancer: a comprehensive review of the literature. Mol Clin Oncol. 2014;2(6):904–908. | ||
Yu B, Tai HC, Xue W, Lee LJ, Lee RJ. Receptor-targeted nanocarriers for therapeutic delivery to cancer. Mol Membr Biol. 2010;27(7):286–298. | ||
Chen PC, Mwakwari SC, Oyelere AK. Gold nanoparticles: from nanomedicine to nanosensing. Nanotechnol Sci Appl. 2008;1:45–65. | ||
Kohli E, Han HY, Zeman AD, Vinogradov SV. Formulations of biodegradable Nanogel carriers with 5′-triphosphates of nucleoside analogs that display a reduced cytotoxicity and enhanced drug activity. J Control Release. 2007;121(1–2):19–27. | ||
Liu J, Li J, Liu N, et al. In vitro studies of phospholipid-modified PAMAM-siMDR1 complexes for the reversal of multidrug resistance in human breast cancer cells. Int J Pharm. 2017;530(1–2):291–299. | ||
Leonessa F, Clarke R. ATP binding cassette transporters and drug resistance in breast cancer. Endocr Relat Cancer. 2003;10(1):43–73. | ||
Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY. Nanomedicine therapeutic approaches to overcome cancer drug resistance. Adv Drug Deliv Rev. 2013;65(13–14):1866–1879. | ||
Lage H. MDR1/P-glycoprotein (ABCB1) as target for RNA interference-mediated reversal of multidrug resistance. Curr Drug Targets. 2006;7(7):813–821. | ||
Navarro G, Sawant RR, Biswas S, Essex S, Tros de Ilarduya C, Torchilin VP. P-glycoprotein silencing with siRNA delivered by DOPE-modified PEI overcomes doxorubicin resistance in breast cancer cells. Nanomedicine. 2012;7(1):65–78. | ||
Willmann L, Erbes T, Halbach S, et al. Exometabolom analysis of breast cancer cell lines: metabolic signature. Sci Rep. 2015;5:13374. | ||
Singh SK, Singh S, Lillard JW, Singh R. Drug delivery approaches for breast cancer. Int J Nanomedicine. 2017;12:6205–6218. | ||
Boddu SHS, Vaishya R, Jwala J, Vadlapudi A, Pal D, Mitra AK. Preparation and characterization of folate conjugated nanoparticles of doxorubicin using Plga-Peg-Fol polymer. Med Chem. 2012;2(4):68–75. | ||
Katsikogianni G, Avgoustakis K. Poly(lactide-co-glycolide)-methoxy-poly(ethylene glycol) nanoparticles: drug loading and release properties. J Nanosci Nanotechnol. 2006;6(9–10):3080–3086. | ||
Kashi TS, Eskandarion S, Esfandyari-Manesh M, et al. Improved drug loading and antibacterial activity of minocycline-loaded PLGA nanoparticles prepared by solid/oil/water ion pairing method. Int J Nanomedicine. 2012;7:221–234. | ||
Tang Y, Wang Y, Kiani MF, Wang B, Classification WB. Classification, treatment strategy, and associated drug resistance in breast cancer. Clin Breast Cancer. 2016;16(5):335–343. | ||
Kura AU, Fakurazi S, Hussein MZ, Arulselvan P. Nanotechnology in drug delivery: the need for more cell culture based studies in screening. Chem Cent J. 2014;8:46. | ||
Kaur G, Dufour JM. Cell lines: valuable tools or useless artifacts. Spermatogenesis. 2012;2(1):1–5. | ||
Capes-Davis A, Theodosopoulos G, Atkin I, et al. Check your cultures! A list of cross-contaminated or misidentified cell lines. Int J Cancer. 2010;127(1):1–8. | ||
de Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3(2):133–149. | ||
Mura S, Hillaireau H, Nicolas J, et al. Influence of surface charge on the potential toxicity of PLGA nanoparticles towards Calu-3 cells. Int J Nanomedicine. 2011;6:2591–2605. | ||
Mathew A, Fukuda T, Nagaoka Y, et al. Curcumin loaded-PLGA nanoparticles conjugated with Tet-1 peptide for potential use in Alzheimer’s disease. PLoS One. 2012;7(3):e32616. | ||
Xu P, Gullotti E, Tong L, et al. Intracellular drug delivery by poly(lactic-co-glycolic acid) nanoparticles, revisited. Mol Pharm. 2009;6(1):190–201. | ||
Hami Z, Rezayat SM, Gilani K, Amini M, Ghazi-Khansari M. In-vitro cytotoxicity and combination effects of the docetaxel-conjugated and doxorubicin-conjugated poly(lactic acid)-poly(ethylene glycol)-folate-based polymeric micelles in human ovarian cancer cells. J Pharm Pharmacol. 2017;69(2):151–160. | ||
Rafiei P, Haddadi A. Pharmacokinetic consequences of PLGA nanoparticles in docetaxel drug delivery. Pharm Nanotechnol. 2017;5(1):3–23. | ||
Zwicke GL, Mansoori GA, Jeffery CJ. Utilizing the folate receptor for active targeting of cancer nanotherapeutics. Nano Rev. 2012;3:18496. |
Supplementary material
 | Table S1 Primers sequences of chemoresistant, apoptotic and tumor suppressor genes |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.